

Abstract
Intraventricular hemorrhage (IVH) of the preterm neonate is a complex developmental disorder, with contributions from both the environment and the genome. IVH, or hemorrhage into the germinal matrix of the developing brain with secondary periventricular infarction, occurs in that critical period of time before the 32nd – 33rd week post-conception and has been attributed to changes in cerebral blood flow to the immature germinal matrix microvasculature. Emerging data suggest that genes subserving coagulation, inflammatory and vascular pathways, and their interactions with environmental triggers may influence both the incidence and severity of cerebral injury and are the subject of this review.
Polymorphisms in the Factor V Leiden gene are associated with the atypical timing of IVH suggesting an as yet unknown environmental trigger. The methylenetetra-hydrofolate reeducates (MTHFR) variants render neonates more vulnerable to cerebral injury in the presence of perinatal hypoxia. The present study demonstrates that the MTHFR 677C>T polymorphism and low 5 minute Apgar score additively increase the risk of IVH. Finally, review of published preclinical data suggests the stressors of delivery result in hemorrhage in the presence of mutations in collagen 4A1 (COL4A1), a major structural protein of the developing cerebral vasculature. Maternal genetics and fetal environment may also play a role.
INTRODUCTION
Converging data suggest that intraventricular hemorrhage (IVH) of the preterm neonate is a complex developmental disorder, with contributions from both the environment and the genome of the child. IVH, or hemorrhage into the germinal matrix of the developing brain with secondary periventricular infarction as shown in Figure 1, occurs in that critical period of time before the 32nd – 33rd week post-conception and has been attributed to changes in cerebral blood flow to the immature germinal matrix microvasculature. Inflammation, coagulation and vascular factors may also play a role. The more severe grades are characterized by acute distension of the cerebral ventricular system with blood (Grade 3) and intraventricular hemorrhage with parenchymal venous infarction (Grade 4) (1). Mortality is high in infants with severe IVH, and one-quarter to one-half of surviving neonates develop cognitive disability and/or cerebral palsy (2,3). In addition, 20% of nondisabled survivors suffer executive function and neuropsychiatric disorders, confirming that severe IVH is a major pediatric public health problem (4,5).
Figure 1.
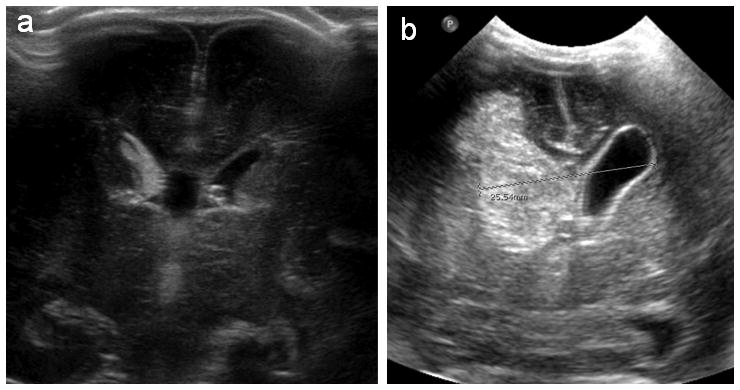
Severe intraventricular hemorrhage. Coronal ultrasounds at postnatal ages 1 and 4 days (Panels A and B, respectively) from a 28 week gestation neonate with IVH. In panel A, blood is seen in the germinal matrix and filling the right lateral ventricle; at postnatal day 4, the ventricular system is dilated, and blood is seen both filling and distending the right lateral ventricle as well as in the parenchyma of the right hemisphere, consistent with Grade 4 IVH.
Multiple lines of clinical data support the hypothesis that, similar to other preterm morbidities (6,7), the etiology of IVH is multifactorial. First, despite the development of sophisticated neonatal intensive care strategies, IVH remains a significant problem of prematurity. Maternal transport, antenatal steroid administration (ANS) and improved resuscitation techniques have become standard of care in neonatal tertiary care units worldwide (8–11), but the incidence of severe IVH has remained 13–15% for almost 20 years (8,12).
Although the incidence of IVH is inversely related to gestational age (GA) at birth, the risk period for hemorrhage is independent of GA (13, 14). The incidence of severe IVH is 7% for those born at 28 weeks and 26% for neonates born 4 weeks earlier, but the critical period for hemorrhage is the first 4 – 5 days of life for both groups. These data suggest that either the transition to extra-uterine life and/or the triggers to which the neonates are exposed contribute to hemorrhage, and both hypoxemia and inflammation have been implicated in severe IVH of the prematurely-born.
Furthermore, both gender and twin studies support the hypothesis that IVH is a complex disorder. Preterm males are more likely than females to experience severe IVH (15). Similarly, studying 450 twin pairs, Bhandari reported that 41.3% of the variance in IVH risk is attributable to familial and environmental factors (16). Candidate gene studies implicate the inflammatory, coagulation and vascular pathways, and recent data suggest that time of hemorrhage may play a role (17–19).
The purpose of this review is to examine preclinical and clinical data supporting the hypothesis that severe IVH is attributable in part to the interaction of the environment with the neonatal genome. Based on the pathogenesis of hemorrhage, factors mediating coagulation, inflammation and vascular pathways have been chosen for review.
Models for interaction between the environment and the genome
Although several investigators have hypothesized that IVH is secondary to the interaction of the environment and the genome (20), the mechanisms by which genetic predisposition and environmental exposures interact are just beginning to be described. Review of published literature interrogating vascular and environmental interactions suggest at least two different mechanisms. These include the impact of an environmental perturbation on a system harboring a known polymorphism, while the other stems from research addressing the influence of fetal programming on adult disorders postulating the role of epigenetics. Underlying both proposed mechanisms is the recognition that IVH is developmental disorder occurring within a critical period of time, and both the polymorphisms and environment events we describe may result in very different or even unremarkable phenotypes in the term infant or older child.
In the first model, a gene confers vulnerability to environmental triggers (21). A common example is hypercarbia. Hypercarbia occurs in association with apneic events, lung disease, pneumothoraces, pulmonary hemorrhage, and other events. Preterm neonates exhibit a narrow range of carbon dioxide over which cerebral blood flow (CBF) remains constant. In response to hypercarbia, CBF to the immature germinal matrix microvasculature markedly increases and, in the presence of a vascular structural polymorphism, may result in hemorrhage (22). Although the same genetic variant that results in germinal matrix vascular instability may predispose the proband to subsequent neurovascular disorders, there is no reported trans-generational change in DNA.
In contrast, epigenetics refers to an alteration in gene function without changes in the underlying DNA sequence (21). Epigenetic mechanisms involve DNA methylation, histone density and posttranslational modifications and the engagement of noncoding RNAs. Some alterations in the epigenome may be hereditable, resulting in trans-generational changes in genotype/phenotype correlations.
The programming of the epigenome is active during gestation, and epigenetic processes respond to environmental stimuli ranging from protein-calorie dietary restriction to hypoxia and fetal inflammatory exposures. Offspring of women experiencing pre-eclampsia, a putative marker for fetal hypoxia, have both hypertension and endothelial dysfunction during young adulthood (23). Similarly, in preclinical models, offspring of mothers exposed to protein-calorie deprivation during pregnancy also have vascular dysfunction, and these findings are reversed by maternal folate supplementation (24). Of note, folate deficiencies have also been associated with abnormalities in DNA methylation (25). Also in preclinical studies, the endothelium-dependent abnormalities in the offspring of restricted diet pregnancies are ameliorated by the administration of histone deacetylase inhibitors, suggesting a trans-generational etiology for the findings (26).
Finally, the interactions between genetic polymorphisms, epigenetic mechanisms such as DNA methylation and expression are complex. Emerging data suggest, however, that genetic variants regulate methylation, and methylation regulates gene expression. Thus a genetic variant that creates or negates a DNA C-phosphate-G (CPG) methylation site in the promoter region of a gene may significantly impact expression of that gene (27).
Pathways for alteration of the IVH epigenome
Studies investigating mechanisms by which fetal or preterm exposures may alter the epigenome to promote or prevent IVH include the effects of hypoxia, inflammation, nutrition and oxidative stress.
IVH has long been associated with hypoxic ischemic events, and putative inflammatory, excitotoxic and apoptotic pathways are involved in the complex cascade following neonatal hypoxia ischemia. The hypoxia-inducible transcription factors (HIFs) are among the endogenous adaptive mechanisms modifying this cascade of events. HIFs are heterodimers of HIF-α and HIF-β subunits that belong to a family of basic helix-loop-helix transcription factors. HIF-1 and HIF-2 are important regulators of oxygen-dependent gene transcription that modulate oxygen and metabolic supply during hypoxia. HIF target genes include those with vasoactive and vasoproliferative effects including vascular endothelial growth factor (VEGF) and inducible NO synthase (iNOS). In a preclinical model of preterm hypoxia, HIF-1α was prominently found in vascular endothelial and glial cells of the subventricular zone (28). Similarly, its target, VEGF, mediates survival and tube stabilization of hypoxic brain microvascular endothelial cells in vitro (29). Finally, possibly acting via acetylation and methylation pathways, chronic hypoxia decreases global transcriptional activity (30). Thus, in preclinical fetal studies, chronic high-altitude hypoxia resulted in reduced histone acetylation and DNA methylation, fetal pulmonary arterial smooth muscle cell proliferation, vessel remodeling and vascular dysfunction (30).
Likewise, biomarkers of inflammation such as interleukin 1-β (IL1- β) and IL6 activate the hypothalamic-pituitary-adrenal (HPA) axis, with putative long-term neurobehavioral sequelae (32). An example of such an epigenetic event is second trimester maternal exposure to type A2/Singapore influenza that significantly increased risk for adult psychiatric disorders (33).
Finally, as discussed above, preclinical studies suggest that, acting via metabolic, vascular and stress-mediated pathways, maternal nutrition may have profound effects on the developing fetus (24, 26, 34).
Pathophysiology of IVH: Preclinical candidates
IVH begins in the germinal matrix (GM), a site of active angiogenesis in the developing brain (20,13) (Figure 2). Endothelial growth and sprouting are critical for angiogenesis, and the emerging blood brain barrier is characterized by endothelial tight junctions, basement membrane proteins, perivascular pericytes and glial endfeet. These processes are regulated by assorted growth factors, cell surface receptors and intracellular signaling pathways.
Figure 2.
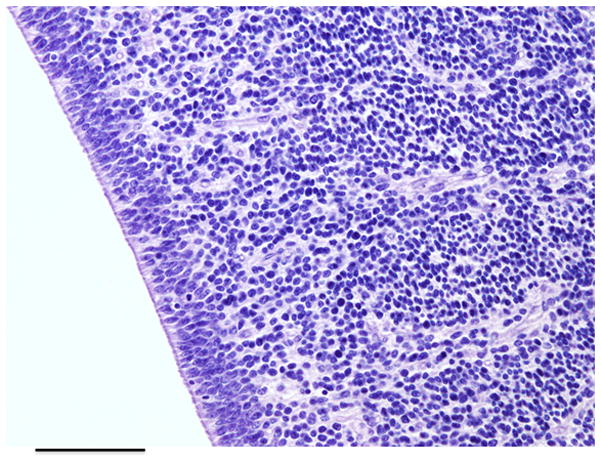
Germinal matrix, a densely cellular region located adjacent to the ependyma of the lateral ventricles. It is composed of immature neural precursor cells and vessels. Magnification 40X; scale bar = 1 mm. (Figure courtesy of A. Huttner, MD, Department of Pathology, Section of Neuropathology, Yale University School of Medicine, New Haven, CT.)
While preclinical studies postulate that it is the developmental stage of the GM microvessels that results in IVH, more recent studies suggest that mutations in one or more microvascular proteins confer vulnerability to environmental triggers (Table 1). Mice with targeted mutations in the basement membrane proteins, fibronectin, laminin, collagen 4A1 (COL4A1) and/or perlecan, demonstrate all are necessary for vascular stabilization. Those with mutations in COL4A1 experience IVH following the stress accompanying vaginal delivery. In the murine model, these hemorrhages are preventable by surgical delivery, suggesting an interaction between an environmental trigger and the genome (35).
Table 1.
Candidate genes for IVH from preclinical studies
| Gene | Function of the gene product | Physiologic process |
|---|---|---|
| Activin receptor-like kinase (Alk5) | αvβ8 integrin-mediated TGFβ type I receptor | Angiogenesis |
| Alpha v integrins | Mediate TGFβ activation | Angiogenesis |
| Annexin 7 (anx7) | Vascular Ca++-activated GTPase supporting Ca++ channel activity; Regulates cerebral blood flow | Cerebral blood flow |
| Collagen 4A1 (COL4A1) | Critical component of developing basement membrane | Angiogenesis |
| CREBa-binding protein (CBP) | Transcriptional co-activator required by many transcription factors | Unknown |
| Death receptor 6 (DR6) | Required for VEGF-mediated endothelial sprouting Drives barrier-genesis in the developing brain |
Angiogenesis |
| Friend leukemia integration (Fli) | Member of Ets family of transcription factors Activator of vascular stabilization |
Unknown |
| Inhibitor of differentiation (Id1, Id3) | Interfere with DNA binding of basic helix-loop-helix transcription factors; essential role in angiogenesis | Angiogenesis |
| TGFb β2 receptor (Tgfbr2) | αvβ8 integrin-mediated TGFβ type II receptor | Angiogenesis |
CREB – cAMP response binding element;
TGF – transforming growth factor
Similarly, although preclinical “risk factor” studies are not available, mice with mutations in activin receptor-like kinase 5 (Alk5) (36), alpha v integrins (37), annexin 7 (anx7) (38), CREB binding protein (CBP)(39), death receptor 6 (DR6) (40), Id proteins 1 and 3 (Id1, Id3, respectively) (41) or Tgfbr2 (36) also develop intracerebral hemorrhage mimicking Gr 4 IVH.
Transforming growth factor- β (TGF-β) activation and signaling is essential for normal blood vessel growth and sprouting in developing brain, and αvβ8 integrin mediates TGF-β activation. Mouse embryos genetically null for integrin β8 develop severe intracerebral hemorrhage beginning at embryonic day E 11.5 (42). Similarly, TGF-β signals are transduced by both TGF-β type II and the TGF-β type I receptors (Tgfbr2 and Alk5, respectively), and in murine systems, selective deletion of Tgfbr2 or Alk5 in endothelial cells results in lethal intracerebral hemorrhage (36). In humans, mutations in Tgfbr2 and Alk5 cause Loeys-Dietz syndrome, characterized by multiple arterial aneurysms and dissections. Men with Alk5 mutations more commonly present with thoracic aortic aneurysm and die earlier than women with this disorder, suggesting a gender predilection for this polymorphism (43).
IVH is also found in mice with genetic alterations in the transcription factors inhibitors of differentiation (Id) 1/3 (41), Friend leukemia integration (Fli) (39,44), and cAMP response element binding protein (CBP) (39). Id1 and Id3 prevent transcription by direct physical interaction with the basic helix-loop-helix transcription factors, are expressed in cerebral endothelial cells and play an essential role in angiogenesis (45). The CREB-binding protein is a transcriptional co-activator and Fli, which is a member of the Ets family of transcription factors, is a key regulator of vascular maturation (46). Finally, DR6 is required for VEGF-mediated endothelial sprouting, is enriched in CNS vasculature and drives barrier-genesis in developing brain (38). Facing environmental triggers including hypoxia, hypercarbia and hypertension, polymorphisms in some or all of these factors may result in hemorrhage.
Likewise, changes in CBF may contribute to hemorrhage. Autoregulation relies on smooth muscle cells, pericytes and proteins ranging from Ca++ and K+ channels, phospholipase A1, arachidonic acid and adenosine to nitric oxide and cytokines among others (20). Notably, mice with mutations in annexin 7 (anx7), a gene encoding a Ca++-activated GTPase supporting Ca++ channel activity, experience IVH, suggesting that, in the presence of environmental perturbations, mutations in genes controlling CBF may contribute to hemorrhage (38).
Evidence for gene-by-environment interactions: Clinical studies of severe hemorrhage
Studies in preterm neonates have implicated an array of candidate genes spanning the coagulation, inflammatory and vascular pathways. For this review, we compare and contrast the studies of Harteman (17), Ryckman (18), and Baier (19) and our Gene Targets study. As shown in Table 2, the numbers of subjects, their BW and GA as well as their racial and ethnic backgrounds were quite varied, as were the years in which they were born and, presumptively, the neonatal intensive care the neonates received (17).
Table 2.
Comparison of the selected study populations
| Reference | Number subjects | Entry criteria | Years of birth | Race/Ethnicity | Variants for comparison |
|---|---|---|---|---|---|
| Baier (19) | Variable numbers of cases & controls | < 1000 g BWa | Not available | African American (AA) | FVL, prothrombin, MTHFR, IL1-β, IL6, TNF-α |
| Harteman (17) | 62 premies 17 atypical (grade 4) PVHI | < 34 weeks GAb | 2005–2010 | Not available | FVL, prothrombin, MTHFR, COL4A1 |
| Ryckman (18) | 64 cases (any grade IVH) 207 controls | < 1500 g BW | 2000–2009 | 77% European (EUR), 7% Hisp, 12% AA, 4% Other | FVL, prothrombin, MTHFR, IL1-β, IL6, TNF-α |
| Gene Targets for IVH Consortium | 316 cases (grade 2–4 IVH) 389 controls | 500–1250 g BW | 2006–2012 | EUR | MTHFR |
BW – Birth weight;
GA – Gestational age
Coagulation Candidates
Coagulation factors have long been considered candidate genes for IVH, both because of the pathophysiology of hemorrhage and because of their putative role in perinatal stroke (47) (Table 3). The most widely studied include the factor V Leiden (F5) variant, polymorphisms of the methylenetetrahydrofolate reductase (MTHFR) gene and the prothrombin 20210G>A variant (F2).
Table 3.
Coagulation, inflammation and vascular candidate genes for IVH from clinical studies
| Gene name, symbol, SNP | Change in Function/Structure | Reported Environment-x-Gene Interactions |
|---|---|---|
| Coagulation | ||
| Factor V Leiden (F5) 1601G>A | Resistance of FV to inactivation by activated protein C; hyper-coagulability | Associated with atypical timing of IVH |
| Methylenetetrahydrofolate reductase (MTHFR) 677C>T; 1298A>C | Decreased conversion of homocysteine to methionine; homocysteine toxic to endothelial cells | Associated with atypical timing of IVH Interactions with maternal diet & folate Interactions with perinatal hypoxia |
| Prothrombin (F2) 97G>A | Elevated prothrombin; hyper-coagulability | |
| Inflammation | ||
| Interleukin 1β (IL1) 87–511T>C 87–31C>T |
Impaired activation of the HPA axis; Altered CNS inflammatory response | Environmental interaction with IL-1β-511T allele and ureaplasma urealytica colonization for PVL but not for severe IVH |
| Interleukin 6 (IL6) 116–121C>G | Altered CNS inflammatory response | |
| Tumor necrosis factor (TNF) 160-31G>A | Impaired acute phase reactant | |
| Vascular | ||
| Collagen 4A1 (COL4A1) | Altered basement membrane protein in the developing cerebrovasculature | Typical and atypical onset of parenchymal IVH |
| Endothelial nitric oxide synthase (eNOS) 786T>C | Decreased endogenous nitric oxide | |
| Superoxide dismutase 3 (SOD3) rs8192287 (−416TG>C) | Impaired oxidative stress response | |
The contribution of the F5 polymorphism to IVH has been interrogated in different populations. A point mutation results in replacement of amino acid 506 arginine to glutamine in an activated protein C cleavage site. Activated protein C cleaves the peptide bonds in activated F5, resulting in inhibition of the coagulation pathway, and the variant presents with hypercoagulability. Gopel (48) reported this polymorphism was associated with Gr 1–2 IVH but protected against parenchymal hemorrhage. In contrast, Ryckman found that the heterozygous genotype was associated with Gr 1–2 but not Gr 3–4 IVH (18).
Similarly, Harteman (17) studied 17 preterm neonates with atypical presentation of Gr 4 IVH; atypical hemorrhages were defined as occurring in the absence of provoking clinical factors more than 96 hours following birth (Figure 3). Seven of 17 were heterozygous for the F5 variant, suggesting an association between this hypercoagulable state and atypical hemorrhage. Recent studies suggest that related or unrelated thrombophilia in the mothers increase the risk of perinatal stroke (47), and 6 of 7 mothers of F5 infants harbored this variant. Although the incidence of variant status in the patients in this manuscript is significantly higher than that for the Dutch population, no data are provided for neonates with typical onset Gr 4 hemorrhage.
Figure 3.
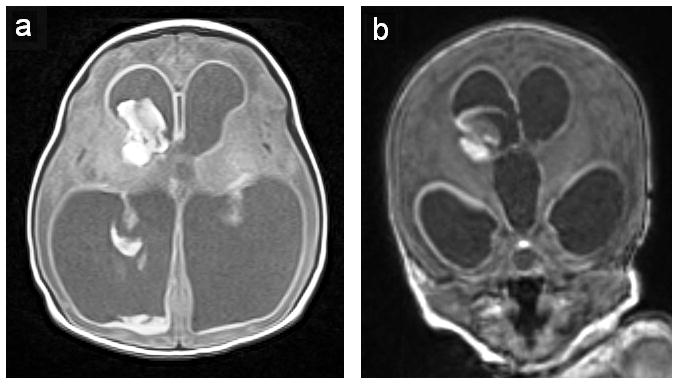
Atypical intraventricular hemorrhage. Axial (Panel A) and coronal (Panel B) images of a one day old 34 week gestation infant with the atypical (fetal) onset of germinal matrix and intraventricular hemorrhage. Note the hemorrhage into the right germinal matrix, intraventricular blood and ventriculomegaly characteristic of posthemorrhagic hydrocephalus. (Figure courtesy of C.C. Duncan, MD, Department of Neurosurgery, Yale University School of Medicine, New Haven, CT.)
Finally, Baier found no association between F5 and IVH in his cohort of 99 mostly African American ELBW neonates (19).
Of note, IVH is more common in male preterm neonates, and although no gender-by-F5 data are currently available for neonates with IVH, males with the F5 polymorphism are more likely than F5 females to experience recurrence of peripheral venous thrombosis suggesting a possible gender effect for this mutation (49).
A second leading candidate is MTHFR. MTHFR catalyzes the reduction of 5,10-methylenetrahydrofolate to 5-methyltetrahydrofolate, which is necessary for the conversion of homocysteine to methionine. Hyperhomocysteinemia is associated with polymorphisms at −677 and −1298, and, especially the 677 TT variant, results in endothelial cell injury and alterations in coagulation including stroke, thrombosis, migraine and vascular disorders (50, 51). Hyperhomocysteinemia is exacerbated under conditions of low folate, and this increases the susceptibility to experimental brain damage.
MTHFR may additionally play an important role in neonatal brain injury. Studying the prevalence of the 677C>T variant in 11 neonates with hypoxic ischemic encephalopathy and their mothers, Dodelson de Kremer (52) found that compared to a 68% incidence of the T allele in the control population, all 11 carried this polymorphism. The variant was more common in mothers of affected offspring and was associated with an increase in maternal homocysteine, suggesting a profound alteration in the fetal environment. The mothers in this study exhibited poor nutrition and the authors postulated that underlying folate deficiency during pregnancy may have exacerbated the influence of hypoxic injury in neonates harboring the MTHFR variant.
More recently, Harteman evaluated 118 infants with hypoxic ischemic encephalopathy and reported that the MRI white matter/watershed pattern of injury was associated with MTHFR CT or TT 677 polymorphisms and plasma homocysteine levels in the upper quartile (53). In this study, neonatal MTHFR polymorphisms were not associated with homocysteine levels, consistent with findings of other observers (54). Of interest, Molloy demonstrated that maternal homocysteine levels are the best predictors of fetal values, emphasizing the importance of the fetal environment (55).
Acute hypoxia ischemia results in thrombosis in subjects lacking known polymorphisms (56), and hypoxia exacerbates the effect of a folate-deficient diet on homocysteine metabolism (57, 58). Blaise demonstrated that in rat pups exposed prenatally to diets deficient in vitamins B12, B2, folate and choline through weaning, hypoxia increased plasma homocysteine levels. MTHFR activity was attenuated by hypoxia. Further, hypoxia enhanced the deficiency-induced drop of the S-adenosylmethionine/S-adenosylhomocysteine ratio, known to influence DNA methylation and gene expression (59). Taken together, these data suggest the potential interaction between maternal and fetal MTHFR polymorphisms, folate and hypoxic ischemic injury to preterm brain.
Co-inheritance of more than one thrombophilia variant is associated with a greater risk of thrombotic events than with a single polymorphism. Thus, in addition to assessing F5, Harteman investigated the 677C>T and 1298A>C polymorphisms in 16/17 preterms with atypical PVHI (Table 3). Six had the −677 T allele, 4 had the −1298 C variant and 4 were compound heterozygous suggesting that 14/16 neonates had potentially deleterious polymorphisms. In contrast, there were no significant differences in genotypes for case and control neonates studied by Ryckman or Baier.
Based on our previous work demonstrating a difference in the MTHFR −1298C polymorphism between severe IVH cases and controls and the putative role of hypoxia in MTHFR-mediated brain injury (60), we tested the hypothesis that there would be a gene-by-environment interaction for these two factors. For this preliminary analysis, we interrogated only the MTHFR 1298A>C variant in the Gene Targets for IVH Consortium (NS053865) database. (Institutional review board approval was obtained from all participating institutions.) This Consortium has both environmental data and DNA from over 1400 inborn appropriate for gestational age preterm neonates with antenatal steroid exposure, BW 500–1250 g and centrally read cranial ultrasounds. Only the 705 European subjects were included in this analysis to avoid racial admixture.
Three hundred sixteen infants had Gr 2– 4 IVH; 389 neonates had no evidence for IVH. Cases had lower BW and GA than controls, and their mothers were more likely to have experienced chorioamnionitis and multiple gestation pregnancies (Table 4). In contrast, case mothers had less preeclampsia and fewer cesarean section deliveries. Cases were more likely to have 5 minute Apgar scores < 3 and require intubation for delivery room resuscitation.
Table 4.
Gene Targets for IVH Consortium Study Subjects
| EUR Gr 2 - 4 IVH | Controls | p value | |
|---|---|---|---|
|
| |||
| Number of subjects | 316 | 389 | |
|
| |||
| Birth weight grams (N, SD) | 821.4 (316, 184.1) | 864.3 (389, 174.0) | <0.0001 |
|
| |||
| GA wks (N, SD) | 25.7 (314, 1.5) | 26.5 (388, 1.5) | <0.0001 |
|
| |||
| Male subjects (%) | 187/316 (59.2%) | 231/389 (59.4%) | 1 |
|
| |||
| IVH status | |||
| Grade 2 | 92 | ||
| Grade 3 | 95 | ||
| Grade 4 | 129 | ||
|
| |||
| Maternal variables | |||
|
| |||
| Maternal prenatal visit (%) | 307/314 (97.8%) | 384/389 (98.7%) | 0.3887 |
|
| |||
| Preeclampsia (%) | 35/314 (11.1%) | 91/388 (23.5%) | <0.0001 |
|
| |||
| Clinical chorioamnionitis (%) | 95/315 (30.2%) | 69/389 (17.7%) | 0.0001 |
|
| |||
| Any ANS w/in 7 d prior to delivery (%) | 277/315 (87.9%) | 336/388 (86.6%) | 0.6504 |
|
| |||
| Complete ANS a within 7 d (%) | 176/316 (55.7%) | 260/389 (66.8%) | 0.003 |
|
| |||
| Multiple gestation (%) | 116/316 (36.7%) | 90/386 (23.3%) | 0.0001 |
|
| |||
| Cesarean section | 187/316 (59.2%) | 281/388 (72.4%) | 0.0002 |
|
| |||
| Delivery room variables | |||
|
| |||
| Apgar 1 minute < 3 (%) | 99/314 (31.5%) | 89/388 (22.9%) | 0.0128 |
|
| |||
| Apgar 5 minutes < 3 (%) | 28/315 (8.9%) | 14/388 (3.6%) | 0.0038 |
|
| |||
| Intubation for resuscitation | 244/295 (82.7%) | 243/351 (69.2%) | <0.0001 |
|
| |||
| Epinephrine for resuscitation | 10/230 (4.3%) | 5/215 (2.3%) | 0.2977 |
|
| |||
| MTHFR polymorphisms | |||
|
| |||
| −1298 CC or CA genotype | 176/316(55.7%) | 176/389(45.2%) | 0.006 |
Antenatal steroid exposure
An analysis of generalized linear mixed model with site as a random effect and all significant variables from Table 4 as fixed effects was performed to measure the relationship between IVH status and those independent variables including GA, preeclampsia, clinical chorioamnionitis, complete ANS within 7 days prior to delivery, multiple gestation, cesarean section, Apgar 1 minute < 3, Apgar 5 minutes < 3, intubation for resuscitation and the MTHFR 1298A>C variant. Similar to previous reports [for review see Volpe, 2008 (61)], this analysis demonstrated that increasing GA, cesarean delivery and a complete course of ANS in the week prior to delivery were protective for Gr 2–4 IVH; in contrast, multiple gestation pregnancy and chorioamnionitis were independent and important risk factors for Gr 2–4 IVH. In addition, the MTHFR variant and the interaction term (Apgar5<3-by-MTHFR allele) were independent and important predictors of Gr 2–4 IVH in our population (Table 5).
Table 5.
Analysis of generalized linear mixed model with random effect from Gene Targets for IVH Consortium
| Effect | Odds Ratio | 95% Confidence Limits | P value | |
|---|---|---|---|---|
| GA | 0.707 | 0.629 | 0.794 | < 0.001 |
| Chorioamnionitis | 1.563 | 1.049 | 2.330 | 0.028 |
| Cesarean delivery | 0.569 | 0.394 | 0.820 | 0.003 |
| Complete course ANS | 0.663 | 0.471 | 0.935 | 0.019 |
| Multiple gestation | 2.445 | 1.685 | 3.576 | < 0.001 |
| MTHFR | 1.427 | 1.022 | 1.992 | 0.037 |
| Apgar5<3-by-MTHFR | 3.210 | 1.008 | 10.225 | 0.048 |
The F2 variant is the last leading coagulation candidate we are discussing. It results in increased thrombin and secondary thrombosis, and in the studies of Baier, Harteman and Ryckman it was not associated with a risk for IVH.
Inflammatory factors
Cytokines are also postulated to play a role in perinatal brain injury, and both Ryckman and Baier explored the role of interleukins in preterm IVH. Hypoxia results in the loss of blood brain barrier function and impaired tight junction protein synthesis (62), permitting cytokines from the peripheral circulation to directly enter preterm brain. In addition, cytokines secreted by cells of the immune system may also be synthesized by CNS glial to act as signal transmitters in the developing brain (63).
Interleukin-1β (Il-1β) is the major cytokine involved in activation of the hypothalamic-pituitary-adrenal (HPA) axis (31). In addition, although the exact mechanisms by which Il-1β is involved in hypoxia ischemia, IVH and perinatal brain injury remain unknown, Il-1β has been implicated in the progression of injury in the developing brain (64). Of importance to the understanding of critical period injuries such as IVH, expression of Il-1β is both developmentally and regionally regulated in the brains of typically developing fetuses and neonates (65). Similarly, in response to hypoxic ischemic injury, Il-1β differentially increases across the brain, suggesting regional vulnerability to cytokine-mediated injury.
Preclinical studies demonstrate that perinatal Il-1β exposure induces acute white matter injury with subsequent ventriculomegaly, loss of mature oligodendrocytes, impaired myelination, decreased myelin basic protein and axonal and dendritic injury (66). In addition, acting via the COX-2 pathway (67), perinatal bacterial infection significantly increases Il-1β, interleukin 6 (IL-6) and corticosterone production in rat pups a few hours after infection, suggesting involvement of both the central inflammatory and HPA pathways (68). Such perinatal immune activation has been associated not only with change in behavior in neonatal animals but also disrupted avoidance learning in male, but not female subjects in adulthood (69). Taken together, these data suggest that early Il-1β-mediated immune activation results in long term changes in both structure and function in developing brain.
When Baier evaluated the role of Il-1β 511C>T polymorphisms in 215 ventilated VLBW infants, the Il-1β -511 T allele was associated with increased risk for IVH. One third of infants with the T allele experienced IVH, compared to 14% with the C allele. There was also a significant difference in Gr 3–4 IVH between the groups. Periventricular leukomalacia (PVL) was also increased, mainly in those infants with the CT genotype. Because of the association of chorioamnionitis and periventricular leukomalacia, Baier interrogated the interaction of ureaplasma urealyticum (UU) colonization and Il-1β 511T allele on the incidence and severity of both IVH and PVL. Consistent with the report of Leviton (70), there was no interaction for these triggers in neonates with IVH. In contrast, infants with both the 511T allele and UU were at greater risk of PVL than infants with one or none of these triggers, suggesting a gene-by-environment interaction.
Ryckman validated this result by finding that the Il-1β-31 C allele was associated with an increased risk for hemorrhage. The C allele of Il-1β-31 is in strong linkage disequilibrium with the T allele of Il-1β -511, and both increase the production of Il-1β in vivo (71).
Similarly, IL-6 has also been implicated in injury in developing brain (65, 72), and has also been shown to activate the HPA axis (73). Thus IL-6 is also believed to be a strong candidate to modify risk for preterm brain injury. Harding reported that in 151 preterm neonates the CC genotype of IL-6–174 significantly increased risk for IVH and neurodevelopmental disability at age 2 years (74). In contrast, interrogating the same polymorphism, neither Baier nor Ryckman found any relationship between IL-6 and IVH in the prematurely-born.
TNF-α plays a pivotal role in the acute phase pro-inflammatory cytokine cascade and is also postulated to be a central mediator of brain injury in the prematurely-born (75). The TNF-α gene is polymorphic, and there are numerous polymorphisms in the promoter region. Studying 178 ventilated VLBW infants, Adcock reported that the −308 A allele in the TNF-α promoter region was associated with IVH in preterm neonates (75). Baier also found that in infants with the TNF-α −308 A allele, the incidence of IVH was 40% compared to 24% in those with GG (19). However, Ryckman found no association between this polymorphism and IVH in her population.
Vascular genes
Proteins both contributing to the integrity of the developing CNS vasculature and those mediating cerebral blood flow are excellent targets for IVH. COL4A1 encodes type IV collagen alpha chain 1. This is one of 6 alpha chains that contribute to type IV collagen, a principal component of basement membranes ubiquitously expressed during development. Truncating mutations in murine Col4a1 result in cerebral hemorrhage in both neonatal and adult mice, and mutations have been reported in infants with congenital porencephaly, fetal IVH and adults with cerebral small vessel disease (35,76). More recently, mutations have also been reported in preterm neonates with IVH (77,78). Studying 41 PT infants with IVH, Bilguvar identified a rare heterozygous duplication within a highly conserved residue in COL4A1 in dizygotic twins with Gr 4 IVH (79).
In addition to inhibiting platelet and leukocyte adhesion to vascular endothelium, NO promotes cerebral vasodilatation (80). Several allelic variants have been reported in promoter of the endothelial NO synthase (eNOS) gene that have been associated with decreased eNOS activity and reductions in NO. Investigating 124 AA preterm neonates, Vannemreddy (81) reported the association of the eNOS gene promoter polymorphism 786T>C with IVH, suggesting that the vascular actions of eNOS are critical for prevention of hemorrhage in the developing brain.
Finally, oxidative stress may also play a role, and Poggi (82) reported that the rs8192287 superoxide dismutase 3 (SOD3) polymorphism is an independent protective factor for IVH in 152 neonates of < 28 weeks GA. Although the mechanism is not yet known, Poggi postulates protection of the cerebral microvessels against oxidative injury.
Implications of the science: Future approaches for decreasing the incidence of IVH
Further understanding of the genetic contributions to IVH, including genome wide association studies and/or whole exome sequencing data, will permit the rationale design of randomized clinical trials. These might include delivery mode trials for fetuses harboring vascular structural polymorphisms and strategies to lower homocysteine in mothers and/or neonates with MTHFR variants. Equally important avenues of molecular investigation might include inhibiting disease-causing pathways, such as the proposed use of rapamycin for subependymal giant cell astrocytomas in children with tuberous sclerosis;(83) upregulating affected proteins from homologous genes as in models of spinal muscular atrophy (84); or counteracting the downstream effects of a deficient protein, such as the proposed use of IGF1 in children with Duchenne’s dystrophy [for review, please see Liew, 2012 (85)].
Available preclinical genetic studies and clinical candidate gene reports suggest that IVH may be attributable to numerous genes with small effect sizes and the environmental factors that interact with them. Because these common variants have small to moderate effects on disease risk, individual risk variants are neither necessary nor sufficient to produce disease, and there are consequently individuals with disease without risk variants and, conversely, individuals without disease who harbor risk variants. A hope is that identification of genes and pathways underlying IVH will permit the development of prenatal diagnostics and/or preventive therapeutics. To address these issues, a large-scale neonatal genomic medicine network must be developed with infrastructural capacity to both host an accessible database of sequence variants and their phenotypic associations and support a framework for defining and cataloging clinically actionable variants (86).
Conclusion
If a major focus of perinatal care is to prevent brain injury and abnormal development (87), then physicians and scientists must better understand those factors that contribute to severe IVH in the prematurely-born. Emerging data suggest an important role of genes subserving coagulation, inflammatory and vascular pathways, and interactions with maternal and neonatal environmental triggers may influence both the incidence and severity of cerebral injury and have long term implications.
Acknowledgments
We are indebted to our medical, nursing, and research colleagues and the infants and their parents who agreed to take part in this study. We are grateful to Deborah Hirtz, MD, for scientific expertise, Linda de Vries, MD, and Praveen Ballabh, MD, for reviewing the manuscript and making important scientific contributions, Jill Maller-Kesselman, MS, for study management, Carol Nelson-Williams for scientific expertise, Walter C. Allan, MD, for ultrasound expertise, and Karen C. Schneider, MPH, for editorial expertise and assistance.
STATEMENT OF FINANCIAL SUPPORT: This work was supported by the National Institutes of Health, Bethesda, MD NS053865. Heping Zhang was also partially supported by R01 DA016750-09.
Footnotes
DISCLOSURES: None of the authors have any disclosures of financial ties to products in the study or potential/perceived conflicts of interest.
References
- 1.Papile LS, Burstein J, Burstein R. Incidence and evolution of the subependymal intraventricular hemorrhage: A study of infants with weights less than 1500 grams. J Pediatr. 1978;92:529–34. doi: 10.1016/s0022-3476(78)80282-0. [DOI] [PubMed] [Google Scholar]
- 2.Nongena P, Ederies A, Azzopardi DV, Edwards AD. Confidence in the prediction of neurodevelopmental outcome by cranial ultrasound and MRI in preterm infants. Arch Dis Child Fetal Neonatal Ed. 2010;95:F388–90. doi: 10.1136/adc.2009.168997. [DOI] [PubMed] [Google Scholar]
- 3.Beaino G, Khoshnood B, Kaminski M, et al. Predictors of cerebral palsy in very preterm infants: the EPIPAGE prospective population-based cohort study. Dev Med Child Neurol. 2010;52:e119–25. doi: 10.1111/j.1469-8749.2010.03612.x. [DOI] [PubMed] [Google Scholar]
- 4.Luu TM, Ment L, Allan W, Schneider K, Vohr BR. Executive and memory function in adolescents born very preterm. Pediatrics. 2011;127:e639–46. doi: 10.1542/peds.2010-1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whitaker AH, Feldman JF, Lorenz JM, et al. Neonatal head ultrasound abnormalities in preterm infants and adolescent psychiatric disorders. Arch Gen Psychiatry. 2011;68:742–52. doi: 10.1001/archgenpsychiatry.2011.62. [DOI] [PubMed] [Google Scholar]
- 6.Hallman M. Premature birth and diseases in premature infants: common genetic background? J Matern Fetal Neonatal Med. 2012;25 (Suppl 1):21–4. doi: 10.3109/14767058.2012.667600. [DOI] [PubMed] [Google Scholar]
- 7.Mohamed S, Schaa K, Cooper ME, et al. Genetic contributions to the development of retinopathy of prematurity. Pediatr Res. 2009;65:193–7. doi: 10.1203/PDR.0b013e31818d1dbd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stoll BJ, Hansen NI, Bell EF, et al. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics. 2010;126:443–56. doi: 10.1542/peds.2009-2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohamed MA, Aly H. Transport of premature infants is associated with increased risk for intraventricular haemorrhage. Arch Dis Child Fetal Neonatal Ed. 2010;95:F403–7. doi: 10.1136/adc.2010.183236. [DOI] [PubMed] [Google Scholar]
- 10.Perlman JM, Wyllie J, Kattwinkel J, et al. Part 11: Neonatal resuscitation: 2010 International Consensus on Cardiopulmonary Resuscitation and Emergency Cardiovascular Care Science With Treatment Recommendations. Circulation. 2010;122:S516–38. doi: 10.1161/CIRCULATIONAHA.110.971127. [DOI] [PubMed] [Google Scholar]
- 11.Sweet DG, Carnielli V, Greisen G, et al. European consensus guidelines on the management of neonatal respiratory distress syndrome in preterm infants - 2010 update. Neonatology. 2010;97:402–17. doi: 10.1159/000297773. [DOI] [PubMed] [Google Scholar]
- 12.Hack M, Wright LL, Shankaran S, et al. Very-low-birth-weight outcomes of the National Institute of Child Health and Human Development Neonatal Network, November 1989 to October 1990. Am J Obstet Gynecol. 1995;172:457–64. doi: 10.1016/0002-9378(95)90557-x. [DOI] [PubMed] [Google Scholar]
- 13.Volpe JJ. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009;8:110–24. doi: 10.1016/S1474-4422(08)70294-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shalak L, Perlman JM. Hemorrhagic-ischemic cerebral injury in the preterm infant. Current concepts Clin Perinatol. 2002;29:745–63. doi: 10.1016/s0095-5108(02)00048-9. [DOI] [PubMed] [Google Scholar]
- 15.Kent AL, Wright IM, Abdel-Latif ME. Mortality and adverse neurologic outcomes are greater in preterm male infants. Pediatrics. 2012;129:124–31. doi: 10.1542/peds.2011-1578. [DOI] [PubMed] [Google Scholar]
- 16.Bhandari V, Bizzarro MJ, Shetta A, et al. Familial and genetic susceptibility to major neonatal morbidities in preterm twins. Pediatrics. 2006;117:1901–6. doi: 10.1542/peds.2005-1414. [DOI] [PubMed] [Google Scholar]
- 17.Harteman JC, Groenendaal F, van Haastert IC, et al. Atypical timing and presentation of periventricular haemorrhagic infarction in preterm infants: the role of thrombophilia. Dev Med Child Neurol. 2012;54:140–7. doi: 10.1111/j.1469-8749.2011.04135.x. [DOI] [PubMed] [Google Scholar]
- 18.Ryckman KK, Dagle JM, Kelsey K, Momany AM, Murray JC. Replication of genetic associations in the inflammation, complement, and coagulation pathways with intraventricular hemorrhage in LBW preterm neonates. Pediatr Res. 2011;70:90–5. doi: 10.1203/PDR.0b013e31821ceb63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baier RJ. Genetics of perinatal brain injury in the preterm infant. Front Biosci. 2006;11:1371–87. doi: 10.2741/1890. [DOI] [PubMed] [Google Scholar]
- 20.Ballabh P. Intraventricular hemorrhage in premature infants: mechanism of disease. Pediatr Res. 2010;67:1–8. doi: 10.1203/PDR.0b013e3181c1b176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–8. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 22.Hambleton G, Wigglesworth JS. Origin of intraventricular haemorrhage in the preterm infant. Arch Dis Child. 1976;51:651–9. doi: 10.1136/adc.51.9.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis EF, Newton L, Lewandowski AJ, et al. Pre-eclampsia and offspring cardiovascular health: mechanistic insights from experimental studies. Clin Sci (Lond) 2012;123:53–72. doi: 10.1042/CS20110627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Torrens C, Brawley L, Anthony FW, et al. Folate supplementation during pregnancy improves offspring cardiovascular dysfunction induced by protein restriction. Hypertension. 2006;47:982–987. doi: 10.1161/01.HYP.0000215580.43711.d1. [DOI] [PubMed] [Google Scholar]
- 25.Shirodkar AV, Marsden PA. Epigenetics in cardiovascular disease. Curr Opin Cardiol. 2011;26:209–15. doi: 10.1097/HCO.0b013e328345986e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rexhaj E, Bloch J, Jayet PY, et al. Fetal programming of pulmonary vascular dysfunction in mice: role of epigenetic mechanisms. Am J Physiol Heart Circ Physiol. 2011;301:H247–52. doi: 10.1152/ajpheart.01309.2010. [DOI] [PubMed] [Google Scholar]
- 27.van Eijk KR, de Jong S, Boks MP, et al. Genetic analysis of DNA methylation and gene expression levels in whole blood of healthy human subjects. BMA genomics. 2012;13:636. doi: 10.1186/1471-2164-13-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trollmann R, Gassmann M. The role of hypoxia-inducible transcription factors in the hypoxic neonatal brain. Brain Dev. 2009;31:503–9. doi: 10.1016/j.braindev.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 29.Chow J, Ogunshola OO, Fan S-Y, Li Y, Ment LR, Madri JA. Astrocyte-derived VEGF mediates survival and tube stabilization of hypoxic brain microvascular endothelial cells in vitro. Brain Res Devel Brain Res. 2001;130:123–32. doi: 10.1016/s0165-3806(01)00220-6. [DOI] [PubMed] [Google Scholar]
- 30.Fish JE, Yan MS, Matouk CC, et al. Hypoxic repression of endothelial nitric-oxide synthase transcription is coupled with eviction of promoter histones. J Biol Chem. 2010;285:810–26. doi: 10.1074/jbc.M109.067868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Q, Lu Z, Ramchandran R, Longo LD, Raj JU. Pulmonary artery smooth muscle cell proliferation and migration in fetal lambs acclimatized to high-altitude long-term hypoxia: role of histone acetylation. AM J Physiol Lung Cell Mol Physiol. 2012;303:L1001–10. doi: 10.1152/ajplung.00092.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gadek-Michalska A, Tadeusz J, Rachwalska P, Spyrka J, Bugajski J. Brain nitric oxide synthases in the interleukin-1beta-induced activation of hypothalamic-pituitary-adrenal axis. Pharmacol Rep. 2012;64:1455–65. doi: 10.1016/s1734-1140(12)70943-x. [DOI] [PubMed] [Google Scholar]
- 33.Machon RA, Mednick SA, Huttunen MO. Adult major affective disorder after prenatal exposure to an influenza epidemic. Arch Gen Psychiatry. 1997;54:322–8. doi: 10.1001/archpsyc.1997.01830160040006. [DOI] [PubMed] [Google Scholar]
- 34.Vieira-Filho LD, Lara LS, Silva PA, et al. Placental oxidative stress in malnourished rats and changes in kidney proximal tubule sodium ATPases in offspring. Clin Exp Pharmacol Physiol. 2009;36:1157–63. doi: 10.1111/j.1440-1681.2009.05212.x. [DOI] [PubMed] [Google Scholar]
- 35.Gould DB, Phalan C, van Mil SE, et al. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med. 2006;354:1489–96. doi: 10.1056/NEJMoa053727. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen HL, Lee YJ, Shin J, et al. TGF-beta signaling in endothelial cells, but not neuroepithelial cells, is essential for cerebral vascular development. Lab Invest. 2011;91:1554–63. doi: 10.1038/labinvest.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCarty JH, Lacy-Hulbert A, Charest A, et al. Selective ablation of alphav integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development. 2005;132:165–76. doi: 10.1242/dev.01551. [DOI] [PubMed] [Google Scholar]
- 38.Srivastava M, Atwater I, Glasman M, et al. Defects in inositol 1,4,5-trisphosphate receptor expression, Ca(2+) signaling, and insulin secretion in the anx7(+/−) knockout mouse. Proc Natl Acad Sci USA. 1999;96:13783–8. doi: 10.1073/pnas.96.24.13783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanaka Y, Naruse I, Hongo T, et al. Extensive brain hemorrhage and embryonic lethality in a mouse null mutant of CREB-binding protein. Mech Dev. 2000;95:133–45. doi: 10.1016/s0925-4773(00)00360-9. [DOI] [PubMed] [Google Scholar]
- 40.Tam SJ, Richmond DL, Kaminker JS, et al. Death receptors DR6 and TROY regulate brain vascular development. Dev Cell. 2012;22:403–17. doi: 10.1016/j.devcel.2011.11.018. [DOI] [PubMed] [Google Scholar]
- 41.Lyden D, Young AZ, Zagzag D, et al. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature. 1999;401:670–7. doi: 10.1038/44334. [DOI] [PubMed] [Google Scholar]
- 42.Zhu J, Motejlek K, Wang D, Zang K, Schmidt A, Reichardt LF. beta8 integrins are required for vascular morphogenesis in mouse embryos. Development. 2002;129:2891–903. doi: 10.1242/dev.129.12.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tran-Fadulu V, Pannu H, Kim DH, et al. Analysis of multigenerational families with thoracic aortic aneurysms and dissections due to TGFBR1 or TGFBR2 mutations. J Med Genet. 2009;46:607–13. doi: 10.1136/jmg.2008.062844. [DOI] [PubMed] [Google Scholar]
- 44.Spyropoulos DD, Pharr PN, Lavenburg KR, et al. Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the Fli1 transcription factor. Mol Cell Biol. 2000;20:5643–52. doi: 10.1128/mcb.20.15.5643-5652.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benezra R, Rafii S, Lyden D. The Id proteins and angiogenesis. Oncogene. 2001;20:8334–41. doi: 10.1038/sj.onc.1205160. [DOI] [PubMed] [Google Scholar]
- 46.Asano Y, Stawski L, Hant F, et al. Endothelial Fli1 deficiency impairs vascular homeostasis: a role in scleroderma vasculopathy. Am J Pathol. 2010;176:1983–98. doi: 10.2353/ajpath.2010.090593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chabrier S, Husson B, Dinomais M, Landrieu P, Nguyen The Tich S. New insights (and new interrogations) in perinatal arterial ischemic stroke. Thromb Res. 2011;127:13–22. doi: 10.1016/j.thromres.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 48.Gopel W, Gortner L, Kohlmann T, Schultz C, Moller J. Low prevalence of large intraventricular haemorrhage in very low birthweight infants carrying the factor V Leiden or prothrombin G20210A mutation. Acta paediatr. 2001;90:1021–4. doi: 10.1080/080352501316978101. [DOI] [PubMed] [Google Scholar]
- 49.Olie V, Zhu T, Martinez I, Scarabin PY, Emmerich J. Sex-specific risk factors for recurrent venous thromboembolism. Thromb Res. 2012;130:16–20. doi: 10.1016/j.thromres.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 50.Kloppenborg RP, Nederkoorn PJ, van der Graaf Y, Geerlings MI. Homocysteine and cerebral small vessel disease in patients with symptomatic atherosclerotic disease. The SMART-MR study. Atherosclerosis. 2011;216:461–6. doi: 10.1016/j.atherosclerosis.2011.02.027. [DOI] [PubMed] [Google Scholar]
- 51.Stuart S, Cox HC, Lea RA, Griffiths LR. The role of the MTHFR gene in migraine. Headache. 2012;52:515–20. doi: 10.1111/j.1526-4610.2012.02106.x. [DOI] [PubMed] [Google Scholar]
- 52.Dodelson de Kremer R, Grosso C. Maternal mutation 677C > T in the methylenetetrahydrofolate reductase gene associated with severe brain injury in offspring. Clin Genet. 2005;67:69–80. doi: 10.1111/j.1399-0004.2004.00373.x. [DOI] [PubMed] [Google Scholar]
- 53.Harteman JC, Groenendaal F, Benders MJ, Huisman A, Blom HJ, de Vries LS. Role of thrombophilic factors in full-term infants with neonatal encephalopathy. Pediatr Res. 2013;73:80–6. doi: 10.1038/pr.2012.150. [DOI] [PubMed] [Google Scholar]
- 54.Refsum H, Grindflek AW, Ueland PM, et al. Screening for serum total homocysteine in newborn children. Clin Chem. 2004;50:1769–84. doi: 10.1373/clinchem.2004.036194. [DOI] [PubMed] [Google Scholar]
- 55.Molloy AM, Mills JL, McPartlin J, Kirke PN, Scott JM, Daly S. Maternal and fetal plasma homocysteine concentrations at birth: the influence of folate, vitamin B12, and the 5,10-methylenetetrahydrofolate reductase 677C-->T variant. Am J Obstet Gynecol. 2002;186:499–503. doi: 10.1067/mob.2002.121105. [DOI] [PubMed] [Google Scholar]
- 56.Bauman ME, Cheung PY, Massicotte MP. Hemostasis and platelet dysfunction in asphyxiated neonates. J Pediatr. 2011;158:e35–9. doi: 10.1016/j.jpeds.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 57.Chawla RK, Watson WH, Jones DP. Effect of hypoxia on hepatic DNA methylation and tRNA methyltransferase in rat: similarities to effects of methyl-deficient diets. J Cell Biochem. 1996;61:72–80. doi: 10.1002/(SICI)1097-4644(19960401)61:1%3C72::AID-JCB9%3E3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 58.Ito K, Miwa N, Hagiwara K, et al. Regulation of methionine adenosyltransferase activity by the glutathione level in rat liver during ischemia-reperfusion. Surg Today. 1999;29:1053–8. doi: 10.1007/PL00010035. [DOI] [PubMed] [Google Scholar]
- 59.Blaise S, Alberto JM, Nedelec E, et al. Mild neonatal hypoxia exacerbates the effects of vitamin-deficient diet on homocysteine metabolism in rats. Pediatr Res. 2005;57:777–82. doi: 10.1203/01.PDR.0000161406.19231.98. [DOI] [PubMed] [Google Scholar]
- 60.Aden U, Line A, Carlo W, et al. Canidate gene analysis: Severe intraventricular hemorrhage in inborn preterm neonates. J Pediatr. 2013 Jul 26; doi: 10.1016/j.jpeds.2013.06.025. 10.1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Volpe JJ. Neurology of the Newborn. 5. Philadephia: Saunders Elsevier; 2008. Intracranial hemorrhage: Germinal matrix-intraventricular hemorrhage of the premature infant; pp. 517–88. [Google Scholar]
- 62.Chen X, Threlkeld SW, Cummings EE, et al. Ischemia-reperfusion impairs blood-brain barrier function and alters tight junction protein expression in the ovine fetus. Neuroscience. 2012;226:89–100. doi: 10.1016/j.neuroscience.2012.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther. 2011;130:226–38. doi: 10.1016/j.pharmthera.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aden U, Favrais G, Plaisant F, et al. Systemic inflammation sensitizes the neonatal brain to excitotoxicity through a pro-/anti-inflammatory imbalance: key role of TNFalpha pathway and protection by etanercept. Brain Behav Immun. 2010;24:747–58. doi: 10.1016/j.bbi.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 65.Sadowska GB, Threlkeld SW, Flangini A, Sharma S, Stonestreet BS. Ontogeny and the effects of in utero brain ischemia on interleukin-1beta and interleukin-6 protein expression in ovine cerebral cortex and white matter. Int J Dev Neurosci. 2012;30:457–63. doi: 10.1016/j.ijdevneu.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fan LW, Tien LT, Zheng B, Pang Y, Rhodes PG, Cai Z. Interleukin-1beta-induced brain injury and neurobehavioral dysfunctions in juvenile rats can be attenuated by alpha-phenyl-n-tert-butyl-nitrone. Neuroscience. 2010;168:240–52. doi: 10.1016/j.neuroscience.2010.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ma Y, Matsuwaki T, Yamanouchi K, Nishihara M. Cyclooxygenase-2-related signaling in the hypothalamus plays differential roles in response to various acute stresses. Brain Res. 2013;1508:23–33. doi: 10.1016/j.brainres.2013.02.042. [DOI] [PubMed] [Google Scholar]
- 68.Cai Z, Pan ZL, Pang Y, Evans OB, Rhodes PG. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr Res. 2000;47:64–72. doi: 10.1203/00006450-200001000-00013. [DOI] [PubMed] [Google Scholar]
- 69.Bilbo SD, Biedenkapp JC, Der-Avakian A, Watkins LR, Rudy JW, Maier SF. Neonatal infection-induced memory impairment after lipopolysaccharide in adulthood is prevented via caspase-1 inhibition. J Neurosci. 2005;25:8000–9. doi: 10.1523/JNEUROSCI.1748-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leviton A, Allred EN, Dammann O, et al. Systemic inflammation, intraventricular hemorrhage, and white matter injury. J Child Neurol. 2012 Oct 30; doi: 10.1177/0883073812463068. epub ahead of print publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hwang IR, Kodama T, Kikuchi S, et al. Effect of interleukin 1 polymorphisms on gastric mucosal interleukin 1beta production in Helicobacter pylori infection. Gastroenterology. 2002;123:1793–803. doi: 10.1053/gast.2002.37043. [DOI] [PubMed] [Google Scholar]
- 72.Wu YW, Croen LA, Torres AR, Van De Water J, Grether JK, Hsu NN. Interleukin-6 genotype and risk for cerebral palsy in term and near-term infants. Ann Neurol. 2009;66:663–70. doi: 10.1002/ana.21766. [DOI] [PubMed] [Google Scholar]
- 73.Girotti M, Donegan JJ, Morilak DA. Influence of hypothalamic IL-6/gp130 receptor signaling on the HPA axis response to chronic stress. Psychoneuroendocrinology. 2013;38:1158–69. doi: 10.1016/j.psyneuen.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harding D, Brull D, Humphries SE, Whitelaw A, Montgomery H, Marlow N. Variation in the interleukin-6 gene is associated with impaired cognitive development in children born prematurely: a preliminary study. Pediatr Res. 2005;58:117–20. doi: 10.1203/01.PDR.0000163523.49021.53. [DOI] [PubMed] [Google Scholar]
- 75.Adcock K, Hedberg C, Loggins J, Kruger T, Baier R. The TNF-alpha - 308, MCP - 1 - 2518 and TGF - beta 1 + 915 polymorphisms are not associated with the development of chronic lung disease in very low birth weight infants. Genes Immun. 2003;4:420–6. doi: 10.1038/sj.gene.6363986. [DOI] [PubMed] [Google Scholar]
- 76.de Vries LS, Koopman C, Groenendaal F, et al. COL4A1 mutation in two preterm siblings with antenatal onset of parenchymal hemorrhage. Ann Neurol. 2009;65:12–8. doi: 10.1002/ana.21525. [DOI] [PubMed] [Google Scholar]
- 77.de Vries LS, Mancini GM. Intracerebral hemorrhage and COL4A1 and COL4A2 mutations, from fetal life into adulthood. Ann Neurol. 2012;71:439–41. doi: 10.1002/ana.23544. [DOI] [PubMed] [Google Scholar]
- 78.Weng YC, Sonni A, Labelle-Dumais C, et al. COL4A1 mutations in patients with sporadic late-onset intracerebral hemorrhage. Ann Neurol. 2012;71:470–7. doi: 10.1002/ana.22682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bilguvar K, DiLuna ML, Bizzarro MJ, et al. COL4A1 Mutation in preterm intraventricular hemorrhage. J Pediatr. 2009;155:743–5. doi: 10.1016/j.jpeds.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ko NU, Rajendran P, Kim H, et al. Endothelial nitric oxide synthase polymorphism (−786T->C) and increased risk of angiographic vasospasm after aneurysmal subarachnoid hemorrhage. Stroke. 2008;39:1103–8. doi: 10.1161/STROKEAHA.107.496596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vannemreddy P, Notarianni C, Yanamandra K, Napper D, Bocchini J. Is an endothelial nitric oxide synthase gene mutation a risk factor in the origin of intraventricular hemorrhage? Neurosurg Focus. 2010;28:E11. doi: 10.3171/2009.10.FOCUS09143. [DOI] [PubMed] [Google Scholar]
- 82.Poggi C, Giusti B, Vestri A, Pasquini E, Abbate R, Dani C. Genetic polymorphisms of antioxidant enzymes in preterm infants. J Matern Fetal Neonatal Med. 2012;25 (Suppl 4):131–4. doi: 10.3109/14767058.2012.714976. [DOI] [PubMed] [Google Scholar]
- 83.Franz DN, Belousova E, Sparangana S, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2013;381:125–32. doi: 10.1016/S0140-6736(12)61134-9. [DOI] [PubMed] [Google Scholar]
- 84.Farooq F, Abadia-Molina F, Mackenzie D, et al. Celecoxib increases SMN and survival in a severe spinal muscular atrophy mouse model via p38 pathway activation. Hum Mol Genet. 2013;22:3415–24. doi: 10.1093/hmg/ddt191. [DOI] [PubMed] [Google Scholar]
- 85.Liew WK, Kang PB. Reent developments in the treatment of Duchenne muscular dystrophy and spinal muscular atrophy. Ther Adv Neurol Disord. 2013;6:147–60. doi: 10.1177/1756285612472386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Manolio TA, Chisholm RL, Ozenberger B, et al. Implementing genomic medicine in the clinic: the feature is here. Genet Med. 2013;15:258–67. doi: 10.1038/gim.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bauer SC, Msall ME. Optimizing neurodevelopmental outcomes after prematurity: lessons in neuroprotection and early intervention. Minerva Pediatr. 2010;62:485–97. [PubMed] [Google Scholar]