

Abstract
Judicious hapten design has been shown to be of importance when trying to generate a viable vaccine against a drug of abuse. Hapten design has typically been predicated upon faithfully emulating the unique chemical architecture that each drug presents. However, the need for drug-hapten congruency may also compromise vaccine immunogenicity if the drug-hapten conjugate possesses epitope instability. There has been no systematic study on the impact of hapten stability as it relates to vaccine immunogenicity. As a starting point, we have probed the stability of a series of cocaine haptens through varying several of its structural elements, including functionality at the C2-position, the nature of the linker and its site of attachment. Accordingly, a hydrolytic stability profile of four cocaine haptens (GNNA, GNNS, GNE and GNC) was produced and these results were compared through each hapten’s immunological properties, which were generated via active vaccination. From this group of four, three of the haptens, GNE, GNNA and GNC were further examined in an animal behavioral model, and findings here were again measured in relationship to hapten stability. We demonstrate a corresponding relationship between the half-life of the hapten and its immunogenicity, wherein haptens presenting a fully representative cocaine framework elicited higher concentrations of cocaine-specific IgG in sera and also conferred better protection against cocaine-induced locomotor activity. Our results indicate that hapten half-life plays an important role in vaccine immunogenicity and this in turn can impact animal behavioral effects when challenged with a drug of abuse.
Keywords: Anti-cocaine vaccine, Hapten, Kinetics, Immunogenicity, Psychomotor stimulant Effects
Introduction
Cocaine abuse and addiction remains a prevalent health and societal problem in the United States with about 2 million users.1 The powerful abuse cycle that cocaine imparts stems from its reinforcing effects, which in part depend on its ability to facilitate dopamine transmission, resulting in heightened alertness and intense feelings of euphoria.2 Currently, there is no therapeutic for the treatment of cocaine addiction. In view of the misuse and lack of treatment, alternative strategies for treating cocaine addiction have been sought. Recent studies have yielded promising results with antibody-based approaches for the treatment of cocaine abuse.3 In contrast to traditional pharmacotherapies, cocaine vaccines act by sequestering cocaine in the circulation prior to its entry into the central nervous system, thus terminating drug-induced rewarding effects without producing unwanted neuroadaptive effects or cognitive impairment.4 To date, only a single cocaine vaccine, termed TA-CD, consisting of succinyl norcocaine (SNC) conjugated to cholera toxin B subunit with aluminum hydroxide gel as the adjuvant, has reached clinical trials.5 However, results observed in clinical trials for this vaccine cocktail have been tempered due to a variation among trial participants in antibody concentrations produced, with high-titer responders achieving abstinence.6 This lack of clinical efficacy has raised renewed interest in cocaine vaccine redesign.4(b),6
A viable vaccine against a drug of abuse, to a great extent, relies on three elements: hapten, carrier protein and adjuvant. While the choice of carrier protein and adjuvant is crucial to ensure a strong immune response, an optimal hapten design is also necessary for antibody specificity and affinity. Thus, how to generate a robust drug-like hapten has been an enduring question in the development of all vaccines against drugs of abuse, including cocaine. While not obvious, the linker and its regiochemical point of hapten attachment should also be carefully considered. For example, a labile chemical bond within the linker could cause the loss of the drug scaffold from the carrier protein, which in turn would diminish the immune response to the hapten. When contemplating cocaine hapten development, the lability of the C2-ester presents another unique challenge;7 hydrolysis at the C2 methyl ester would skew the immune response away from cocaine to its degradation product benzoyl ecgonine.8 An immune response compromised by unwanted reactivity to inactive metabolites would clearly downgrade the value of any cocaine vaccine.
Accordingly, to both compliment and expand upon the previous haptens GNC/GND/GNE, two new antigens, termed GNNA and GNNS, were designed and synthesized (Figure 1). These new cocaine-haptens were designed based on the following: (1) An amine linker was selected for attachment at the tropane nitrogen for both molecules. Our reasoning here was our belief that carrier protein attachment through a bridgehead linker might allow for a more effective hapten presentation of its ester dominant epitopes. (2) Compared to the amide linker utilized within SNC, an amine linker should better mirror the structure of cocaine in terms of chemical identity and electronic properties. (3) To further probe the effect of the C2-ester, both from the standpoint of stability and epitope presentation, we replaced the ester with an amide in GNNA while maintaining the methyl ester in GNNS.
Figure 1.

The structure of (−)-cocaine and cocaine-like haptens.
To systematically investigate each of these haptens’ stability, we developed a strategy for in vitro assessment through the labeling of each hapten with a chromophore at the position of carrier protein attachment. Thus, the stability profile of GNC, GNE, GNNA and GNNS could now be readily accessed. Next, to gauge hapten stability as it relates to vaccine efficacy, we also present the serological analysis of Swiss Webster mice following vaccination with GNC-KLH, GNE-KLH, GNNA-KLH and GNNS-KLH. Lastly, active immunization with GNNA-KLH, GNE-KLH and GNC-KLH was also examined in an animal behavioral model and these results were analyzed from the perspective of hapten stability.
Results
Synthesis of cocaine haptens and hapten-protein immunoconjugates
The synthesis of cocaine haptens GNNS and GNNA is illustrated in Scheme 1. The synthesis started with the demethylation of (−)-cocaine hydrochloride by forming a carbamate intermediate followed by treatment with zinc dust. The obtained (−)-norcocaine was coupled with 4-bromobutyric benzyl ester in the presence of Bu4NI and Et3N in CH3CN to afford compound 1 in 43% yield. Hydrogenolysis of 1 in the presence of 1 atm of H2 and 10% Pd-C in MeOH generated the desired compound 2 (GNNS). To obtain compound 5 (GNNA), the synthesis was initiated from compound 1. The methyl ester group of 1 was hydrolyzed by microwave irradiation in 1:1 dioxane–H2O at 160 °C to provide compound 3 in 98% yield. Amidation of acid 3 was achieved in 83% yield using methylamine hydrochloride, EDC and DMAP in DCM. Finally, compound 5 (GNNA) was generated by catalytic hydrogenolysis in excellent yield.
Scheme 1.

Following hapten synthesis, the corresponding immunoconjugates were prepared by coupling haptens GNC, GNE, GNNA and GNNS to the carrier protein keyhole limpet hemocyanin (KLH) under standard protein conjugation conditions.12 Prior to administration, each protein conjugate as well as the control vehicle, KLH, were formulated with SAS (Sigma Adjuvant System®), a stable oil-in-water emulsion derived from bacterial and mycobacterial cell walls.
In addition, all haptens studied were also coupled to bovine serum albumin (BSA) for ELISA microtiter plate coating as well as monitoring the coupling efficiency using MALDI-TOF mass spectrometry.
Hapten stability study
To examine the stability profile of the cocaine haptens, each compound was coupled to 7-methoxycoumarin-4-acetic acid (MCA) through an ethylenediamine spacer. As shown in Scheme 2, MCA was coupled with N-Boc-ethylenediamine in the presence of EDC and DMAP in DCM, followed by the treatment of 50% TFA in DCM to afford amine 6 in 60% yield. Finally, amine 6 was coupled with GNNS, GNNA, GNC and GNE to afford MCA-labeled products 7–10 in yields that varied from 42% to 90%. The individual labeled cocaine-haptens were then dissolved in normal rat sera, and incubated at 37 °C. The degradation of each MCA-conjugated hapten was determined using an HPLC-based assay with monitoring at 325 nm.
Scheme 2.

The half-lives (T1/2) of GNC-MCA, GNE-MCA and GNNS-MCA in rat sera are shown in Figure 2. Rat sera allowed for the interrogation of each of these haptens for esterolytic stability. Our choice of rat sera reflected its availability in our laboratory and we note that hydrolase activity levels differ among rodent models; however, we felt these differences would not impact the overall concept we set out to explore, namely how hapten stability impacts immunogenicity. GNE-MCA proved to be the most stable with a T1/2 = 60.3 hrs, followed by GNNA-MCA, which displayed a half-life of 34.7 hrs. Unsurprisingly, GNC-MCA and GNNS-MCA were the least stable, with half-lives in sera of 26.1 hrs and 16.5 hrs, respectively. Notably, for GNE-MCA and GNNA-MCA, the only degradation product observed resulted from loss of the benzyl ester at the C3 position, and no C2 amide degradation product was detected during the course of the study (data not shown).
Figure 2.

Half-life (T1/2) of MCA-labeled cocaine haptens (GNNS-MCA, GNC-MCA, GNNA-MCA and GNE-MCA) as measured in rat sera. The degradation of each MCA-labeled hapten was monitored using an HPLC-based assay.
Titers and affinities of cocaine-specific antibodies generated by active immunization in mice
The efficacy of the prepared immunoconjugates was assessed by vaccination of groups of 5 Swiss Webster mice. We selected Swiss Webster mice as they have been reported to show significant cocaine-induced locomotor hyperactivity.13 Test groups included KLH negative control, GNC-KLH, GNE-KLH, GNNS-KLH and GNNA-KLH. Each vaccine was administered on days 0, 21, and 42 with bleeds taken one week post-vaccination. This vaccination schedule has previously been reported by our laboratory.8,9 The success of active vaccination is contingent upon both the magnitude of the response as well as the affinity and specificity of the antibodies generated. The magnitude of the immune response was assessed by ELISA on microtiter plates coated with the corresponding hapten-BSA conjugates. As shown in Table 1, GNE-KLH evoked the highest-titer antibodies (~1:30000) followed by GNNA-KLH, GNC-KLH and GNNS-KLH, all of which elicited antibody titers in the ~1:20000 level.
Table 1.
Average ELISA titer and average relative affinity of antisera from immunized mice against cocaine as determined by equilibrium dialysis.a
| Immunoconjugate | titer | Kd (mean ± SD, nM) | [Ab] (mean ± SD, μg/mL) |
|---|---|---|---|
| GNNS-KLH | 23000 | 2.05 ± 0.96 | 65.43 ± 24.22 |
| GNC-KLH | 21000 | 0.79 ± 0.16 | 47.53 ± 9.54 |
| GNNA-KLH | 19800 | 3.59 ± 1.47 | 73.15 ± 14.17 |
| GNE-KLH | 28000 | 5.68 ± 0.39 | 123.47 ± 26.69 |
All assays were performed in triplicate.
ND, not detectable.
All binding constants, as well as antibody concentrations, were calculated from a soluble radioimmunoassay (RIA) and normalized to allow for direct comparison of values between test groups. As shown in Table 1, the polyclonal responses of all four vaccines had low nanomolar affinities. The anti-cocaine antibody with the highest affinity was observed with GNC (Kd ~ 0.97 nM), followed by GNNS and GNNA with a comparable affinity between 2.0 and 4.0 nM, and finally GNE, which showed a cocaine-binding affinity about 5.7 nM.
Cocaine-specific IgG concentrations were also calculated from the RIA analysis,14 which are shown in Table 1. Among the four investigated vaccines, GNE-KLH provided the highest cocaine-specific IgG concentration in sera of approximately 123 μg/mL, followed by the GNNA-KLH (~ 73 μg/ml) and GNNS-KLH (~ 65 μg/mL). GNC-KLH demonstrated the lowest immunogenicity among the four immunoconjugates, with an antibody concentration in sera of about half that produced by GNE-KLH.
Psychomotor activation effects of cocaine in mice
Four groups of 8 vaccinated mice (KLH negative control, GNNA-KLH, GNE-KLH and GNC-KLH) were assessed for psychomotor stimulant effects of cocaine seven days after the second immunization boost. Following a session of habituation to the test chambers and intraperitoneal (i.p.) injections of saline, the mice were challenged with injections of 5 mg/kg cocaine (i.p.) and evaluated for drug-induced locomotor activity (Figures 3S and 4S). In response to 5 mg/kg cocaine challenge, mice vaccinated with KLH showed significant increases in the number of beam breaks (Figure 3A and Figure 4S, treatment effect F (2,23)=7.63, p<0.01) compared to saline injection. The anti-cocaine antibodies elicited by GNE-KLH vaccination completely prevented the drug-induced locomotor hyperactivity in response to the administration of cocaine during the entire testing period (Figure 3A and Figure 4S). Similarly, the anti-cocaine antibodies elicited by GNNA-KLH vaccination blocked the drug-induced locomotor hyperactivity in response to the administration of cocaine during the testing period (Figure 3A and Figure 4S). For mice vaccinated with GNC-KLH, the anti-cocaine antibodies showed no significant protection against 5 mg/kg cocaine during the test session in beam breaks (Figure 3A and Figure 4S; treatment effect F (2,23)=13.04, p<0.001). Crossovers were not significantly elevated by 5 mg/kg cocaine challenge for any of the groups (Figure 3B).
Figure 3.
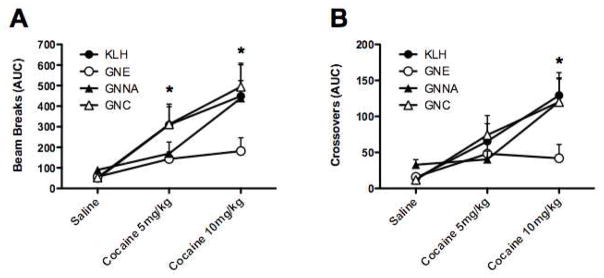
Efficacies of GNE-KLH, GNNA-KLH and GNC-KLH in suppressing the psychomotor stimulating effects (beam breaks: Figure 3A; crossovers: Figure 3B) of repeated cocaine treatment in mice. *p < 0.05.
Subsequently, all mice were habituated and injected with saline, then challenged with 10 mg/kg cocaine on the next day (Figures 5S and 6S). In response to this higher dose cocaine challenge, mice vaccinated with KLH showed significant increases in the number of beam breaks (Figure 3A, treatment effect F (2,23)=7.63, p<0.01) and crossovers (Figure 3B, treatment effect F (2,23)=10.3, p<0.01) compared to saline injection. Again, the anti-cocaine antibodies elicited by GNE-KLH vaccination completely prevented the drug-induced locomotor hyperactivity in response to the administration of 10 mg/kg cocaine during the entire testing period (Figure 3A and B, Figure 6S). However, mice vaccinated with GNNA-KLH no longer demonstrated significant protection against the psychomotor-stimulating effects of a higher cocaine dose, and showed comparable drug-induced locomotor hyperactivity during the testing period as control mice (Figure 3A and Figure 6S). As was demonstrated previously, mice vaccinated with GNC-KLH showed no significant protection against 10 mg/kg cocaine during the test session (Figure 6S), and exhibited significant increases in the number of beam breaks (Figure 3A, treatment effect F (2,23)=7.63, p<0.01) and crossovers (Figure 3B, treatment effect F (2,23)=10.3, p<0.01) compared to saline injection.
Discussion
A potent titer coupled to a highly specific immune response are two facets thought to be important in developing a successful vaccine. Typically, an immune response occurs in four sequential phases, i.e., innate immunity (0–4 hrs), early adaptive response (4–96 hrs), adaptive immune response (4–7 days) and finally immunological memory (several years or more).15 In developing vaccines against drugs of abuse, a naive T cell must first encounter the antigen in the early adaptive response, and then be induced to proliferate and differentiate into armed effector T cells that contribute to the activation of antibody-secreting B cells. Taking these core immunological tenets into account, we posit that retention of the complete cocaine scaffold during the antigen presentation pathway (4–96 hrs) is imperative to ensure maximal cocaine-specific immune response. From the standpoint of developing a cocaine vaccine, hydrolysis of one of the two major epitopes or scaffold elimination due to linker hydrolysis would result in either non-specific or ineffective anti-cocaine immune response. As a means to test this hypothesis, we examined the chemical stability profile of four cocaine haptens labeled with 7-methoxycoumarin-4-acetic acid (MCA). The conjugation of this chromophore allows tracking of the hydrolysis at the more labile C2-ester position and mirrors the position of carrier protein attachment. The main metabolic pathway of cocaine is hydrolysis to first benzoylecgonine and then ecgonine methyl ester. Similarly, the major degradation products observed in our in vitro assay were from the hydrolysis of C2 and/or C3 ester.
As shown in Figure 2, among the studied haptens GNE displayed the longest half-life (T1/2 ~ 60.3 hrs) followed by GNNA (T1/2 ~ 34.7 hrs) then GNC (T1/2 ~ 26.1 hrs) with GNNS being the least stable (T1/2 ~ 16.5 hrs). This observed order of stability is readily explained by the structural features incorporated within each of the haptens. With a C2-amide functionality, GNE and GNNA should be more stable than the other two haptens possessing a C2-ester group. Indeed, HPLC analysis confirmed that the only hydrolysis product detected for GNE-MCA and GNNA-MCA during the course of the study was at the C3 position. In addition, we note that the site of linker attachment seems to have an impact on the degradation of the haptens studied. With constant functionality installed at the C2-position, haptens also containing a linker appendage at this position demonstrated a longer half-life than those with a linker at the tropane nitrogen (T1/2 ~ 60.3 hrs for GNE versus T1/2 ~ 34.7 hrs for GNNA, or T1/2 ~ 26.1 hrs for GNC versus T1/2 ~ 16.5 hrs for GNNS). A plausible explanation for these findings is that steric hindrance from the C2-linker impedes the hydrolysis of the C3-benzoyl ester, thus prolonging hapten half-life.
The serological analysis of Swiss Webster mice vaccinated with our four immunoconjugates provides an additional metric that hapten stability plays a substantial role in vaccine immunogenicity (Table 1). Among the haptens studied, GNE, the most stable, produced the highest concentration of cocaine-specific IgG in sera (~ 123 μg/mL). GNNA, which exhibited a shorter half-life than GNE, demonstrated a lower immunogenicity; the antibody concentration in sera was about 40% less than that elicited by GNE. Compared to GNE and GNNA, GNC and GNNS, both with shorter half-lives, were also less immunogenic. Interestingly, GNNS, which exhibited the shortest in vitro half-life, was still moderately immunogenic. We view this result from the perspective that while the C-2 ester on this hapten can be readily hydrolyzed the full tropane scaffold would still remain intact on the carrier protein, wherein with GNC hydrolysis of this bond would result in complete hapten loss. Taken collectively, hapten half-life is a key determinant of vaccine immunogenicity.
As stated, the success of active vaccination is contingent upon both the magnitude of the response as well as the affinity and specificity of the antibodies generated. Gratifyingly, polyclonal antibodies elicited by all four of these haptens displayed high affinity to cocaine with Kd values between 0.9 and 6.0 nM. By comparing the structural features and the affinities, it was apparent that the nature of the linker played an important role in determining the cocaine-binding affinity. The results suggest that the C2-ester may better mimic the cocaine structure than the amide bond (Kd ~ 0.97 nM for GNC and Kd ~ 2.05 nM for GNNS versus Kd ~ 5.68 nM for GNE).
As a means to correlate hapten stability with behavioral studies, three of the immunoconjugates, GNC-KLH, GNE-KLH and GNNA-KLH, were assessed for their protective effects in a mouse model of acute cocaine-induced locomotor activity. These three haptens were chosen due to their distinct structural stability and immunogenicity. As demonstrated in Figure 3, active immunization with GNE-KLH resulted in a significantly blunted behavioral response to cocaine during the 5 mg/kg and 10 mg/kg testing periods. For GNNA-KLH, the protective effect was only seen at low drug doses and was surmounted at higher doses of cocaine. GNC-KLH showed no significant protection against either 5 mg/kg or 10 mg/kg cocaine challenge. These results are consistent with antibody concentration as the guiding factor, where the highest cocaine-specific IgG concentration in sera was observed with antibodies induced by GNE (~ 123 μg/mL) followed by GNNA (~ 73 μg/mL), with GNC having the lowest concentration (~ 47 μg/mL, Table 1). Furthermore, the superior behavioral effects observed with GNE-KLH corroborate our previous reports,11 which indicate that replacement of the C2-ester with the corresponding C2-amide affords a stable hapten with sustainable and robust immunogenicity. Thus, the GNE hapten represents a promising candidate for anti-cocaine vaccination development.
Conclusions
In summary, a series of experiments were conducted to investigate whether hapten stability would have an impact on cocaine vaccine immunogenicity. To probe this premise, four haptens were engaged, each with unique structural characteristics ranging from hapten stability epitope-display to linker regiochemical placement/stability. Within this context, we demonstrated a corresponding relationship between the half-life of the hapten and its immunogenicity, wherein haptens presenting a fully representative cocaine framework elicited higher concentrations of cocaine-specific IgG in sera and also conferred better protection against cocaine-induced locomotor activity. We suggest that drug-hapten stability is a crucial determinant for elicitation of potent immunogenicity, and that “hapten-structure-tailoring” represents a unique and powerful approach for developing vaccines against drugs of abuse.
Experimental Section
All reactions involving air or moisture-sensitive reagents or intermediates were performed under an argon atmosphere. Chemicals and solvents were of reagent grade and were used without further purification. The (−)-cocaine hydrochloride was supplied by the National Institute on Drug Abuse (NIDA). Flash chromatography was performed on silica gel 60 (230–400 mesh) and analytical TLC was carried out on glass plates coated with a 0.25-mm layer of silica gel 60 F-254. HPLC separations were performed on a Vydac 218TP C18 reversed phase preparative (10–15 μm) HPLC column using a gradient of acetonitrile and water. The LC/MS analysis was performed using an Agilent G-1956D single quadrupole mass spectrometer equipped with an 1100 Series LC system from Agilent Technologies. 1H and 13C NMR spectra were obtained using Bruker 500 MHz or 600 MHz instruments. Chemical shifts are reported in parts per million (ppm, δ) referenced to the residual 1H resonance of the solvent (CDCl3, 7.26 ppm) or (CD3OD, 3.31 ppm). 13C spectra are referenced to the residual 13C resonance of the solvent (CDCl3, 77.0 ppm) or (CD3OD, 49.0 ppm). Splitting patterns were designated as follows: s, singlet; br, broad; d, doublet; dd, doublet of doublets; t, triplet; q, quartet; m, multiplet. High resolution mass spectra were obtained in the Scripps Center for Mass Spectrometry. Hapten protein conjugates were analyzed using MALDI-TOF MS. The purity of all synthetic compounds was higher than 95% as determined by HPLC and/or LC/MS.
(1R,2R,3S,5S)-methyl 3-(benzoyloxy)-8-(4-(benzyloxy)-4-oxobutyl)-8-azabicyclo[3.2.1]octane-2-carboxylate (1)
To a solution of 58.3 mg (0.20 mmol) of norcocaine in 2 mL of CH3CN was added 62.0 mg (0.24 mmol) of benzyl 4-bromobutanoate and 39.3 μL (0.28 mmol) of Et3N, followed by 5.6 mg (0.015 mmol) of Bu4NI. The reaction mixture was refluxed at 46 °C overnight. After cooling to room temperature, the mixture was diluted with sat aq NH4Cl. The mixture was extracted with EtOAc. The combined organic layer was washed with brine, dried (MgSO4) and concentrated under diminished pressure. The residue was purified by flash chromatography on a silica gel column (20 ×1.7 cm). Elution with 10:1 hexanes/EtOAc gave the product 1 as a yellowish oil: yield 40.3 mg (43.0%); silica gel TLC Rf 0.41 (7:3 hexanes/EtOAc); 1H NMR (CDCl3) δ 1.66–1.73 (m, 4H), 1.84–1.85 (m, 1H), 1.94–2.02 (m, 2H), 2.26 (t, 2H, J = 6.0 Hz), 2.38–2.50 (m, 3H), 3.01 (t, 1H, J = 6.0 Hz), 3.28 (br, 1H), 3.62 (s, 3H), 3.63–3.65 (m, 1H), 5.09–5.13 (m, 2H), 5.22 (quin, 1H, J = 6.0 Hz), 7.29–7.33 (m, 1H), 7.34–7.35 (m, 4H), 7.41 (t, 2H, J = 6.0 Hz), 7.52 (t, 1H, J = 6.0 Hz) and 8.01–8.03 (m, 2H); 13C NMR (CDCl3) δ 25.23, 26.36, 26.98, 32.53, 36.52, 51.13, 52.09, 52.71, 61.48, 63.51, 66.91, 68.17, 129.01, 129.06, 129.17, 129.17, 129.37, 130.55, 131.22, 133.76, 136.97, 167.02, 171.46 and 174.54; mass spectrum (ESI), m/z 466.2224 (M+H)+ (C27H32NO6 requires m/z 466.2224).
4-((1R,2R,3S,5S)-3-(benzoyloxy)-2-(methoxycarbonyl)-8-azabicyclo[3.2.1]octan-8- yl)butanoic acid (2)
A mixture of 19.0 mg (0.04 mmol) of 1 and 9.5 mg of 10% Pd/C in 1 mL of MeOH was stirred under a H2 atm at room temperature for 2 hrs. The catalyst was removed by filtration and the crude products were purified on a VYDAC® C18 reversed phase semi-preparative (250 × 22 mm, 10–15 μm) HPLC column using water and 0.1% TFA in CH3CN mobile phases. A linear gradient was employed (90:10 H2O/0.1%TFA in CH3CN → 10:90 H2O/0.1%TFA in CH3CN) over a period of 40 min at a flow rate of 10 mL/min. Fractions containing the desired product were collected, frozen and lyophilized to give 2 as colorless oil: yield 14.4 mg (94.1%); −57.90° (c 1.38, MeOH); 1H NMR (CD3OD) δ δ 1.87–1.98 (m, 2H), 2.07–2.14 (m, 2H), 2.26–2.48 (m, 6H), 2.96–2.99 (m, 2H), 3.50 (dd, J = 6.0 Hz, 12.0 Hz), 3.96 (s, 3H), 3.94–3.96 (m, 1H), 4.27 (d, 1H, J = 6.0 Hz), 5.53 (quin, 1H, J = 6.0 Hz), 7.49 (t, 2H, J = 6.0 Hz), 7.62 (t, 1H, J = 6.0 Hz) and 7.65 (d, 2H, J = 6.0 Hz); 13C NMR (CD3OD) δ 22.48, 24.13, 24.63, 33.96, 34.00, 48.01, 52.55, 52.90, 62.40, 63.12, 65.63, 129.15, 129.98, 130.18, 134.15, 166.14, 172.94 and 178.70; mass spectrum (ESI), m/z 376.1754 (M+H)+ (C20H26NO6 requires m/z 376.1755).
(1R,2R,3S,5S)-3-(benzoyloxy)-8-(4-(benzyloxy)-4-oxobutyl)-8-azabicyclo[3.2.1]octane-2-carboxylic acid (3)
To a solution of 19 mg (0.40 mmol) of 1 in 1 mL of 1.2 mL of dioxane was added 1.2 mL of water in a 10-mL microwave reaction tube. The reaction vial was then placed in the microwave heater and heated to 160 °C with the following settings: maximum pressure 250 psi, 300 watts, and medium stirring. Reaction progress was monitored by TLC and upon disappearance of 1, the mixture was transferred to a round-bottomed flask and concentrated under diminished pressure. The crude product was used for the next step promptly without any purification or characterization.
(1R,2R,3S,5S)-8-(4-(benzyloxy)-4-oxobutyl)-2-(methylcarbamoyl)-8- azabicyclo[3.2.1]octan-3-yl benzoate (4)
To a solution of 12.0 mg (0.027 mmol) of 3 in 1 mL of DCM was added 8.8 μL (0.08 mmol) of N-methylmorpholine and 7.2 mg (0.04 mmol) of EDC, followed by 53.2 μL (0.11 mmol) of methylamine (2.0 M in THF) in 1mL of DCM and 1.0 mg (0.008 mmol) of DMAP at 0 °C. The reaction mixture was stirred at room temperature overnight before the solvent was removed under diminished pressure. The crude product was purified on a VYDAC® C18 reversed phase semi-preparative (250 × 22 mm, 10–15 μm) HPLC column using water and 0.1% TFA in CH3CN mobile phases. A linear gradient was employed (90:10 H2O/0.1%TFA in CH3CN → 10:90 H2O/0.1%TFA in CH3CN) over a period of 40 min at a flow rate of 10 mL/min. Fractions containing the desired product were collected, frozen and lyophilized to give 4 as colorless oil: yield 10.2 mg (82.6%); 1H NMR (CD3OD) δ 2.02–2.09 (m, 2H), 2.10–2.22 (m, 2H), 2.33–2.39 (m, 3H), 2.47–2.50 (m, 1H), 2.51–2.63 (m, 2H), 3.73 (d, 3H, J = 6.0 Hz), 3.03–3.14 (m, 2H), 3.17 (d, 1H, J = 6.0 Hz), 4.07–4.08 (m, 1H), 4.29–4.31 (m, 1H), 5.16 (s, 2H), 5.54–5.59 (m, 1H), 7.31–7.39 (m, 4H), 7.49 (t, 2H, J = 6.0 Hz), 7.63 (d, 1H, J = 6.0 Hz), 7.95 (d, 2H, J = 6.0 Hz) and 8.45–8.47 (m, 1H); 13C NMR (CD3OD) δ 20.92, 24.15, 24.16, 25.98, 30.87, 33.82, 46.36, 51.29, 63.22, 63.42, 65.08, 67.11, 128.79, 128.85, 129.03, 129.18, 130.00, 134.27, 136.88, 165.88, 173.15 and 173.18; mass spectrum (ESI), m/z 465.2383 (M+H)+ (C27H33N2O5 requires m/z 465.2384).
4-((1R,2R,3S,5S)-3-(benzoyloxy)-2-(methylcarbamoyl)-8-azabicyclo[3.2.1]octan-8- yl)butanoic acid (5)
A mixture of 10.0 mg (0.02 mmol) of 4 and 5 mg of 10% Pd/C in 1 mL of MeOH was stirred under a H2 atm at room temperature for 2 hrs. The catalyst was removed by filtration and the crude products were purified on a VYDAC® C18 reversed phase semi-preparative (250 × 22 mm, 10–15 μm) HPLC column using water and 0.1% TFA in CH3CN mobile phases. A linear gradient was employed (90:10 H2O/0.1%TFA in CH3CN → 10:90 H2O/0.1%TFA in CH3CN) over a period of 40 min at a flow rate of 10 mL/min. Fractions containing the desired product were collected, frozen and lyophilized to give 5 as colorless oil: yield 8.0 mg (99%); −26.13° (c 0.8, MeOH); 1H NMR (CD3OD) δ 2.00–2.06 (m, 2H), 2.14–2.24 (m, 2H), 2.36–2.42 (m, 2H), 2.43–2.54 (m, 4H), 2.74 (d, 3H, J = 6.0 Hz), 3.06–3.16 (m, 2H), 3.19 (d, 1H, J = 6.0, 12.0 Hz), 4.09–4.11 (m, 1H), 4.34 (d, 1H, J = 12.0 Hz), 5.56–5.60 (m, 1H), 7.49 (t, 2H, J = 6.0 Hz), 7.63 (t, 1H, J = 6.0 Hz), 7.95 (d, 2H, J = 6.0 Hz) and 8.46 (br, 1H); 13C NMR (CD3OD) δ 20.98, 24.17, 24.20, 25.83, 25.97, 30.62, 33.84, 46.35, 46.39, 51.40, 63.26, 63.36, 65.11, 129.18, 130.00, 134.27, 165.89, 173.09, 173.18 and 175.18; mass spectrum (ESI), m/z 375.1915 (M+H)+ (C27H32NO6 requires m/z 375.1914).
N-(2-aminoethyl)-2-(7-methoxy-2-oxo-2H-chromen-4-yl)acetamide (6)
To a solution of 190.0 mg (0.811 mmol) of 7-methoxycoumarin-4-acetic acid in 2.5 mL of DCM was added 179.0 mg (0.94 mmol) of EDC, 98.0 μL (0.62 mmol) of N-Boc-ethylenediamine in DCM followed by 23.0 mg (0.19 mmol) of DMAP at 0 °C. After stirring at room temperature overnight, the reaction mixture was quenched with saturated NH4Cl aqueous solution and extracted with EtOAc. The combined organic layer was washed with brine, dried (MgSO4) and concentrated under diminished pressure. The remaining residue was placed under high vacuum for 2 hrs, followed by the addition of 1 mL of DCM and 1mL of TFA at 0 °C. The resultant mixture was stirred at room temperature for 2 hrs before the solvent was removed under diminished pressure. The crude product was purified on a VYDAC® C18 reversed phase semi-preparative (250 × 22 mm, 10–15 μm) HPLC column using water and 0.1% TFA in CH3CN mobile phases. A linear gradient was employed (90:10 H2O/0.1%TFA in CH3CN → 10:90 H2O/0.1%TFA in CH3CN) over a period of 40 min at a flow rate of 10 mL/min. Fractions containing the desired product were collected, frozen and lyophilized to give 6 as colorless oil: yield 103.4 mg (60%); 1H NMR (CD3OD) δ 3.15 (t, 2H, J = 6.0 Hz), 3.56 (t, 2H, J = 6.0 Hz), 3.87 (s, 2H), 3.97 (s, 3H), 6.33 (s, 1H), 6.99–7.04 (m, 2H), and 7.75 (d, 1H, J = 12.0 Hz); 13C NMR (CD3OD) δ 37.49, 38.80, 39.67, 55.44, 100.92, 112.72, 112.98, 113.05, 126.35, 150.91, 155.79, 162.13, 163.68 and 171.08; mass spectrum (ESI), m/z 277.1189 (M+H)+ (C27H32NO6 requires m/z 277.1183).
(1R,2R,3S,5S)-methyl 3-(benzoyloxy)-8-(4-((2-(2-(7-methoxy-2-oxo-2H-chromen-4- yl)acetamido)ethyl)amino)-4-oxobutyl)-8-azabicyclo[3.2.1]octane-2-carboxylate (7)
To a solution of 4.8 mg (0.013 mmol) of 2 in 1 mL of DMF was added 8.5 μL (0.076 mmol) of N-methylmorpholine and 27.7 mg (0.14 mmol) of EDC, followed by 10.0 mg (0.036 mmol) of 7 in 1mL of DCM and 0.8 mg (0.0063 mmol) of DMAP at 0 °C. The reaction mixture was stirred at room temperature overnight before the solvent was removed under diminished pressure. The crude product was purified on a VYDAC® C18 reversed phase semi-preparative (250 × 22 mm, 10–15 μm) HPLC column using water and 0.1% TFA in CH3CN mobile phases. A linear gradient was employed (90:10 H2O/0.1%TFA in CH3CN → 10:90 H2O/0.1%TFA in CH3CN) over a period of 40 min at a flow rate of 10 mL/min. Fractions containing the desired product were collected, frozen and lyophilized to give 7 as colorless oil: yield 6.9 mg (85%); 1H NMR (CD3OD) δ 1.93–2.06 (m, 2H), 2.14–2.22 (m, 2H), 2.33 (t, 3H, J = 6.0 Hz), 2.35–2.48 (m, 3H), 3.10 (t, 2H, J = 6.0 Hz), 3.28–3.30 (m, 3H), 3.34–3.39 (m, 3H), 3.58–3.60 (m, 4H), 3.72–3.76 (m, 2H), 3.88 (s, 3H), 4.06–4.08 (m, 1H), 5.57–5.61 (m, 1H), 6.24 (s, 1H), 6.92–6.97 (m, 2H), 7.50 (t, 2H, J = 6.0 Hz), 7.62–7.68 (m, 2H) and 7.94 (d, 2H J =12.0 Hz); 13C NMR (CD3OD) δ 21.25, 23.58, 24.27, 32.60, 33.50, 39.52, 39.69, 39.85, 46.51, 52.28, 52.91, 55.92, 62.06, 63.74, 64.94, 101.36, 113.19, 113.20, 113.48, 126.83, 129.23, 129.88, 129.94, 134.31, 151.89, 156.21, 162.62, 164.11, 165.88, 170.61, 173.80 and 173.97; mass spectrum (ESI), m/z 634.2755 (M+H)+ (C34H40N3O9 requires m/z 634.2759).
(1R,2R,3S,5S)-methyl 3-(benzoyloxy)-8-(4-((2-(2-(7-methoxy-2-oxo-2H-chromen-4- yl)acetamido)ethyl)amino)-4-oxobutyl)-8-azabicyclo[3.2.1]octane-2-carboxylate (8)
To a solution of 1.8 mg (0.0048 mmol) of 5 in 1 mL of DMF was added 3.2 μL (0.029 mmol) of N-methylmorpholine and 9.2 mg (0.048 mmol) of EDC, followed by 9.0 mg (0.033 mmol) of 7 in 1mL of DCM and 0.3 mg (0.0023 mmol) of DMAP at 0 °C. The reaction mixture was stirred at room temperature overnight before the solvent was removed under diminished pressure. The crude product was purified on a VYDAC® C18 reversed phase semi-preparative (250 × 22 mm, 10–15 μm) HPLC column using water and 0.1% TFA in CH3CN mobile phases. A linear gradient was employed (90:10 H2O/0.1%TFA in CH3CN → 10:90 H2O/0.1%TFA in CH3CN) over a period of 40 min at a flow rate of 10 mL/min. Fractions containing the desired product were collected, frozen and lyophilized to give 8 as colorless oil: yield 2.7 mg (90%); 1H NMR (CD3OD) δ 1.90–2.21 (m, 2H), 2.29–2.53 (m, 2H), 2.70–2.71 (m, 3H), 2.98–3.09 (m, 2H), 3.22 (s, 3H), 3.45 (s, 3H), 3.73–3.79 (m, 3H), 3.76–3.78 (m, 3H), 3.88–3.89 (m, 3H), 4.06–4.07 (m, 1H), 4.28–4.29 (m, 1H), 5.54–5.58 (m, 1H), 6.23 (s, 1H), 6.91–6.96 (m, 2H), 7.48 (t, 1H, J = 6.0 Hz), 7.62–7.68 (m, 3H) and 7.92–1.94 (m, 2H); 13C NMR (CD3OD) δ 21.48, 24.11, 24.24, 25.99, 32.41, 32.46, 33.83, 39.30, 39.52, 39.71, 46.44, 51.55, 55.85, 55.92, 63.27, 63.31, 65.12, 101.34, 101.38, 113.14, 113.25, 113.50, 126.82, 129.18, 130.00, 134.27, 151.54, 151.84, 156.21, 156.22, 162.63, 164.08, 164.10 and 165.91; mass spectrum (ESI), m/z 633.2919 (M+H)+ (C34H41N4O8 requires m/z 633.2919).
(1R,2R,3S,5S)-6-((2-(2-(7-methoxy-2-oxo-2H-chromen-4-yl)acetamido)ethyl)amino)-6- oxohexyl 3-(benzoyloxy)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate (9)
To a solution of 5.0 mg (0.012 mmol) of GNC in 1 mL of DMF was added 2.6 μL (0.024 mmol) of N-methylmorpholine and 7.2 mg (0.037 mmol) of EDC, followed by 6.8 mg (0.024 mmol) of 7 in 1mL of DCM and 0.45 mg (0.0037 mmol) of DMAP at 0 °C. The reaction mixture was stirred at room temperature overnight before the solvent was removed under diminished pressure. The crude product was purified on a VYDAC® C18 reversed phase semi-preparative (250 × 22 mm, 10–15 μm) HPLC column using water and 0.1% TFA in CH3CN mobile phases. A linear gradient was employed (90:10 H2O/0.1%TFA in CH3CN → 10:90 H2O/0.1%TFA in CH3CN) over a period of 40 min at a flow rate of 10 mL/min. Fractions containing the desired product were collected, frozen and lyophilized to give 9 as colorless oil: yield 3.3 mg (42%); 1H NMR (CD3OD) δ 1.00–1.06 (m, 2H), 1.18–1.35 (m, 4H), 1.93 (t, 3H, J = 6.0 Hz), 2.18–2.26 (m, 2H), 2.37–2.51 (m, 4H), 2.88 (s, 3H), 3.24–3.27 (m, 3H), 3.59–3.60 (m, 1H), 3.72 (s, 2H), 3.88 (2, 3H), 3.97–4.07 (m, 3H), 4.22–4.24 (m, 1H), 5.54–5.58 (m, 2H), 6.24 (s, 1H), 6.92–6.95 (m, 2H), 7.47–7.50 (m, 2H), 7.62–7.67 (m, 2H) and 7.94–7.96 (m, 2H); 13C NMR (CD3OD) δ 23.20, 24.29, 25.56, 25.72, 28.25, 33.33, 36.14, 38.87, 39.42, 39.56, 39.83, 46.70, 55.90, 63.97, 64.68, 64.89, 66.74, 100.08, 101.39, 113.15, 113.48, 126.81, 129.30, 129.89, 130.06, 134.43, 151.66, 156.23, 162.58, 164.07, 165.85, 170.53,173.35 and 175.55; mass spectrum (ESI), m/z 662.3086 (M+H)+ (C36H44N3O9 requires m/z 662.3072).
(1R,2R,3S,5S)-2-((6-((2-(2-(7-methoxy-2-oxo-2H-chromen-4-yl)acetamido)ethyl)amino)-6- oxohexyl)carbamoyl)-8-methyl-8-azabicyclo[3.2.1]octan-3-yl benzoate (10)
To a solution of 7.0 mg (0.01 mmol) of GNE in 1 mL of DMF was added 8.5 μL (0.08 mmol) of N-methylmorpholine and 27.7 mg (0.14 mmol) of EDC, followed by 10.0 mg (0.04 mmol) of 7 in 1mL of DCM and 0.8 mg (0.006 mmol) of DMAP at 0 °C. The reaction mixture was stirred at room temperature overnight before the solvent was removed under diminished pressure. The crude product was purified on a VYDAC® C18 reversed phase semi-preparative (250 × 22 mm, 10–15 μm) HPLC column using water and 0.1% TFA in CH3CN mobile phases. A linear gradient was employed (90:10 H2O/0.1%TFA in CH3CN → 10:90 H2O/0.1%TFA in CH3CN) over a period of 40 min at a flow rate of 10 mL/min. Fractions containing the desired product were collected, frozen and lyophilized to give 10 as colorless oil: yield 10.2 mg (61%); silica gel TLC Rf 0.41 (7:3 hexanes/EtOAc); 1H NMR (CD3OD) δ 1.10–1.15 (m, 6H), 1.25–1.41 (m, 4H), 1.99 (t, 2H, J = 6.0 Hz), 2.14–2.36 (m, 2H), 2.41–2.58 (m, 4H), 3.06–3.09 (m, 1H), 3.18–3.21 (m, 2H), 3.25–3.26 (m, 2H), 3.72 (s, 3H), 3.88 (s, 3H), 3.98–4.01 (m, 1H), 4.14–4.16 (m, 1H), 5.51–5.55 (m, 2H), 6.24 (s, 1H), 6.90–6.95 (m, 2H), 7.45–7.48 (m, 2H), 7.60–7.66 (m, 2H) and 7.94–7.96 (m, 2H); 13C NMR (CD3OD) δ 23.72, 24.25, 25.65, 26.88, 29.35, 33.69, 36.23, 38.15, 39.44, 39.61, 39.90, 46.64, 48.99, 55.90, 63.70, 64.94, 65.87, 90.98, 101.38, 113.16, 113.45, 113.61, 126.78, 129.19, 129.99, 130.10, 134.34, 151.67, 156.23, 162.59, 164.08, 165.99, 170.65, 172.39,173.61 and 175.6; mass spectrum (ESI), m/z 661.3237 (M+H)+ (C36H45N4O8 requires m/z 661.3232).
Hapten-Protein Immunoconjugates
Each hapten was activated at room temperature for 6 h using standard EDC/sulfo-NHS (2 equivalent each) coupling procedures in DMF. After DMF removal under reduced pressure, to the residue was slowly added the corresponding amount of KLH or BSA protein (1 mg of hapten versus 1 mg of protein) in 1 mL of 100 mM PBS, pH 7.2. The resultant solution was allowed to stand for 12 h at 4 °C. Coupling efficiencies were monitored using MALDI-TOF MS, save for KLH, which cannot be directly analyzed.
Hapten Stability Study
Each labeled hapten (~ 0.5 mg) was dissolved in 1 mL of normal rat sera (Sigma-Aldrich), and incubated at 37 °C. At the time points of approximately 6, 23, 45, 68, 76, 94, 117, 146 and 214 hrs, 100 μL aliquots were removed and mixed with 400 μL of cold methanol and 50 μL of acetic acid. After vortexing and sonication, the mixture was incubated at 0 °C for 10 min. The suspension was centrifuged at 13000 rpm for 5 min, and then evaporated under diminished pressure to dryness. After adding 70 μL of methanol, the resultant mixture was vortexed for 5 min and then centrifuged at 13000 rpm for 3–5 min. Fifty μL of the resultant supernatant was analyzed by HPLC with monitoring at 325 nm. All assays were performed in duplicate. The amounts of labeled hapten and the corresponding hydrolysis products were determined by interpolation of peak height and area relative to standard curves. The half-life was calculated by fitting plots of the degradation percentage versus time to the one-phase-decay equation using Prism Version 5.0b software (GraphPad Software, USA).
Enzyme-Linked Immunosorbent Assay (ELISA)
Production of cocaine-specific IgG was monitored by ELISA using the immunoconjugate of each hapten as the coating antigen. Titers were calculated from the plot of absorbance versus log dilution, as the dilution corresponding to an absorbance reading 50% of the maximal value. BSA conjugate was added to COSTAR 3690 microtiter plates and allowed to dry at 37 °C overnight. Following methanol fixation, nonspecific binding was blocked with a solution of 5% nonfat powdered milk in PBS for 0.5 hr at 37 °C. Next, mouse serum was serially diluted in a 1% BSA solution across the plate and allowed to incubate for 1–2 h at 37 °C in a moist chamber. Plates were then washed with DI H2O and treated with goat anti-mouse-HRP antibody for 0.5 hr at 37 °C. Following another wash cycle, plates were developed with the TMB 2-step kit (Pierce; Rockford, IL).
Radioimmunoassay (Equilibrium Dialysis)
Refined values of antibody affinity and cocaine binding capacity were determined via a soluble radioimmunoassay (RIA). A modified version of Müller’s method19 was followed as it allows for determination of both the affinity constant and concentration of specific antibody in serum. The RIA was carried out in a 96-well equilibrium dialyzer MWCO 5000 Da (Harvard Apparatus, Holliston, MA) to allow easy separation of bound and free L-[benzoyl 1-3,4-3H(N)]-cocaine tracer; specific activity = 26 Ci/mmol (PerkinElmer, Boston, MA). Mouse serum was diluted in RIA buffer (sterile filtered 2% BSA in 1× PBS, pH 7.4) to a concentration that would bind 50% of ~ 30,000 decays/min of 3H-cocaine tracer. A 75 μL aliquot of serum was combined with 75 μL of radiolabeled tracer (~ 30,000 decays/min); 150 μL of unlabeled cocaine at varying concentrations in RIA buffer (sterile filtered 1% BSA in 1× PBS, pH 7.4) was added to the solvent chamber and the samples were allowed to reach equilibrium on a plate rotator (Harvard Apparatus, Holliston, MA) at room temperature for at least 22 h. A 75 μL aliquot from each sample/solvent chamber was slowly aspirated and suspended in 5 mL of scintillation fluid (Ecolite, ICN, Irvine, CA), and the radioactivity of each sample was determined by liquid scintillation assay.
Locomotor Testing
Sixteen identical hanging wire cages (26 cm wide × 35 cm long × 20 cm high) were used to monitor locomotor activity following daily intraperitoneal injections of saline (1 mg/mL) or cocaine (5 or 10mg/kg) in the testing context (n = 7–8 per group). Each cage was equipped with two pairs of infrared emitter–detector photocells that were positioned along the long axis 1 cm from the floor and 8 cm from the front and back of the cage. Photocell interruptions served as a measure of locomotor activity. Beam breaks and cage crossovers were recorded by a PC in 10-minute bins (Figures 3S–6S). Area under the curve (AUC) was calculated using the 30 minutes pre-injection as the threshold, then determining the total peak area in the 60 minutes post-injection. AUCs were compared for each vaccine group using a repeated-measure one-way ANOVA, and a Dunnett’s test was used for post-hoc comparisons.
Supplementary Material
Acknowledgments
We acknowledge the Skaggs Institute for Chemical Biology and the National Institute on Drug Abuse (DA08590 to K.D.J.) for financial support.
ABBREVIATIONS
- Abs
antibodies
- DMAP
dimethylaminepyridine
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- i.p
intraperitoneal
- KLH
keyhole limpet hemocyannin
- MCA
7-methoxycoumarin-4-acetic acid
- pAbs
polyclonal antibodies
- SAS
Sigma Adjuvant System®
- SNC
succinyl norcocaine
- sulfo-NHS
N-hydroxysulfosuccinimide sodium salt
Footnotes
Supporting Information. Additional experimental details as discussed in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.National Institute of Drug Abuse. National Household Survey on Drug Abuse, Population Estimates. U.S. Dept. of Health and Human Services; Rockville, MD: 2001. [Google Scholar]
- 2.(a) Carrera MRA, Meijler MM, Janda KD. Cocaine Pharmacology and Current Pharmacotherapies for Its Abuse. Bioorg Med Chem. 2004;12:5019–5030. doi: 10.1016/j.bmc.2004.06.018. [DOI] [PubMed] [Google Scholar]; (b) Gawin FH. Cocaine Addiction, Psychology, and Neurophysiology. Science. 1991;251:1580–1586. doi: 10.1126/science.2011738. [DOI] [PubMed] [Google Scholar]
- 3.(a) Kinsey BM, Kosten TR, Orson FM. Anti-cocaine Vaccine Development. Expert Rev Vaccine. 2010;9:1109–1114. doi: 10.1586/erv.10.102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Moreno A, Janda KD. Immunopharmacotherapy: Vaccination Strategies as a Treatment for Drug Abuse and Dependence. Pharmacol Biochem Behav. 2009;92:199–205. doi: 10.1016/j.pbb.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Moreno A, Janda KD. Current Challenges for the Creation of Effective Vaccines against Drugs of abuse. Expert Rev Vaccines. 2011;10:1637–1639. doi: 10.1586/erv.11.145. [DOI] [PubMed] [Google Scholar]; (b) Janda KD, Treweek JB. Vaccines Targeting Drugs of Abuse: Is the Glass Half-Empty or Half-Full? Nat Rev Immunol. 2012;12:67–72. doi: 10.1038/nri3130. [DOI] [PubMed] [Google Scholar]
- 5.(a) Haney M, Gunderson EW, Jiang H, Collins ED, Foltin RW. Cocaine-Specific Antibodies Blunt the Subjective Effects of Smoked Cocaine in Human. Biol Psychiatry. 2010;67:59–65. doi: 10.1016/j.biopsych.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kosten TR, Rosen M, Bond J, Settles M, Roberts JSC, Shields J, Jack L, Fox B. Human Therapeutic Cocaine Vaccine: Safety and Immunogenicity. Vaccine. 2002;20:1196–1204. doi: 10.1016/s0264-410x(01)00425-x. [DOI] [PubMed] [Google Scholar]; (c) Martell BB, Mitchell E, Poling J, Gonsai K, Kosten TR. Vaccine Pharmacotherapy for the Treatment of Cocaine Dependence. Biol Psychiatry. 2005;58:158–164. doi: 10.1016/j.biopsych.2005.04.032. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kinsey BM, Jackon DC, Orson FM. Anti-Drug Vaccines to Treat Substance Abuse. Immun Cell Biol. 2009;87:309–314. doi: 10.1038/icb.2009.17. [DOI] [PubMed] [Google Scholar]; (b) Martell BA, Orson FM, Poling J, Mitchell E, Rossen RD, Gardner T, Kosten TR. Cocaine Vaccine for the Treatment of Cocaine Dependence in Methadone-Maintained Patients: A Randomized, Double-Blind, Placebo-Controlled Efficacy Trial. Arch Gen Psychiatry. 2009;66:1116–1123. doi: 10.1001/archgenpsychiatry.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Li P, Zhao K, Deng S-X, Landry DW. Nonenzymatic Hydrolysis of Cocaine via Intramolecular Acid Catalysis. Helv Chim Acta. 1999;82:85–89. [Google Scholar]; (b) Deng SX, Bharat N, Fischman MC, Landry DW. Covalent Modification of Proteins by Cocaine. Proc Natl Acad Sci USA. 2002;99:3412–3416. doi: 10.1073/pnas.042700599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrera MRA, Ashley JA, Wirsching P, Koob GF, Janda KD. A Second-Generation Vaccine Protects against the Psychoactive Effects of Cocaine. Proc Natl Acad Sci USA. 2001;98:1988–1992. doi: 10.1073/pnas.041610998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Carrera MRA, Ashley JA, Parsons LH, Wirsching P, Koob GF, Janda KD. Suppression of Psychoactive Effects of Cocaine by Active Immunization. Nature. 1995;378:727–730. doi: 10.1038/378727a0. [DOI] [PubMed] [Google Scholar]; (b) Carrera MRA, Ashley JA, Zhou B, Wirsching P, Koob GF, Janda KD. Cocaine Vaccines: Antibody Protection Against Relapse in a Rat Model. Proc Natl Acad Sci USA. 2000;32:6202–6207. doi: 10.1073/pnas.97.11.6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakurai M, Wirsching P, Janda KD. Design and synthesis of a cocaine-diamide hapten for vaccine development. Tetrahedron Lett. 1996;37:5479–5482. [Google Scholar]
- 11.(a) Koob G, Hicks MJ, Wee S, Rosenburg JB, De BP, Kaminsky SM, Moreno A, Janda KD, Crystal RG. Anti-Cocaine Analog to a Disrupted Adenovirus. CNS Neurol Disord Drug targets. 2011;10:899–1992. doi: 10.2174/187152711799219334. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hicks MJ, Bishnu PD, Rosenberg JB, Davidson JT, Moreno AY, Janda KD, Wee S, Koob GF, Hackett NR, Kaminsky SM, Worgall S, Toth M, Mezey JG, Crystal RG. Cocaine Analog Coupled to Disrupted Adenovirus: A vaccine Strategy to Evoke High-Titer Immunity Against Addictive Drugs. Mol Ther. 2011;19:612–619. doi: 10.1038/mt.2010.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moreno A, Janda KD. Impact of Distinct Chemical Structures for the Development of a Methamphetamine Vaccine. Impact of Distinct Chemical Structures for the Development of a Methamphetamine. J Am Chem Soc. 2011;133:6587–6595. doi: 10.1021/ja108807j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomsen M, Caine SB. Psychomotor Stimulant Effects of Cocaine in Rats and 15 Mouse Strains. Exp Clin Psychopharm. 2011;19:321–341. doi: 10.1037/a0024798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Müller R. Determination of Affinity and Specificity of Anti-Hapten Antibodies by Competitive Radioimmunoassay. Methods Enzymol. 1983;92:589–601. doi: 10.1016/0076-6879(83)92046-3. [DOI] [PubMed] [Google Scholar]
- 15.Janeway C, Travers P, Walport M, Shlomchik M. Immunobiology: the Immune System in Health and Disease. Garland Science Publishing; New York: 2004. pp. 409–455. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.