

Abstract
Label-free quantitation of proteins analyzed by tandem mass spectrometry uses either integrated peak intensity from the parent-ion mass analysis (MS1) or features from fragment-ion analysis (MS2), such as spectral counts or summed fragment-ion intensity. We directly compared MS1 and MS2 quantitation by analyzing human protein standards diluted into Escherichia coli extracts on an Orbitrap mass spectrometer. We found that summed MS2 intensities were nearly as accurate as integrated MS1 intensities, and both outperformed MS2 spectral counting in accuracy and linearity. We compared these results to those obtained from two low-resolution ion-trap mass spectrometers; summed MS2 intensities from LTQ and LTQ Velos instruments were similar in accuracy to those from the Orbitrap. Data from all three instruments are available via ProteomeXchange with identifier PXD000602. Abundance measurements using MS1 or MS2 intensities had limitations, however. While measured protein concentration was on average well correlated with the known concentration, there was considerable protein-to-protein variation. Moreover, not all human proteins diluted to a mole fraction of 10−3 or lower were detected, with a strong fall-off below 10−4 mole fraction. These results show that MS1 and MS2 intensities are simple measures of protein abundance that are on average accurate, but should be limited to quantitation of proteins of intermediate to higher fractional abundance.
Keywords: Intensity, spectral counts, Orbitrap, ion trap
Introduction
Quantitative proteomics experiments are carried out using shotgun (discovery), directed, and targeted methods1. While directed and targeted proteomics using internal standards can offer greater precision and higher accuracy, shotgun proteomics remains a workhorse method when a global picture of the proteome is required. When co-translational or post-translational stable isotopic labeling methods are used, shotgun proteomics can offer excellent quantitative power, particularly when comparing two samples. These methods are more limited for characterization of proteins from scarce tissues, however, either because co-translational methods like SILAC2 are not possible—or, in the case of the SILAC mouse3, impractical—or because post-translational methods require extra sample manipulations that decrease sensitivity and reduce coverage.
Label-free methods for quantitative proteomics are simple and practical4,5. Two basic approaches to label-free quantitation have been used. In the first, integrated peak intensities (ion currents) of parent peptides are used to compare expression levels between samples. Individual peptide expression can be measured or peptide signals can be combined for protein expression comparisons. The accuracy and sensitivity of these MS1 methods have been improved by use of high-resolution instruments, including those with orbitrap detectors, which allow great precision in measurement of precursor m/z values. Popular MS1 methods include the iBAQ algorithm6–8, where a protein's total intensity is divided by the number of tryptic peptides between 6 and 30 amino acids in length, and the “top three” method9,10, where the three peptides with highest intensity are used for quantitation.
The second set of methods uses features of the MS2 spectra, which are collected after peptide fragmentation. Spectral counts, the number of MS2 spectra matched to a given protein, have proven very popular11, although the precision and accuracy of this method have been questioned4,5. More recently, several groups have used the summed ion intensities of the MS2 spectra in attempts to improve the accuracy of MS2 methods12–16. All MS2 methods have been criticized because collection of MS2 spectra is data-dependent, biased toward more abundant peptides, and subject to saturation5. Notably, MS2 spectra are not necessarily acquired at the peak of a peptide's elution profile and acquisition varies from peptide to peptide, so the perception is that spectral counts and MS2 intensities are not necessarily linearly related to protein abundance (Fig. 1).
Figure 1.

Illustration of MS1 and MS2 quantitation. (A) Chromatogram illustrating elution of three peptides (blue, red, green). MS1 spectra are sampled at regular intervals (gray and black arrows); black arrows indicate example spectra a, b, and c in panel B. Because of this regular sampling, each peak is accurately measured. By contrast, MS2 spectra are acquired in a data-dependent manner. For example, a MS2 spectrum is acquired for the blue peptide early in its peak (point d); because of dynamic exclusion, another spectrum is not acquired until point f, well past the blue peptide's peak. In this case, because of the size of the peak, two spectral counts are recorded; MS2 spectra for the red and green are only acquired once during each peak, although they are taken near the each peptide's peak (e and g). Although these discrepancies reduce the accuracy of measurement of any single peak using MS2 methods, these discrepancies average out when the same peptides are measured multiple times and different peptides are analyzed. (B) Example MS1 spectra from panel A; peptides in A are indicated by colored peaks. Additional peptides are indicated by gray peaks. In spectrum a, the blue peptide is more abundant than the red peptide; the reverse is true in spectrum b. (C) Example MS2 spectra from panel A. The blue peptide fragments are either dark blue (for d) or light blue (for f). While one spectral count is recorded for the red and green peptides, the sum of all MS2 peaks is different for them, reflecting peptide abundance, sampling time, ionization efficiency, and other factors.
Despite practical and theoretical objections to label-free methods, empirical evaluation in our laboratories has suggested that they can be quite accurate, at least on average. We found that when using data from an Orbitrap mass spectrometer, iBAQ accurately predicts protein abundance from MS1 spectra over a range of concentrations17. In addition, using a low-resolution ion trap mass spectrometer, we showed that MS2 intensities accurately quantified—over a substantial dynamic range—a collection of standard proteins diluted in a complex protein mixture, with significantly better accuracy than spectral counting18. In each of these cases, we assessed the detectability of known proteins spiked into complex protein mixtures, which should mimic detection under normal conditions.
Given the ubiquity of relatively inexpensive ion-trap mass spectrometers, we wondered how label-free quantitation with them compared directly to that using MS1 quantitation. We therefore ran the same standard protein mixtures on Orbitrap, Velos, and LTQ instruments, analyzing both MS1 and MS2 data for the Orbitrap and MS2 data for the Velos and LTQ instruments. We found that while iBAQ and Top3 methods were on average the most accurate, MS2 quantitation either with the Orbitrap or with one of the two ion traps nearly matched those MS1 results. These results show that for appropriate samples, MS2 quantitation on a low-resolution ion trap mass spectrometer can be applicable for proteomics quantitation.
Experimental Procedures
Preparation of UPS samples
For the UPS1 experiments, the entire Universal Proteomics Standard Set (Sigma-Aldrich) was diluted into 50 μl of 1x SDS-PAGE sample buffer. Two parts by volume of an E. coli protein extract of 2 mg/ml was added to two parts of 2x sample buffer to yield a 1 mg/ml E. coli extract. Because the average E. coli protein molecular mass was 31.2 kDa, the average protein concentration was 32 μM. An aliquot of 4 μl of UPS1 solution (each protein at 400 fmol) was added to 25 μg (800 pmol) of the diluted E. coli extract. Each UPS1 protein was thus present at a mole fraction of 5 × 10−4. Note that while the Sigma website indicates 48 proteins are present in this set, we detected at approximately the same molar concentration a 49th protein (P07339; cathepsin D), formerly present in the UPS1 mixture but that was said to have been replaced.
For the UPS2 experiments, the entire Proteomics Dynamic Range Standard Set (Sigma-Aldrich) was diluted into 50 μl of 1x SDS-PAGE sample buffer. For each 31.3 μg sample of E. coli protein (1000 pmol), 10 μl of the UPS2 solution was added (UPS2 proteins present at 10,000, 1000, 100, 10, 1, and 0.1 fmoles per sample). UPS2 proteins were thus present at mole fraction of 10−2 to 10−7.
One experiment consists of a single sample of protein processed as below and run on one of the three mass spectrometers. For each experiment, the entire sample (37–43 μl) was run on SDS-PAGE; the dye front was allowed to move ~1 cm into the gel. The gel lane at and above the dye front was cut into six slices, which were subjected to reduction, alkylation, trypsin digestion, and extraction. SDS-PAGE and peptide preparation were described in detail elsewhere17–19. For each mass spectrometer, 24 different LC-MS/MS runs were conducted: four separate experiments (quadruplicates) were carried out with the same protein mixture (i.e., technical replicates), each processed separately by SDS-PAGE and each generating six peptide samples for LC-MS/MS.
UPS and E. coli database
The 50 UPS1 and UPS2 protein sequences in FASTA format were downloaded 5/9/2012 from www.sigmaaldrich.com/life-science/proteomics/mass-spectrometry/ups1-and-ups2-proteomic.html. The sequences were added to a non-redundant RefSeq database of E. coli BL21(DE3), taxonomy number 469008, with 4228 entries extracted from a 6/22/2009 NCBI nr download. The combined databases includes 179 contaminants, 50 UPS sequences, 4228 E. coli sequences, and reversed versions of all three (8914 total sequences). The NCBI sequences were downloaded, taxon 469008 extracted, RefSeq filtered, and sequences reversed using utilities available at www.ProteomicAnalysisWorkbench.com.
Orbitrap mass spectrometry
The LC-MS/MS system consisted of a Thermo Electron Orbitrap Velos ETD mass spectrometer system with a Protana nanospray ion source interfaced to a self-packed 8 cm × 75 μm internal-diameter Phenomenex Jupiter 10 μm C18 reversed-phase capillary column. Half of each UPS/E. coli sample was injected. The peptides were eluted from the column by an acetonitrile/0.1 M acetic acid gradient at a flow rate of 0.5 μl/min over 1.2 hours. The nanospray ion source was operated at 2.5 kV. The digest was analyzed using the double play capability of the instrument acquiring full scan mass spectra to determine peptide molecular weights and 20 product ion spectra to determine amino acid sequence in sequential scans. The UPS2 analysis was also described elsewhere17.
Ion-trap mass spectrometry
For analysis on the mass spectrometer, each protein digest was analyzed by LC-MS/MS using an Agilent 1100 series capillary LC system (Agilent Technologies Inc., Santa Clara, CA) and Velos or LTQ linear ion trap mass spectrometer (ThermoFisher, San Jose, CA). Electrospray ionization was performed with an ion max source fitted with a 34 gauge metal needle (ThermoFisher, cat. no. 97144-20040) and 2.7 kV (Velos) or 2.8 kV (LTQ) source voltage. Samples were applied at 20 μl/min to a trap cartridge (Michrom BioResources, Auburn, CA), and then switched onto a 0.5 × 250 mm Zorbax SB-C18 column with 5 μm particles (Agilent Technologies) using a mobile phase containing 0.1% formic acid, 7–30% acetonitrile gradient over 195 min, and 10 μl/min flow rate. Data-dependent collection of MS2 spectra were of the five (Velos) or three (LTQ) most abundant parent ions following each survey scan from m/z 400–2000. Dynamic exclusion was used. For the Velos, the repeat count was equal to 1, exclusion list size was 100, exclusion duration was 30 sec, and exclusion mass width was −1 to +4. For the LTQ, the repeat count was equal to 1, exclusion list size was 50, exclusion duration was 30 sec, and exclusion mass width was −1 to +1.5. The tune file was configured with no averaging of microscans, a maximum inject time of 200 msec, and AGC targets of 3 × 104 in MS1 mode and 1 × 104 in MSN mode.
MS1 MaxQuant analysis
MaxQuant version 1.2.2.5 software was used for protein identification and quantitation. Using Andromeda20, mass spectrometry data were searched against the database containing UPS and E. coli proteins. MaxQuant reports summed intensity for each protein, as well as its iBAQ value. In the iBAQ algorithm6,7, the intensities of the precursor peptides that map to each protein are summed together and divided by the number of theoretically observable peptides, which is considered to be all tryptic peptides between 6 and 30 amino acids in length. This operation converts a measure that is expected to be proportional to mass (intensity) into one that is proportional to molar amount (iBAQ).
To determine relative molar abundances, for each E. coli and human protein, we determined its relative iBAQ (riBAQ), a normalized measure of molar abundance17. We removed from the analysis all contaminant proteins that entered our sample-preparation workflow, for example keratins and trypsin, and then divided each remaining protein's iBAQ value by the sum of all non-contaminant iBAQ values:
The mean ± SD for the four runs are reported in Tables S1 (UPS1 data) and S2 (UPS2 data).
Top three analysis
The average intensity of the three (or fewer, if fewer peptides were detected) peptides with the highest intensity, Top3, was determined for each protein detected9. Peptides were chosen separately for each experiment, so the same peptides were not necessarily used across the four experiments. We generated a normalized “top three” abundance factor:
where Top3 is the average intensity for the three most abundant peptides of an individual protein and the denominator is the sum of all Top3 values in an experiment. Peptide intensities were summed for all charge states and variable modifications. The mean ± SD for the four runs are reported in Tables S1 and S2.
MS2 intensity and spectral count analysis
Using SEQUEST, MS2 spectra were searched against the database described above. Setting to 1% the peptide false-discovery rate, estimated using a decoy (reversed) database, proteins were identified using the PAW pipeline18,21. Normalized molar intensity (im) was calculated from:
where i is the summed intensity for an individual protein, mr is its molecular mass, and the denominator is the sum of all i/mr values. Normalized molar counts (cm) were calculated similarly, except the summed spectral counts for a protein (c) was used instead:
The mean ± SD for the four runs each on Orbitrap, Velos, and LTQ mass spectrometers are reported in Tables S1 and S2.
Results
To evaluate the accuracy of label-free methods for detection of proteins of widely varying abundance in complex mixtures, we diluted two sets of human proteins into a protein extract of Escherichia coli. The UPS1 set includes 49 proteins, all at the same molar abundance, allowing us to compare detectability of proteins at the same mole fraction; the UPS2 set contains the same proteins, but in subsets of eight that vary 105 fold in molar abundance. By knowing the weighted average molecular mass of all detected E. coli proteins (31.2 kD), we could calculate the mole fraction for each human protein that was spiked into the protein mixture. To mimic the situation we use for analyzing small amounts of proteins isolated from hair bundles of the inner ear17–19, we separated proteins using very short SDS-PAGE runs, then digested proteins in the gel using trypsin. We used LC-MS/MS to analyze the resulting peptide mixtures and generate MS1 and MS2 data.
From the raw data, we calculated MS1 intensities, MS2 intensities, or MS2 spectral counts, then determined relative molar abundances by dividing each protein's abundance by the sum of all mass-adjusted protein abundances (Tables S1 and S2). While the iBAQ and Top3 methods inherently correct for molecular mass, MS2 intensities and counts were divided by protein mass to generate a molar abundance factor. If a given abundance measure is accurate, points representing the dependence of normalized molar abundance on known mole fraction—plotted on a log-log plot—will fall on a line of slope 1 with an intercept of 0 (the unity line).
Relationship between mole fraction and MS1 quantitation
We analyzed the data collected on an Orbitrap mass spectrometer with MaxQuant, which couples measurement of MS1 intensities with peptide identification using the search engine Andromeda20,22. We used two MS1 analysis methods. In the first, we used the iBAQ abundance measure6,7, which converts intensities to a value proportional to molar abundance, dividing the summed MS1 intensities by a protein's number of theoretically observable peptides. For each protein, we generated a relative iBAQ value (riBAQ) by dividing a protein's iBAQ by the sum of all iBAQ values for quantified proteins, excluding contaminants. In the second method, we averaged intensities of the three (or fewer) peptides with the highest intensity, then divided this Top3 value9 by the sum of all Top3 values. This operation generated a relative Top3 abundance measure (rTop3), which should be correlated with the mole fraction of the protein of interest.
We first examined the 49 proteins of the UPS1 protein set, all diluted to a log mole fraction of −3.30. The riBAQ and rTop3 values were determined for each; after binning, the distributions of the UPS1 abundance estimates were plotted (Fig. 2). All 49 proteins were detected by the Andromeda search engine, and the average riBAQ and rTop3 values were not significantly different from the known mole fraction. The distribution of riBAQ values (Fig. 2A) was slightly narrower than that of the rTop3 values (Fig. 2B), but otherwise the two methods were comparable. Many more E. coli proteins were detected (Fig. S1).
Figure 2.

Distributions of estimated mole fraction for UPS1 proteins. All 49 proteins were diluted to a log10 mole fraction of −3.30 in an E. coli extract. The average (± SD) estimated mole fraction for each of the quantitation methods is indicated, as is the number of proteins detected. Data were binned and fit with a single Gaussian distribution. (A) riBAQ; MaxQuant analysis of Orbitrap MS1 data. (B) Top three most intense peptides; MaxQuant analysis of Orbitrap MS1 data. (C) Normalized molar counts; PAW analysis of Orbitrap MS2 data. (D) Normalized molar intensity; PAW analysis of Orbitrap MS2 data. (E) Normalized molar counts; PAW analysis of Velos MS2 data. (F) Normalized molar intensity; PAW analysis of Velos MS2 data. (G) Normalized molar counts; PAW analysis of LTQ MS2 data. (H) Normalized molar intensity; PAW analysis of LTQ MS2 data.
When then examined the UPS2 protein set, which was diluted to a wide range of final mole fraction values. All UPS1 proteins diluted to mole fractions of 10−2 and 10−3 were detected, as were six of eight at 10−4 and two of eight at 10−5. The fits for the relationship between known mole fraction and riBAQ or rTop3 were not significantly different from the unity line on a log-log plot (Fig. 3A,B).
Figure 3.

Comparison of label-free quantitation of UPS2 proteins to known mole fraction. UPS2 human protein standards were spiked into an E. coli extract. Identical samples were run on Orbitrap, Velos, and LTQ mass spectrometers. Points from dilutions at 10−4, 10−3, and 10−2 were fit (red line). The unity line is indicated by a dashed line. The fraction of spiked proteins detected for each dilution level is also indicated. (A) Orbitrap riBAQ, fit with the unity line. (B) Orbitrap rTop3, fit with the unity line. (C) Orbitrap normalized molar counts, fit with y=1.09x+0.062x2. (D) Orbitrap normalized molar intensity, fit with y=0.95x. (E) Velos normalized molar counts, fit with y=1.17x+0.090x2. (F) Velos normalized molar intensity, fit with y=0.98x. (G) LTQ normalized molar counts, fit with y=1.07x+0.034x2. (H) LTQ normalized molar intensity, fit with y=1.08x.
The relationship between riBAQ and rTop3 for all proteins detected in the summed experiments, including E. coli proteins, indicated near linearity over 5 log units (Fig. 4F).
Figure 4.

Comparison of label-free quantitation of UPS2 and E. coli proteins on the Orbitrap. Lavender points, E. coli proteins; black points, UPS2 proteins. Mean ± SD is indicated for each; fits are second-order polynomials with equations indicated. (A) riBAQ and normalized molar intensity. (B) riBAQ and normalized molar counts. (C) Normalized molar intensity and normalized molar counts. (C) rTop3 and normalized molar intensity. (D) rTop3 and normalized molar counts. (E) riBAQ and rTop3.
Relationship between mole fraction and MS2 quantitation
To directly compare these MS1 results with MS2 counts and intensity, we subjected the Orbitrap RAW data files used for MaxQuant analysis to our standard workflow, which uses SEQUEST searches and a linear discriminant score transformation18,21 to identify peptides and quantify proteins. Counts or intensity from peptides shared by two or more proteins were divided amongst the proteins based on relative proportion of counts or intensity from unique peptides23. We calculated relative molar counts (cm) or relative molar intensity (im) by dividing a protein's counts or intensity by its molecular mass, then normalized by dividing by the sum of all counts/mass or intensity/mass18. SEQUEST detected fewer human proteins than did Andromeda; in the UPS1 dataset, only 47 proteins were detected.
When compared directly to MS1 quantitation, spectral counts were less accurate than riBAQ values; log cm values averaged only −3.02, substantially different from the known mole fraction (Fig. 3C). In contrast, normalized molar intensities were nearly as accurate as riBAQ values in estimating the mole fraction of UPS1 proteins (Fig. 3D). im values were not normally distributed, however.
In UPS2 experiments, only 7 of 8 proteins at 10−3 and 5/8 at 10−4 mole fraction were quantified by SEQUEST, with none below the latter dilution (Fig. 2B,C). cm values systematically diverged from a linear relationship with log mole fraction, with elevated cm values for proteins diluted to a high mole fraction (Fig. 3C). Fitting the cm values to known mole fractions was statistically better with a second-order polynomial fit compared to a linear fit. Compared to im, however, protein-to-protein variability was substantially less for cm calculations.
A fit of riBAQ and im for all proteins detected in the summed experiments, including E. coli proteins, was almost linear over a wide concentration range (Fig. 4A). By contrast, cm showed the same systematic variation from either riBAQ or im values (Fig. 4B,C) that was apparent with just the UPS2 proteins (Fig. 3). The relationships between rTop3 and either im (Fig. 4D) or cm (Fig. 4E) were similar to those seen with riBAQ. Together, the UPS1 and UPS2 experiments show that when using the same mass spectrometer, MS2 intensities perform nearly as well in quantitation as do summed MS1 intensities.
Comparison of quantitation using Orbitrap and ion-trap mass spectrometers
We ran additional samples of UPS1 or UPS2 proteins diluted with E. coli proteins on Velos and LTQ mass spectrometers. In the UPS1 experiments, cm data from either instrument were substantially different from the known mole fraction (Fig. 2E,G). The Velos im data accurately reflected the known mole fraction (Fig. 2F). The LTQ im values were not distributed normally, however, and im was less accurate than other intensity measurements (Fig. 2H).
As in experiments with the Orbitrap instrument, accurately fitting the relationship between cm and the known mole fraction in the ion-trap UPS2 experiments required a second-order polynomial (Fig. 3E,G). By contrast, the im data could be fit linearly through the 0,0 point with slopes near 1.0 (Fig. 3F,H). These data show that the accuracy of im quantitation on the ion-trap mass spectrometers is nearly as good as that on the Orbitrap.
When comparing all E. coli and human proteins, the relationship between Orbitrap riBAQ and ion-trap cm or im was less accurate; there was considerable scatter in the data (Fig. S2A,B,D,E). The relationship between cm and im calculated from the ion trap data was scattered considerably less but was not fit well with a straight line through the 0,0 point (Fig. S2C,F).
Dependence of accuracy on protein mass
In the UPS2 data, we noticed that for proteins with many spectral counts, their abundances were systematically overestimated by riBAQ (Fig. 5A). Likewise, the relationship between counts per replicate and the error in estimation of mole fraction were very similar for the UPS1 proteins (Fig. 5B), even though they are present in a narrower concentration range.
Figure 5.
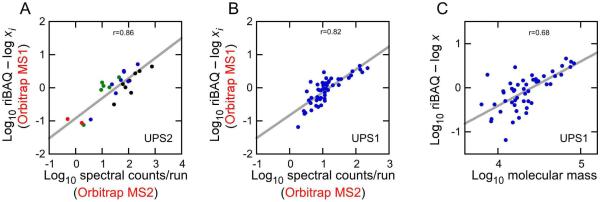
A systematic error in abundance estimation. (A) Relationship between the log10 spectral counts per run and the error in log10 mole fraction estimated for UPS2 proteins. Red symbols, 5 fmol injected; green symbols, 50 fmol; blue symbols, 500 fmol; black symbols, 5000 fmol. (B) Relationship between the log10 spectral counts per run and the error in log10 mole fraction estimated for UPS1 proteins. The same amount of each protein (200 fmol, corresponding to log10 mole fraction of −3.3) was injected. (C) Relationship between the log10 molecular mass and the error in log10 mole fraction estimated for UPS1 proteins.
A strong predictor of the number of spectral counts and therefore the error in abundance estimation was simply the protein's molecular mass. As with counts, as the mass of a protein increased, its molar abundance was systemically overestimated (Fig. 5C). This relationship was more scattered than the one with peptide identifications, although as the molecular mass increased past ~30 kD, the correlation between mass and error became more predictable (Fig. 5C). The correlation was not improved by substituting molecular mass with the number of tryptic peptides between 6 and 30 amino acids long, the factor used to convert intensity to iBAQ.
A peptide's MS2 spectral count frequency, determined by MaxQuant, was loosely correlated with its MS1 intensity per observation (Fig. S3). In contrast to the report of Silva et al.9, we found considerable variation in the intensity per peptide for the proteins we quantified, including the three most intense peptides (Fig. 6). Distributions of spectral counts per peptide varied substantially between proteins, even between those of similar molecular mass (Fig. 7). As with the intensity distributions, only the largest proteins had peptides with particularly large numbers of spectral counts.
Figure 6.

MS1 signal responses (intensity) for peptides from UPS1 proteins. For each protein, peptides were organized by decreasing intensity. In upper right of each panel, the protein symbol, molecular mass, and average log10 intensity for the top three peptides are indicated. At right side of peptide distribution, the total number of peptides detected is indicated. All panels use the same horizontal and vertical scales.
Figure 7.

MS2 spectral count totals for peptides from UPS1 proteins. For each protein, peptides were organized by decreasing numbers of spectral counts. Call-outs are as in Fig. 5.
Discussion
We demonstrate here that normalized MS2 intensities are nearly as accurate at predicting protein abundance as are two MS1 summed intensity methods, relative iBAQ and relative Top3. Moreover, quantitation using MS2 intensities obtained with low-resolution ion-trap mass spectrometers is comparable to either MS1 or MS2 quantitation using data from an Orbitrap instrument. There are limitations with using intensities, however; relatively few proteins are detected of mole fraction less than 10−4, and estimations of protein abundance are skewed by molecular mass, where abundances of high-mass proteins are overestimated. Nevertheless, both MS1 and MS2 intensities consistently produced more accurate relative molar fractions than MS2 spectral counting, perhaps the most popular label-free quantitation method.
Quantitation with MS1 intensities
The iBAQ method, which sums all identified peptide intensities and normalizes them against detectable tryptic peptide number, has proven useful in a number of contexts6,7,17. This method was recently shown to be superior to other label-free methods for accurate protein quantitation8. In our hands, the relative iBAQ measure accurately reported the abundance of proteins spiked into a complex protein mixture.
With our samples and instrument configuration, the “top three” method for protein quantitation9,10 was no more accurate than the iBAQ method. This comparison was drawn from the same dataset, so neither peptide identification algorithms nor instrument settings entered into the comparison. We saw a much larger variation in the intensity per observation for the three most intense peptides detected (CV of ~100% for non-log transformed data) than the variation reported in the original paper (CV values of ~10%)9. Our experiments differed substantially, however, as we diluted UPS1 and UPS2 proteins into complex protein mixtures rather than diluting them with buffer as did Silva et al. Moreover, because of the complex peptide mixture, we used data-dependent MS2 acquisition in our experiments, which led to only the more abundant peptides being identified in MS2 scans. Finally, Silva et al. acquired MS1 data with every other scan, which theoretically provides more accurate intensity values.
The basic premise of the “top three” method does not hold under our experimental conditions; the average MS signal response of the three best ionizing peptides was not shared between proteins. Apparently high signal response peptides are rare, and thus are more often detected in large proteins with many tryptic peptides. These peptides also dominate the summed intensity, so the iBAQ algorithm yields quantitation results similar to that of the “top three” method.
Influence of SDS-PAGE separation
In our experiments with miniscule inner ear tissues13,17–19,24–26, we use SDS-PAGE protein separation and in-gel protease digestion to generate tryptic peptides for analysis. Many of our samples are dilute (<0.1 Mg of total protein in ~50 μl) and contain a substantial amount of agarose, used for isolation of the sensory hair bundles of the inner ear. SDS-PAGE is ideal for these samples because it allows protein concentration and elimination of agarose, which is not mobile in the electric field.
Use of SDS-PAGE to separate proteins prior to trypsin digestion likely affected our results, however, as protein behavior in the gel may vary substantially27. Indeed, some of the dependence we see on protein molecular mass (Fig. 5) likely stems from preferential loss of small proteins during processing of the gel for protease digestion. Peptide recovery following SDS-PAGE and in-gel digestion varies considerably28, so stochastic appearance of efficiently extracted peptides also likely contributes to the large variance seen with small molecular mass proteins. That said, SDS-PAGE followed by in-gel digestion has been shown to allow much greater protein identification numbers than other second-dimension separation methods29–31.
Quantitation with MS2 intensities
Using MS1 and MS2 data collected from the same mass spectrometry run, we found that normalized molar intensities, derived from MS2 scans, behaved nearly identically to the MS1-derived riBAQ (Figs. 2D and 3A). Given the stochastic nature of MS2 data collection, this result is rather surprising; peptide peaks that are sampled frequently in MS1 scans may have only a single MS2 spectrum acquired, not necessarily at the peak of the peptide elution profile (Fig. 1). Because relatively abundant proteins were quantified using MS2 scans of many different peptides, and because we used four replicates to improve reliability, the effects of stochastic sampling were dampened in our experiments.
While more UPS2 proteins were detected from MS1 data analyzed with MaxQuant, this discrepancy likely represents differences in the data analyses rather than the MS1 to MS2 comparison. Andromeda's slightly superior identification performance was apparent with the Orbitrap UPS1 data, where it detected all 49 proteins, while SEQUEST detected only 47 proteins.
Quantitation with low-resolution mass spectrometers
Our results show that under the conditions we used, quantitation using MS2 data acquired with ion-trap mass spectrometers was as accurate as MS2 (or even MS1) quantitation on the Orbitrap (compare Figs. 2F and 2H with Fig. 2C). This important result shows that even in the era of high-resolution instruments like Orbitraps, workhorse ion-trap mass spectrometers still have utility. Protein quantitation is accurate with these instruments for medium- and high-abundance proteins, with normalized molar intensity performing better than normalized molar counts. Normalized molar counts were more precise, however, readily seen by comparing standard deviations for abundance estimates in the UPS1 (Fig. 2) and UPS2 (Fig. 3) experiments.
As expected, the Orbitrap detected more proteins than either ion trap, although the difference was not profound. There are too few UPS2 proteins at each dilution for comparisons to be robust, while we used the UPS1 proteins at a mole fraction where all instruments were relatively reliable. A more telling comparison would be to compare the mass spectrometers with UPS1 proteins diluted to 10−5, where the Orbitrap detected two UPS2 proteins (of eight) but neither ion-trap instrument detected any components of the UPS2 mix. Nevertheless, these results highlight the stark fact that shotgun mass spectrometry samples a small fraction of the proteins present at low abundances.
Protein-to-protein variability
There are substantial limitations of the UPS1 and UPS2 protein standards that may reduce the generalizability of these results. First, the UPS protein sets are dominated by proteins of low molecular mass (average mass ~27 kD); eukaryotic tissue extracts we routinely study have weighted averages of >40 kD. Second, most of the UPS proteins are soluble proteins, while many interesting proteins in other tissues are membrane proteins. Finally, the UPS proteins are readily isolated proteins, and the physicochemical properties of their digested peptides may favor separation and detection in proteomics experiments.
While the distribution of riBAQ values was fit well by a Gaussian curve (Fig. 2A), normalized molar intensities, whether using an Orbitrap or the ion-trap instruments, were not distributed normally (Fig. 2D,F,H). The LTQ normalized molar intensity data were particularly diversely distributed (Fig. 2H). The reason for this non ideal distribution is not yet clear.
All intensity quantitation approaches, whether based on MS1 or MS2 data, suffer substantial variance due to the high variability in the intensity per detected peptide. This intensity variance is closely correlated with peptide observation variance. For medium to higher abundance proteins, the wide individual peptide-to-peptide variability is largely reduced due to the substantial number of peptides detected per protein. Protein variability would be expected to increase as the numbers of detected peptides decrease, effectively limiting the applicable range of these quantitative methods. Processing of an appropriate sample, such as UPS2 proteins in a complex background, under similar conditions to biological samples of interest, should allow estimation of the relevant reliable range of quantitation provided by these methods.
Detection of rare proteins
Although we detected ~1200 E. coli proteins, the genome contains over 4000 open reading frames32. As E. coli are estimated to have 2 × 106 total protein copies per cell8, it is likely that many proteins present at 1–10 copies per cell (log10 mole fraction of −6.3 to −5.3) were not detected in our experiments. Proteins in that abundance range are indeed relatively rare (Fig. S1). Likewise, the UPS2 experiments showed that few proteins present at log10 mole fraction of −5 and below were detected. Presumably the large number of peptides derived from proteins present at higher levels prevented most of these low-abundance peptides from being detected. Proteins present at less than one part in 10,000 are thus very inefficiently detected using shotgun mass spectrometry under the conditions we use.
Conclusions
Our experiments show that MS2 intensities, while imperfect, report protein abundance nearly as accurately as does quantitation based on MS1 intensities. This result is somewhat surprising, given that MS1 methods sample entire peptide liquid chromatography elution peaks, while MS2 scans are obtained stochastically and infrequently within those peaks (Fig. 1). Clearly, the more abundant the protein and the larger the number of replicates, the more reproducible the abundance measurement is. Vagaries of peptide quantitation are minimized by the intrinsic averaging that occurs with all peptides contributing to detection of each protein.
The equivalence of MS1 and MS2 intensities (Fig. 4A) indicates that a relatively small fraction of the variation seen in MS2 intensities is due to stochastic sampling. Instead, the extreme variation in the propensity of individual peptides to be ionized by electrospray (e.g., Fig. 6) is the predominant source of variability in these methods; larger proteins are more accurately measured, however, as responses of many peptides are averaged.
In our hands, the iBAQ and Top3 methods are equivalent in quantifying proteins, although the accuracy of each is reduced by the dependence on molecular mass we observed (Fig. 5). Although this relationship may depend on our use of SDS-PAGE runs and in gel digests, the practicality of those methods for scarce protein detection demands their use.
Our results also show that MS2 intensities measured on ion-trap mass spectrometers reasonably accurately correlate with protein abundance. While somewhat less accurate than abundances determined using an Orbitrap mass spectrometer, ion-trap MS2 intensities are readily measured and the widespread availability of these instruments will ensure that this method is useful for many future studies. While absolute accuracy and precision require targeted proteomics methods, label-free quantitation is simple, on average accurate, and reasonably precise, guaranteeing a place in the proteomics quantitation repertoire for some time to come.
Supplementary Material
Acknowledgements
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository33 with the dataset identifier PXD000602. We thank the PRIDE team for assistance in this submission. This work was supported by National Institutes of Health grants F32 DC012455, R01 DC011034 (PGBG), and R01 EY007755 (LLD).
Footnotes
Supporting information Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Domon B, Aebersold R. Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 2010;28:710–721. doi: 10.1038/nbt.1661. [DOI] [PubMed] [Google Scholar]
- (2).Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- (3).Kruger M, Moser M, Ussar S, Thievessen I, Luber CA, Forner F, Schmidt S, Zanivan S, Fassler R, Mann M. SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell. 2008;134:353–364. doi: 10.1016/j.cell.2008.05.033. [DOI] [PubMed] [Google Scholar]
- (4).Schulze WX, Usadel B. Quantitation in mass spectrometry-based proteomics. Annu. Rev. Plant Biol. 2010;61:491–516. doi: 10.1146/annurev-arplant-042809-112132. [DOI] [PubMed] [Google Scholar]
- (5).Bantscheff M, Lemeer S, Savitski MM, Kuster B. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal. Bioanal. Chem. 2012;404:939–965. doi: 10.1007/s00216-012-6203-4. [DOI] [PubMed] [Google Scholar]
- (6).Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- (7).Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Paabo S, Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 2011;7:548. doi: 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Arike L, Valgepea K, Peil L, Nahku R, Adamberg K, Vilu R. Comparison and applications of label free absolute proteome quantification methods on Escherichia coli. J Proteomics. 2012;75:5437–5448. doi: 10.1016/j.jprot.2012.06.020. [DOI] [PubMed] [Google Scholar]
- (9).Silva JC, Gorenstein MV, Li GZ, Vissers JP, Geromanos SJ. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol. Cell. Proteomics. 2006;5:144–156. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- (10).Grossmann J, Roschitzki B, Panse C, Fortes C, Barkow-Oesterreicher S, Rutishauser D, Schlapbach R. Implementation and evaluation of relative and absolute quantification in shotgun proteomics with label-free methods. J Proteomics. 2010;73:1740–1746. doi: 10.1016/j.jprot.2010.05.011. [DOI] [PubMed] [Google Scholar]
- (11).Liu H, Sadygov RG, Yates JR. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- (12).Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 2007;25:117–124. doi: 10.1038/nbt1270. [DOI] [PubMed] [Google Scholar]
- (13).Shin JB, Streijger F, Beynon A, Peters T, Gadzala L, McMillen D, Bystrom C, Van der Zee CE, Wallimann T, Gillespie PG. Hair bundles are specialized for ATP delivery via creatine kinase. Neuron. 2007;53:371–386. doi: 10.1016/j.neuron.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Asara JM, Christofk HR, Freimark LM, Cantley LC. A label-free quantification method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen. Proteomics. 2008;8:994–999. doi: 10.1002/pmic.200700426. [DOI] [PubMed] [Google Scholar]
- (15).Griffin NM, Yu J, Long F, Oh P, Shore S, Li Y, Koziol JA, Schnitzer JE. Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis. Nat. Biotechnol. 2010;28:83–89. doi: 10.1038/nbt.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Colaert N, Vandekerckhove J, Gevaert K, Martens L. A comparison of MS2-based label-free quantitative proteomic techniques with regards to accuracy and precision. Proteomics. 2011;11:1110–1113. doi: 10.1002/pmic.201000521. [DOI] [PubMed] [Google Scholar]
- (17).Shin JB, Krey JF, Hassan A, Metlagel Z, Tauscher AN, Pagana JM, Sherman NE, Jeffery ED, Spinelli KJ, Zhao H, Wilmarth PA, Choi D, David LL, Auer M, Barr-Gillespie PG. Molecular architecture of the chick vestibular hair bundle. Nat. Neurosci. 2013;16:365–374. doi: 10.1038/nn.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Spinelli KJ, Klimek JE, Wilmarth PA, Shin JB, Choi D, David LL, Gillespie PG. Distinct energy metabolism of auditory and vestibular sensory epithelia revealed by quantitative mass spectrometry using MS2 intensity. Proc. Natl. Acad. Sci. U. S. A. 2012;109:E268–E277. doi: 10.1073/pnas.1115866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Shin JB, Longo Guess CM, Gagnon LH, Saylor KW, Dumont RA, Spinelli KJ, Pagana JM, Wilmarth PA, David LL, Gillespie PG, Johnson KR. The R109H variant of fascin-2, a developmentally regulated actin crosslinker in hair-cell stereocilia, underlies early-onset hearing loss of DBA/2J mice. J. Neurosci. 2010;30:9683–9694. doi: 10.1523/JNEUROSCI.1541-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- (21).Wilmarth PA, Riviere MA, David LL. Techniques for accurate protein identification in shotgun proteomic studies of human, mouse, bovine, and chicken lenses. J. Ocul. Biol. Dis. Infor. 2009;2:223–234. doi: 10.1007/s12177-009-9042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- (23).Fei SS, Wilmarth PA, Hitzemann RJ, McWeeney SK, Belknap JK, David LL. Protein database and quantitative analysis considerations when integrating genetics and proteomics to compare mouse strains. J. Proteome Res. 2011;10:2905–2912. doi: 10.1021/pr200133p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gagnon LH, Longo-Guess CM, Berryman M, Shin JB, Saylor KW, Yu H, Gillespie PG, Johnson KR. The chloride intracellular channel protein CLIC5 is expressed at high levels in hair cell stereocilia and is essential for normal inner ear function. J. Neurosci. 2006;26:10188–10198. doi: 10.1523/JNEUROSCI.2166-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhao H, Williams DE, Shin JB, Brugger B, Gillespie PG. Large membrane domains in hair bundles specify spatially constricted radixin activation. J. Neurosci. 2012;32:4600–4609. doi: 10.1523/JNEUROSCI.6184-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Grati M, Shin JB, Weston MD, Green J, Bhat MA, Gillespie PG, Kachar B. Localization of PDZD7 to the stereocilia ankle-link associates this scaffolding protein with the Usher syndrome protein network. J. Neurosci. 2012;32:14288–14293. doi: 10.1523/JNEUROSCI.3071-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Steck G, Leuthard P, Burk RR. Detection of basic proteins and low molecular weight peptides in polyacrylamide gels by formaldehyde fixation. Anal Biochem. 1980;107:21–24. doi: 10.1016/0003-2697(80)90486-8. [DOI] [PubMed] [Google Scholar]
- (28).Havlis J, Shevchenko A. Absolute quantification of proteins in solutions and in polyacrylamide gels by mass spectrometry. Anal. Chem. 2004;76:3029–3036. doi: 10.1021/ac035286f. [DOI] [PubMed] [Google Scholar]
- (29).Fang Y, Robinson DP, Foster LJ. Quantitative analysis of proteome coverage and recovery rates for upstream fractionation methods in proteomics. J. Proteome Res. 2010;9:1902–1912. doi: 10.1021/pr901063t. [DOI] [PubMed] [Google Scholar]
- (30).Piersma SR, Fiedler U, Span S, Lingnau A, Pham TV, Hoffmann S, Kubbutat MH, Jimenez CR. Workflow comparison for label-free, quantitative secretome proteomics for cancer biomarker discovery: method evaluation, differential analysis, and verification in serum. J. Proteome Res. 2010;9:1913–1922. doi: 10.1021/pr901072h. [DOI] [PubMed] [Google Scholar]
- (31).Antberg L, Cifani P, Sandin M, Levander F, James P. Critical comparison of multidimensional separation methods for increasing protein expression coverage. J. Proteome Res. 2012;11:2644–2652. doi: 10.1021/pr201257y. [DOI] [PubMed] [Google Scholar]
- (32).Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- (33).Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J, O'Kelly G, Schoenegger A, Ovelleiro D, Perez-Riverol Y, Reisinger F, Rios D, Wang R, Hermjakob H. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;41:D1063–D1069. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.