

Abstract
Joubert syndrome (JS) is characterized by a distinctive cerebellar structural defect, namely the « molar tooth sign ». JS is genetically heterogeneous, involving 18 genes identified to date, which are all required for cilia biogenesis and/or function. In a consanguineous family with JS associated with optic nerve coloboma, kidney hypoplasia and polydactyly, combined exome sequencing and mapping identified a homozygous splice site mutation in PDE6D, encoding a prenyl-binding protein. We found that pde6d depletion in zebrafish leads to renal and retinal developmental anomalies and wild-type but not mutant PDE6D is able to rescue this phenotype. Proteomic analysis identified INPP5E, whose mutations also lead to JS or MORM syndromes, as novel prenyl-dependent cargo of PDE6D. Mutant PDE6D shows reduced binding to INPP5E, which fails to localize to primary cilia in patient fibroblasts and tissues. Furthermore, mutant PDE6D is unable to bind to GTP-bound ARL3, which acts as a cargo-release factor for PDE6D-bound INPP5E. Altogether, these results indicate that PDE6D is required for INPP5E ciliary targeting and suggest a broader role for PDE6D in targeting other prenylated proteins to the cilia. This study identifies PDE6D as a novel JS disease gene and provides the first evidence of prenyl-binding dependent trafficking in ciliopathies.
Keywords: Joubert syndrome, primary cilia, PDE6D, INPP5E, prenylation
INTRODUCTION
Primary cilia are highly conserved organelles consisting of a microtubule-based axoneme emerging from a basal body derived from the mother centriole and ensheathed by the ciliary membrane. They are required for the transduction of various extracellular signals and their importance has become increasingly clear as defective ciliary biogenesis and/or function has been shown to underlie human genetic diseases collectively termed ciliopathies that include Joubert syndrome (JS; MIM# 213300, http://www.omim.org/). JS is characterized by the absence or underdevelopment of the cerebellar vermis and accompanying brainstem abnormalities resulting in the “molar tooth sign” visualized by brain MRI. Neurologic symptoms include ataxia, oculomotor apraxia, dysregulation of breathing pattern and developmental delay (Joubert et al. 1969). Other associated features include retinal dystrophy, renal disease, liver fibrosis and polydactyly. JS is genetically heterogeneous with 18 genes identified to date, all encoding proteins localized within or near the primary cilium, including JS in the growing group of ciliopathies. Known mutations account for almost 50% of cases, indicating that additional genetic lesions exist. Therefore, in order to identify novel JS genes, we combined exome sequencing and mapping of consanguineous families excluding known JS loci. In one of those families (JS-18) with 3 affected sibs, we identified a homozygous splice site mutation in PDE6D (MIM# 602676), a gene encoding a protein originally described as the fourth subunit of the rod specific cGMP phosphodiesterase (PDE6) and thus termed PDE6D (Li et al. 1998; Li and Baehr 1998; Zhang et al. 2004). PDE6D was subsequently shown to be a prenyl-binding protein as evidenced by its crystal structure that revealed an immunoglobulin-like fold in which two beta sheets form a hydrophobic pocket into which prenyl groups can insert (Hanzal-Bayer et al. 2002). Prenylation is a lipid modification with covalent addition of farnesyl or geranylgeranyl isoprenoids to the cystein of the CaaX box, a four amino-acid motif at the C-terminus of target proteins. This lipid modification promotes membrane interactions of most of the prenylated proteins (Marshall 1993). Targeted disruption of Pde6d in mice leads to slowly progressing rod/cone dystrophy associated with mislocalization of key prenylated components of phototransduction consistent with a role of PDE6D as a cargo adaptor targeting prenylated proteins from their site of synthesis through the connecting cilium to the outer segment (Zhang et al. 2007). In an effort to understand the pathophysiological mechanisms underlying PDE6D mutation in JS, we identified INPP5E (MIM# 613037), mutations in which also cause JS as well as another ciliopathy, Mental retardation, Obesity, congenital Retinal dystrophy and Micropenis syndrome (MORM syndrome; MIM# 610156), as a farnesylated cargo of PDE6D. We found that proper INPP5E trafficking to the primary cilia requires both PDE6D and farnesylation, thus uncovering a mechanism used to target prenylated proteins to the primary cilium and elucidating a pathophysiological mechanism underlying two ciliopathies, Joubert and MORM syndromes.
MATERIALS AND METHODS
Research subjects
We used standard methods to isolate genomic DNA from peripheral blood of the affected children and family members or from frozen fetal tissue or amniocytes. The fetus had a complete autopsy and karyotyping after genetic counseling and we obtained parental consent in conformity with French law. Informed consent for molecular analysis was obtained from all participating families, and the study was approved by the Ethical committee of Paris Ile de France II.
Genome-wide scan analysis
Genomic DNA was isolated by phenol/chloroform extraction and purified using Microcon YM-30 filters (Millipore). Genotyping was performed on the 250K NspI array of the Affymetrix 250K system, which consists of 262,000 SNPs (Affymetrix), according to the manufacturerÆs instructions. Chips and data were processed on the Affymetrix platform with the Command Console Software. SNP genotypes were called using the Affymetrix BRLMM algorithm in the Genotyping Console 3.0.2 Software (Affymetrix). We performed multipoint linkage analysis using MERLIN software (http://www.sph.umich.edu/csg/abecasis/Merlin/) assuming a fully penetrant recessive model and a disease allele frequency of 0.001. Areas of homozygosity on chromosome 2 were confirmed through high-resolution haplotype analysis in the family.
Exome sequencing
DNA (3 μg) was extracted from leukocyte cells from JS-18a and was sheared with a Covaris S2 Ultrasonicator. An adaptor-ligated library was prepared with the TruSeq DNA Sample Preparation Kit (Illumina). Exome capture was performed with the SureSelect Human All Exon kit (Agilent) (Bolze et al. 2010; Byun et al. 2010; Liu et al. 2011; Bogunovic et al. 2012). Paired-end sequencing was carried out on an Illumina HiSeq 2000 that generated 100-bp reads. For sequence alignment, variant calling and annotation, the sequences were aligned to the human genome reference sequence (hg19 build) using the Burrows-Wheeler Aligner (BWA) (Li and Durbin 2009). Downstream processing was carried out with the Genome analysis toolkit (GATK) (McKenna et al. 2010), SAMtools (Li et al. 2009) and Picard [http://picard.sourceforge.net]. Variant calls were made with a GATK Unified Genotyper. All calls with a read coverage <2x and a Phred-scaled SNP quality of ≤20 were removed from consideration. All variants were annotated using an annotation software system that was developed in-house.
Mutational screening
Mutational screening of the PDE6D gene was performed by direct sequencing of PCR products of the 5 coding exons and the adjacent intronic junctions in individuals with JS/MKS, SLS, or LCA various ciliopathies. PCR primers (Supp. Table S1) were selected with the Primer3 program (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/) according to reference sequence NM_002601.2. PCR products were purified with the Exo-SAP cleanup kit (USB) and sequenced with BigDye chemistry and the ABI 3100 (Applied Biosystems) automated sequencer. Sequences were analyzed with SeqScape software (Applied Biosystems). The DNA mutation numbering system we used is based on cDNA sequence with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence, according to journal guidelines (www.hgvs.org/mutnomen). The PDE6D mutation reported has been submitted to the LOVD database (www.lovd.nl/PDE6D).
PDE6D RT-PCR
RNA from fibroblasts of the sib 2 and one age-matched control were extracted with the QIAGEN RNeasy Kit, including on-column DNase digestion. Complementary DNA (cDNA) synthesis from total RNA was conducted using the GeneAmp RNA PCR Core Kit (Applied Biosystems) with random hexamer primers. Primers were selected in exons 1, 2 and 3 (forward), 4–5 (reverse), preventing potential contaminating genomic DNA amplification (Supp. Table S1).
DNA constructs
C-terminal Myc-DDK-tagged ORF clone of PDE6D (NM_002601.2) was obtained from Origene (RC203172). Deletion of exon 3 was performed using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer’s instructions and were confirmed by direct sequencing (Primers used are listed in Supp. Table S1). The WT and Exon 3 deleted inserts were then transferred into the pCS2+ expression vector.
Immunocytochemistry
After two days of serum starvation, confluent fibroblast primary cultures or RPE1 cells were fixed in 4% paraformaldehyde, treated in 50 mM NH4Cl, 0.3% Triton X-100 followed by 1 h in BSA blocking solution. Cells were then incubated with mouse monoclonal acetylated a-tubulin (Sigma, clone 6-11B-1), rabbit polyclonal pericentrin (AbCam, ab4448), INPP5E (Proteintech, 17797-1-AP) and PDE6D (Sigma, HPA037434) antibodies for 1 h and with alexa Fluor 488 goat anti-rabbit IgG and alexa Fluor 555 donkey anti-mouse IgG (Invitrogen). To measure cilium length, confocal images were taken using Leica SP5. We performed three-dimensional reconstruction of cilia using Imaris software, which allowed for length assessment irrespective of angle of orientation. At least 100 control and patient cells were analyzed per experiment. Three independent experiments were performed.
Tandem affinity purification
Tandem affinity purifications and mass spectrometry were performed as previously described (Wright et al. 2011).
Recombinant proteins
GST-ARL2 TN, -ARL2 QL, -ARL3 TN, and -ARL3 QL were expressed and purified as previously described (Wright et al. 2011).
GST Pulldown assays
GST pulldown assays were performed as previously described (Wright et al. 2011).
Coimmunoprecipiation assays
RPE cells were transfected with equal amounts of LAP6-PDE6D constructs and pCS2+6xMyc-INPP5E constructs and harvested 48 hours later. Cells were lysed in 50 mM HEPES pH 7.6, 300 mM KCl, 1 mM EGTA, 1 mM MgCl2, 0.5 mM DTT, 0.3% NP-40, 10% glycerol, and 1X Roche Complete Protease Inhibitor cocktail. After pelleting cellular debris, anti-GFP antibodies conjugated to Protein A-Affiprep (BioRad, Hercules, CA) were added and washed 5 times with lysis buffer. Eluted proteins were separated by SDS-PAGE and analyzed by immunoblotting with anti-Myc (GeneTex) or anti-GFP (Abcam) HRP conjugated antibodies.
Protein blot analysis
Cell lysates were prepared from fibroblasts using ice-cold RIPA buffer plus proteases and phosphatases inhibitor cocktail (Roche). Immunoblotting was performed using INPP5E (Proteintech, 17797-1-AP, specifity previously tested (Humbert et al. 2012) followed by HRP-conjugated antibody and ECL-Plus detection (Amersham). An antibody against β-actin (Santa Cruz Biotechnology; sc-81178) was used as a loading control.
Bioinformatics
Genomic locations are according to the hg19 human genome assembly. The ciliary proteome was searched using web-based tools (Gherman et al. 2006; Inglis et al. 2006). Protein sequence conservation was determined using ClustalW multiple amino acid sequence alignment (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Predicted and known prenylated proteins are listed within PrenBase. PrePS stands for Prenylation Prediction Suite and combines three predictors for protein CaaX farnesylation, CaaX geranylgeranylation and Rab geranylgeranylation in one webinterface.
Zebrafish experiments
Zebrafish maintenance, embryo generation, and staging. Wild-type (WT) zebrafish of AB, TUAB, TL, and hybrid strains were maintained as previously described (Solnica-Krezel et al. 1994). Embryos were obtained from natural matings, kept at 28.5°C in E3 solution, and staged according to morphology and/or age, as indicated (Kimmel et al. 1995). RNA and morpholino injections. Capped sense RNA encoding PDE6D was in vitro synthesized (Ambion mMessage mMachine) and purified using G-50 Sephadex Quick Spin Columns (Roche) and suspended in DEPC water. Microinjections (0.5 ng) were performed on 1–4 cell embryos using a Nanoliter 2000 (World Precision Instruments, Inc.). An exon 2 splice donor-blocking morpholino oligonucleotide (MO), (Gene-Tools, LLC) with sequence 5′-TAATGCTGAGAACCACAAACCTTCA-3′, or a translation blocking MO 5′-TTCGTCCGAAGACATCTTTTTCCTT-3′ were resuspended in 0.1M KCl, 0.1% phenol red to a concentration of 0.25 mM and a volume of 4.6 nl was injected per embryo. A control random sequence morpholino did not cause a phenotype at similar injection concentration. RTPCR primers flanking exon 2 were 5′-CGGACGAAGACAGAGCGAAGGAG-3′ (forward) and 5′-GGAATCACAAAGCCAAACTCAAAA-3′ (reverse).
Rescue experiments
After co-injection of pde6d morpholino and PDE6D RNA, we quantified the edema phenotype and considered the embryos as rescued when they had no pericardial edema. The retinal phenotype was considered rescued when no apoptosis was observed in the eye and when the development of the retinal cell layers could clearly be discerned in histological sections. T test was performed to evaluate the difference between the experimental groups. *P < 0.05.
RESULTS
In a consanguineous family with three affected and 2 healthy sibs (Fig. 1A), the first female sib (JS-18a) presents with intrauterine growth retardation, facial dysmorphism, postaxial polydactyly of feet and syndactyly II–III, renal hypoplasia, microphthlamia and an extinguished electroretinogram. The diagnosis of JS was confirmed upon brain MRI (Fig. 1B, C). Her brother (JS-18b) has JS with polydactyly, microphthalmia and coloboma. The third sib (JS-18c) is a male fetus terminated at 14gw following findings of brain anomalies and polydactyly. Fetal examination showed additional severe retinal dysplasia (Fig. 1D). A microarray-based genome wide scan analysis identified a unique 17 Mb homozygous region on chromosome 2 defining a novel JS locus containing 208 genes, 15 of which are present in ciliary gene databases (Fig. 1E).
Figure 1. Homozygosity mapping combining exome sequencing identified a splice site mutation in PDE6D gene in a Joubert family.

(A) JS-18 consists of a consanguineous Joubert family from Pakistan (first cousin) with 3 affected sibs and 2 healthy children. Electropherogram of the c.140-1G>A PDE6D mutation. All three affected cases (JS-18a, b, and c) showed a homozygous change, whereas the parents and unaffected children were heterozygous carriers. (B) Axial view of brainstem abnormalities from first affected sib JS-18a with deep interpedoncular fossa and stretched cerebellar peduncles resulting in the molar tooth sign as compared to a control brain (C). (D) Section of the eye from third sib JS-18c showing severe retinal dysplasia. Both normal (top) and altered (down) retina area have been enlarged for comparison. (E) Homozygosity mapping was performed with Affymetrix 250k SNPs array and identified a unique region of homozygosity on chromosome 2, with a maximal lodscore of this family of 2.6 reached at this locus and thus defining a novel JBTS locus. Among the 208 genes present in this region, 15 are recorded in ciliary proteome database (www.ciliaproteome.org) comprising PDE6D (F) in which the c.140-1G>A mutation was identified by exome sequencing. Deleted exon 3 is colored in green. (G) RT-PCR amplification and sequencing of PDE6D mRNA from control and patient fibroblasts showing an in frame deletion of exon 3 in affected patients confirming a splicing defect. (H) Ribbon representation of PDE6D (PDB code 3T5G) using UCSF Chimera package (Pettersen et al. 2004) showing exon-3 encoded region in green containing Ile53 and Leu63 hydrophobic residues (red) constituting the nonpolar binding pocket of PDE6D with the 7 others shown in yellow.
We then performed whole exome sequencing and after using sequential filtering of the variations and mapping data only one truncating variation remained in a gene linked to ciliary function, namely PDE6D (Supp. Figure S1). PDE6D contains 5 exons and encodes a 150 amino acid protein (NM_002601.2, Fig. 1F). DNA sequencing confirmed the c.140-1G>A mutation present in all 3 affected sibs at the homozygous state and in parents at the heterozygous state (Fig. 1A). Sequencing of cDNA from JS-18b fibroblasts showed that the c.140-1G>A homozygous change leads to an in frame deletion of exon 3 (Fig. 1G), predicting a 108 amino acid truncated protein. The crystal structure of human PDE6D features an immunoglobulin-like β-sandwich fold composed of two β-sheets comprising four antiparallel strands. Exon 3 encodes amino acids A46 to E88 forming two entire antiparallel β-strands (β4 and β5) and part of β3 and β6. Furthermore, two of the nine hydrophobic residues constituting the nonpolar binding pocket (Ismail et al. 2011) are encoded by PDE6D exon 3, namely I53 and L63 (Fig.1H). These observations suggest that exon 3 deletion disrupts PDE6D hydrophobic pocket conformation and its subsequent binding to its prenylated interactors.
In order to evaluate the involvement of PDE6D in ciliopathies, we screened a cohort of 940 various ciliopathy conditions, including 782 patients with JS/Meckel (MKS; MIM# 249000), 20 with Senior-Løken (SLSN; MIM266900) and 80 with Leber congenital amaurosis (LCA; MIM# 204000), as well as 58 patients displaying kidney hypodysplasia comprising 17 with syndromic coloboma. No additional mutations were found, indicating that PDE6D mutations represent a rare cause of ciliopathy in human.
Unlike the mouse model, disruption of PDE6D in humans leads to multiorgan involvement. We therefore analyzed PDE6D expression during human embryogenesis and found a ubiquitous localization of PDE6D in accordance with the pleiotropic effect of human PDE6D mutation. Interestingly highest levels of PDE6D protein were observed in the central nervous system, kidney tubules and epithelial cells of the respiratory tract (Supp. Figure S2), organs generally affected in ciliopathies.
We then analyzed the subcellular localization of PDE6D in control and patient fibroblast primary cultures and observed that both wild type and truncated PDE6D are localized predominantly to the basal body of primary cilia (Fig. 2A), indicating correct targeting of the truncated protein. We also observed that both the number and gross morphology of primary cilia appear normal in JS-18b fibroblasts (n=100 cells analyzed in 3 independent experiments) and in kidney sections from JS-18c as compared to age-matched controls (Fig. 2A and 2B) indicating that PDE6D is not involved in ciliary biogenesis.
Figure 2. PDE6D mutation does not alter ciliogenesis and truncated PDE6D is properly localized to the base of primary cilia in patient fibroblast primary cultures and kidneys.

(A) Immunocytochemistry on patient and control fibroblast primary cultures using PDE6D and acetylated alpha tubulin, a ciliary axoneme marker (α-ac-Tubulin) antibodies showing that truncated PDE6D is translated and normally localized within patient fibroblast primary cilia (n=100 control and patient cells analyzed in 3 independent experiments). (B) Immunohistochemistry on PDE6D mutated fetal kidney as compared to aged-matched control using, antibodies raised against α-ac-Tubulin and pericentrin, a basal body marker. Primary cilia from patient kidney tubules appear normal in number and morphology.
To confirm the pathogenicity of PDE6D mutation we generated a knockdown model of PDE6D deficiency in zebrafish and used it to test the function of human PDE6D alleles as described previously (Solnica-Krezel et al. 1994; Kimmel et al. 1995). Compared to controls, pde6d morphant embryos consistently exhibited microphthalmia and pericardial edema (Fig. 3A, B and Supp. Figure S3). Closer examination of pde6d morphant embryos revealed kidney morphogenesis defects including distended, blocked pronephric openings (Fig. 3C) and proximal tubule cysts (Fig. 3D). Histological sections of the eyes revealed disorganized retinal cell layers at 3dpf (Fig. 3E, F). Co-injection of wild-type PDE6D mRNA with pde6d-morpholino significantly reduced the percent of embryos showing edema while the effect of mutated PDE6D mRNA was not significantly different from morpholino alone (Fig. 3H). Moreover, the co-injection also rescued eye development. PDE6D mutant RNA rescued the phenotypes at a lower efficiency suggesting that, with regard to microphthalmia, the mutation causes a partial loss of gene function. These data show that PDE6D function is critical for retinal and renal development in zebrafish in accordance with patient phenotype and that the in frame deletion of exon 3 constitutes a pathogenic mutation.
Figure 3. pde6d depletion in zebrafish.
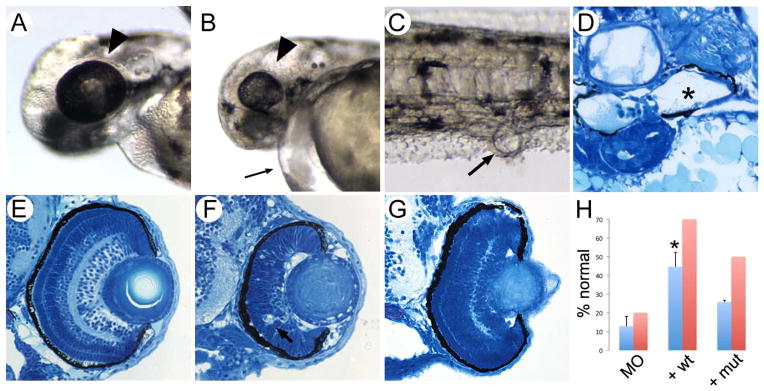
Eye size in control 3dpf embryo (A; arrowhead) compared to exon 2 pde6d morphant eyes at the same stage (B; arrowhead) revealed severe microphthalmia in the morphants. Exon 2 pde6d morphants also consistently exhibited pericardial edema (B; small arrow). (C) Cloacal cysts present in pde6d morphants were associated with distended pronephric tubules (D; asterisk marks distended lumenal space). (E, F) Histological sections of the wild-type 3 dpf eye showed retinal lamination while the exon 2 pde6d morphant eye showed absence of lamination and probable apoptosis (arrow in F). A translation blocking pde6d ATG morpholino produced similar results. (G) Co-injection of wild-type human PDE6D mRNA with morpholinos (+ wt) rescued retinal lamination and apoptosis phenotypes. (H) Quantification shows that co-injection of wild-type human PDE6D mRNA with morpholinos (+ wt) significantly increased the number of pde6d morphant embryos lacking pericardial edema (p < 0.02) (blue bars) as well as rescuing normal retinal lamination (red bars; n=10 embryos). Although the exon 3 deletion mutant human PDE6D mRNA (+ mut) also partially rescued the pde6d morphant phenotypes, the effect was not significant for cardiac edema and less efficient than wt for retinal lamination (n=8 embryos). pde6d mRNA injection by itself did not induce any phenotype in wild type embryos.
To identify novel PDE6D interactors that might shed light on the role of PDE6D in JS, we performed tandem affinity purifications followed by mass spectrometry from RPE cells stably expressing LAP-tagged PDE6D (Supp. Figure S4A). In addition to the well known interacting proteins RPGR (Linari et al. 1999b) and ARL2/3 (Linari et al. 1999a; Hanzal-Bayer et al. 2005), we identified INPP5E that has been also recently identified as a novel PDE6D interactor (Humbert et al. 2012) (Supp. Figure S4A, B). INPP5E is an inositol polyphosphate 5-phosphatase that is localized to primary cilia and predicted to be C-terminally farnesylated (http://mendel.imp.ac.at/PrePS/PRENbase/). Interestingly, mutations impairing its catalytic lipid phosphatase activity cause JS (Bielas et al. 2009). In addition, a homozygous nonsense mutation removing the last 18 residues, including the prenylation motif (CaaX-box), was found to cause another ciliopathy with a broader clinical spectrum, the MORM syndrome (Jacoby et al. 2009).
We next asked whether the interaction between INPP5E and PDE6D is mediated by prenylation, as would be predicted from earlier work on PDE6D (Zhang et al. 2004; Zhang et al. 2007; Ismail et al. 2011). To address this question we performed co-immunoprecipitation assays using a GFP-tagged PDE6D construct along with wild type (WT), MORM (c.1879C4T; p.Q627STOP) or cysteine to alanine CaaX-box mutant Myc-tagged INPP5E constructs in RPE1 cells. PDE6D efficiently co-immunoprecipitated WT-INPP5E but not the MORM-INPP5E or the cysteine to alanine CaaX-box mutant protein (Fig. 4A), indicating that the interaction is indeed mediated by farnesylation. This result suggests that INPP5E is likely a cargo of PDE6D. Consistent with this notion, co-expression of GFP-tagged PDE6D and mCherry-tagged INPP5E showed partial co-localization, with PDE6D localized to the transition zone and proximal end of the cilium while INPP5E localized uniformly along the length of the axoneme (Supp. Figure S5). This partial co-localization is reminiscent of the ciliary cargo/cargo adaptor pair, namely the myristoylated ciliary protein NPHP3 and the myristoyl-binding, PDE6D homolog UNC119B(Wright et al. 2011). However, this PDE6D localization differs slightly from our immunostaining of PDE6D in fibroblasts and kidney sections (Fig. 2A, 2B). We presume that, similar to UNC119B, PDE6D is only transiently present in the cilium where it subsequently releases its cargo (see below), and may explain why we fail to observe transition zone/ciliary staining of PDE6D in fibroblasts and kidney sections. Taken together, these data suggest a role for PDE6D similar to that of UNC119b in targeting farnesylated, rather than myristoylated, proteins to the cilium.
Figure 4. INPP5E ciliary targeting and binding to PDE6D are dependent upon farnesylation of INPP5E C-terminal CaaX-box.
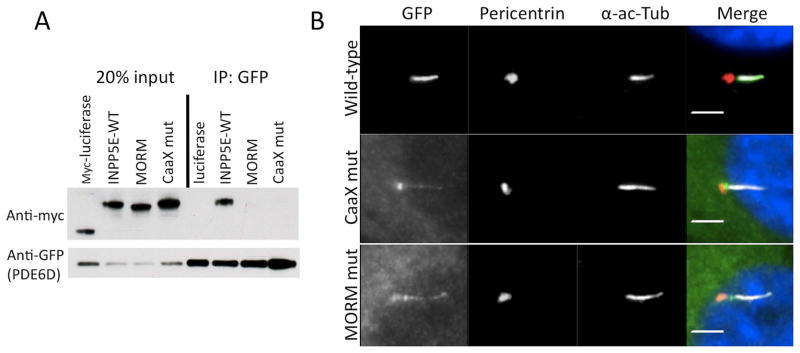
(A) Co-immunoprecipitations (Co-IP) were performed using Myc-tagged wild-type (WT), MORM, or cysteine to alanine CaaX-box mutant INPP5E and GFP-tagged PDE6D. PDE6D binds to WT INPP5E but not to MORM or CaaX-box mutant INPP5E indicating that PDE6D binds to INPP5E in a farnesyl-dependent manner. (B) RPE cells were transfected with the indicated GFP-tagged INPP5E constructs and stained with pericentrin and acetylated alpha tubulin antibodies to visualize basal bodies and cilia, respectively. These data show that both the Caax-box and MORM mutant constrcuts fail to normally accumulate whithin primary cilia axoneme as compared to the wild-type construct.
We next tested whether prenylation is required for proper ciliary localization of INPP5E. By overexpressing GFP-tagged constructs in RPE1 cells we found that in contrast to wild-type INPP5E, the MORM mutant INPP5E fails to properly accumulate in the cilium, and similarly, a cysteine to alanine mutation in the INPP5E CaaX-box also disrupted proper ciliary accumulation (Fig. 4B). Interestingly, while a small amount of both mutant proteins are able to enter the cilium, a significant amount appears to accumulate in the transition zone, which is thought to act as a diffusion barrier between the cilium and the cytoplasm (Fig. 4B). These data indicate that PDE6D binds to INPP5E in a farnesyl-dependent manner, and that loss of PDE6D-binding may contribute to the mislocalization of the MORM and CaaX-box mutant proteins.
We next tested whether deletion of exon 3 of PDE6D (Δexon3-PDE6D) would disrupt binding to INPP5E, as predicted by structural analysis. Indeed we found that Δexon3-PDE6D shows reduced binding to INPP5E by co-immunoprecipitation assays (Fig. 5A), indicating that exon 3- encoded residues are critical for the conformation of the PDE6D hydrophobic pocket. Having determined that both the PDE6D-INPP5E interaction and proper INPP5E ciliary localization requires prenylation, we then asked whether PDE6D is required for INPP5E ciliary localization. As shown in Supp. Figure S6, INPP5E is localized to ~80% of cilia in control RPE1 cells, while depletion of PDE6D with two independent siRNAs led to a virtually complete loss of ciliary INPP5E, suggesting that PDE6D is indispensible for proper INPP5E ciliary targeting. We then analyzed by immunofluoresence INPP5E localization in JS-18 patient fibroblasts and kidneys. Consistent with our results in RPE cells, INPP5E localizes along the length of the cilium in control fibroblasts (>90% of INPP5E positive cilia, n=100 in 3 independent experiments), but is absent from primary cilia in JS-18b patient fibroblasts (<1% INPP5E positive cilia, n=100 in 3 independent experiments) (Fig. 5B). Importantly, INPP5E protein is expressed in JS-18b fibroblasts as determined by immunoblotting (Supp. Figure S7). Consistently, immunohistochemistry analysis of JS-18c kidney sections shows an accumulation of INPP5E at the apical pole of epithelial tubule cells (Fig. 5C) indicating that in the absence of functional PDE6D, INPP5E is unable to enter the primary cilium. Taken together, these data indicate that PDE6D plays a role in trafficking INPP5E to the cilium, and that a defect in INPP5E trafficking may underlie the pathology of JS.
Figure 5. PDE6D truncation leads to INPP5E ciliary mislocalization in patient fibroblast cells and renal tissue.

(A) Co-IP were performed using GFP-tagged WT or the Δexon3-PDE6D and Myc-tagged WT INPP5E. Δexon3-PDE6D binding to INPP5E is impaired. (B) Immunocytochemistry on control and JS-18b patient fibroblasts using INPP5E and acetylated alpha tubulin (α-ac-Tubulin) antibodies showing mislocalization of INPP5E in patient fibroblasts where it is found diffuse within the cytoplasm whereas it is concentrated within the ciliary axoneme in control fibroblasts (n=100 control and patient cells analyzed in 3 independent experiments). (C) Immunohistochemistry on control and JS-18c kidney sections using INPP5E and α-ac-Tubulin antibodies showing accumulation of INPP5E at the apical pole of epithelial tubule cells from patient JS-18c and within the primary cilium of an age-matched control kidney.
The Arf-like small GTPases ARL2 (MIM# 601175), which is localized throughout the cytoplasm, and ARL3 (MIM# 604695), which is both cytoplasmic and ciliary, each bind to PDE6D in a GTP-dependent manner and serve to release farnesylated cargoes from PDE6D (Ismail et al. 2011). Biochemically, ARL2 interacts with β4 and β7 of PDE6D at one face and with β6 on the opposite face of the β-sandwich (Hanzal-Bayer et al. 2002; Ismail et al. 2011). As discussed above, exon-3 deletion leads to deletion of β4 and part of β6 suggesting that the interaction between PDE6D and ARL2 or ARL3 might be disrupted and thus lead to altered PDE6D cargo release. To test this hypothesis, we performed GST pulldown assays with GST-ARL2 or -ARL3 comparing WT versus Δexon3-PDE6D to test ARL2 and ARL3 binding capacities. As shown in Supp. Figure S8A, while the WT-PDE6D efficiently bound to ARL2 and ARL3 in a GTP-specific manner, the Δexon3-PDE6D failed to bind. We next tested the ability of ARL2 and ARL3 to release INPP5E from PDE6D and found that ARL3-binding, but not ARL2-binding, efficiently released INPP5E from PDE6D (Supp. Figure S8B). We next wondered whether ARL2, ARL3 and RP2, the ARL3-specific GAP, are necessary for INPP5E ciliary localization. As expected, depletion of ARL2 had no effect on INPP5E localization. However, to our surprise, depletion of ARL3 or RP2 had no effect on INPP5E localization (Supp. Figure S8C). This differs from our previous work on the myristoyl-binding PDE6D homolog UNC119B (MIM# 604011), which is required along with ARL3 and RP2 for proper targeting of myristoylated ciliary proteins (Wright et al. 2011). It is currently unclear why INPP5E ciliary targeting does not strictly require ARL3. One possibility is that we are unable to distinguish between properly unloaded, ciliary membrane-bound INPP5E and INPP5E that may be in complex with PDE6D.
DISCUSSION
In this study we identified PDE6D, encoding a prenyl-binding protein, as a novel JS disease-causing gene associated with eye defects, polydactyly and kidney hypoplasia. We showed that pde6d knockdown in zebrafish alters both eye and kidney development, phenotypes that are partially rescued by wild-type human PDE6D but not by JS truncated PDE6D, demonstrating both the highly conserved function of PDE6D and the pathogenicity of the mutant allele. Using an unbiased proteomic approach we identified INPP5E, which is also mutated in JS, as a ciliary farnesyl-dependent interactor of PDE6D, and show that PDE6D activity is required both in tissue culture cells as well as patient fibroblasts and kidney tubules. These results suggest that a defect in the targeting of a subset of prenylated proteins, including INPP5E, to the primary cilium underlies the clinical manifestation of PDE6D mutation.
While we observe multi-organ involvement in our JS patients, in accordance with PDE6D expression pattern during human development, the effects of Pde6d gene disruption in mice appears limited to the eye, likely due to its role in helping to localize a variety of prenylated proteins involved in phototransduction, including PDE6 subunits as well as the rhodopsin kinases GRK1 and GRK7, to the outer segments of retinal rods and cones (Zhang et al. 2004; Zhang et al. 2007), though a detailed analysis of kidney and brain might reveal subtle abnormalities. Mistargeting of these phototransduction components might explain the eye phenotype of PDE6D mutated patients but not limb, renal and cerebral anomalies also observed. We show here that PDE6D function is required for ciliary targeting of INPP5E, whose mutations also cause JS or MORM (Bielas et al. 2009; Jacoby et al. 2009). In zebrafish, pde6d depletion leads to a phenotype similar to the PDE6D mutated patients, including severe eye developmental anomalies and renal cyst formation. Interestingly, inpp5e morphant zebrafish were recently reported with a similiar phenotype (Luo et al. 2012) and Inpp5e−/− mice present bilateral anophthalmia, polydactyly, kidney cysts and cerebral developmental defects (Jacoby et al. 2009) consistent with the clinical spectrum of anomalies observed in PDE6D mutated patients. Our study also provides insight into the pathophysiological mechanism underlying MORM syndrome where patients bear a non-sense mutation leading to the loss of INPP5E CaaX-box with subsequent loss of PDE6D binding and likely loss of INPP5E targeting to the primary cilium.
As we were preparing our manuscript, (Humbert et al. 2012) published a study reporting a functional network for INPP5E ciliary targeting involving PDE6D, ARL13B and CEP164. While largely consistent with our results, we do find several notable differences. Whereas we and others (Jacoby et al. 2009) find that the MORM mutant INPP5E fails to traffic properly to the primary cilium, Humbert et al. report proper ciliary localization and conclude that C-terminal prenylation is not required for cililary targeting. We tested whether prenylation is required directly by making the cysteine to alanine mutation in the CaaX box and found that, similarly to the C-terminally truncated MORM mutant, prenylation is required for proper ciliary accumulation. While it is unclear why we see differences, one possibility is that different constructs were used leading to different expression levels of the transfected constructs. The sensitivity of the antibodies used may also be involved in this discrepancy. However, because we find that prenylation is required for proper ciliary accumulation, that PDE6D co-localizes with INPP5E in the proximal end of the primary cilium, and because we do not observe obvious Golgi, ER, or vesicular membrane accumulation of INPP5E upon PDE6D depletion in tissue culture cells or in our JS patient samples, we believe PDE6D is directly required for delivery of its farnesylated ciliary cargo and not indirectly required due to a proposed role in extracting farnesylated proteins from donor membranes as suggested by (Humbert et al. 2012).
Consistent with our results, Humbert and colleagues also found that despite binding to PDE6D and stimulating cargo release, ARL3 depletion seems not to perturb INPP5E ciliary localization. This is surprising given that ARL3 is an important cargo-release factor for UNC119B, which shares extensive sequence and functional homologies with PDE6D and that targets a subset of myristoylated proteins to the cilium (Wright et al. 2011). This observation could be due to our inability to distinguish between PDE6D-bound INPP5E and ciliary membrane-bound INPP5E, which would have been correctly unloaded from PDE6D, or reflect a difference between PDE6D and UNC119B in their dependency on a release factor for unloading cargo. Humbert and colleagues reported the involvement of another ARL family small GTPase, ARL13B in the INPP5E ciliary targeting network (Humbert et al. 2012). The authors suggest that ARL13B releases INPP5E from PDE6D by binding to INPP5E rather than PDE6D, which differs from the cargo release mechanism described biochemically and structurally used by ARL3 for both PDE6D and UNC119B (Ismail et al. 2011; Wright et al. 2011; Ismail et al. 2012). It will be of interest to further determine the role of ARL13B in PDE6D-dependent protein targeting.
Altogether, our results demonstrate a prenyl-dependent interaction between PDE6D and INPP5E and that this interaction is required for correct addressing of INPP5E to the primary cilia. While mislocalization of INPP5E seems sufficient to explain the PDE6D-associated phenotype in human, one cannot exclude that other ciliary prenylated PDE6D targets largely involved in primary cilia function such as RAB8A and RHOA (Nancy et al. 2002; Norton et al. 2005) might also be involved in the pathophysiological mechanism, as well as other ras-related GTP-binding proteins such as RAB28 we identified in this study. Overall, this study provides the first evidence of prenyl-binding dependent trafficking in the pathogenesis of two human ciliopathies namely Joubert and MORM and highlights a specific primary cilium membrane targeting mechanism for prenylated proteins mediated by PDE6D.
Supplementary Material
Acknowledgments
We thank the patients and their families for participation. We thank M. Nicouleau for technical help. This work, ST and EF were supported by grants from ANR 2010 FOETOCILPATH N° 1122 01 and Fondation IMAGINE, grants from the Fondation pour la Recherche Médicale (FRM DEQ20071210558 to S.S.), grants DK053093 and DK070263 to I.A.D and NS048453 to J.G.G. from the National Institutes of Health, grants from the European Research Council (ERC Starting Grant 260888) and the Italian Ministry of Health (Ricerca Corrente 2012 and Ricerca Finalizzata Malattie Rare 2008) to EMV.
Footnotes
CONFLICT OF INTEREST STATEMENT. None declared.
References
- Bielas SL, Silhavy JL, Brancati F, Kisseleva MV, Al-Gazali L, Sztriha L, Bayoumi RA, Zaki MS, Abdel-Aleem A, Rosti RO, Kayserili H, Swistun D, et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet. 2009;41:1032–1036. doi: 10.1038/ng.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, Salem S, Radovanovic I, Grant AV, Adimi P, Mansouri N, Okada S, et al. Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science. 2012;337:1684–1688. doi: 10.1126/science.1224026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolze A, Byun M, McDonald D, Morgan NV, Abhyankar A, Premkumar L, Puel A, Bacon CM, Rieux-Laucat F, Pang K, Britland A, Abel L, et al. Whole-exome-sequencing-based discovery of human FADD deficiency. Am J Hum Genet. 2010;87:873–881. doi: 10.1016/j.ajhg.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun M, Abhyankar A, Lelarge V, Plancoulaine S, Palanduz A, Telhan L, Boisson B, Picard C, Dewell S, Zhao C, Jouanguy E, Feske S, et al. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J Exp Med. 2010;207:2307–2312. doi: 10.1084/jem.20101597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherman A, Davis EE, Katsanis N. The ciliary proteome database: an integrated community resource for the genetic and functional dissection of cilia. Nat Genet. 2006;38:961–962. doi: 10.1038/ng0906-961. [DOI] [PubMed] [Google Scholar]
- Hanzal-Bayer M, Linari M, Wittinghofer A. Properties of the interaction of Arf-like protein 2 with PDEdelta. J Mol Biol. 2005;350:1074–1082. doi: 10.1016/j.jmb.2005.05.036. [DOI] [PubMed] [Google Scholar]
- Hanzal-Bayer M, Renault L, Roversi P, Wittinghofer A, Hillig RC. The complex of Arl2-GTP and PDE delta: from structure to function. EMBO J. 2002;21:2095–2106. doi: 10.1093/emboj/21.9.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert MC, Weihbrecht K, Searby CC, Li Y, Pope RM, Sheffield VC, Seo S. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc Natl Acad Sci U S A. 2012;109:19691–19696. doi: 10.1073/pnas.1210916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglis PN, Boroevich KA, Leroux MR. Piecing together a ciliome. Trends Genet TIG. 2006;22:491–500. doi: 10.1016/j.tig.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Ismail SA, Chen Y-X, Miertzschke M, Vetter IR, Koerner C, Wittinghofer A. Structural basis for Arl3-specific release of myristoylated ciliary cargo from UNC119. EMBO J. 2012;31:4085–4094. doi: 10.1038/emboj.2012.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail SA, Chen Y-X, Rusinova A, Chandra A, Bierbaum M, Gremer L, Triola G, Waldmann H, Bastiaens PIH, Wittinghofer A. Arl2-GTP and Arl3-GTP regulate a GDI-like transport system for farnesylated cargo. Nat Chem Biol. 2011;7:942–949. doi: 10.1038/nchembio.686. [DOI] [PubMed] [Google Scholar]
- Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, Pernot E, Kisseleva MV, Compère P, Schiffmann SN, Gergely F, Riley JH, et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet. 2009;41:1027–1031. doi: 10.1038/ng.427. [DOI] [PubMed] [Google Scholar]
- Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19:813–825. doi: 10.1212/wnl.19.9.813. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn Off Publ Am Assoc Anat. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma Oxf Engl. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinforma Oxf Engl. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Baehr W. Expression and characterization of human PDEdelta and its Caenorhabditis elegans ortholog CEdelta. FEBS Lett. 1998;440:454–457. doi: 10.1016/s0014-5793(98)01501-4. [DOI] [PubMed] [Google Scholar]
- Li N, Florio SK, Pettenati MJ, Rao PN, Beavo JA, Baehr W. Characterization of human and mouse rod cGMP phosphodiesterase delta subunit (PDE6D) and chromosomal localization of the human gene. Genomics. 1998;49:76–82. doi: 10.1006/geno.1998.5210. [DOI] [PubMed] [Google Scholar]
- Linari M, Hanzal-Bayer M, Becker J. The delta subunit of rod specific cyclic GMP phosphodiesterase, PDE delta, interacts with the Arf-like protein Arl3 in a GTP specific manner. FEBS Lett. 1999a;458:55–59. doi: 10.1016/s0014-5793(99)01117-5. [DOI] [PubMed] [Google Scholar]
- Linari M, Ueffing M, Manson F, Wright A, Meitinger T, Becker J. The retinitis pigmentosa GTPase regulator, RPGR, interacts with the delta subunit of rod cyclic GMP phosphodiesterase. Proc Natl Acad Sci U S A. 1999b;96:1315–1320. doi: 10.1073/pnas.96.4.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Okada S, Kong X-F, Kreins AY, Cypowyj S, Abhyankar A, Toubiana J, Itan Y, Audry M, Nitschke P, Masson C, Toth B, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. 2011;208:1635–1648. doi: 10.1084/jem.20110958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo N, Lu J, Sun Y. Evidence of a role of inositol polyphosphate 5-phosphatase INPP5E in cilia formation in zebrafish. Vision Res. 2012;75:98–107. doi: 10.1016/j.visres.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CJ. Protein prenylation: a mediator of protein-protein interactions. Science. 1993;259:1865–1866. doi: 10.1126/science.8456312. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nancy V, Callebaut I, El Marjou A, de Gunzburg J. The delta subunit of retinal rod cGMP phosphodiesterase regulates the membrane association of Ras and Rap GTPases. J Biol Chem. 2002;277:15076–15084. doi: 10.1074/jbc.M109983200. [DOI] [PubMed] [Google Scholar]
- Norton AW, Hosier S, Terew JM, Li N, Dhingra A, Vardi N, Baehr W, Cote RH. Evaluation of the 17-kDa prenyl-binding protein as a regulatory protein for phototransduction in retinal photoreceptors. J Biol Chem. 2005;280:1248–1256. doi: 10.1074/jbc.M410475200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Solnica-Krezel L, Schier AF, Driever W. Efficient recovery of ENU-induced mutations from the zebrafish germline. Genetics. 1994;136:1401–1420. doi: 10.1093/genetics/136.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright KJ, Baye LM, Olivier-Mason A, Mukhopadhyay S, Sang L, Kwong M, Wang W, Pretorius PR, Sheffield VC, Sengupta P, Slusarski DC, Jackson PK. An ARL3-UNC119-RP2 GTPase cycle targets myristoylated NPHP3 to the primary cilium. Genes Dev. 2011;25:2347–2360. doi: 10.1101/gad.173443.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li S, Doan T, Rieke F, Detwiler PB, Frederick JM, Baehr W. Deletion of PrBP/delta impedes transport of GRK1 and PDE6 catalytic subunits to photoreceptor outer segments. Proc Natl Acad Sci U S A. 2007;104:8857–8862. doi: 10.1073/pnas.0701681104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liu X, Zhang K, Chen C-K, Frederick JM, Prestwich GD, Baehr W. Photoreceptor cGMP phosphodiesterase delta subunit (PDEdelta) functions as a prenyl-binding protein. J Biol Chem. 2004;279:407–413. doi: 10.1074/jbc.M306559200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.