

Abstract
An unresolved problem in biological signal transduction is how particular branches of highly interconnected signaling networks can be decoupled, allowing activation of specific circuits within complex signaling architectures. Although signaling dynamics and spatiotemporal mechanisms serve critical roles, it remains unclear if these are the only ways cells achieve specificity within networks. The transcription factor Steroidogenic Factor-1 (SF-1) is an excellent model to address this question, as it forms dynamic complexes with several chemically distinct lipid species (phosphatidylinositols, phosphatidylcholines and sphingolipids). This property is important since lipids bound to SF-1 are modified by lipid signaling enzymes (IPMK & PTEN), regulating SF-1 biological activity in gene expression. Thus, a particular SF-1/lipid complex can interface with a lipid signaling enzyme only if SF-1 has been loaded with a chemically compatible lipid substrate. This mechanism permits dynamic downstream responsiveness to constant upstream input, disentangling specific pathways from the full network. The potential of this paradigm to apply generally to nuclear lipid signaling is discussed, with particular attention given to the nuclear receptor superfamily of transcription factors and their phospholipid ligands.
Signaling Networks are highly interconnected
A major discovery in biological signaling over the past two decades has been that intracellular signal transduction occurs through highly integrated networks, rather than through isolated, linear signaling pathways (Fig 1) (Dutkowski et al., 2013; Guruharsha et al., 2012; Kapp et al., 2012; van Wageningen et al., 2010; Weng et al., 1999). These networks permit signaling crosstalk and other interactions that give rise to emergent network properties which potentially deliver far greater richness to biological signaling than had been realized (Bhalla and Iyengar, 1999; Helikar et al., 2008). Specifically, networks provide the cell with the capacity to store information within the network, outside the genome (Bhalla and Iyengar, 1999; Helikar et al., 2008), cluster standardized signaling output responses in the presence of high levels of background noise (Bhalla and Iyengar, 1999; Fritsche-Guenther et al., 2011; Helikar et al., 2008; Levy and Siegal, 2012), provide robustness to fate decisions and responsiveness to environmental change (Ku et al., 2012; Levy and Siegal, 2012), among other advantages (Bhalla et al., 2002). Given that signaling network crosstalk has been described in organisms from E. coli (Antiqueira et al., 2012) through metazoans (Natarajan et al., 2006) and humans (Ku et al., 2012), it seems clear that evolution has chosen complex, highly integrated networks as the preferred mode of intracellular information transfer (Kulkarni, 2013).
Fig. 1.

Biological networks are highly interconnected. Network interaction maps for the human proteins A) Inositol polyphosphate multikinase (IPMK), B) SF-1, and C. both IPMK and SF-1. Network connections represent Co-expression (Purple), Co-localization (Blue), Genetic interactions (Green), Shared Pathway (Cyan), Physical Interaction (Red), Predicted Interaction (Orange), or Shared Protein Domains (Moss). Maps generated using GeneMANIA (http://genemania.org, University of Toronto).
However, networks are not without at least one significant problem, which is also a great strength – the ability to cluster multiple signals into a few uniform outputs (Ku et al., 2012; Levy and Siegal, 2012). This property gives rise to the idea that “everything regulates everything else” and fuels the question of what mechanisms cells have evolved to selectively activate particular signaling molecules and pathways (Kiel and Serrano, 2012b), without inevitably having broad pleiotropic effects on the entire signaling architecture (Granek et al., 2011; Guruharsha et al., 2012; Kiel and Serrano, 2012a).
Known mechanisms of network disentanglement
There are several solutions to this problem that have already been characterized, and the field of signaling dynamics has realized some of the most elegant and effective answers to this basic problem. Altering the frequency or amplitude of signaling events can disentangle signaling networks in many ways (Ganesan and Zhang, 2012), however an excellent illustration is the Crz1 transcription factor (Cai et al., 2008). When the amplitude of Crz1 activation (the concentration of active Crz1) is steadily increased, Crz1 target promoters are non-proportionally activated according to the affinity of Crz1 for each promoter. However, when the frequency of Crz1 activation is increased instead of the amplitude, all Crz1 target promoters are activated proportionally, regardless of the affinity of Crz1 for each promoter (Cai et al., 2008). In other words, target promoter activation is non-proportional across Crz1 amplitudes, but proportional across Crz1 frequencies. In this way downstream effectors can be differentially regulated while still integrated into the network, simply by altering the amplitude or frequency of signaling (Purvis and Lahav, 2013). This highlights how dynamics can use an intrinsic network property to impart specificity and control within a complex system.
There are also less abstract, spatiotemporal mechanisms that confer specificity to signaling networks (Scott and Pawson, 2009; Steinacher and Soyer, 2012). These mechanisms generally rely on organizing signaling molecules in time and space to gain specificity by putting the right enzymes and substrates together at the right time. This can be accomplished through controlling enzyme/substrate concentration by post-translational modification, expression or degradation (Buchler and Cross, 2009; Rocks et al., 2005; Yu et al., 2011), sub-cellular localization and compartmentalization (Cohen and Cohen, 1989; Grecco et al., 2011; Kurosaki, 2002; Rocks et al., 2005), regulating the formation of multi-subunit complexes (Good et al., 2011; von Kriegsheim et al., 2009), and signal channeling through properly oriented and positioned signaling enzymes (Good et al., 2009; Rajakulendran et al., 2009; Whitmarsh et al., 1998). There are many examples of these well-characterized mechanisms throughout biology, which have been very well reviewed (Good et al., 2011; Kholodenko et al., 2010; Scott and Pawson, 2009).
Here, I attempt to present a new way cells can activate specific branches of complex networks that does not rely on time, space or signaling dynamics. This mechanism uses the chemical specificity encoded within lipid headgroups to interface between signaling enzymes and protein effectors. It is not exclusive of the above-mentioned strategies, and often may synergize with other mechanisms to enhance their effectiveness. The first example of this type of regulation centers on the phospholipid-regulated transcription factor Steroidogenic Factor-1.
Steroidogenic Factor-1 (SF-1) as a model system
Steroidogenic Factor-1 (SF-1, NR5A1) is a member of the nuclear receptor superfamily of ligand-regulated transcription factors, and is expressed in humans almost exclusively in tissues involved in steroid production (Morohashi et al., 1995). SF-1 binds specific DNA sequences in chromatin and recruits other transcriptional co-regulators to the promoters of genes (Lund et al., 2002). This action activates SF-1 target genes, which mainly encode steroidogenic enzymes (Omura and Morohashi, 1995). Many nuclear receptors (NRs) bind hydrophobic ligands that regulate their gene expression activity (Issemann and Green, 1990), and NRs are often regulated by several chemical variations on these hydrophobic molecules (Burris et al., 2013), making them attractive targets for pharmaceutical intervention.
SF-1 binds many different lipid species
Crystallographic structure analysis of SF-1 from several labs (Krylova et al., 2005; Li et al., 2005; Ortlund et al., 2005) unexpectedly demonstrated electron density attributed to a co-purifying bacterial phospholipid in the ligand-binding pocket of SF-1 (Fig. 2A) (Krylova et al., 2005). These studies also showed that SF-1 binds more physiologically relevant metazoan lipids in vitro, including important signaling lipids such as sphingosines, phosphatidylcholines and phosphatidylinositols (Dammer et al., 2007; Krylova et al., 2005; Li et al., 2005; Urs et al., 2007; Urs et al., 2006). The bacterial phospholipid structures were supported by an additional crystal structure of SF-1 bound to phosphatidylcholine (Fig. 2B) (Sablin et al., 2009), one of the most abundant phospholipids in eukaryotic nuclei (Hunt, 2006). Designed mutations in SF-1 that perturb phospholipid binding (Sablin et al., 2009; Sablin et al., 2003) also decrease SF-1 activation of every target gene that has been examined (Blind et al., 2012; Sablin et al., 2009), indicating that lipid binding is important for SF-1 biological activity. All current PDB crystal structures of SF-1 bound to various phospholipids indicate a similar mode of stoichiometric 1:1 binding, in which the hydrophobic acyl chains of the phospholipid are buried deep in the ligand-binding pocket of SF-1, while the hydrophilic headgroups are exposed to solvent (Fig 2C and 2D). The physiological reason phospholipids activate SF-1, a master regulator of steroid metabolism, remains unaddressed in the literature, although a strong connection exists between steroidogenesis and sphingolipid metabolism (Lucki and Sewer, 2008). In any case, what is clear is that SF-1 binds several chemically distinct lipid species whose headgroups are structurally exposed to solvent (Fig 2C), and SF-1 needs these lipids for full biological activity.
Fig. 2.

SF-1 binds many chemically distinct phospholipid species. A) Crystal structure of mouse SF-1 (Blue), Cofactor peptide (Cyan) and bacterial phosphatidylglycerol (sticks) (PDB: 1YMT). B) Crystal structure of mouse SF-1 bound to eukaryotic phosphatidylcholine (PC), same coloring scheme as A (PDB: 3F7D). C) Superposition of A (Yellow), B (Blue) and mouse SF-1 bound to bacterial phosphatidylethanolamine (Green, PDB: 1YP0). D) Cutaway of C, demonstrating tight superposition of lipids and exposure of lipid headgroups to solvent. E) Several of the lipids known to bind SF-1, demonstrating different headgroup chemistry.
PIP2 in SF-1 is phosphorylated by IPMK, but not by p110 PI3Ks
The structures of SF-1 bound to phospholipids indicate that the headgroups are solvent accessible (Fig 2). Since PIP2 and PIP3 are important membrane signaling lipids that have failed to yield crystal structures complexed to SF-1, we simulated how these lipids might bind SF-1 (Blind et al., 2012; Sablin et al., 2009). As expected, these simulations revealed the inositol headgroup to be solvent exposed (Fig 3), and led us to hypothesize that PIP2 bound by SF-1 might be accessible to lipid signaling enzymes. The inositol polyphosphate multi-kinase (IPMK) demonstrates this activity, and the kinetics of IPMK activity on SF-1/PIP2 differ greatly from PIP2 in membrane systems (a 6 fold increases in KCAT/KM, (Blind et al., 2012)), showing IPMK favors phosphorylation of SF-1/PIP2 in vitro (Blind et al., 2012). Chemically or genetically impeding IPMK in cells decreases SF-1 gene activation, and is dependent on SF-1 binding to PIPs. The lipid phosphatase PTEN de-phosphorylates PIP3 bound to SF-1, and oppositely regulates SF-1 gene expression in cells compared to IPMK (Blind et al., 2012). Importantly, PI3-kinases of the p110 class (PI3Kα, PI3Kγ and PI3Kδ) fail to phosphorylate PIP2 bound to SF-1, suggesting that IPMK somehow has unique access to PIP2 held by SF-1, as simulated in figure 4 (Fig 4) using the structures of yeast IPMK (IPK2) and the simulated mouse SF-1 bound to PIP2. These data demonstrate that lipids bound to a non-membrane protein can be modified by lipid signaling enzymes, regulating the biological activity of the protein. Importantly, lipid substrate properties are altered when bound by non-membrane proteins, as p110 PI3-kinases are inactive on SF-1/PIP2 (Blind et al., 2012). The structural elements specific to IPMK that allow phosphorylation of SF-1/PIP2, and/or those of the p110-class of PI3-kinases that prevent activity, await further investigation.
Fig. 3.
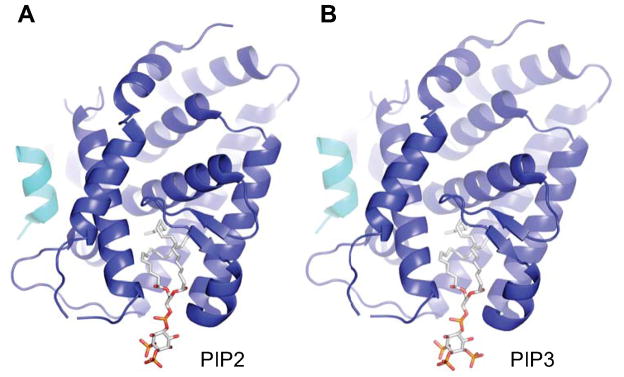
SF-1/PIP2 and SF-1/PIP3 simulations suggest a solvent accessible headgroup. A) Simulation of mouse SF-1 bound by PIP2 and B) PIP3, based on mouse SF-1/PC structure (3F7D), predicting high solvent accessibility of lipid headgroups, suggesting they may be available to lipid signaling kinases. Coloring scheme same as Fig. 2.
Fig. 4.

Simulation of mouse SF-1/PIP2 interaction with IPMK bound with ATP. Simulation of how mouse SF-1 bound to PIP2 (Blue, simulation from Figure 3A) may interact with yeast IPMK (IPK2, Red PDB: 2IF8), showing bound PIP2 and ATP as sticks, and IPMK-bound magnesium ion (Purple).
Nuclear Lipid Signaling and SF-1
The physicochemical arrangement of SF-1 as a non-membrane nuclear protein/lipid complex provides a model system that may begin to explain a large body of literature that has documented several interesting biochemical and cell-biological phenomena. The nucleus of eukaryotic cells seems to contain a significant fraction of lipids that do not localize to any known bilayer structure (Follo et al., 2012; Hunt, 2006; Irvine, 2003; Maraldi et al., 1994). These lipids are not only visualized within the nucleoplasm (Barlow et al., 2010; Boronenkov et al., 1998; Hammond et al., 2009) but are also metabolically distinct from membrane lipids (Follo et al., 2013; Keune et al., 2013; Lindsay et al., 2006; Rose and Frenster, 1965; Vann et al., 1997). Further, many lipid signaling and metabolic enzymes shuttle into the nucleus (Bassi et al., 2013; Boronenkov et al., 1998; Schramp et al., 2012), however it is unknown if these enzymes perform the same functions within the nucleoplasm as they perform at cytoplasmic membranes. The simplest explanation of these data is that non-membrane nuclear lipids are bound by soluble nuclear proteins, and that this association alters which enzymes can modify/metabolize them (Barlow et al., 2010; Lindsay et al., 2006). That SF-1 binds phospholipids, and is a soluble, non-membrane protein acted on by lipid signaling enzymes (Blind et al., 2012), begins to shed light on these phenomena. It also provides a model system to further interrogate the signaling functions of nuclear lipids, and to ask what evolutionary advantage signaling enzyme activity on protein/lipid complexes might confer to networks and systems (Poyatos, 2012).
Each SF-1 phospholipid presents unique chemistry to signaling enzymes
Although the kinase activity of IPMK on PIP2 held by SF-1 is new in itself, it raises the interesting question of what advantage phosphorylating a lipid bound to a protein, rather than the protein itself, grants the cell. The new phosphate incorporated into PIP3 may well have evolved to be incorporated into a structurally analogous serine or threonine residue, so why phosphorylate a small molecule associated with a protein rather than the protein itself? The answer may lie with the inherent ability of SF-1 to accommodate several different phospholipid species (Sablin et al., 2009), each presenting a unique chemical interface for signaling enzymes on the SF-1 protein (Blind et al., 2012). An amino acid encoded by the genome would be present in every SF-1 molecule made by the cell (save alternative splice variations). However, by dynamically altering which lipid occupies SF-1, the cell gains the potential to chemically couple and/or decouple SF-1 from particular signaling enzymes, independent of space, time or signaling dynamics.
Phospholipids can chemically decouple SF-1 from particular signaling enzymes
To illustrate this mechanism, take the examples of PIP2 and PC (Fig 2E) bound to SF-1 (Fig 5A). It is well established that both these chemically distinct phospholipids bind SF-1 (and the close SF-1 homolog LRH-1) to regulate gene expression programs (Blind et al., 2012; Lee et al., 2011; Musille et al., 2013; Musille et al., 2012; Sablin et al., 2009; Sablin et al., 2003). However, kinases like IPMK require a free hydroxyl group to catalyze phosphorylation. Since PC lacks this hydroxyl, it consequently lacks the headgroup chemistry necessary to allow phosphorylation by IPMK (Fig 2E). SF-1 occupancy by PC must therefore decouple SF-1 from direct IPMK signaling, allowing SF-1 activities to occur independent of IPMK kinase activation (Fig 5A). Similarly, PTEN catalyzes de-phosphorylation of phosphorylated inositol lipid species in (Maehama and Dixon, 1999), so when SF-1 is occupied by PC, PTEN signaling is decoupled from SF-1 activity (Fig 5B), enabling the SF-1 circuit to operate independently of PTEN-sensitive branches of the full signaling network. It should also be noted that since PI3-kinases of the p110-class do not act on SF-1/PIP2, SF-1 also represents a signaling molecule that is coupled to PTEN signaling, but inherently de-coupled from p110 PI3-kinase signaling (Blind et al., 2012).
Fig. 5.

Chemically distinct phospholipids can decouple SF-1 from signaling enzymes. A) SF-1 occupied by PIP2 is phosphorylated by IPMK, coupling SF-1 to IPMK signaling. SF-1 occupied by PC cannot be phosphorylated by IPMK, as PC lacks the required headgroup chemistry, de-coupling SF-1 from IPMK signaling. B) SF-1 occupied by PIP3 is de-phosphorylated by PTEN, coupling SF-1 to PTEN signaling, while PTEN has no effect on PC bound to SF-1, decoupling SF-1 from PTEN signaling.
This dynamic responsiveness to signaling based on SF-1 lipid occupancy may not be limited to phospholipid-class lipids. Although only IPMK and PTEN have been demonstrated to act directly on SF-1/lipid complexes, simulations of SF-1 binding other lipids suggest they may act through similar mechanisms (Fig. 6). The sphingolipid signaling enzyme acid ceramidase catalyzes hydrolysis of the unique N-acyl bond in ceramide to generate sphingosine and a free fatty acid (Fig 6A) (Lucki and Sewer, 2012). While only small amounts of ceramide have been found associated with SF-1 in the cell lines that have been examined (Urs et al., 2007), relatively high amounts of sphingosine have been found bound to SF-1, which inhibits SF-1 gene expression programs in cells (Fig 2E) (Urs et al., 2006). Acid ceramidase physically interacts with SF-1 and co-localizes to the nucleus in living cells (Lucki et al., 2012). Since lipids of the phospholipid-class chemically lack the N-acyl bond ceramidases hydrolyze (Fig 2E) (Bhabak et al., 2012), ceramidases cannot act on phospholipids such as PIP2, PIP3 or PC. Phospholipids may therefore decouple SF-1 from direct sphingolipid signaling pathways (Lucki et al., 2012; Urs et al., 2006), enabling the SF-1 circuit to operate independently of that branch of the full network. Diacylglycerol kinase theta (DGKθ) is another lipid signaling enzyme known to generate phosphatidic acid from diacylglycerol (DAG) (Raben and Tu-Sekine, 2008). DGKθ interacts with SF-1 (Li et al., 2007) and DGKθ activity stimulates SF-1 gene expression programs in living cells (Cai and Sewer, 2013). Again, since IPMK requires an inositol headgroup to catalyze phosphorylation (Blind et al., 2012; Resnick et al., 2005), when DAG occupies SF-1, SF-1 must be decoupled from direct IPMK signaling.
Fig. 6.

Chemically distinct lipids have the potential to decouple other effectors. A) SF-1 occupied by ceramide may be sensitive to ceramidase signaling, but SF-1 occupied by PIP2 will be insensitive, decoupling SF-1 from ceramide signaling. B) LRH-1 occupied by PIP2 may be sensitive to IPMK/PTEN signaling, but LRH-1 occupied by PC will be insensitive. C) PPAR occupied by DAG may be sensitive to DAG kinase signaling, PPAR occupied by other lipids would not.
Are other nuclear receptors regulated by the same mechanism?
Some other NRs that may be regulated similarly as SF-1 are Liver Receptor Homolog-1 (LRH-1) (Lee et al., 2011; Musille et al., 2013; Ortlund et al., 2005), and the Peroxisome Proliferator Activated Receptor (PPAR) members of the nuclear receptor superfamily (Wahli and Michalik, 2012). LRH-1 is known to bind PIP2 and PIP3 in vitro (Krylova et al., 2005), making IPMK and PTEN activity possible in cells. LRH-1 and IPMK are both highly expressed in the human liver (Chang et al., 2002), and both reside in the nucleus (Resnick et al., 2005), however it remains to be determined if IPMK and/or PTEN directly regulate LRH-1 (Fig 6B).
The PPARs (alpha, beta/delta and gamma) are also nuclear receptors that are well established to bind lipids and be regulated by them (Baker et al., 2005; Issemann and Green, 1990; Wahli and Michalik, 2012). It is also clear PPARs are regulated by many lipid signaling enzymes in living cells (Huang et al., 2011; Pandey et al., 2008; Tsukahara et al., 2010), however the mechanism of that regulation has not been addressed. Two simple mechanisms exist for regulation of PPARs, one of which is commonly assumed to occur - that PPARs dynamically exchange different lipids with membrane systems, not unlike phosphoinositide transfer proteins. But another non-exclusive mechanism is that lipids bound to PPARs are modified by lipid signaling enzymes while bound to the PPAR protein. For example, phosphatidic acid is known to bind and activate PPARα in cells, and DAG kinase ζ, an enzyme that phosphorylates DAG in membrane systems to generate phosphatidic acid, is also known to activate PPARs (Huang et al., 2011). Thus, DAG kinase ζ may be directly phosphorylating DAG held by PPARs to generate phosphatidic acid within PPAR (Fig 5C). A second example may be Phospholipase D2 (PLD2), a lipid signaling enzyme that generates cyclic phosphatidic acid (cPA) (Tsukahara et al., 2010) from phosphatidic acid in membrane systems, which is known to inhibit PPARγ gene expression (Tsukahara et al., 2010). Could PLD2 be acting directly on PPAR/PA complexes to generate PPAR/cPA? Third, linoleic acid binds PPARα, and is generated by cytosolic Phospholipase A2 (cPLA2) in membrane systems. cPLA2 is known to activate PPAR alpha in cells (Pandey et al., 2008). Should these lipid signaling enzymes prove to act directly on PPAR/lipid complexes, these chemically distinct lipids provide a new mechanism that can connect or de-connect these important transcription factors to lipid signaling pathways, providing specificity to signaling within the network.
How are signaling lipids loaded into non-membrane proteins?
Although lipids regulate the biological activity of the above mentioned non-membrane proteins, there is almost no data describing how these proteins acquire phospholipids from cellular membrane systems. Among many hypotheses, three appear plausible as mechanisms describing how this may happen: direct, entropy-driven phospholipid transfer from membrane systems to the non-membrane proteins, like phosphoinositide transfer proteins (PITPs) (Schaaf et al., 2008); phospholipid-transfer protein mediated exchange between non-membrane proteins (Lev, 2012); and/or co-translational loading of non-membrane proteins at the endoplasmic reticulum.
It is clear that at least some NRs such as SF-1 and LRH-1 are capable of non-enzymatic, entropy-driven phospholipid exchange directly with membrane systems in vitro (Blind et al., 2012; Krylova et al., 2005; Sablin et al., 2009), not unlike the phospholipid transfer activities of PITPs (Schaaf et al., 2008). Indeed, the recombinant phospholipid-binding domain of SF-1 could be classified as a phospholipid transfer protein based on its ability to exchange phospholipids between membrane systems in vitro (Blind et al., 2012; Cockcroft and Carvou, 2007; Krylova et al., 2005). Phosphoinositide transfer protein (PITP) mediates lipid transfer between membrane compartments (Cockcroft, 2012; Phillips et al., 2006), yet whether or not these same proteins facilitate loading of lipids into non-membrane proteins, or between non-membrane proteins, remains unknown. The Sec14 family of phospholipid transfer proteins is the best characterized family of transfer proteins, with a crystal structure of the human homolog of yeast Sec14 (Sfh1) informing biochemical experiments showing that Sec14 can act as a lipid nanoreactor to facilitate the interaction of particular membrane phospholipids with lipid kinases and phosphatases (Bankaitis et al., 2012; Schaaf et al., 2008). It is important to clarify that whether or not proteins of the nuclear receptor superfamily are capable of dynamic phospholipid exchange with membrane systems in living cells remains an open and very challenging mechanistic question, although it is often simply assumed to occur.
Another way non-membrane lipid binding proteins could acquire lipids is through co-translationally sensing the lipid content at the endoplasmic reticulum (ER, Fig 7). Signal sequences direct mRNA messages that encode secretory and transmembrane proteins to the rough endoplasmic reticulum through the signal recognition particle (SRP). Messages encoding cytoplasmic or nuclear proteins were classically thought to be translated on free ribosomes in the cytosol, independent of SRP and membranes. However, several studies have used unbiased approaches to demonstrate that the subset of actively translating ribosomal/RNA complexes associated with membranes does not require the presence of a classic signal sequence (Diehn et al., 2000; Lerner et al., 2003; Reid and Nicchitta, 2012). In fact, many members of the nuclear receptor superfamily have been identified as being translated on membrane-bound ribosomes (Diehn et al., 2000), along with many other proteins. One hypothesis explaining this phenomenon is that membrane-translated proteins are co-translationally sensing the ER lipid content by acquiring lipids from the ER as they fold in close proximity to this membrane system. Dissecting how lipids are loaded into non-membrane proteins represents a challenging problem, as lipid exchange is a process that does not chemically alter the substrate, and therefore is difficult to quantify, particularly in living cells (Cockcroft, 1997).
Fig. 7.

Models of lipid loading into non-membrane proteins, like SF-1. Steps labeled with “?” are not known to occur, all other steps are well established to occur. Classically, 1A) only signal-containing nascent peptides are translated on rough ER, however 1B) many nuclear proteins are translated on membrane-bound ribosomes, where 2) folding nuclear proteins may sense the ER lipid content while attaining mature structure 3) then carry that information into the nucleus 4). SF-1 is able to exchange lipids with membrane systems in vitro 5) this may happen in cells, although no data exist. Equilibrium concentration fluctuations in membrane lipids 6) may allow varying sensitivity to signaling enzymes such as IPMK and PTEN. Phosphoinositide transfer proteins 7) mediate lipid movement between membrane systems, but it is not known if they aid in loading lipids into non-membrane proteins, aiding in equilibration 8).
Structural genomics and the future of non-membrane lipid signaling
It was structural analyses that first linked phospholipids to SF-1 (Blind et al., 2012; Krylova et al., 2005; Sablin et al., 2009), and which drove the hypothesis that lipid signaling enzymes might directly act on non-membrane protein/lipid complexes. Other intracellular small signaling molecules have been unexpectedly identified by crystallography in a similar manner, such as Inositol hexakisphosphate (Ins(1,2,3,4,5,6)P6) discovered in the catalytic core of the ADAR2 RNA editing enzyme (Macbeth et al., 2005) and inositol tetrakisphosphate (Ins(1,4,5,6)P4) discovered in the interface between HDAC3 and NCOR2 (Watson et al., 2012). Star-PAP is a nuclear poly-A polymerase that binds PIP2, and whose activity is regulated by PIPK1α to selectively regulate mRNAs (Li et al., 2012; Mellman et al., 2008), however no structural data is available describing the position of the lipid headgroup or its solvent accessibility. Whether or not these complexes are directly coupled to signaling enzymes remains to be determined, but the fortuitous discovery of these small signaling molecules by crystallography highlights the potential of structural genomics to uncover new biology (Telesco and Radhakrishnan, 2012). By applying high-throughput techniques to structure determination we undoubtedly will discover more complexes of small signaling molecules and proteins (Telesco and Radhakrishnan, 2012).
The potential of nuclear protein/lipid complexes to disentangle signaling networks reveals how biology can evolve innovative approaches to take advantage of complex systems like networks, while still maintaining tight regulatory control of individual molecules. The combination of structural genomics to discover new protein/lipid complexes, classic enzymology to demonstrate enzyme activity on these complex substrates, with innovative nuclear & cell biological analyses will provide more insight into signal transduction, nuclear biology and ultimately the design of nature (Poyatos, 2012).
Acknowledgments
The author would like to thank Holly A. Ingraham for her encouragement and support, along with Vytas Bankaitis, John York and Lucio Cocco for all their help and fantastic science.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antiqueira L, Janga SC, da Costa LF. Extensive cross-talk and global regulators identified from an analysis of the integrated transcriptional and signaling network in Escherichia coli. Molecular bioSystems. 2012;8:3028–3035. doi: 10.1039/c2mb25279a. [DOI] [PubMed] [Google Scholar]
- Baker PR, Lin Y, Schopfer FJ, Woodcock SR, Groeger AL, Batthyany C, Sweeney S, Long MH, Iles KE, Baker LM, et al. Fatty acid transduction of nitric oxide signaling: multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. The Journal of biological chemistry. 2005;280:42464–42475. doi: 10.1074/jbc.M504212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankaitis VA, Ile KE, Nile AH, Ren J, Ghosh R, Schaaf G. Thoughts on Sec14-like nanoreactors and phosphoinositide signaling. Advances in biological regulation. 2012;52:115–121. doi: 10.1016/j.jbior.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow CA, Laishram RS, Anderson RA. Nuclear phosphoinositides: a signaling enigma wrapped in a compartmental conundrum. Trends in cell biology. 2010;20:25–35. doi: 10.1016/j.tcb.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassi C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, Mak TW, Neel BG, Raught B, Stambolic V. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science. 2013;341:395–399. doi: 10.1126/science.1236188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhabak KP, Proksch D, Redmer S, Arenz C. Novel fluorescent ceramide derivatives for probing ceramidase substrate specificity. Bioorganic & medicinal chemistry. 2012;20:6154–6161. doi: 10.1016/j.bmc.2012.08.035. [DOI] [PubMed] [Google Scholar]
- Bhalla US, Iyengar R. Emergent properties of networks of biological signaling pathways. Science. 1999;283:381–387. doi: 10.1126/science.283.5400.381. [DOI] [PubMed] [Google Scholar]
- Bhalla US, Ram PT, Iyengar R. MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science. 2002;297:1018–1023. doi: 10.1126/science.1068873. [DOI] [PubMed] [Google Scholar]
- Blind RD, Suzawa M, Ingraham HA. Direct modification and activation of a nuclear receptor-PIP(2) complex by the inositol lipid kinase IPMK. Science signaling. 2012;5:ra44. doi: 10.1126/scisignal.2003111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boronenkov IV, Loijens JC, Umeda M, Anderson RA. Phosphoinositide signaling pathways in nuclei are associated with nuclear speckles containing pre-mRNA processing factors. Molecular biology of the cell. 1998;9:3547–3560. doi: 10.1091/mbc.9.12.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchler NE, Cross FR. Protein sequestration generates a flexible ultrasensitive response in a genetic network. Molecular systems biology. 2009;5:272. doi: 10.1038/msb.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris TP, Solt LA, Wang Y, Crumbley C, Banerjee S, Griffett K, Lundasen T, Hughes T, Kojetin DJ. Nuclear receptors and their selective pharmacologic modulators. Pharmacological reviews. 2013;65:710–778. doi: 10.1124/pr.112.006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai K, Sewer MB. cAMP-stimulated transcription of DGKtheta requires steroidogenic factor 1 and sterol regulatory element binding protein 1. Journal of lipid research. 2013;54:2121–2132. doi: 10.1194/jlr.M035634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Dalal CK, Elowitz MB. Frequency-modulated nuclear localization bursts coordinate gene regulation. Nature. 2008;455:485–490. doi: 10.1038/nature07292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SC, Miller AL, Feng Y, Wente SR, Majerus PW. The human homolog of the rat inositol phosphate multikinase is an inositol 1,3,4,6-tetrakisphosphate 5-kinase. The Journal of biological chemistry. 2002;277:43836–43843. doi: 10.1074/jbc.M206134200. [DOI] [PubMed] [Google Scholar]
- Cockcroft S. Phosphatidylinositol transfer proteins: requirements in phospholipase C signaling and in regulated exocytosis. FEBS letters. 1997;410:44–48. doi: 10.1016/s0014-5793(97)00414-6. [DOI] [PubMed] [Google Scholar]
- Cockcroft S. The diverse functions of phosphatidylinositol transfer proteins. Current topics in microbiology and immunology. 2012;362:185–208. doi: 10.1007/978-94-007-5025-8_9. [DOI] [PubMed] [Google Scholar]
- Cockcroft S, Carvou N. Biochemical and biological functions of class I phosphatidylinositol transfer proteins. Biochimica et biophysica acta. 2007;1771:677–691. doi: 10.1016/j.bbalip.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Cohen P, Cohen PT. Protein phosphatases come of age. The Journal of biological chemistry. 1989;264:21435–21438. [PubMed] [Google Scholar]
- Dammer EB, Leon A, Sewer MB. Coregulator exchange and sphingosine-sensitive cooperativity of steroidogenic factor-1, general control nonderepressed 5, p54, and p160 coactivators regulate cyclic adenosine 3′,5′-monophosphate-dependent cytochrome P450c17 transcription rate. Molecular endocrinology. 2007;21:415–438. doi: 10.1210/me.2006-0361. [DOI] [PubMed] [Google Scholar]
- Diehn M, Eisen MB, Botstein D, Brown PO. Large-scale identification of secreted and membrane-associated gene products using DNA microarrays. Nature genetics. 2000;25:58–62. doi: 10.1038/75603. [DOI] [PubMed] [Google Scholar]
- Dutkowski J, Kramer M, Surma MA, Balakrishnan R, Cherry JM, Krogan NJ, Ideker T. A gene ontology inferred from molecular networks. Nature biotechnology. 2013;31:38–45. doi: 10.1038/nbt.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follo MY, Faenza I, Fiume R, Ramazzotti G, McCubrey JA, Martelli AM, Manzoli FA, Cocco L. Revisiting nuclear phospholipase C signalling in MDS. Advances in biological regulation. 2012;52:2–6. doi: 10.1016/j.advenzreg.2011.09.018. [DOI] [PubMed] [Google Scholar]
- Follo MY, Marmiroli S, Faenza I, Fiume R, Ramazzotti G, Martelli AM, Gobbi P, McCubrey JA, Finelli C, Manzoli FA, et al. Nuclear phospholipase C beta1 signaling, epigenetics and treatments in MDS. Advances in biological regulation. 2013;53:2–7. doi: 10.1016/j.jbior.2012.09.009. [DOI] [PubMed] [Google Scholar]
- Fritsche-Guenther R, Witzel F, Sieber A, Herr R, Schmidt N, Braun S, Brummer T, Sers C, Bluthgen N. Strong negative feedback from Erk to Raf confers robustness to MAPK signalling. Molecular systems biology. 2011;7:489. doi: 10.1038/msb.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan A, Zhang J. How cells process information: quantification of spatiotemporal signaling dynamics. Protein science: a publication of the Protein Society. 2012;21:918–928. doi: 10.1002/pro.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good M, Tang G, Singleton J, Remenyi A, Lim WA. The Ste5 scaffold directs mating signaling by catalytically unlocking the Fus3 MAP kinase for activation. Cell. 2009;136:1085–1097. doi: 10.1016/j.cell.2009.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good MC, Zalatan JG, Lim WA. Scaffold proteins: hubs for controlling the flow of cellular information. Science. 2011;332:680–686. doi: 10.1126/science.1198701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granek JA, Kayikci O, Magwene PM. Pleiotropic signaling pathways orchestrate yeast development. Current opinion in microbiology. 2011;14:676–681. doi: 10.1016/j.mib.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grecco HE, Schmick M, Bastiaens PI. Signaling from the living plasma membrane. Cell. 2011;144:897–909. doi: 10.1016/j.cell.2011.01.029. [DOI] [PubMed] [Google Scholar]
- Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nature reviews. Genetics. 2012;13:654–666. doi: 10.1038/nrg3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond GR, Schiavo G, Irvine RF. Immunocytochemical techniques reveal multiple, distinct cellular pools of PtdIns4P and PtdIns(4,5)P(2) The Biochemical journal. 2009;422:23–35. doi: 10.1042/BJ20090428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helikar T, Konvalina J, Heidel J, Rogers JA. Emergent decision-making in biological signal transduction networks. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:1913–1918. doi: 10.1073/pnas.0705088105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Zhang H, Shao Z, O’Hara KA, Kopilas MA, Yu L, Netticadan T, Anderson HD. Suppression of endothelin-1-induced cardiac myocyte hypertrophy by PPAR agonists: role of diacylglycerol kinase zeta. Cardiovascular research. 2011;90:267–275. doi: 10.1093/cvr/cvq401. [DOI] [PubMed] [Google Scholar]
- Hunt AN. Dynamic lipidomics of the nucleus. Journal of cellular biochemistry. 2006;97:244–251. doi: 10.1002/jcb.20691. [DOI] [PubMed] [Google Scholar]
- Irvine RF. Nuclear lipid signalling. Nature reviews. Molecular cell biology. 2003;4:349–360. doi: 10.1038/nrm1100. [DOI] [PubMed] [Google Scholar]
- Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- Kapp GT, Liu S, Stein A, Wong DT, Remenyi A, Yeh BJ, Fraser JS, Taunton J, Lim WA, Kortemme T. Control of protein signaling using a computationally designed GTPase/GEF orthogonal pair. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:5277–5282. doi: 10.1073/pnas.1114487109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keune WJ, Jones DR, Divecha N. PtdIns5P and Pin1 in oxidative stress signaling. Advances in biological regulation. 2013;53:179–189. doi: 10.1016/j.jbior.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Kholodenko BN, Hancock JF, Kolch W. Signalling ballet in space and time. Nature reviews. Molecular cell biology. 2010;11:414–426. doi: 10.1038/nrm2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel C, Serrano L. Challenges ahead in signal transduction: MAPK as an example. Current opinion in biotechnology. 2012a;23:305–314. doi: 10.1016/j.copbio.2011.10.004. [DOI] [PubMed] [Google Scholar]
- Kiel C, Serrano L. Structural data in synthetic biology approaches for studying general design principles of cellular signaling networks. Structure. 2012b;20:1806–1813. doi: 10.1016/j.str.2012.10.002. [DOI] [PubMed] [Google Scholar]
- Krylova IN, Sablin EP, Moore J, Xu RX, Waitt GM, MacKay JA, Juzumiene D, Bynum JM, Madauss K, Montana V, et al. Structural analyses reveal phosphatidyl inositols as ligands for the NR5 orphan receptors SF-1 and LRH-1. Cell. 2005;120:343–355. doi: 10.1016/j.cell.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Ku CJ, Wang Y, Weiner OD, Altschuler SJ, Wu LF. Network crosstalk dynamically changes during neutrophil polarization. Cell. 2012;149:1073–1083. doi: 10.1016/j.cell.2012.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni MM, Perrimon N. Handbook of Systems Biology Concepts and Insights. Elsevier; 2013. Analyzing the structure, function and information flow in singaling networks using quantitative cellular signatures; pp. 89–113. [Google Scholar]
- Kurosaki T. Regulation of B-cell signal transduction by adaptor proteins. Nature reviews. Immunology. 2002;2:354–363. doi: 10.1038/nri801. [DOI] [PubMed] [Google Scholar]
- Lee JM, Lee YK, Mamrosh JL, Busby SA, Griffin PR, Pathak MC, Ortlund EA, Moore DD. A nuclear-receptor-dependent phosphatidylcholine pathway with antidiabetic effects. Nature. 2011;474:506–510. doi: 10.1038/nature10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner RS, Seiser RM, Zheng T, Lager PJ, Reedy MC, Keene JD, Nicchitta CV. Partitioning and translation of mRNAs encoding soluble proteins on membrane-bound ribosomes. RNA. 2003;9:1123–1137. doi: 10.1261/rna.5610403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev S. Nonvesicular lipid transfer from the endoplasmic reticulum. Cold Spring Harbor perspectives in biology. 2012:4. doi: 10.1101/cshperspect.a013300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy SF, Siegal ML. The robustness continuum. Advances in experimental medicine and biology. 2012;751:431–452. doi: 10.1007/978-1-4614-3567-9_20. [DOI] [PubMed] [Google Scholar]
- Li D, Urs AN, Allegood J, Leon A, Merrill AH, Jr, Sewer MB. Cyclic AMP-stimulated interaction between steroidogenic factor 1 and diacylglycerol kinase theta facilitates induction of CYP17. Molecular and cellular biology. 2007;27:6669–6685. doi: 10.1128/MCB.00355-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Laishram RS, Ji Z, Barlow CA, Tian B, Anderson RA. Star-PAP control of BIK expression and apoptosis is regulated by nuclear PIPKIalpha and PKCdelta signaling. Molecular cell. 2012;45:25–37. doi: 10.1016/j.molcel.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Choi M, Cavey G, Daugherty J, Suino K, Kovach A, Bingham NC, Kliewer SA, Xu HE. Crystallographic identification and functional characterization of phospholipids as ligands for the orphan nuclear receptor steroidogenic factor-1. Molecular cell. 2005;17:491–502. doi: 10.1016/j.molcel.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Lindsay Y, McCoull D, Davidson L, Leslie NR, Fairservice A, Gray A, Lucocq J, Downes CP. Localization of agonist-sensitive PtdIns(3,4,5)P3 reveals a nuclear pool that is insensitive to PTEN expression. Journal of cell science. 2006;119:5160–5168. doi: 10.1242/jcs.000133. [DOI] [PubMed] [Google Scholar]
- Lucki NC, Li D, Bandyopadhyay S, Wang E, Merrill AH, Sewer MB. Acid ceramidase (ASAH1) represses steroidogenic factor 1-dependent gene transcription in H295R human adrenocortical cells by binding to the receptor. Molecular and cellular biology. 2012;32:4419–4431. doi: 10.1128/MCB.00378-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucki NC, Sewer MB. Multiple roles for sphingolipids in steroid hormone biosynthesis. Sub-cellular biochemistry. 2008;49:387–412. doi: 10.1007/978-1-4020-8831-5_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucki NC, Sewer MB. Nuclear sphingolipid metabolism. Annual review of physiology. 2012;74:131–151. doi: 10.1146/annurev-physiol-020911-153321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund J, Borud B, Mellgren G, Aesoy R, Hoang T, Jacob AL, Bakke M. Differential regulation of SF-1-cofactor interactions. Endocrine research. 2002;28:505–513. doi: 10.1081/erc-120016830. [DOI] [PubMed] [Google Scholar]
- Macbeth MR, Schubert HL, Vandemark AP, Lingam AT, Hill CP, Bass BL. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science. 2005;309:1534–1539. doi: 10.1126/science.1113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends in cell biology. 1999;9:125–128. doi: 10.1016/s0962-8924(99)01519-6. [DOI] [PubMed] [Google Scholar]
- Maraldi NM, Cocco L, Capitani S, Mazzotti G, Barnabei O, Manzoli FA. Lipid-dependent nuclear signalling: morphological and functional features. Advances in enzyme regulation. 1994;34:129–143. doi: 10.1016/0065-2571(94)90013-2. [DOI] [PubMed] [Google Scholar]
- Mellman DL, Gonzales ML, Song C, Barlow CA, Wang P, Kendziorski C, Anderson RA. A PtdIns4,5P2-regulated nuclear poly(A) polymerase controls expression of select mRNAs. Nature. 2008;451:1013–1017. doi: 10.1038/nature06666. [DOI] [PubMed] [Google Scholar]
- Morohashi K, Hatano O, Nomura M, Takayama K, Hara M, Yoshii H, Takakusu A, Omura T. Function and distribution of a steroidogenic cell-specific transcription factor, Ad4BP. The Journal of steroid biochemistry and molecular biology. 1995;53:81–88. doi: 10.1016/0960-0760(95)00041-w. [DOI] [PubMed] [Google Scholar]
- Musille PM, Kohn JA, Ortlund EA. Phospholipid--driven gene regulation. FEBS letters. 2013;587:1238–1246. doi: 10.1016/j.febslet.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musille PM, Pathak MC, Lauer JL, Hudson WH, Griffin PR, Ortlund EA. Antidiabetic phospholipid-nuclear receptor complex reveals the mechanism for phospholipid-driven gene regulation. Nature structural & molecular biology. 2012;19:532–537. S531–532. doi: 10.1038/nsmb.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan M, Lin KM, Hsueh RC, Sternweis PC, Ranganathan R. A global analysis of cross-talk in a mammalian cellular signalling network. Nature cell biology. 2006;8:571–580. doi: 10.1038/ncb1418. [DOI] [PubMed] [Google Scholar]
- Omura T, Morohashi K. Gene regulation of steroidogenesis. The Journal of steroid biochemistry and molecular biology. 1995;53:19–25. doi: 10.1016/0960-0760(95)00036-y. [DOI] [PubMed] [Google Scholar]
- Ortlund EA, Lee Y, Solomon IH, Hager JM, Safi R, Choi Y, Guan Z, Tripathy A, Raetz CR, McDonnell DP, et al. Modulation of human nuclear receptor LRH-1 activity by phospholipids and SHP. Nature structural & molecular biology. 2005;12:357–363. doi: 10.1038/nsmb910. [DOI] [PubMed] [Google Scholar]
- Pandey NR, Renwick J, Misquith A, Sokoll K, Sparks DL. Linoleic acid-enriched phospholipids act through peroxisome proliferator-activated receptors alpha to stimulate hepatic apolipoprotein A-I secretion. Biochemistry. 2008;47:1579–1587. doi: 10.1021/bi702148f. [DOI] [PubMed] [Google Scholar]
- Phillips SE, Vincent P, Rizzieri KE, Schaaf G, Bankaitis VA, Gaucher EA. The diverse biological functions of phosphatidylinositol transfer proteins in eukaryotes. Critical reviews in biochemistry and molecular biology. 2006;41:21–49. doi: 10.1080/10409230500519573. [DOI] [PubMed] [Google Scholar]
- Poyatos JF. On the search for design principles in biological systems. Advances in experimental medicine and biology. 2012;751:183–193. doi: 10.1007/978-1-4614-3567-9_9. [DOI] [PubMed] [Google Scholar]
- Purvis JE, Lahav G. Encoding and decoding cellular information through signaling dynamics. Cell. 2013;152:945–956. doi: 10.1016/j.cell.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben DM, Tu-Sekine B. Nuclear diacylglycerol kinases: regulation and roles. Frontiers in bioscience: a journal and virtual library. 2008;13:590–597. doi: 10.2741/2704. [DOI] [PubMed] [Google Scholar]
- Rajakulendran T, Sahmi M, Lefrancois M, Sicheri F, Therrien M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature. 2009;461:542–545. doi: 10.1038/nature08314. [DOI] [PubMed] [Google Scholar]
- Reid DW, Nicchitta CV. Primary role for endoplasmic reticulum-bound ribosomes in cellular translation identified by ribosome profiling. The Journal of biological chemistry. 2012;287:5518–5527. doi: 10.1074/jbc.M111.312280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick AC, Snowman AM, Kang BN, Hurt KJ, Snyder SH, Saiardi A. Inositol polyphosphate multikinase is a nuclear PI3-kinase with transcriptional regulatory activity. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:12783–12788. doi: 10.1073/pnas.0506184102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocks O, Peyker A, Kahms M, Verveer PJ, Koerner C, Lumbierres M, Kuhlmann J, Waldmann H, Wittinghofer A, Bastiaens PI. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science. 2005;307:1746–1752. doi: 10.1126/science.1105654. [DOI] [PubMed] [Google Scholar]
- Rose HG, Frenster JH. Composition and metabolism of lipids within repressed and active chromatin of interphase lymphocytes. Biochimica et biophysica acta. 1965;106:577–591. doi: 10.1016/0005-2760(65)90073-1. [DOI] [PubMed] [Google Scholar]
- Sablin EP, Blind RD, Krylova IN, Ingraham JG, Cai F, Williams JD, Fletterick RJ, Ingraham HA. Structure of SF-1 bound by different phospholipids: evidence for regulatory ligands. Molecular endocrinology. 2009;23:25–34. doi: 10.1210/me.2007-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sablin EP, Krylova IN, Fletterick RJ, Ingraham HA. Structural basis for ligand-independent activation of the orphan nuclear receptor LRH-1. Molecular cell. 2003;11:1575–1585. doi: 10.1016/s1097-2765(03)00236-3. [DOI] [PubMed] [Google Scholar]
- Schaaf G, Ortlund EA, Tyeryar KR, Mousley CJ, Ile KE, Garrett TA, Ren J, Woolls MJ, Raetz CR, Redinbo MR, et al. Functional anatomy of phospholipid binding and regulation of phosphoinositide homeostasis by proteins of the sec14 superfamily. Molecular cell. 2008;29:191–206. doi: 10.1016/j.molcel.2007.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramp M, Hedman A, Li W, Tan X, Anderson R. PIP Kinases from the Cell Membrane to the Nucleus. Sub-cellular biochemistry. 2012;58:25–59. doi: 10.1007/978-94-007-3012-0_2. [DOI] [PubMed] [Google Scholar]
- Scott JD, Pawson T. Cell signaling in space and time: where proteins come together and when they’re apart. Science. 2009;326:1220–1224. doi: 10.1126/science.1175668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinacher A, Soyer OS. Evolutionary principles underlying structure and response dynamics of cellular networks. Advances in experimental medicine and biology. 2012;751:225–247. doi: 10.1007/978-1-4614-3567-9_11. [DOI] [PubMed] [Google Scholar]
- Telesco SE, Radhakrishnan R. Structural systems biology and multiscale signaling models. Annals of biomedical engineering. 2012;40:2295–2306. doi: 10.1007/s10439-012-0576-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukahara T, Tsukahara R, Fujiwara Y, Yue J, Cheng Y, Guo H, Bolen A, Zhang C, Balazs L, Re F, et al. Phospholipase D2-dependent inhibition of the nuclear hormone receptor PPARgamma by cyclic phosphatidic acid. Molecular cell. 2010;39:421–432. doi: 10.1016/j.molcel.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs AN, Dammer E, Kelly S, Wang E, Merrill AH, Jr, Sewer MB. Steroidogenic factor-1 is a sphingolipid binding protein. Molecular and cellular endocrinology. 2007;265–266:174–178. doi: 10.1016/j.mce.2006.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs AN, Dammer E, Sewer MB. Sphingosine regulates the transcription of CYP17 by binding to steroidogenic factor-1. Endocrinology. 2006;147:5249–5258. doi: 10.1210/en.2006-0355. [DOI] [PubMed] [Google Scholar]
- van Wageningen S, Kemmeren P, Lijnzaad P, Margaritis T, Benschop JJ, de Castro IJ, van Leenen D, Groot Koerkamp MJ, Ko CW, Miles AJ, et al. Functional overlap and regulatory links shape genetic interactions between signaling pathways. Cell. 2010;143:991–1004. doi: 10.1016/j.cell.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vann LR, Wooding FB, Irvine RF, Divecha N. Metabolism and possible compartmentalization of inositol lipids in isolated rat-liver nuclei. The Biochemical journal. 1997;327(Pt 2):569–576. doi: 10.1042/bj3270569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kriegsheim A, Baiocchi D, Birtwistle M, Sumpton D, Bienvenut W, Morrice N, Yamada K, Lamond A, Kalna G, Orton R, et al. Cell fate decisions are specified by the dynamic ERK interactome. Nature cell biology. 2009;11:1458–1464. doi: 10.1038/ncb1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends in endocrinology and metabolism: TEM. 2012;23:351–363. doi: 10.1016/j.tem.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Watson PJ, Fairall L, Santos GM, Schwabe JW. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature. 2012;481:335–340. doi: 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng G, Bhalla US, Iyengar R. Complexity in biological signaling systems. Science. 1999;284:92–96. doi: 10.1126/science.284.5411.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science. 1998;281:1671–1674. doi: 10.1126/science.281.5383.1671. [DOI] [PubMed] [Google Scholar]
- Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J, Kubica N, Hoffman GR, Cantley LC, Gygi SP, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332:1322–1326. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]