

Abstract
Myeloid-derived suppressor cells (MDSC) are present in most cancer patients where they inhibit natural anti-tumor immunity and are an obstacle to anti-cancer immunotherapies. They mediate immune suppression through their production of proteins and soluble mediators that prevent the activation of tumor-reactive T lymphyocytes, polarize macrophages towards a tumor-promoting phenotype, and facilitate angiogenesis. The accumulation and suppressive potency of MDSC is regulated by inflammation within the tumor microenvironment. Recently exosomes have been proposed to act as intercellular communicators, carrying active proteins and other molecules between sender cells and receiver cells. In this report we describe the proteome of exosomes shed by MDSC induced in BALB/c mice by the 4T1 mammary carcinoma. Using bottom-up proteomics, we have identified 412 proteins. Spectral counting identified 63 proteins whose abundance was altered > 2-fold in the inflammatory environment. The pro-inflammatory proteins S100A8 and S100A9, previously shown to be secreted by MDSC and to be chemotactic for MDSC, are abundant in MDSC-derived exosomes. Bioassays reveal that MDSC-derived exosomes polarize macrophages towards a tumor-promoting type 2 phenotype, in addition to possessing S100A8/A9 chemotactic activity. These results suggest that some of the tumor-promoting functions of MDSC are implemented by MDSC-shed exosomes.
Keywords: extracellular vesicles, exosomes, myeloid-derived suppressor cells, chemotaxis, macrophages, proteomics, spectral counting, tumors, protein S100A8, immune suppression
Introduction
Exosomes (1–3) are present in high abundance in the tumor microenvironment where they transfer information between cells (4). Exosomes of tumor origin stimulate apoptosis of tumor-reactive T cells, induce immune suppressive myeloid-derived suppressor cells (MDSC), promote angiogenesis, and exchange genetic material between cells (5–10). Increased understanding of the mechanisms that activate anti-tumor immunity, combined with promising therapeutic strategies in some cancer patients and experimental animals have led to enthusiasm for immunotherapy as a treatment for established cancers. Despite limited recent successes, active immunotherapy has not been widely effective (11). The lack of efficacy is attributed in large part to immune suppressive cells present in most cancer patients (12, 13). MDSC are present in virtually all cancer patients and experimental animals with cancer and are considered one of the dominant cell populations that obstructs immunotherapy (14, 15). MDSC inhibit anti-tumor immunity by preventing the activation of tumor-reactive T lymphocytes, by inhibiting T lymphocyte trafficking to sites where they could be activated (16), and by polarizing macrophages towards a tumor-promoting phenotype (17). Some of these mechanisms require cell-to-cell-interactions between MDSC and the target cells, and in some cases, the release of soluble mediators. Identification of the molecules regulating these processes could lead to drug interventions for preventing MDSC-mediated suppression. In vivo, the development of MDSC tracks with the level of inflammation, with increasing inflammation enhancing the potency and quantity of MDSC (18, 19). Exosomes, as extracellular messengers, may contribute to the differences in MDSC abundance and suppressive activity under heightened inflammatory conditions
We now report that MDSC isolated from BALB/c mice carrying 4T1 mammary carcinomas shed exosomes that contain proteins derived from many subcellular compartments and are associated with diverse functions. The protein cargo of MDSC-derived exosomes appears to be regulated by the extent of inflammation in which the MDSC develop in vivo. Importantly, MDSC-shed exosomes polarize macrophages towards a tumor-promoting phenotype and drive MDSC chemotaxis, suggesting that MDSC-derived exosomes play an important role as communicators in the tumor microenvironment.
Experimental
Myeloid-derived suppressor cells
BALB/c mice were injected in the mammary fat pad with 7000 wild type 4T1 mammary carcinoma cells or 4T1 cells stably transfected and expressing interleukin-1β (IL-1β) as described. When tumors were greater than ~8 mm in diameter (~3–4 weeks after initial inoculation), MDSC were harvested from the blood and monitored by immunofluorescence and flow cytometry for purity by expression of the MDSC markers Gr1 and CD11b (Figure 1) (18). MDSC used in experiments were >90% Gr1+CD11b+. MDSC induced by wild type 4T1 and 4T1/IL-1β tumor cells are termed “conventional” and “inflammatory” MDSC, respectively. All procedures with animals and animal-derived materials were approved by the UMBC and UMCP Institutional Aminal Care and Use Committees.
Figure 1.

A: Flow cytometry profile of MDSC expression for Gr1 and CD11b. B: Sucrose density (g/mL) and optical density (OD 280) plots of fractions from sucrose density gradients containing exosomes from conventional (left) and inflammatory (right) MDSC.
Exosomes
Purified MDSC obtained from 2–3 mice (~1 × 108 MDSC for each experiment) were plated at 4×106 cells/ml in serum-free HL-1 medium (BioWhittaker, Walkersville, MD) and maintained at 37° C with 5% A CO2. After 16 hrs the cultures were centrifuged at 805 g for 5 min (Eppendorf 5810R centrifuge), the pellets discarded, and the supernatants centrifuged at 2090 g for 30 min (Sorvall RC5C, SS34 rotor). The supernatants were then ultracentrifuged (Beckman L8 ultracentrifuge) at 100,000 g for 20 hrs at 10° C using an SW40Ti rotor. Supernatants were discarded and the pellets containing the exosomes were resuspended in PBS and absorbances were measured at 260 and 280 nm. Protein content was assayed by Bradford Quick Start according to the manufacturer’s directions (Biorad). Exosomes were stored at −80° C until used. For migration experiments, exosomes were resuspended to the original volume of the conditioned medium from which they were obtained so a direct comparison of the effects of exosomes versus conditioned medium could be made. For the MDSC-macrophage cross-talk experiments, exosomes were used at 1x, 2.5x, and 5x concentrations. On average, one ml of conditioned medium contained 714 μg of exosomal protein.
Sucrose density gradient fractionation of exosomes
Freshly prepared ultracentrifuged exosomes were resuspended in 1.8 to 2.0 ml of 2.5M sucrose/0.020 M Hepes and layered on the bottom of 9/16″ × 3 ¾″ polyallomer ultracentrifuge tubes (Beckman). A 10 ml gradient of 0.25 M to 2.0 M sucrose in 0.020M Hepes was then layered over the 2 ml containing the exosomes for a total volume of 11.8 to 12 ml. Gradients were ultracentrifuged (Beckman L8 ultracentrifuge) at 10° C for 16 hrs at 100,000g in an SW40Ti rotor. One half ml fractions were collected and assessed by optical density (Figure 1). Density of the fractions was confirmed by refractometry.
Transmission electron micrographs
An aliquot containing 0.03–0.3 pg exosomes suspended in 2% glutaraldehyde was applied to a Formvar-coated grid and negatively stained with uranyl acetate. Electron micographs were acquired using a Zeiss EM10 transmission electron microscope at an accelerating voltage of 80keV.
Protein analysis
Aliquots of conventional and inflammatory exosomes containing 25 μg of total protein were lysed in 8 M urea. Fifty mM ammonium bicarbonate was added to each sample to dilute the final urea concentration to 0.8 M, which is compatible with tryptic digestion. Each sample was then reduced in 20 mM DTT at 56 °C for 30 minutes followed by alkylation in 40 mM iodoacetamide at room temperature in the dark for 30 minutes. Three technical replicates of 7 μg total protein were analyzed by LC-MS/MS (using HPLC MS/MS parameters outlined below) for three biological replicates each for conventional and inflammatory exosomes.
HPLC MS/MS Analysis
LC-MS/MS analyses were performed on a Shimadzu Prominent nanoHPLC (Shimadzu BioSciences, Columbia MD) in-line with an LTQ-orbitrap XL (Thermo Fisher Scientific, San Jose, CA). Peptides prepared via tryptic digestion were injected onto an Acclaim PepMap 300 C18 precolumn (Dionex, Sunnyvale, CA) followed by desalting by 10% Solvent A (97.5% H2O, 2.5% ACN, and 0.1% formic acid) for 20 minutes. Peptides were fractionated on a C-18 analytical column (Grace Vydak, Deerfield IL) with a linear gradient increasing from 0 to 40% solvent B (97.5% ACN, 2.5% H2O, and 0.1% formic acid) in 170 minutes, followed by an increase from 40 to 85% solvent B in 40 minutes. Flow rate was 500 nL/min. Precursor scans were acquired in the orbitrap with a resolution of 30,000 at m/z 400. In each cycle the nine most abundant ions were selected for fragmentation by collisional induced dissociation (CID), and product ion scans were acquired in the LTQ. A dynamic exclusion of 1 repeat count over 180 seconds was used.
Bioinformatics
Peptides and proteins were identified by the PepArML meta-search engine (20) using the mouse reference proteome of the UniProtKnowledgeBase (June 2013) containing 50,807 sequences. Carbamidomethylation of cysteine was selected as a fixed modification and oxidation of methionine and deamidation of asparagine and glutamine residues were selected as variable modifications. Peptide identifications from technical and biological replicates were pooled and filtered at 1% (spectral) FDR, as computed by PepArML using the method of Elias and Gygi (21), and identified proteins required to contain at least two distinct, unshared peptides, ensuring protein FDR of at most 0.01%. Spectral counts for identified proteins, spectral count ratios (RSC) as described in Old et al. (22), and statistical significance for differential spectral counts determined using in house software. Differential spectral count p-values were computed using the Fisher exact-test, and corrected for multiple testing by transformation to false-discovery-rate (FDR) using the method of Benjamini and Hochberg (23). Identified proteins are assigned to cellular components according to their Gene Ontology annotations and the PIR GO Slim.
MDSC chemotaxis
Cells used in the chemotaxis assay were >90% Gr1+CD11b+ conventional MDSC as assessed by flow cytometry (24). Five hundred μl of fresh media, media from MDSC cultures (conditioned media), or MDSC-derived exosomes from the equivalent amount of conditioned medium in fresh media were placed in individual wells of 24 well plates (lower compartment). Monoclonal antibodies to S100A8, S100A9, or irrelevant control isotype matched antibodies (10 μg/500 μl; Santa Cruz Biotech) were included in some wells. Transwells with an 8μm polycarbonate semi-permeable membrane were then inserted in each well and 1×106 MDSC in 100 μl of serum-free IMDM medium were placed in the transwells (upper compartment). Assembled transwells were incubated at 37° C in 5% CO2 for 3hrs, and the MDSC in the bottom chamber were then quantified by hemocytometer. Values for each sample are the average results of duplicate samples and three independent hemocytometer counts per well (25).
MDSC-macrophage cross-talk
BALB/c mice were injected intraperitoneally with 1 ml of 3% thioglycolate and peritoneal exudate cells (PEC) harvested 4 days later. Percent of macrophages in the exudate was determined by flow cytometry analysis of the macrophage markers F4/80 and CD11b. PEC were plated in 24 well plates at 7.5 × 105 F4/80+CD11b+ cells/well/500 μl DMEM medium supplemented with 10% fetal bovine serum and incubated at 37° C in 5% CO2 for 3hrs. Non-adherent cells (non-macrophages) were then removed and the attached macrophages were washed with macrophage medium (DMEM/5% serum). Five hundred μl of macrophage medium containing 7.5 × 105 conventional MDSC (>90% Gr1+CD11b+ cells) or MDSC-derived exosomes from 7.5 × 105 (1X), 18.7 × 105 (2.5X), or 37.5 × 105 (5X) MDSC were then added to each well. MDSC and macrophages were activated with IFNγ and LPS and co-cultured at a ratio of 1:1 (5×105 cells of each type/200 μl/well) for 18 hours. Supernatants were harvested and assayed by ELISA for IL-12 (19).
Results and Discussion
The techniques most widely used for characterization of exosomes are density measurements and imaging by transmission electron microscopy. The vesicles shed by both conventional and inflammatory MDSC (Figure 1A) exhibited densities between 1.2 and 1.3 g/mL (Figure 1B) (1, 3, 26). Transmission electron micrographs of this material revealed vesicles with diameters 25–30 nm (Figure 2), within the range ascribed to exosomes (1–3, 26). It should be noted that the MDSC that shed these small exosomes are smaller than most eukaryotic cells (35). The negative control shown in Figure 2 indicates that structures of 15 nm in diameter are artifacts of TEM sample preparation.
Figure 2.

Transmission electron microscope images of (left) exosomes shed by conventional MDSC; (middle) exosomes shed by inflammatory MDSC; the TEM stain itself (right).
Exosomal protein was used as an indicator of the number of exosomes, and was normalized for the number of MDSC cells used in each preparation in order to compare the amount of exosomes shed by conventional MDSC and inflammatory MDSC. The ratio, expressed as exosomal protein (μg) per MDSC cell, at the end of 16 hr was [1.1 ± 0.1] × 10−6 (n=4) and [1.2 ± 0.2] × 10−6 (n=4) for conventional and inflammatory MDSC, respectively. The similar ratios demonstrate that conventional and inflammatory MDSC shed exosomes with approximately the same frequency.
Three hundred and eighty seven proteins were identified (from 2528 peptides) in exosomes from conventional MDSC (Supplemental Table 1), and 374 proteins were identified (from 2280 peptides) in exosomes from inflammatory MDSC (Supplemental Table 1). When the two inventories are combined, 412 proteins were identified. Mathivanan and Simpson (27) established ExoCarta as a database of proteins reported from analyses of different exosome proteomes. About 83% of the proteins identified in exosomes from MDSC are in ExoCarta (August 2013). A searchable website EVpedia has been created by Kim et al (28) for exosomal proteins. About 87% of the proteins identified here are also listed in EVpedia (August 2013). A third database, Vesiclepedia, has recently been established for all extracellular vesicles (29). Among proteins identified in our exosomes, 93% are listed in Vesiclepedia (August 2013). Exosomal proteins from the inventory that have not previously been reported in ExoCarta, EVpedia, or Vesiclepedia are shown in Supplemental Table 2.
Annexins (A1, A2, A3, A6, A7, A11) and tetraspanins, including CD177, were identified along with GTPases, NCK microfibrils (NCK associated protein 1 like), cytoskeletal proteins, VSP35 (a member of the ESCRT complex), heat shock cognate 71 kDa protein, heat shock 70 kDa protein 4, and HSP90 alpha and beta, all proteins reported to be characteristic of exosomes (30).
A suite of subunits from the 26S proteasome was present, including 26S protease regulatory subunit 6A, 26S proteasome non-ATPase regulatory subunits 1,2,5,6,7,11, and 13, proteasome subunit beta type-2 and proteasome subunit alpha type-6. This raises the possibilities that the proteasome may be carried by these exosomes, or that the disassembled proteasome is removed from the cell by these exosomes.
More than a dozen histone variants were detected, as well as several elongation factors (1-gamma, 1-alpha 1, and 2), DNA topoisomerase, RNA helicase (ATP-dependent RNA helicase DDX39A) and a zinc finger (DBF-type zinc finger-containing protein 2 homolog). The presence of these and other nucleic acid binding proteins is consistent with reports that exosomes can alter protein expression in receiver cells (5,7, 9–11,12, 31,32).
Metabolic enzymes were identified from the pentose phosphate pathway responsible for synthesis of NADPH and ribose-5-phosphate including: glucose-6-phosphate 1-dehydrogenase X, 6-phosphogluconate dehydrogenase and transketolase. Most of the enzymes in the glycolysis and gluconeogenesis pathways were present: pyruvate kinase PKM, glucose-6-phosphate isomerase, glyceraldehyde-3-phosphosphate dehydrogenase, alpha-enolase, L-lactate dehydrogenase A chain, fructose-bisphosphate aldolase, phosphoglycerate kinase 1, glycogen phosphorylase liver form and glycogenin-1. These proteins are also listed in databases of proteins identified in various kinds of exosomes.
Several proteins relevant to the immune system were also identified. S100A8 and S100A9 are known (25, 33) to drive chronic and acute inflammation. Myeloid bactecin strongly interacts with immune cells and participates in the inflammatory response (34).
Figure 3 summarizes the sub-cellular locations of the exosomal proteins identified, and compares them with a set of 305 proteins identified (35) in a whole cell lysate of conventional MDSC. Exosomes from both conventional and inflammatory MDSC contain proteins residing in the major intracellular organelles and the distribution of proteins in both samples is similar. Compared to the whole cell lysate, a lower percentage of exosomal proteins are assigned to the parental cell nucleus and endoplasmic reticulum.
Figure 3.

Intracellular protein locations assigned by Gene Ontology annotations and the PIR GO Slim. Green: from exosomes from conventional MDSC; Red: from exosomes from inflammatory MDSC; Blue: from lysate (35) of conventional MDSC.
Chronic inflammation in the tumor microenvironment increases the quantity and suppressive potency of the parental MDSC (18, 19), and is associated with altered concentrations of some proteins in MDSC (35). In the exosomes shed by MDSC spectral counting was used to compare protein abundances. Out of 412 proteins quantified in conventional and inflammatory samples (See Supplementary Table 1) the abundances of 63 proteins differed by more than two fold with FDR less than 0.05. Heightened inflammation is associated with decreased abundances of 33 proteins in exosomes, including several proteins that participate in the innate immune response: ficolin-1, C4b-binding protein, chitinase-3-like protein 3, complement C3, and CD5 antigen-like. Other proteins that decreased with heightened inflammation include cytoskeletal proteins (spectrin beta 1, ankyrin-1, tubulin beta-1 chain and nesprin-1) and chemotactic proteins (myeloid cysteine-rich protein and platelet factor 4), suggesting a change in exosome migration.
Thirty proteins increased in abundance under heightened inflammatory conditions, including GTP and ATP binding proteins (ATP-citrate synthase, ADP-ribosylation factor 1, and phosphatidylinositol 4-phosphate 3-kinase C2 domain containing subunit gamma). Interestingly, the coatomer subunit beta, recruited by ADP-ribosylation factors that participate in membrane curvature and budding (36) is increased more than 2 fold with inflammation. Other proteins that increase include biosynthetic proteins (serine-tRNA ligase cytoplasmic, valine-tRNA ligase, aminopeptidase B, fatty acid synthase, ATP-citrate synthase, and elongation factor 1-gamma) and the retromer component vacuolar protein sorting-associated protein 35, which participates in protein sorting (37).
The relative abundance of proteins S100 A8 and A9 did not change in exosomes shed from MDSC due to inflammation. These are usually present as the A8/A9 heterodimer, a pro-inflammatory mediator that is present at sites of inflammation. It was previously shown to be chemotactic and induce migration of MDSC to tumor sites (25, 33). It also stimulates polarization of macrophages towards the tumor-promoting M2 phenotype (17). These functions have previously been attributed to the extracellular secretion of S100A8/A9 by MDSC themselves and by tumor cells. To test the possibility that exosomes carrying S100A8/A9 are responsible for these effects, bioassays were conducted with intact MDSC, MDSC-derived exosomes, and macrophages.
Chemotactic activity was assessed by co-culturing intact MDSC and MDSC-derived exosomes on opposite sides of a semi-permeable membrane and measuring the number of MDSC migrating through the membrane towards the exosomes (Figure 4). Exosomes were chemotactic for intact MDSC and chemotaxis was significantly inhibited if antibodies to S100A8 or S100A9 were included with the exosomes. The chemotaxis was specific since inclusion of an irrelevant isotype matched antibody did not prevent migration. These results indicate that MDSC-derived exosomes mediate chemotaxis through their content of S100A8 and A9. The same assay has been previously applied to conditioned media (25), where antibodies to S100 A8 and A100 A9 reduced migration by amounts similar to what is reported here for exosomes. In the current assay, exosomes were >90% as chemotactic as the equivalent amount of conditioned medium, indicating that most of the S100 A8/A9 activity is contained in exosomes.
Figure 4.
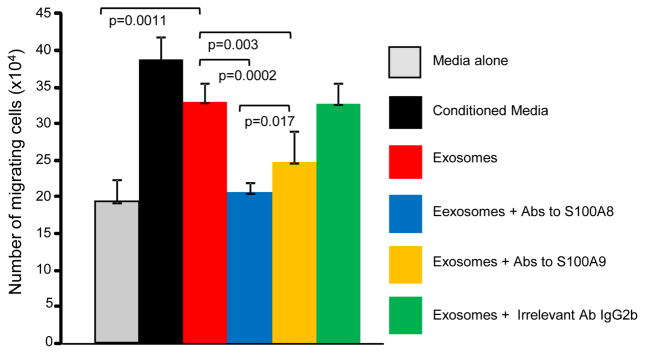
Exosomes shed by MDSC contain S100A8 and S100A9 proteins that are chemotactic for MDSC. MDSC were placed in the upper compartment of transwells and either tumor-conditioned medium or MDSC shed exosomes ± antibodies to S100A8 or S100A9 were placed in the lower compartment. The number of MDSC migrating to the lower compartment was determined after 3 hrs of incubation. Values are the average ± SD of 3 independent cell counts of duplicate samples. The concentration of purified exosomes is equivalent to the concentration of exosomes in conditioned medium.
One of the suppressive mechanisms used by MDSC to promote tumor progression is their conversion of macrophages from tumoricidal cells (so-called M1 macrophages) to cells that facilitate tumor growth (so-called M2 macrophages). This conversion involves the switching off of macrophage production of IL-12, a cytokine that drives the development of tumoricidal T lymphocytes and natural killer cells (17). To determine if this function is mediated by MDSC-derived exosomes, M1 macrophages were co-cultured with either intact MDSC or exosomes derived from an equal number of MDSC or multiples thereof (2.5X, 5X). Macrophage production of IL-12 was measured by ELISA (Figure 5). MDSC-derived exosomes decreased IL-12 production; however, on a per cell basis they are less effective than intact MDSC. These results indicate that exosomes contribute to the ability of MDSC to polarize macrophages; however non-exosome mechanisms are also involved.
Figure 5.
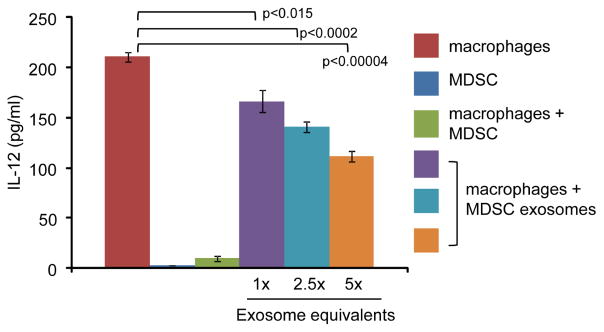
Exosomes shed by MDSC polarize macrophages from a tumoricidal M1 phenotype to a tumor-promoting M2 phenotype by inhibiting macrophage production of IL-12. Type 1 macrophages were co-cultured alone, or in the presence of intact MDSC or exosomes derived from either an equal number of MDSC (1X) or multiples thereof. shed by MDSC. IL-12 production by macrophages was measured by ELISA.
Conclusions
Exosomes shed by myeloid-derived suppressor cells are rich in proteins. Exosomal proteins, unlike RNA, can have an immediate effect on the target cell, making the exosomal proteome a valuable source of information about intracellular communication in the tumor microenvironment. Their cargo includes many histones, enzymes active in energy metabolism, proteasome subunits and the pro-inflammatory mediators S100A8 and S100A9. Neither the number of exosomes shed per cell nor the concentration of protein S100 A8 in the exosomes varies significantly between MDSC isolated from low and high inflammatory murine environments. Biological assays demonstrate both autocrine and paracrine activities for these exosomes and demonstrate causality for S100A8 and S100A9. These results demonstrate that some of the immune suppressive activity of MDSC is mediated by MDSC-shed exosomes. Since MDSC infiltrate solid tumors and also circulate in the blood and are present in the bone marrow, MDSC-derived exosomes may function as short and long-range mediators to regulate anti-tumor immunity and facilitate tumor growth.
Supplementary Material
Supplemental Table 1. Peptides and proteins identified in exosomes shed by conventional and inflammatory MDSC. Rsc is reported as the log2 ratio of inflammatory versus conventional exosomes.
Supplemental Table 2. Proteins identified in the combined inventory of exosomes shed by conventional and inflammatory MDSC that were not present in exosomal compendiums ExoCarta, Vesiclepedia, and EVpedia (as of August 2013).
Table 1.
Proteins with significantly greater abundance in conventional exosomes (Rsc ≥1 and FDR ≤0.05). Rsc is reported as the log2 ratio of conventional versus inflammatory exosomes.
| Accession | Protein | Rsc | FDR |
|---|---|---|---|
| P70390 | Short stature homeobox protein 2 | 8.6 | 6.26E-157 |
| P04919 | Band 3 anion transport protein | 6.4 | 5.05E-32 |
| P08032 | Spectrin alpha chain, erythrocytic 1 | 5.6 | 7.16E-19 |
| Q8CIZ8 | von Willebrand factor | 4.8 | 3.56E-10 |
| P08226 | Apolipoprotein E | 4.6 | 5.86E-09 |
| Q61171 | Peroxiredoxin-2 | 4.4 | 1.53E-07 |
| Q9QUM0 | Integrin alpha-IIb | 4.3 | 6.14E-07 |
| P01837 | Ig kappa chain C region | 3.7 | 1.28E-04 |
| P29788 | Vitronectin | 3.3 | 1.50E-03 |
| P49722 | Proteasome subunit alpha type-2 | 3.3 | 1.50E-03 |
| Q02357 | Ankyrin-1 | 3.3 | 1.50E-03 |
| Q3UGX2 | Spectrin beta 1 | 3.3 | 1.50E-03 |
| Q8K482 | EMILIN-2 | 3.3 | 1.50E-03 |
| Q9QWK4 | CD5 antigen-like | 3.0 | 9.63E-03 |
| Q07797 | Galectin-3-binding protein | 2.8 | 1.74E-02 |
| P11276 | Fibronectin | 2.7 | 3.08E-124 |
| O70165 | Ficolin-1 | 2.7 | 2.95E-02 |
| P08607 | C4b-binding protein | 2.6 | 4.68E-03 |
| P10605 | Cathepsin B | 2.5 | 8.26E-03 |
| P07724 | Serum albumin | 2.5 | 7.87E-22 |
| Q61646 | Haptoglobin | 2.1 | 1.08E-03 |
| P01872 | Ig mu chain C region secreted form | 2.1 | 7.19E-13 |
| O35744 | Chitinase-3-like protein 3 | 2.0 | 8.90E-05 |
| P35441 | Thrombospondin-1 | 1.9 | 4.55E-14 |
| Q6ZWR6 | Nesprin-1 | 1.9 | 1.54E-02 |
| P62259 | 14-3-3 protein epsilon | 1.5 | 6.12E-03 |
| P82198 | Transforming growth factor-beta-induced protein ig-h3 | 1.5 | 1.04E-02 |
| Q8C2Q7 | Heterogeneous nuclear ribonucleoprotein H | 1.5 | 4.89E-02 |
| A2AQ07 | Tubulin beta-1 chain | 1.5 | 1.88E-03 |
| P01027 | Complement C3 | 1.3 | 4.62E-08 |
| Q8K426 | Myeloid cysteine-rich protein | 1.3 | 2.59E-02 |
| Q9Z126 | Platelet factor 4 | 1.1 | 4.71E-03 |
| Q9R1P3 | Proteasome subunit beta type-2 | 1.0 | 2.41E-02 |
Table 2.
Proteins with significantly greater abundance in inflammatory exosomes (Rsc ≥1 and FDR ≤0.05). Rsc is reported as the log2 ratio of inflammatory versus conventional exosomes.
| Accession | Protein | RSC | FDR |
|---|---|---|---|
| Q5SS00 | DBF4-type zinc finger-containing protein 2 homolog | 3.6 | 1.48E-03 |
| Q8VDP4 | DBIRD complex subunit KIAA1967 homolog | 3.1 | 1.39E-02 |
| E9QQ35 | Phosphatidylinositol 4-phosphate 3-kinase C2 domain- containing subunit gamma | 3.1 | 1.10E-03 |
| P12970 | 60S ribosomal protein L7a | 3.0 | 2.29E-02 |
| P84078 | ADP-ribosylation factor 1 | 3.0 | 2.29E-02 |
| P62908 | 40S ribosomal protein S3 | 2.8 | 4.49E-03 |
| P08730 | Keratin, type I cytoskeletal 13 | 2.8 | 3.76E-02 |
| Q6ZQA0 | Neurobeachin-like protein 2 | 2.8 | 3.76E-02 |
| P26638 | Serine--tRNA ligase, cytoplasmic | 2.4 | 3.10E-02 |
| P42227 | Signal transducer and activator of transcription 3 | 2.4 | 3.10E-02 |
| Q9D154 | Leukocyte elastase inhibitor A | 2.4 | 2.75E-05 |
| Q9Z1Q9 | Valine--tRNA ligase | 2.2 | 7.61E-05 |
| Q8VCT3 | Aminopeptidase B | 2.0 | 1.48E-03 |
| P84096 | Rho-related GTP-binding protein RhoG | 1.9 | 1.91E-03 |
| P42932 | T-complex protein 1 subunit theta | 1.8 | 3.28E-02 |
| O88593 | Peptidoglycan recognition protein 1 | 1.8 | 4.31E-03 |
| Q99KE1 | NAD-dependent malic enzyme, mitochondrial | 1.8 | 2.29E-02 |
| O55029 | Coatomer subunit beta′ | 1.7 | 1.49E-02 |
| Q9EQH3 | Vacuolar protein sorting-associated protein 35 | 1.7 | 9.63E-03 |
| Q9CWJ9 | Bifunctional purine biosynthesis protein PURH | 1.6 | 1.48E-03 |
| P80315 | T-complex protein 1 subunit delta | 1.5 | 2.40E-02 |
| P19096 | Fatty acid synthase | 1.4 | 4.22E-03 |
| P28293 | Cathepsin G | 1.3 | 1.72E-04 |
| Q63844 | Mitogen-activated protein kinase 3 | 1.2 | 3.10E-02 |
| P60766 | Cell division control protein 42 homolog | 1.2 | 1.49E-02 |
| Q8CCK0 | Core histone macro-H2A.2 | 1.2 | 1.91E-03 |
| Q9Z1Q5 | Chloride intracellular channel protein 1 | 1.2 | 1.01E-02 |
| Q9D8N0 | Elongation factor 1-gamma | 1.2 | 6.62E-03 |
| Q91V92 | ATP-citrate synthase | 1.1 | 2.40E-02 |
| P62827 | GTP-binding nuclear protein Ran | 1.0 | 1.15E-03 |
Acknowledgments
We acknowledge Virginia Clements for preparing the exosomes and Lisa Birkheimer for husbandry of the mice. We thank Dr. Yan Wang, Director of Proteomic Core Facility, Maryland Pathogen Research Institute, University of Maryland, College Park, for advice about the LC-MS/MS analysis, and Tim Maugel, Director of the Laboratory for Biological Ultrastructure, University of Maryland, College Park, for advice about TEM imaging. The work was supported by a grant from the National Institutes of Health GM021248. WC acknowledges a Royal Thai Government Fellowship.
Footnotes
This information is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Shen B, Wu N, Yang JM, Gould SJ. Protein targeting to exosomes/microvesicles by plasma membrane anchors. J Biol Chem. 2011;286:14383–14395. doi: 10.1074/jbc.M110.208660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi DS, Yang JS, Choi EJ, Jang SC, Park S, Kim OY, Hwang D, Kim KP, Kim YK, Kim S, Gho YS. The Protein Interaction Network of Extracellular Vesicles Derived from Human Colorectal Cancer Cells. J Prot Res. 2012;11:1144–1151. doi: 10.1021/pr200842h. [DOI] [PubMed] [Google Scholar]
- 3.Gould SJ, Raposo G. As we wait: coping with an imperfect nomenclature for extracellular vesicles. J Extracellular Vesicles. 2013;2:1–3. doi: 10.3402/jev.v2i0.20389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnstone RM. Exosomes biological significance: A concise review. Blood Cells, Molecules and Diseases. 2006;36:315–321. doi: 10.1016/j.bcmd.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Simpson RJ, Lim JW, Moritz RL, Mathivanan S. Exosomes: proteomic insights and diagnostic potential. Expert Reviews. 2009;6:267–283. doi: 10.1586/epr.09.17. [DOI] [PubMed] [Google Scholar]
- 6.Iero M, Valenti R, Huber V, Filipazzi P, Parmiani G, Fais S, Rivoltini L. Tumour-released exosomes and their implications in cancer immunity. Cell Death and Differentiation. 2008;15:80–88. doi: 10.1038/sj.cdd.4402237. [DOI] [PubMed] [Google Scholar]
- 7.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 8.Xiang X, Poliakov A, Liu C, Liu Y, Deng Z, Wang J, Cheng Z, Shah SV, Wang G, Zhang L, Grizzle WE, Mobley J, Zhang H. Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer. 2009;124:22621–2633. doi: 10.1002/ijc.24249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skog J, Wurdinger T, van Rijn S, Meijer D, Gainche L, Sena-Esteves M, Curry ST, Carter RS, Krichevesky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and protein that promote tumor growth and provide diagnostic biomarkers. Nat Cell Bio. 2008;10:1470–1476. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balaj L, Lessard R, Dai L, Cho Y, Pomeroy SL, Breakefield XO, Skog J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nature Commun. 2011;2:180. doi: 10.1038/ncomms1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lesterhuis WJ, Haanen JB, Punt CJ. Cancer immunotherapy--revisited. Nature Rev Drug Discovery. 2011;10:591–600. doi: 10.1038/nrd3500. [DOI] [PubMed] [Google Scholar]
- 12.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 13.Poschke I, Mougiakakos D, Kiessling R. Camouflage and sabotage: tumor escape from the immune system. Cancer Immunol Immunother. 2011;60:1161–1171. doi: 10.1007/s00262-011-1012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nature Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marx J. Cancer immunology. Cancer’s bulwark against immune attack: MDS cells. Science. 2008;319:154–156. doi: 10.1126/science.319.5860.154. [DOI] [PubMed] [Google Scholar]
- 16.Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand-Rosenberg S. Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8+ T cells. J Immunol. 2009;183:937–944. doi: 10.4049/jimmunol.0804253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ostrand-Rosenberg S. Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother. 2010;59:1593–1600. doi: 10.1007/s00262-010-0855-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol. 2006;176:284–290. doi: 10.4049/jimmunol.176.1.284. [DOI] [PubMed] [Google Scholar]
- 19.Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007;67:10019–10026. doi: 10.1158/0008-5472.CAN-07-2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edwards N, Wu X, Tseng CW. An unsupervised, model-free, machine-learning combiner for peptide identifications from tandem mass spectra. Clinical Proteomics. 2009;5:23–36. [Google Scholar]
- 21.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 22.Old WM, Meyer-Arendt K, Aveline-Wolf L, Pierce KG, Mendoza A, Sevinsky JR, Resing KA, Ahn NG. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol Cell Prot. 2005;4:1487–1502. doi: 10.1074/mcp.M500084-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J Royal Statistical Soc Series B (Methodological) 1995;57:289–300. [Google Scholar]
- 24.Sinha P, Clements VK, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol. 2005;174:636–645. doi: 10.4049/jimmunol.174.2.636. [DOI] [PubMed] [Google Scholar]
- 25.Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181:4666–4675. doi: 10.4049/jimmunol.181.7.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thery C, Zitvogel L, Amigorene S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 27.Mathivanan S, Simpson RJ. ExoCarta: A compendium of exosomal proteins and RNA. Proteomics. 2009;9:4997–5000. doi: 10.1002/pmic.200900351. [DOI] [PubMed] [Google Scholar]
- 28.Kim D, Kang B, Kin O, Choi D, Lee J, Kim SR, Go G, Yoon YJ, Kim JH, Jang SC, Park K, Choe E, Kim KP, Desiderio DM, Kim Y, Lotvall J, Hwang D, Gho YS. EVpedia: an integrated database of high-throughput data for systemaic analyses of extracellular vesicles. J Extracellular Vesicles. 2013;2:20384. doi: 10.3402/jev.v2i0.20384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalra H, Simpson RJ, Ji H, Aikawa E, Altevogt P, Askenase P, Bond VC, Borràs FE, Breakefield X, Budnik V, Buzas E, Camussi G, Clayton A, Cocucci E, Falcon-Perez JM, Gabrielsson S, Gho YS, Gupta D, Harsha HC, Hendrix A, Hill AF, Inaal JM, Jenster G, Kiang LS, Krämer-Albers EM, Llorente A, Lötvall J, Mincheva-Nilsson L, Nazarenko I, Nieuwland R, Nolte-‘t Hoen ENM, Pandey A, Patel T, Piper MG, Pluchino S, Prasad TSK, Rajendran L, Raposo G, Record M, Reid GE, Sánchez-Madrid F, Schiffelers RM, Siljander P, Stoorvogel W, Taylor D, Thery C, Valadi H, van Balkom BWM, Vázquez J, Vidal M, Yáñez-Mó M, Zoeller M, Mathivanan S. Vesiclepedia: A compendium for extracellular vesicles with continuous community annotation. PLoS Biology. 2012;12:e1001450. doi: 10.1371/journal.pbio.1001450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mathivanan S, Ji H, Simpson RJ. Exosomes: Extracellular organelles important in intercellular communication. J Proteomics. 2010;73:1907–1920. doi: 10.1016/j.jprot.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 31.Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT, Jr, Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–1476. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor DD, Gercel-Taylor CG. Tumour-derived exosomes and their role in cancer-associated T-cell signaling defects. Brit J Cancer. 2005;92:305–311. doi: 10.1038/sj.bjc.6602316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T, Roth J, Gabrilovich DI. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205:2235–2249. doi: 10.1084/jem.20080132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soehnlein O, Weber C, Lindbom L. Neutrophil granule proteins tune monocytic cell function. Trends Immunol. 2009;30:538–546. doi: 10.1016/j.it.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 35.Choksangakarn W. PhD thesis. University of Maryland; 2009. Development of a Proteomic Strategy for Analysis of Plasma Membrane Proteins. [Google Scholar]
- 36.Graham TR, Kozlov MM. Interplay of proteins and lipids in generating membrane curvature. Curr Opin Cell Bio. 2010;22:430–436. doi: 10.1016/j.ceb.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nature Rev Mol Cell Biol. 2009;10:623–635. doi: 10.1038/nrm2745. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Peptides and proteins identified in exosomes shed by conventional and inflammatory MDSC. Rsc is reported as the log2 ratio of inflammatory versus conventional exosomes.
Supplemental Table 2. Proteins identified in the combined inventory of exosomes shed by conventional and inflammatory MDSC that were not present in exosomal compendiums ExoCarta, Vesiclepedia, and EVpedia (as of August 2013).