

Abstract
We report a complex with a rare CuII–O–CuII structural motif that is stable at room temperature, which allows its in-depth characterization by a variety of spectroscopic methods. Interest in such compounds is fueled by the recent discovery that a CuII–O–CuII species on the surface of Cu-ZSM-5 is capable of oxidizing methane to methanol and this in turn ties into mechanistic discussions on the methane oxidation at the dicopper site within the particulate methane monooxygenase. For the synthesis of our Cu2O complex we have developed a novel, neutral ligand system, FurNeu, exhibiting two N-(N',N'-dimethylaminoethyl)(2-pyridylmethyl)amino binding pockets connected by a dibenzofuran spacer. The reaction of FurNeu with CuCl yielded [FurNeu](Cu2(μ-Cl))(CuCl2), 1, demonstrating the geometric potential of the ligand to stabilize Cu–X–Cu moieties. A CuI precursor with weakly coordinating anions was chosen in the next step, namely [Cu(NCCH3)4]OTf, which led to the formation of [FurNeu](Cu(NCCH3))2(OTf)2, 3. Treatment of 3 with O2 or PhIO led to identical green solutions, whose UV/Vis spectra were markedly different from the one displayed by [FurNeu](Cu)2(OTf)4, 4, prepared independently from FurNeu and Cu(OTf)2. Further investigations including PhIO consumption experiments, NMR and UV/Vis spectroscopy, HR-ESI mass spectrometry and protonation studies led to the identification of the green product as [FurNeu](Cu2(μ-O))(OTf)2, 5. DOSY NMR spectroscopy confirmed its monomeric character. Over longer periods of time 5 decomposes to give [Cu(picoloyl)2], formed through an oxidative N-dealkylation reaction followed by further oxidation of the ligand. Due to its slow decomposition reaction all attempts to crystallize 5 failed. However, its structure in solution could be determined by EXAFS analysis in combination with DFT calculations, which revealed a Cu–O–Cu angle that amounts to 105.17°. Moreover, TDDFT calculations helped to rationalize the UV/Vis absorptions of 5. The reactivity of complex 5 with 2,4-di-tert-butylphenol, DTBP, was also investigated; the initially formed biphenol product, TBBP, was found to further react in the presence of excessive O2 to yield 2,4,7,9-tetra-tert-butyloxepino[2,3-b]benzofuran, TBOBF, via an intermediate diphenoquinone, TBDQ. It turned out that 5, or its precursor 3, can even be employed as a catalyst for the oxidation of DTBP to TBBP or for the oxidation of TBBP to TBOBF.
1. Introduction
In the last decades significant efforts have been dedicated to systems composed of ligated CuI and O2, which were fueled by the biological relevance and the quest for efficient oxidation catalysts.1–9 Accordingly, several Cux/Oy species have been characterized over the years, and detailed analysis of their electronic structures have helped to establish a structure-function correlation for this important class of compounds.1,2,8–14 Their composition (nuclearity) and stability have been found to be mainly dependent on the nature of the supporting ligands employed.3,15
Dioxygen activation at two copper(I) centers mostly yields either a (μ-1,2)peroxo dicopper(II) adduct TP, a(μ-η2:η2)peroxo complex P or a bis(μ-oxo)dicopper(III) complex O with a fully cleaved O–O bond (Chart 1). Especially the binding motif P was found to be relevant to the biological dioxygen fixation and activation by copper enzymes like the oxygen carrier protein hemocyanin (Hr), the monooxygenase tyrosinase (Tyr) and catechol oxidase.11,16 Moreover, studies on bioinorganic model compounds mimicking these active sites indicated that the energy barriers for the interconversion between P and O complexes are very low,17 and, therefore, factors like electronic and steric properties of the supporting ligands8,15,18,19, temperature13,20, solvent15,21, concentration13,20, and identity of the counterion19,22 may have a strong impact on the equilibrium position between P and O.3,5,6,23
Chart 1.
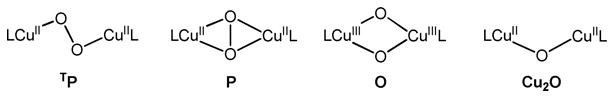
Possible products after dioxygen activation at two copper(I) centers. L = ligand.
Compared to the Cu2O2 systems mentioned above, only few (μ-oxo)dicopper(II) complexes,Cu2O, have been described in the literature24–26 and only a small number of these compounds are characterized unequivocally.27–32 Mostly, they have been obtained via a cooperative 4e- reduction of one dioxygen molecule by a total of four CuI ions (Scheme 1), and they were reported as undesired products in connection with oxyHc modeling chemistry in the 1980s.33 Alternatively, they can be formed by the reaction of two equivalents of a CuI compound with one equivalent of an oxygen atom transfer reagent like iodosobenzene, PhIO.27,29,32
Scheme 1.

Formation and reactivity of (μ-oxo)dicopper(II) complexes, Cu2O.
The rarity of the Cu2O cores can possibly be explained by the electron density a single oxido ligand experiences between two electron rich CuII centers, which makes it reactive, for instance, towards water: the presence of H2O in the reaction system was found to lead to the formation of the corresponding bis(μ-hydroxo)dicopper(II) compounds.27–29,32 This basic/nucleophilic character of the oxo group has also been held responsible for the ability of the Cu2O complexes to react with CO2 forming the corresponding (μ-carbonato)dicopper(II) complexes.32,34 Surprisingly, some of the representatives also exhibit a rather effective oxygen atom transfer capability onto phosphines27,29,30,35 – a reactivity that is usually reserved for electrophilic metal oxygen units (Scheme 1).4,5,36
While Cu2O complexes have lived a shadowy existence during the last few decades, recently they moved into the spot light: An oxygen-activated Cu-ZSM-5 (Figure 1, A) has been shown to selectively oxidize methane to methanol at [CuII–O–CuII]2+ cores.37–39 Furthermore, contemporaneous crystallographic analyses (Figure 1, B) combined with activity studies on particulate methane monooxygenase (pMMO) and engineered protein variants have indicated that the catalytic function is also based on a dicopper site,40–44 and (among others) a Cu2O motif is discussed as a very likely candidate for the methane oxidizing species in the enzyme active site.24–26,45 Accordingly, the synthesis and investigation of Cu2O complexes advanced to one of the major goals in the current Cu/O2 chemistry:24–26,45 Any progress in Cu2O model chemistry could set the basis for a better understanding of biological as well as heterogeneous catalysis.46
Figure 1.
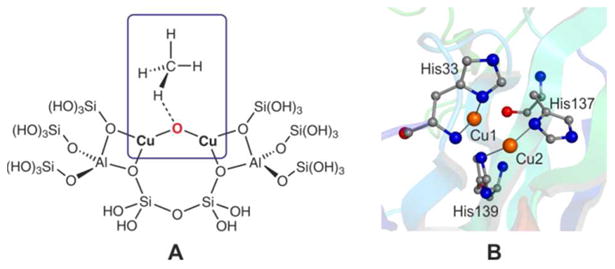
A) Simplified model of the methane oxidizing active site in Cu-ZSM-5.38 B) Structure of the active site in pMMO from Methylococcus capsulatus (Bath), PDB code:1YEW.41
Recently we have described a procedure that allowed for the synthesis and isolation of novel dinuclear CuI complexes based on a bis(diiminato) framework provided by the ligand [Xanthdim]2- (Chart 2). One of them, [Xanthdim](Cu(NCCH3))2, was shown to react with O2 or a soluble derivative of iodosobenzene at −80 °C to give a green intermediate, which was identified as a Cu2O species based on the results of UV/Vis and vibrational studies.47 However, the reported instability of the Cu2O core precluded any structural assignment on the basis of EXAFS or X-ray diffraction studies.
Chart 2.
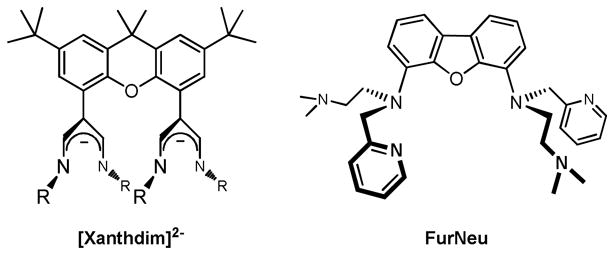
Comparison of the two ligand systems Xanthdim48 and FurNeu (this work). R = 2,3-Dimethylphenyl.
For the further development of this dicopper chemistry we have now designed a novel ligand system, FurNeu, which differs from the Xanthdim ligand48 in the following features: (i) denticity and structure of the two N-donor binding pockets, (ii) overall charge of the ligand and (iii) nature of the ligand backbone (Chart 2).
We show that FurNeu provides access to a rare room temperature stable Cu2O core, which enables for the first time an extensive characterization by NMR, DOSY NMR, EPR, UV/Vis spectroscopy, HR-ESI mass spectrometry, extensive theoretical considerations as well as by EXAFS spectroscopy. The reactivity of the Cu2O core in presence of externally added substrates has also been studied in considerable detail. In particular, we demonstrate an unprecedented reactivity for a Cu2O core towards sterically hindered phenols under aerobic conditions.
2. Results and Discussion
2.1. Ligand Synthesis
FurNeu
Compared to the Xanthdim system, the completely different framework of the two potential metal coordinating sites within the FurNeu ligand suggested an extension of the distance between these binding pockets. From Pacman porphyrin chemistry it is known that the space between two cofacially located porphyrin units is increased, if a xanthene derived spacer is replaced by a dibenzofuran moiety.49 With this background and inspired by related contributions of Hagadorn et al. FurNeu was synthesized by a direct tethering of N-(N',N'-dimethylaminoethyl)(2-pyridylmethyl)amine to 4,6-diiododibenzofuran, FurI2, mediated by CuI (Scheme 2, see supporting information (SI, experimental section) for details).50 FurNeu was isolated in 21 % yield and its identity was proved with the aid of NMR and IR spectroscopy, high-resolution electrospray ionization (HR-ESI) mass spectrometry and a CHN analysis.
Scheme 2.
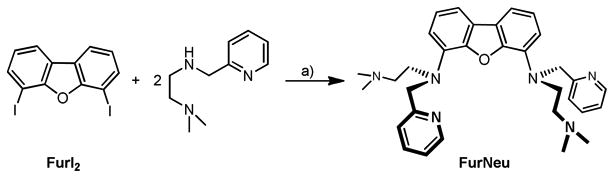
Synthesis of FurNeu. a) CuI (cat.), (CH2OH)2, K3PO4, iPrOH.
2.2. Synthesis and Characterization of FurNeu Copper Chloride Complexes
[FurNeu](Cu2(μ-Cl))(CuCl2), 1
The addition of two equivalents of CuCl to a solution of FurNeu in tetrahydrofuran (thf) and appropriate workup led to a yellow product, which after crystallization could be investigated by single-crystal X-ray diffraction analysis. The molecular structure of [FurNeu](Cu2(μ-Cl))(CuCl2), 1, is shown in Figure 2.
Figure 2.
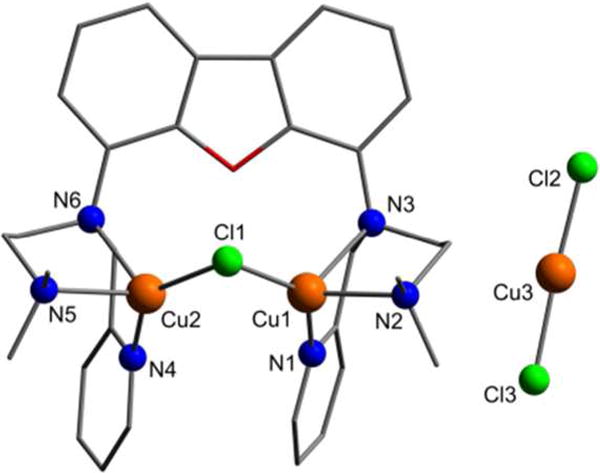
Molecular structure of [FurNeu](Cu2(μ-Cl))(CuCl2), 1. All hydrogen atoms have been omitted for clarity. Selected bond lengths [Å] and angles [°]: Cu1⋯Cu2 3.119(1), N3⋯N6 5.431(2), Pycenter–Pycenter ≈ 4.8, Cu1– Cl1 2.2430(4), Cu1–N1 1.9949(14), Cu1–N2 2.1813(14), Cu1– N3 2.3803(14), Cu2-Cl1 2.2122(14), Cu2–N4 1.9955(14), Cu2–N5 2.1892(13), Cu2–N6 2.3510(13), Cu3–Cl2 2.1097(5), Cu3-Cl3 2.1086(5), Cu1–Cl2–Cu2 88.883(16), Cl1–Cu1–N1 129.19(4), Cl1– Cu2–N4 139.88(4), Cl2–Cu3–Cl3 172.16(2).
As expected the two N3-binding sites of FurNeu coordinate one copper(I) ion each. Both binding pockets are arranged in a vis-à-vis mode and the two copper atoms (Cu1, Cu2) are bridged by one chloride ligand (Cl1). Considering the plane spanned by the ligand backbone the pyridylmethyl and dimethylaminoethylene units, respectively, exhibit a cis-configuration. This arrangement might be favored due to a π-π stacking interaction between the two pyridyl units. However, as judged by the distance of these aromatic units (4.8 Å) the strength of this interaction is rather weak.51 The atoms Cu1 and Cu2 are located at a distance of 3.119(1) Å from each other, and the Cu1–Cl1–Cu2 angle amounts to 88.883(16)°. All of the Cu–N bond lengths are similar to those in comparable systems.52 The single charge of the complex cation [FurNeu](Cu2(μ-Cl))+ in complex 1 is compensated by an almost linear dichlorocuprate anion (Cl2–Cu3–Cl3:172.26°), CuCl2-, which must have been formed in the course of the reaction, and hence the FurNeu/CuCl ratio amounts to 1:3 within [FurNeu](Cu2(μ-Cl))(CuCl2), 1. Consequently, employment of three equivalents of CuCl gave 1 in 59 % yield after workup (Scheme 3). 1 has been fully characterized with the aid of NMR (see Figure S1, SI) and IR spectroscopy, HR-ESI mass spectrometry and an elemental analysis. The structure of 1 clearly shows that with respect to geometric arguments the FurNeu system allows for an intermolecular activation of small substrates between the coordinated metal ions.
Scheme 3.
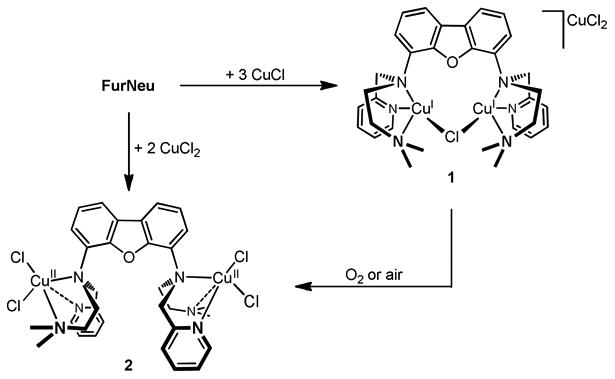
Syntheses of [FurNeu](Cu2(μ-Cl))(CuCl2), 1, and [FurNeu](CuCl2)2, 2, and oxidation of 1 yielding 2 (in this case the exact stoichiometry remains unconsidered).
[FurNeu](CuCl2)2, 2
Upon exposure to air or dioxygen the color of a yellow solution of 1 in acetonitrile turned green within seconds. NMR spectroscopic investigations pointed to the formation of a paramagnetic product containing CuII ions. Slow evaporation of the solvent from the reaction mixture at room temperature led to the formation of single crystals and [FurNeu](CuCl2)2, 2, could be clearly identified as an oxidation product of 1 through an X-ray diffraction analysis (Scheme 3, for further details see SI). Alternatively, complex 2 could be synthesized in about 78% yield by the reaction of FurNeu with two equivalents of CuCl2 in acetonitrile (Scheme 3). Complete characterization of 2 was performed by a variety of spectroscopic methods as well as an elemental analysis (for further details see SI).
2.3. Synthesis and Characterization of FurNeu Copper Triflate Complexes
The potential binding/activation of molecules by metal complexes can be hampered by the presence of halide anions. These anions can be bound to the metal centers very tightly and thus block the access of substrates due to the lack of any free coordination site. Consequently, for activation studies with FurNeu copper complexes the synthesis of a corresponding dinuclear precursor containing weakly coordinating anions was pursued.
[FurNeu](Cu(NCCH3))2(OTf)2, 3
To a thf solution of FurNeu two equivalents of [Cu(NCCH3)4]OTf were added to obtain [FurNeu](Cu(NCCH3))2(OTf)2, 3, as a beige solid in 61 % yield. Although the molecular structure of 3 could not be determined by X-ray crystallography, it was characterized in detail by NMR, UV/Vis (Figure 3, yellow line) and IR spectroscopy, mass spectrometry and an elemental analysis (Scheme 4).
Figure 3.
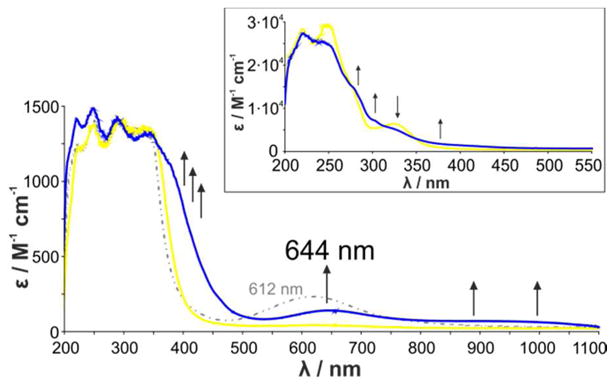
UV/Vis spectra of a solution of [FurNeu](Cu(NCCH3))2(OTf)2, 3, dissolved in acetonitrile (2 mM) before (yellow line) and 20 min after the reaction with O2 (blue line); employing PhIO as the oxidant an analogous spectrum with a somewhat higher absorbance is obtained (see SI, Figure S5). The dotted grey line represents the UV/Vis absorption behavior of a solution of [FurNeu](Cu)2(OTf)4, 4, in acetonitrile (2 mM). The reported extinction coefficient of the oxygenation product of 3 is based on the PhIO reaction. The inset shows the spectral changes associated with the oxidation of 3 in the ultra-violet region, the investigation of which required a reduction of the concentration to 0.1 mM.
Scheme 4.
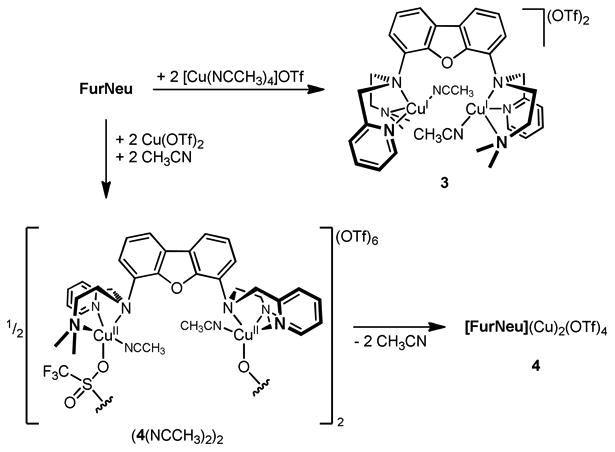
Syntheses of [FurNeu](Cu(NCCH3))2(OTf)2, 3 and [FurNeu](Cu)2(OTf)4, 4.
Complex 3 is readily soluble in acetonitrile and shows a distinct sensitivity against air and dioxygen. A 1H NMR spectrum of an acetonitrile-d3 solution of 3 showed the complete set of signals expected for the FurNeu ligand. Moreover, a singlet at 1.96 ppm was observed – a shift identical to free acetonitrile, suggesting a dynamic exchange of the coordinated CH3CN ligands with the corresponding trideuterated CD3CN solvent molecules.53
In the solid state, in contrast, the acetonitrile ligands are found to be comparatively tightly bound to the copperions: even after drying 3 for several days in vacuum no decrease of the intensity of the corresponding singlet signal at 1.96 ppm was noted upon re-dissolution in acetonitrile-d3. Compared to the 1H NMR spectrum of [FurNeu](Cu(μ-Cl))(CuCl2), 1, some signals corresponding to the ligand within 3 (for example in the aromatic region, see Figure S1, SI) did not show the expected coupling pattern and appeared broadened (compare Figure 4, b)): Either the fast exchange of the acetonitrile co-ligands in solution or a lack of fixation of the metal coordination sites by a bridging ligand may cause this line broadening.
Figure 4.
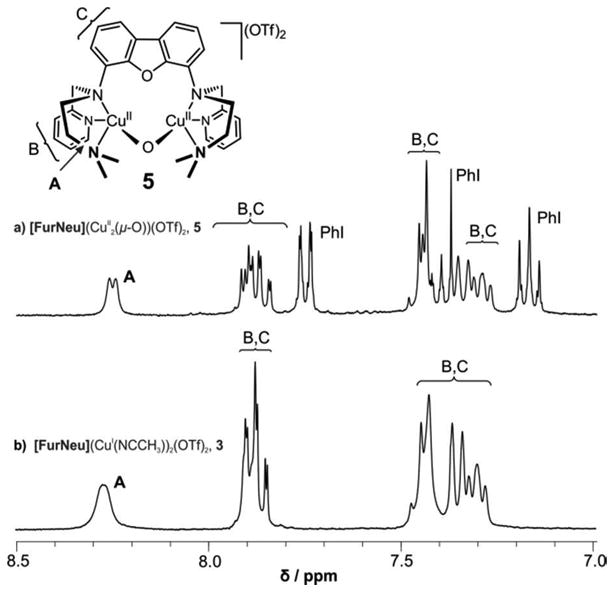
Comparison of the aromatic regions in the 1H NMR spectra of solutions of a) [FurNeu](Cu2(μ-O))(OTf)2, 5, and b) [FurNeu](Cu(NCCH3))2(OTf)2, 3, respectively, in acetonitrile-d3. The signals detected and the positions of the corresponding H atoms within the molecule are denoted with the help of the capital letters. Moreover, spectrum a) contains the signal set of the by-product iodobenzene (PhI).
[FurNeu](Cu)2(OTf)4, 4
It was conceivable that the reaction of 3 with dioxygen either leads to copper-oxygen products associated with dioxygen activation or to a dicopper(II) triflate species arising from simple one electron oxidation, as was observed in case of 1 (giving 2). In order to be able to distinguish between these possibilities the analogous copper(II) triflate compound was synthesized as a reference compound directly via the reaction of FurNeu with two equivalents of Cu(OTf)2 in acetonitrile as the solvent. Complexation of the copper(II) ions was indicated by an immediate color change of the reaction mixture from light beige to deep blue. Workup gave blue [FurNeu](Cu)2(OTf)4, 4, in 50 % yield and it was fully characterized (Scheme 4, for further details see SI) by a variety of spectroscopic methods and an elemental analysis.
2.4. Access to a CuII–O–CuII Motif (Cu2O) and its characterization
UV/Vis Spectroscopy
To investigate the reactivity of [FurNeu](Cu(NCCH3))2(OTf)2, 3, towards dioxygen it was dissolved in acetonitrile, and after treatment with excessive O2 at -40 °C the initially formed pale yellow solution slowly changed its color to green. Annealing of the reaction mixture to room temperature accelerated this process and repetition of the experiment without any cooling immediately gave a green solution. In both cases UV/Vis measurements showed that during the reaction of 3 with dioxygen three new absorption features evolved (Figure 3, blue line): two broad absorptions between 400 and 500 nm and between 800 and 1100 nm, and the appearance of a distinct peak at 644 nm. At room temperature the formation of the 644 nm band was completed after 20 minutes, but at −40 °C the reaction was not finished even after 90 minutes.
In particular the absorption band at 644 nm is reminiscent of the UV/Vis spectrum reported previously for the temperature sensitive xanthene based Cu2O species.47 On the other hand the absorption features inherent to the product of the oxygenation of 3 is clearly distinct from the absorption features of the simple one-electron oxidation product 4. For comparison, the UV/Vis spectrum of an acetonitrile solution of [FurNeu](Cu)2(OTf)4, 4 (Figure 3, dotted grey line) exhibits an absorption feature at 612 nm (ε = 260 M-1 cm-1) and does not show the broad features between 400 to 500 nm and between 800 to 1100 nm that are present in the product of the oxygenation of 3. Accordingly, the formation of 4 in course of the reaction of 3 with dioxygen can be clearly ruled out. The reaction of 3 with iodosobenzene, PhIO, resulted in absorption features identical to what was obtained for the 3/O2 system.
Titration experiments confirmed that only one equivalent of PhIO is necessary for the quantitative oxygenation of 3, and ε = 140 M-1 cm-1 was determined for the absorption band at 644 nm. The addition of more than one equivalent of PhIO did not lead to any further changes in the absorption spectrum, while the reaction between 3 and 0.5 equivalents of iodosobenzene generated the oxidized product in only 50% yield (Figure S9, SI). Accordingly, we tentatively assign the oxygenation product of 3 to a room temperature stable Cu2O complex, [FurNeu](Cu2(μ-O))(OTf)2, 5 (Scheme 5), which in the following is further supported by subsequent ESI-MS, NMR, EXAFS and DFT studies (see below).
Scheme 5.
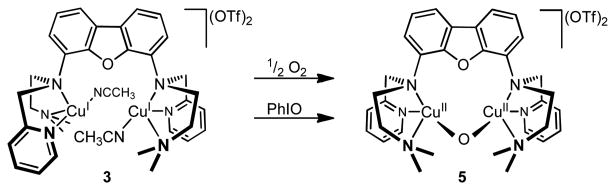
Access to [FurNeu](Cu2(μ-O))(OTf)2, 5, starting from [FurNeu](Cu(NCCH3))2(OTf)2, 3, through the reaction with O2 and PhIO, respectively.
Notably, while 5 is generated in near-quantitative yield in the reaction of 3 with PhIO, the oxidation of 3 with dioxygen yielded 5 in only 85% yield (Figure S10). The lower efficiency of the dioxygen reaction can be attributed to the more complicated steps associated with the dioxygen activation and O-O bond cleavage processes involving the 4 electrons - delivered by a total of 4 CuI ions - that are necessary for the generation of 5 from 3. The iodosobenzene reaction, in contrast, involves a simple oxygen-atom transfer to the dinuclear copper(I) precursor, which may explain the near-quantitative conversion of 3 to 5.
EPR Spectroscopy
EPR experiments are also consistent with the formation of 5 as a result of oxygenation of 3. Similar to the previously reported Cu2O complexes [(TpMeCuII)2(μ-O)]30,31 (TpMe = tris(3,5-dimethylpyrazolyl)borate) and [MePy2(CuII)]2(μ-O)(X)2, (X= ClO4-, [B[3,5-(CF3)2C6H3]4]-, MePy2 = N-methyl-N,N-bis[2-(2-pyridyl)ethyl]amine)27,29 the product of the reaction between 3 and one equivalent of PhIO was found to be EPR silent, suggesting strong antiferromagnetic coupling of the two copper(II) centers mediated by the bridging oxo ligand. This finding again excludes the formation of [FurNeu](Cu)2(OTf)4, 4, whose EPR spectrum showed the typical signals associated with a copper(II) center, thereby revealing that the copper centers in a [[FurNeu](Cu)2]4+ entity are not interacting with each other in the absence of any bridging ligand.
NMR Spectroscopy
The 1H-NMR spectrum of 5 also corroborates our Cu2O assignment. In Figure 4 we show the 1H NMR spectra of 5 and 3, which are found to be nearly identical to each other. Only small differences in the chemical shifts are observed, and altogether the signals of 5 are somewhat sharper, comparable to those of the chloro bridged dicopper(I) complex 1 (see Figure S1, SI). As outlined above we have rationalized the decreased line widths in comparison with 3 in case of 1 with the rigidity provided by the chloro (Cl-) bridge and consistently it can be attributed to the oxo (O2-) bridge for the case of 5. In general, these findings are comparable to those of a previous study by Karlin et al, who also reported that the 1H NMR spectra of [MePy2(CuII)]2(μ-O)(X)2,27,29 containing a Cu2O core, and its mononuclear CuI precursor are very similar to each other. Moreover, the absence of paramagnetic line broadening and shifts in the 1H NMR spectrum of 5 is a further indication of its diamagnetic ground state. In addition to the signals associated with 5, the 1H-NMR spectrum of the reaction mixture of 3 and PhIO exhibits a set of signals associated with iodobenzene (PhI); relative integration of the corresponding signals established a 1:1 stoichiometry for 5 and PhI, thereby suggesting a complete conversion of the PhIO employed.
DOSY NMR measurements were also performed, which indicated the existence of 5 predominantly in its monomeric form (see SI, Figures S20-S22), thus excluding the possibility of any oligomerization processes associated with the formation of 5, as observed in the comparable system supported by the Xanthdim ligand.47
Mass spectrometry
While IR (Figures S11 and S12) and Raman (λexc = 442 nm, 514 nm, 647 nm, 1064 nm) experiments, unfortunately, did not lead to the observation of isotope sensitive signals, HR-ESI mass spectrometry strongly supported the formation of the FurNeu based Cu2O complex 5. The reaction of 3 with either O2 or PhIO led to the development of a new signal (the intensity of which varied, depending on the mass spectrometer used) in the mass spectrum, which can be assigned to the doubly charged complex cation [FurNeu](Cu2(μ-16O))2+ (experiment with PhI16O: 332.0864, calcd 332.0919, Figure 5, a)). As expected, upon usage of the corresponding 18O containing O atom sources (18O2 or PhI18O) the characteristic peak shifted by one mass unit (experiment with PhI18O: 333.0833, calcd 333.0944, Figure 5, b)) and in all cases the experimentally observed isotopic patterns matched the calculated ones excellently. An additional isotope sensitive signal was observed in the ESI-MS spectrum at m/z = 362.0980, which also shifted by one unit when an 18O source was employed in the reaction. The intensity of this peak was found to be much higher as compared to the previously mentioned peak at m/z = 332.0864 (see also SI, Figures S13 and S14). With the aid of tandem mass spectrometry experiments the origin of this signal could be assigned to an adduct between the complex cation [FurNeu](Cu2(μ-O))2+ and acetic acid (Figure 5, c) and d)). The adduct formation presumably involves the protonation of the oxo ligand in 5, followed by the binding of the acetate ligand to one of the CuII centers, which would be consistent with the expected basicity of the oxido ligand in 5 (see below in the Reactivity part: Protons). The origin of the acetic acid in our reaction mixture is not clear at this point and possibly arises from a contamination of the acetonitrile that we employed as the solvent.
Figure 5.
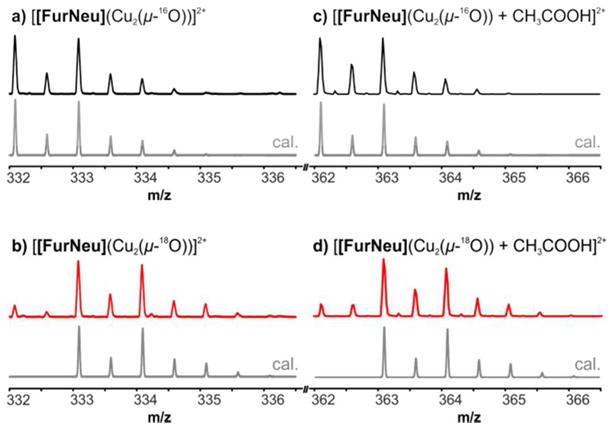
Characteristic peaks in the HR-ESI mass spectra of solutions of [FurNeu](Cu2(μ-16O))(OTf)2, 5 (black lines), and [FurNeu](Cu2(μ-18O))(OTf)2, 518O (red lines), respectively, in acetonitrile. Signals of the corresponding dications [[FurNeu](Cu2(μ-16/18O))]2+ and [[FurNeu](Cu2(μ-16/18O)) + CH3COOH]2+ were also observed after the reaction of 3 with both 16O2 (18O2) and PhI16O (PhI18O). The grey lines represent the calculated peak patterns. For clarity reasons the intensities of the signals shown were equalized. The full spectra are given in the SI (Figure S13 and S14).
To further confirm the adduct formation between a carboxylic acid and 5 under the conditions of the ESI-MS measurements we repeated our studies in presence of different externally added carboxylic acids. The results of these studies are summarized in Figure 6. After exposing an acetonitrile solution of 3 to an atmosphere of dioxygen in presence of externally added formic acid at room temperature a new peak at m/z = 355.0875 indicated the formation of the corresponding adduct [[FurNeu](Cu2(μ-16O)) + HCOOH]2+ (calcd 355.0946, Figure 6, a)). Its intensity was slightly higher than the acetic acid pendant, [[FurNeu](Cu2(μ-16O)) + CH3COOH]2+, which presumably originates from the trace amount of the acetic acid impurity in the solvent. In a further experiment the presence of additional acetic acid only led to the detection of the known signal at m/z = 362.0970 ([[FurNeu](Cu2(μ-16O)) + CH3COOH]2+, calcd 362.1024, Figure 6, b)). The usage of propionic acid afforded the formation of the dication [[FurNeu](Cu2(μ -16O)) + C2H5COOH]2+, as evidenced by a new signal at m/z = 369.1024 (calcd 369.1102, Figure 6, c)).
Figure 6.
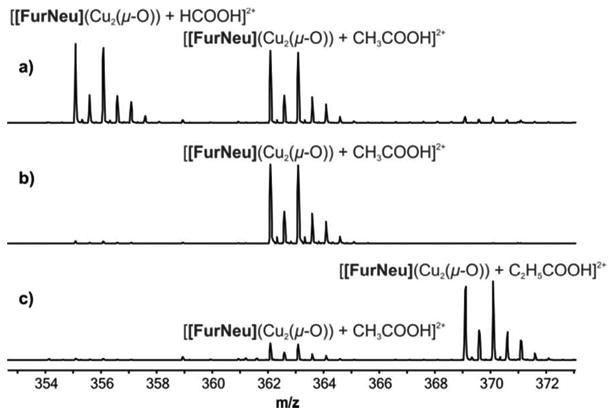
Cutouts of the HR-ESI mass spectra of solutions of 5 in acetonitrile in the presence a) formic acid, b) acetic acid, c) propionic acid.
X-ray Absorption Spectroscopy
X-ray absorption spectroscopic studies were performed at Cu K–edge in order to directly probe the metal oxidation state in 5. The X-ray absorption near edge spectral (XANES) feature of complex 5 was found to be typical of a copper(II) center (edge = 8987.7 eV; Figure S23, SI).54 In particular, the absence of a low energy peak maximum due to the 1s to 4p transition in the region between 8983 and 8986 eV excludes the possibility of the presence of any Cu(I) center in 5.
Extended X–ray absorption fine structure (EXAFS) analysis revealed further structural details. Figure 7 shows the Fourier transforms (r space) of the Cu K-edge EXAFS data and the best-fit.
Figure 7.
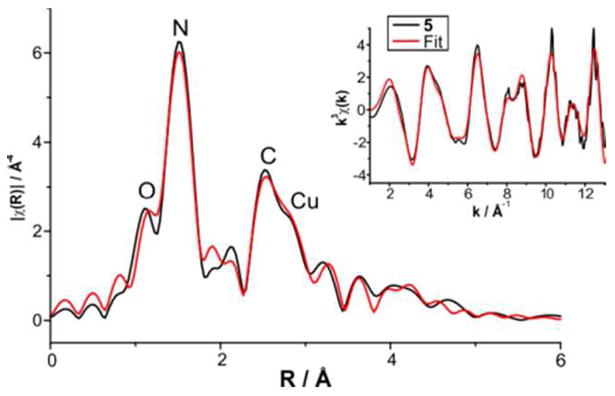
Overlay of the k3 weighted EXAFS of 5 and the best fit (No. 8 in Table S2, SI) in R and k space, respectively. Main figure: Black line shows the Fourier transformed EXAFS data and the red line represents the best fit (No. 8 in Table S2, SI) in R space. Inset: Black line shows the weighted RAW data and the red line the back-Fourier transformed fit (range R = 2-13 Å) in k space.
The first coordination sphere can be fit well by two sub-shells of N/O atoms at absorber-scatterer distances (r) of 1.79 and 1.95 Å. Splitting of the first coordination shell into two sub-shells was necessary in order to significantly improve the quality of the fit (fits 8-10 in Table S2, SI). The shorter-distance shell is attributed to the oxygen scatterer of the CuII–O–CuII moiety, which is also supported by DFT calculation (vide infra, Table 1, Table S3, SI). The shell at 1.95 Å with 3 scatterers represents the donor nitrogen atoms of the polydentate ligand FurNeu. Notably, this distance in 5 is significantly shorter than the average CuII–N distances of 2.18 and 2.07 Å, as obtained by X-ray crystallography for complexes 1 and 2, respectively; this points toward more covalent Cu–N bonds in 5. Cu-EXAFS of 5 also showed a peak at 2.91 Å corresponding to the Cu scatterer, thereby strongly supporting the presence of a homodinuclear Cu⋯Cu center in 5. Fits 11-15 show that the inclusion of a Cu scatterer is an absolute necessity for obtaining a reasonable simulation (fit 8) of the experimental data; no good simulation could be obtained by considering outer shell contributions from C and N only. Although the additional outer-shell features could be satisfactorily accounted for by considering single scattering paths involving 8 carbon and 3 N/O scatters, the fit could be significantly improved by introducing multiple-scattering pathways (fits 7 and 8 in Table S2, SI).
Table 1.
Summary of the structural and spectroscopic properties of [FurNeu](Cu2(μ-O))(OTf)2, 5, and oxygen-activated Cu-ZSM-538 determined theoretically and experimentally.
| 5 | Cu-ZSM-5/O2 | |
|---|---|---|
| Cu⋯Cu / Å | 2.844 | 3.29 |
| 2.91 (EXAFS) | ≈2.9 (EXAFS, not significant) | |
| Cu–O / Å | 1.791 | 1.75/1.76 |
| 1.79 (Cu-O/N, EXAFS) | ||
| Cu–O–Cu / ° | 105.17 | 139 |
| νs / cm−1 | 565 | 456 |
| 456 (exp.) | ||
| νas / cm−1 | 619 | 852 |
| 870 (exp.) | ||
| Cu–O–Cu bend / cm−1 | 236 | 253 |
| 237 (exp.) | ||
| λ (CuII d-d) / nm | 648 | |
| 644 (exp.) | 752 (exp.) | |
| λ (O→ CuII LMCT / nm | <450 | |
| <450 (exp.) | 441 (exp.) |
Theoretical Considerations
DFT analysis of a rare molecular entity like the dicopper μ-oxido species reported here cannot rely on a specific validation of the level of theory to be adopted, due to the obvious lack of benchmark. However, the vast literature available on the assessment of density functional can be used to define reliable and stringent approaches for the calculation of molecular geometries, relative stabilities and spectroscopic properties using DFT.
In the case of geometry optimization of 5, we decided to adopt the B3LYP functional.55 In fact, such hybrid functional was shown to be capable of excellent performances in the reproduction of experimentally-derived metal-ligand bond lengths (see ref. 56, a benchmark study in which metal oxides had a prominent role). The latter are clearly key geometric parameters for a comparison with experimental EXAFS data. Energy minimization of 5 were thus carried out at the B3LYP/def2-TZVP level,57 and broken-symmetry58 was applied to represent the antiferromagnetic coupling between the two Cu centers that results in the experimentally observed S=0 ground state (see also SI for further details). Based on the crystal structure of 1 we set out with a model featuring a cis arrangement of the pyridylmethyl and dimethylaminoethylene units of the ligand backbone. The minimum geometry obtained is shown in Figure 8. In this model the Cu⋯Cu interatomic distance is 2.844 Å, while the Cu–O bonds are 1.791 Å long and the Cu–N bonds lengths vary between 2.415 and 2.022 Å. These results are in reasonable agreement with the EXAFS data reported above (see also Table 1). The calculated Cu2-O1–Cu3 angle in 5 amounts to 105.17°. Vibrational frequency calculations evidenced three bands that involve the [CuII–O–CuII]2+ motif: the Cu–O–Cu bending mode is calculated at 236 cm-1, while the symmetric and antisymmetric metal-oxo stretches are predicted at 565 (νs) and 619 cm-1 (νas), respectively, in the theoretical spectrum. Taking all calculated geometric and vibrational information together significant differences between the Cu2O core in complex 5 and the [CuII–O–CuII]2+ entity in the oxygen-activated Cu-ZSM-5 species (see Table 1) become evident: For the latter the Cu–O–Cu angle was calculated to be much larger (139°38 in Cu-ZSM-5 vs. 105.17° in 5), and the angle in turn has a significant influence on the respective positions of νs and νas.
Figure 8.
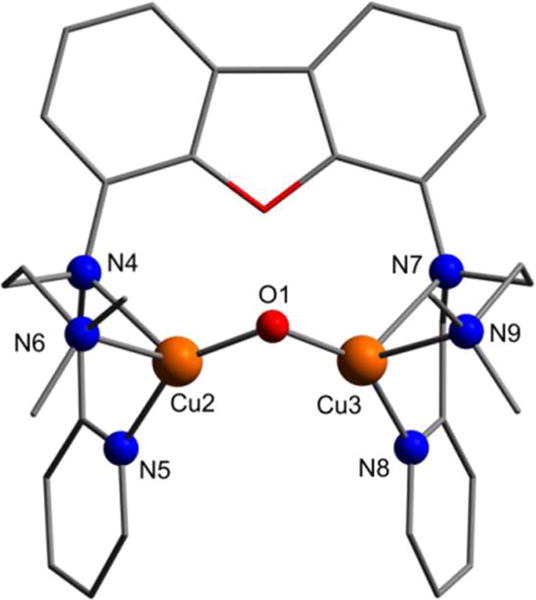
Structure of the cis isomer of the complex cation [[FurNeu](Cu2(μ-O))]2+ of [FurNeu](Cu2(μ-O))(OTf)2, 5, as optimized at the B3LYP/def2-TZVP level. All hydrogen atoms have been omitted for clarity. Selected bond lengths [Å] and angles [°]: Cu2⋯Cu3 2.844, Cu2–O1 1.791, Cu3–O1 1.791, Cu2–N4 2.407, Cu2-N5 2.022, Cu2–N6 2.163, Cu3–N7 2.415, Cu3-N8 2.024, Cu3-N9 2.157, Cu2–O1–Cu3 105.17.
Next, we decided to explore the potential energy surfaces (PES) of model 5, in terms of calculation of singlet-triplet energy splitting, as well as cis-trans isomerisation. Due to known limitations of B3LYP in computing spin-state splitting (an effect of the Hartree-Fock exchange contribution to the total B3LYP energy), we decided to perform energy minimizations also with an additional functional to check coherence of computed energies.59, In particular, we chose the BP86 functional,60 as it completely lacks Hartree-Fock exchange, and was previously extensively and successfully used in our laboratories for studies on first-row transition metal complexes.61 All B3LYP/def2-TZVP and BP86/def2-TZVP energy differences are reported in Table 2 (see SI for further methodological details). Surprisingly, such values evidence discrepancies between the selected levels of theory which are unusually large in the case of cis-trans isomerization (ΔΔEBP86-B3LYP up to 10.2 kcal/mol). As far as singlet-triplet splitting is concerned, results are much more consistent (ΔΔEBP86-B3LYP up to 1.8 kcal/mol) but for both BP86 and B3LYP the values of the energy splitting are smaller than 2 kcal/mol, which is within the uncertainty of DFT. Such a picture prevented us to safely draw conclusions as far as the general features of the PES of 5 is concerned. Clearly, the above reported experimental results already provide conclusive demonstration of the S=0 state of 5, while the cis-trans isomerisation issue still requires further insights, which are provided below by means of Time-dependent DFT (TDDFT) analysis.
Table 2.
Relative energies computed for the cis and trans isomers of [FurNeu](Cu2(μ-O))(OTf)2, 5 (singlet and triplet states), at either the BP86/def2-TZVP or at the B3LYP/def2-TZVP levels (see also the Theory section in SI for details). All values are given in kcal/mol.
| model | B3LYP/def2-TZVP | BP86/def-TZVP |
|---|---|---|
| trans (singlet) | 0.0 | +5.1 |
| trans (triplet) | +1.0 | +4.3 |
| cis (singlet) | +6.9 | +1.8 |
| cis (triplet) | +5.2 | 0.0 |
TDDFT calculation on UV/Vis absorption features of the model – both with cis and trans arrangement – was carried out using the CAM-B3LYP62 functional and a smaller basis set (def2-SVP) or a larger one (def2-TZVP) as explained in Methods (SI). The choice of the CAM-B3LYP functional stems from the improvement it allows in the treatment of charge-transfer states, as compared to conventional functionals.62,63 Let us consider the case of the cis ligand arrangement first. As expected in case of correct structural assignment, the larger basis allowed for the calculation of a spectrum closer to the experimental spectra recorded for 5 (see below and Figure S28, SI). Figure 9 illustrates the outcome of CAM-B3LYP/def2-TZVP calculations: it becomes obvious that TDDFT results reproduce the dominant absorption features derived from experiments. In particular, the band centered at 648 nm in Figure 9 closely matches the experimentally observed 644 nm band (Figure 3 (blue line) and Figure S5 (green line)). Moreover, two predicted signals at 837 and 843 nm are also in reasonable agreement with the experimental spectrum, which reveals a broad absorption in the 800 – 1100 nm region. Finally, TDDFT bands around and below 450 nm nicely fit the experimental data in the same spectral region. As far as relative intensities of the theoretical bands are concerned, the overall picture appears satisfactory notwithstanding the well-known limitations of TDDFT in computing oscillator strengths.64 It is interesting to note that calculations on the UV/Vis absorption features of the corresponding trans isomer of 5 led to a distinctly different spectrum (see SI, Figure S31) thus supporting a cis configuration of the ligand in 5, as had been found also in case of 1 (vide infra).
Figure 9.
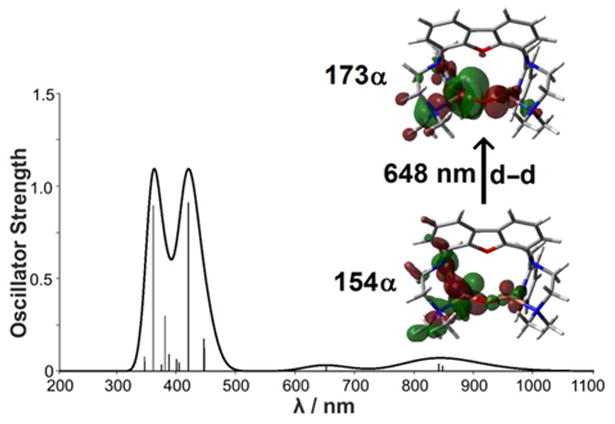
Calculated absorption spectra for 5 using TDDFT at the CAM-B3LYP/def2-TZVP level (compare experimental spectrum, Figure 3 (blue line) and Figure S5 (SI, green line), respectively. The molecular orbitals involved in leading excitations for the transition at 648 nm are also shown (see also Table S6 in the SI for a detailed analysis of the other main transitions). The thicker line represents the Gaussian-broadened spectrum with s = 900 cm-1.
An analysis of the nature of transitions was also performed, based on the individuation of dominant molecular orbital (MO) contributions to the excitations (Table S6 in the SI) and on the evaluation of fractional contribution of basis set functions in the various MOs (Table S7 in the SI). It turned out that the band at 648 nm originates predominantly from a CuII d-d transition. The band around 450 nm, in contrast, mainly derives from a ligand to metal (LM) charge transfer with prominent involvement of the oxo ligand; the one at around 850 nm originates from several transitions of similar relevance, which also correspond to LM charge transfers. The good agreement between the computed and experimental absorption spectra also provides a strong support for the Cu2O assignment of 5.
It is important to note that the position of the CuII d-d transition absorption band at 644 nm in 5 differs substantially from the value of 752 nm, reported for the Cu-ZSM-5/O2 system (see also Table 1),26 while the temperature-sensitive Cu2O core supported by the Xanthdim ligand showed absorption features comparable to those of 5.47 The deviation of the Cu-ZSM-5/O2 system can be possibly rationalized by the different environment the [CuII–O– CuII]2+ entity is embedded in: In Cu-ZSM-5 it is linked to the oxido platform of the zeolite surface, while the FurNeu ligand system in [FurNeu](Cu2(μ-O))(OTf)2, 5, provides an N-based donor sphere. As pyridine and alkyl amine units are comparatively strong donors, FurNeu leads to a much stronger ligand field than the zeolite surface and thus to higher energy d-d excited states for 5 as compared to the Cu–O–Cu units in the Cu-ZSM-5/O2 system. The nature of the coordination sphere, however, does not have a decisive influence on the positions of the band resulting from the corresponding O→CuII LM charge transfer transition (5: <450 nm, Cu-ZSM-5/O2: 441 nm).
2.5. Reactivity of the Cu2O Complex 5
Decomposition of 5
Compared to the Xanthdim based Cu2O,47 which was only stable at −80 °C, [FurNeu](Cu2(μ-O))(OTf)2, 5, is far less sensitive to temperature: Although temperatures higher than 30 °C led to the discoloration of the reaction mixture within two hours, there was no evidence for the decomposition of the deep green acetonitrile solutions of 5 at room temperature. However, from solutions of 5 in CH3CN, when stored for more than three days at room temperature, reproducibly purple crystals precipitated, the molecular structure of which, as determined by single-crystal X-ray diffraction analysis, revealed the formation of [Cu(picoloyl)2] (see Scheme 6, and Figure S24, SI).65 Due to its low solubility in common organic solvents and H2O further characterization of [Cu(picoloyl)2] was possible only by means of IR spectroscopy.66 On the basis of the determined low yield (7%) of [Cu(picoloyl)2] and the unsuccessful identification of the other by-products (e. g. hydroxylated species) of the reaction we can only speculate on the relevant decomposition mechanism of 5. The conversion of 5 to [Cu(picoloyl)2] involves the dioxygenation of a methyl pyridyl residue of FurNeu proceeding via cleavage of the bond to the amino N atom to which it had been connected originally. Reactions of this type have already been reported for a small number of transition metal complexes exhibiting 2-pyridylmethyl units under aerobic conditions: For example the collapse of the monoanionic mebpena ligand (mebpena- = N-methyl-N,N'-bis(2-pyridylmethyl)ethylendiamine-N'-acetate) was reported to proceed via C–N bond scission in the presence of cobalt(II) perchlorate, dioxygen and water.67 In this context a radical mechanism was assumed, initialized by an H atom abstraction from the CH2 group of the pyridylmethyl unit. Also O complexes (Chart 1) have been shown to undergo N-dealkylation reactions, wherein a tertiary amine is cleaved into a secondary one and an aldehyde species.68 In the further course of such reactions the aldehyde gets converted into the corresponding carboxylic acid and carboxylate, respectively, by a transient oxidant, which was, however, not experimentally detected. A similar reaction sequence is also conceivable for the decomposition of the dicopper(II) complex [FurNeu](Cu2(μ-O))(OTf)2, 5 (Scheme 6), although specific experiments with added substrates (like 1-benzyl-1,4-dihydronicotinamide and 9,10-dihydroanthracene) did not lead (over several hours at room temperature) to evidences for a pronounced H atom abstraction reactivity of 5. However, as mentioned above, formation of [Cu(picoloyl)2] takes days and is highly reproducible so that a decomposition of 5 initiated by a slow H atom abstraction step is feasible, nevertheless. In any case, this behavior is unique to 5 since other FurNeu based copper(II) complexes ([FurNeu](CuCl2)2, 2, and [FurNeu](Cu(NCCH3))2(OTf)2, 3) did not form [Cu(picoloyl)2] under the same conditions.
Scheme 6.
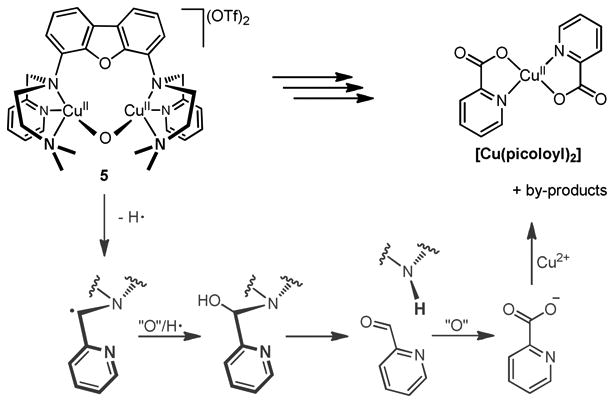
Proposed mechanism for the decomposition route of [FurNeu](Cu2(μ-O))(OTf)2, 5, giving [Cu(picoloyl)2].
Reactivity of the Cu2O Complex 5 Towards Exogenous Substrates: Protons
Many of the previously reported Cu2O compounds possessed highly basic oxo groups,27–29,32 which made them extremely susceptible to proton that often led to formation of the corresponding [CuII–(OH) –CuII] cores, even in the presence of weak proton donors like water (pKa =14). The higher thermal stability of 5, however, already demonstrates a reduced basicity of its oxo group, which is also reflected in its inability to react with water. Thus no significant changes in the absorption spectrum was observed upon addition of 1 – 20 equivalents of water to a CH3CN solution of 5 at 25 °C. In contrast, addition of 1.2 equivalents of a stronger proton donor like acetic acid (pKa = 4.5), that had found to react already in the ESI-MS studies described above, led to the disappearance of the characterestic absorption features associated with the Cu2O core; the broad bands in the region between 350 and 500 nm (assigned as an O→CuII LM CT transitions) and between 800 and 1100 nm (assigned to LMCT transitions) vanished, and the 644 nm band got broadened and shifted to 690 nm (ε = 100 M-1 cm-1; Figures S17-19, SI). Notably, the absorption spectrum of the resultant solution formed upon protonation of 5 is distinct from that of 4. This helps us to exclude an alternative CuII–(OH)–CuII assignment of 5, whose protonation in such case would have resulted in the formation 4 with concomitant loss of a water molecule. The reaction of acetic acid with 5 also led to the disappearance of all the signals of the originally diamagneticCu2O compound in the 1H NMR spectrum, which, together with the UV/Vis data, may suggest a proton induced modification with respect to the oxo bridge resulting in a paramagnetic compound (Figures S15 and S16, SI).
Reactivity of the Cu2O Complex 5 Towards Exogenous Substrates: PPh3
After the addition of five equivalents of PPh3 a solution of [FurNeu](Cu2(μ-O))(OTf)2, 5, in acetonitrile changed its color from green to light brown within three hours under anaerobic conditions. Monitoring the reaction with the help of UV/Vis spectroscopy indicated a decrease of the characteristic absorption bands associated with 5, and the investigation of the reaction mixture by 31P NMR spectroscopy proved the formation of (O)PPh3 in 8 % yield (with respect to the amount of 3 employed for the generation of 5). The low efficiency of this oxygen atom transfer may either be rationalized by a steric clash between 5 and the substrate or by a potentially high nucleophilicity of the bridging oxo ligand.4,5 Reactivity towards carbon dioxide was not observed and comparable studies employing carbon disulfide as a substrate did not lead to the detection of any dithiocarbonate species.
Reactivity of the Cu2O complex, 5, Towards Exogenous Substrates: 2,4-Di-tert-butylphenol
As phenols are often employed as test systems to explore the behavior of novel copper complexes, the reactivity of 5 towards the sterically hindered phenol derivative 2,4-di-tert-butylphenol, DTBP, was investigated. A green solution of 5 in acetonitrile or acetonitrile-d3 was found to get decolorized within one hour at room temperature in presence of two equivalents of DTBP. Again the characteristic UV/Vis spectroscopic features of 5 vanished, and after three hours the product of the oxidative coupling of DTBP, 3,3′,5,5′-tetra-tert-butyl 2,2′-biphenol, TBBP, was identified by means of 1H and 13C NMR spectroscopy with a yield of 56 % (based on DTBP). In addition, equimolar amounts of water and an additional set of signals belonging to a FurNeu containing species were detected by 1H NMR spectroscopy (see SI, Scheme S4, top and Figure S26, A)). The latter showed an almost identical signal pattern and similar signal shifts compared to the ones caused by [FurNeu](Cu2(μ-O))(OTf)2, 5, and [FurNeu](Cu(NCCH3))2(OTf)2, 3, respectively. Based on the absence of a copper(II) d-d transition in the recorded UV/Vis spectrum of the reaction solution the identity of this unknown species was assigned to a copper(I) species, {[FurNeu](CuI)2}, derived from 3. Furthermore, the 1H NMR spectra measured for the reaction solution with acetonitrile-d3 as the solvent exhibited four low intensity singlet signals of equal integrals in the aliphatic region (Figures S26, A) and S27, A), SI). The positions of these signals (δ = 1.20, 1.25, 1.35, 1.44 ppm) suggested the presence of four chemically and magnetically different tert-butyl groups within one compound featuring a chemical environment resembling the ones in DTBP and TBBP, respectively. Surprisingly, after exposing this reaction mixture to an atmosphere of dioxygen the color of the solution immediately changed to purple, and an accompanying UV/Vis experiment led to the observation of a new broad absorption feature at 573 nm (ε = 260 M-1 cm-1, see SI, Scheme S4, bottom). In the course of one hour at room temperature the color of the solution changed to green and 1H NMR spectra recorded subsequently contained only the set of signals belonging to the species that had been minor within the original spectrum: Beside the four singlet signals, which exhibited now the highest intensity, two additional singlet signals at δ = 5.61 ppm and 6.50 ppm and two doublet signals at δ =7.20 ppm and 7.40 ppm could be detected (see Figures S26, B) and S27, B), SI). The absence of signals belonging to the FurNeu ligand was attributed to the paramagnetism of the resulting FurNeu based copper(II) species formed in the dioxygen atmosphere, and based on the lack of signals belonging to TBBP or the starting material DTBP the product was anticipated to derive from these compounds. Only an X-ray diffraction analysis conducted for crystals that could be grown revealed the identity of this product as 2,4,7,9-tetra-tert-butyloxepino[2,3-b]benzofuran, TBOBF, which to our knowledge so far has not been structurally characterized. Its molecular structure is shown in Scheme 7 and Figure S25 (SI).
Scheme 7.
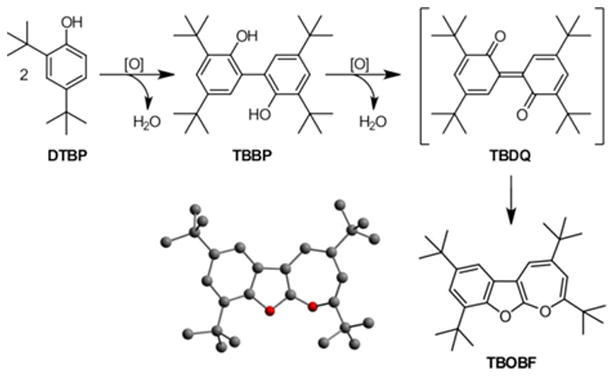
Formation of TBOBF starting from DTBP in the presence of a suitable oxidant ([O]).
Müller and co-workers had already reported the observation of a temporary color change of reaction mixtures to purple, though, while investigating dehydrogenation studies setting out from sterically hindered phenol and biphenol derivatives. Formation of a quinone-derived end-product was suspected, but a variety of valence isomers had to be discussed.69 Several of those could be excluded with the aid of IR and NMR spectroscopic investigations, but, as in our case, even the knowledge of the exact sum formula did not allow for the determination of the correct molecular structure. Almost two decades later, in 1978, the group of Meier was able to identify the unknown oxidation product as TBOBF combining experimental and theoretical studies,70 but even without this knowledge a formation mechanism starting from mono-phenol derivatives was discussed:69,71 With DTBP as a starting material an oxidative coupling reaction mediated by suitable oxidants under anaerobic conditions first leads to TBBP. In the presence of excessive oxidant or after re-activation of the oxidant employed by dioxygen TBBP gets oxidized again forming the corresponding quinonic form 3,3′,5,5′-tetra-tert-butyl 2,2′-diphenoquinone, TBDQ, which causes the characteristic purple color. Subsequently, spontaneous isomerization of TBDQ results in the formation of pale yellow TBOBF (Scheme 7).
Even though numerous examples of the copper mediated oxidative coupling giving TBBP are existing4,5,12,14,19,31,36,72 the formation of TBOBF starting from DTBP is only rarely reported.73–75
Catalytic Studies
Having found that 5 (which is produced upon treatment of 3 with O2) oxidizes DTBP to TBBP with the concomitant formation of a FurNeu copper(I) complex, which apparently activates O2 for the conversion of TBBP to TBOBF, the question occurred whether the copper(I) complex 3 can be employed directly to promote the DTBP→TBOBF conversion in the presence of O2, perhaps even as a catalyst. Indeed, while no reaction between 3 and DTBP was observed under inert conditions the exposure of a solution of this mixture in acetonitrile as the solvent to dioxygen again led to an immediate color change from pale yellow to purple. After 15 minutes the complete conversion of four equivalents DTBP to TBOBF and four equivalents H2O was ensured with the aid of 1H NMR spectroscopy. This finding suggests that 3 acts as a pre-catalyst which in contact with dioxygen forms the active catalyst 5 (although we cannot rule out other active species like superoxide complexes). In a dioxygen atmosphere 5 is able to mediate the catalytic oxidative coupling of DTBP as well as the further catalytic oxidation of the resulting TBBP giving TBOBF (Scheme 8).
Scheme 8.
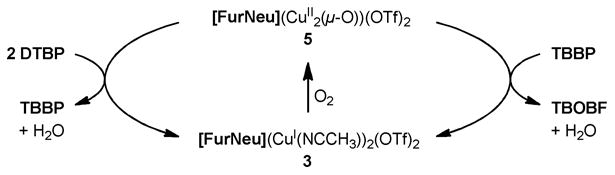
Summary of the catalytic cycle relevant to the oxidative coupling of DTBP giving TBBP and to the oxidation of TBBP giving TBOBF mediated by 5.
In order to evaluate the influence of water, which is a by-product of the phenol oxidation reaction, on the efficiency of the catalytic system an excess of degassed water (23 equivalents with respect to the amount of 3) was added to an acetonitrile solution of 3 and four equivalents of DTBP. A 1H NMR spectrum recorded again indicated inertness of the starting materials, and after exposure to O2 once more full conversion to TBOBF via TBDQ was evidenced by 1H NMR spectroscopy (after three hours), that is, no detrimental effect of water became evident. Hence, 3 was now reacted with 20 equivalents of DTBP in a dioxygen atmosphere. After three hours the formation of 66 % TBBP and 15 % TBOBF (based on DTBP) was detected. No further conversion of either DTBP or TBBP was noted pointing to a deactivation of the catalyst within the initial three hours. The difference between the two TONs (TBBP: 16.2, TBOBF: 3.0, with respect to 3) is consistent with the fact that the formation of TBOBF starting from DTBP proceeds via TBBP as an intermediate product.73,74 Further proof for that came from two orienting experiments: i) In the presence of substoichiometric amounts of O23 mediated the oxidative coupling of DTBP basically giving TBBP as had been observed for the anaerobic reaction of preformed 5 (see above). ii) Employing TBBP instead of DTBP led to the complete conversion to TBOBF in the presence of 3 and an atmosphere of excessive dioxygen.
With the aim of evaluating the relevance of the bridging oxo ligand in [FurNeu](Cu2(μ-O))(OTf)2, 5, for its reactivity towards phenols [FurNeu](Cu)2(OTf)4, 4, a copper(II) complex lacking an oxido ligand, was reacted with two equivalents of DTBP for comparison: After the addition of the phenol derivative the initially blue solution of 4 in acetonitrile or acetonitrile-d3 got slowly decolorized, and the UV/Vis spectrum of the reaction solution recorded a decrease of the characteristic absorption band of 4 at 612 nm. After three hours the coupling product TBBP was detected in 50 % yield by 1H NMR spectroscopy. Additionally, the 1H NMR spectrum contained a set of signals, which could be assigned to a FurNeu based compound as well as one further broad singlet signal at 12.04 ppm. By comparison with the 1H NMR spectra obtained after the reactions of FurNeu and 3, respectively, with two equivalents of a proton donating compound (for example 2,6-lutidium salts) the origin of the new signal at 12.04 ppm could be ascribed to a doubly protonated 3, [3(H)2]2+. Due to the position of the additional signal it was concluded, that the protons are trapped by the N-donor units previously bound to the copper(I) ions. After exposure of the reaction mixture to an atmosphere of dioxygen all signals associated with [3(H)2]2+ vanished possibly due to the formation of a paramagnetic FurNeu based copper(II) species. Moreover, neither additional oxidative coupling of DTBP nor further oxidation of TBBP giving TBOBF was observed. This leads to the conclusion that 3 under acidic conditions does not mediate the oxidation of phenol-based substrates with O2. To further confirm the inhibiting influence of acid, 3 was dissolved together with proton donors (2,6-lutidinum salts) in acetonitrile prior to the addition of DTBP and after subsequent exposure to dioxygen indeed no coupling product was detected. All the experiments made are summarized in the SI (Scheme S5, Table S4).
Thus, both copper(II) complexes, 4 and 5, do have the ability to mediate the oxidative coupling of DTBP. In the case of [FurNeu](Cu2(μ-O))(OTf)2, 5, H2O is formed as a by-product, so that the further oxygenation of the resulting [FurNeu](Cu(NCCH3))2(OTf)2, 3, closes the catalytic cycle. Upon usage of [FurNeu](Cu)2(OTf)4, 4, as the oxidant the protons formed in the course of the coupling reaction get bound by N-donors of the resulting copper(I) complex 3 (instead of getting trapped by the basic oxido ligand) and, hence, it gets deactivated with respect to further oxidation chemistry.
3. Conclusion
CuII–O–CuII cores have recently been proposed as the potential active species responsible for the challenging oxidation of methane to methanol realized at the surface of a Cu-grafted zeolithe24,26,38,39 as well as in the active center of the copper-enzyme pMMO.40–44 Hence, a fundamental understanding of the electronic structure and behavior of such cores could aid the development of more general systems for C–H bond functionalization. However, reports of CuII–O–CuII cores are extremely rare in the literature, possibly due to the high electron density that should be inherent to an oxo ligand forming the sole bridge between two copper centers. Moreover, such complexes are often thermally unstable. The lack of sufficient kinetic and thermodynamic stability of the CuII–O–CuII cores has previously precluded their in-depth spectroscopic characterization, thereby, making a definite structural assignment difficult.
Consequently, this report represents a significant progress in the field. A novel ligand system FurNeu has been developed to preorientate two CuI ions in close proximity for the activation of O2. This has been realized within the precursor complex 3 containing weakly coordinating triflate anions and acetonitrile co-ligands that are readily replaced by O2. Treatment of [FurNeu](Cu(NCCH3))2(OTf)2, 3, with O2 or PhIO leads to green solutions of the oxygenation product that could be identified as a Cu2O complex [FurNeu](Cu2(μ-O))(OTf)2, 5, by means of UV/Vis spectroscopy, PhIO consumption experiments, NMR as well as DOSY NMR spectroscopy, HR-ESI mass spectroscopy, and protonation studies. EXAFS analysis in combination with DFT has revealed the solution structure of 5, and DFT studies could also rationalize the UV/Vis spectrum. The CuII–O–CuII core in 5 is surprisingly stable at room temperature, and decays only slowly over an extended period of time to a [Cu(picoloyl)2] species, possibly by an N-dealkylation mechanism. The thermal stability of 5 can be attributed to a reduced basicity of its oxo goup, which is also reflected in its inability to react with water. In contrast to the CuII–O–CuII entities in Cu-ZSM-5 5 does not show a pronounced H atom abstraction reactivity. This may be explained by the marked difference in the structures, as revealed in our investigation: While the CuII–O–CuII unit in 5 is in a relaxed state, the situation within ZSM-5 may be regarded as strained favoring formation of a SOMO that acts as the abstracting site.38 Investigations concerning the reactivity of 5 showed that it catalyzes the oxidation of 2,4-di-tert-butylphenol to first give the corresponding biphenol (TBBP), which then is oxidized further via a diphenoquinone (TBDQ) intermediate to yield 2,4,7,9-tetra-tert-butyloxepino[2,3-b]benzofuran, TBOBF, which has rarely been observed as an oxidation product in copper mediated oxidations. As 5 can be generated in-situ from the precursor compound 3 via reaction with O2, 3 shows the same catalytic activity.
Supplementary Material
Acknowledgments
We are grateful to the Humboldt-Universität zu Berlin for financial support as well as to the Cluster of Excellence, Unifying Concepts in Catalysis, for valuable discussions and financial support. We also thank Tobias Spranger and Prof. Dr. Clemens Mügge for DOSY NMR measurements, Dr. Erik R. Farquhar for the help with the collection and analysis of the X-ray absorption spectroscopic data, and Ramona Metzinger for the crystal structure analysis of compound 1. XAS data were obtained on beamline X3B of the National Synchrotron Light Source (Brookhaven National Laboratory, Upton, NY, USA). Beamline X3B is operated by the Case Western Reserve University Center for Synchrotron Biosciences, supported by NIH Grant P30–EB–009998. NSLS is supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE–AC02–98CH10886.
Footnotes
Supporting Information. Experimental Section, Molecular structures of 2·1.5(CH3CN), 2·2(CH3CN), (4(NCCH3)2)2, [Cu(picoloyl)2], and TBOBF, detailed discussions of 2 and 4, X-ray crystallographic data, HR-ESI mass spectra of 5 and 518O, additional UV/Vis and NMR data, DOSY NMR spectra, XAS details and data of 5, summary of the reactivity studies of 3, 4 and 5, additional TDDFT results. This material is available free of charge via the Internet at http://pubs.acs.org.
Dedication: This work is dedicated to Prof. Dr. Dr. h.c. mult. W.A. Herrmann on the occasion of his 65th birthday.
References
- 1.Holland PL. Dalton Trans. 2010:5415. doi: 10.1039/c001397h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cramer CJ, Tolman WB. Acc Chem Res. 2007;40:601. doi: 10.1021/ar700008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mirica LM, Ottenwaelder X, Stack TDP. Chem Rev. 2004;104:1013. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- 4.Lewis EA, Tolman WB. Chem Rev. 2004;104:1047. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]
- 5.Hatcher LQ, Karlin KD. J Biol Inorg Chem. 2004;9:669. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]
- 6.Que L, Tolman WB. Angew Chem Int Ed. 2002;41:1114. doi: 10.1002/1521-3773(20020402)41:7<1114::aid-anie1114>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 7.Schindler S. Eur J Inorg Chem. 2000:2311. [Google Scholar]
- 8.Holland PL, Tolman WB. Coord Chem Rev. 1999:190–192. 855. [Google Scholar]
- 9.Kitajima N, Moro-oka Y. Chem Rev. 1994;94:737. [Google Scholar]
- 10.a Que L, Tolman WB. Nature. 2008;455:333. doi: 10.1038/nature07371. [DOI] [PubMed] [Google Scholar]; b Koval IA, Gamez P, Belle C, Selmeczi K, Reedijk J. Chem Soc Rev. 2006;35:814. doi: 10.1039/b516250p. [DOI] [PubMed] [Google Scholar]; c Klinman JP. Chem Rev. 1996;96:2541. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]; d Mahadevan V, Klein Gebbink RJM, Daniel T, Stack P. Curr Opin Chem Biol. 2000;4:228. doi: 10.1016/s1367-5931(99)00080-0. [DOI] [PubMed] [Google Scholar]; e Itoh S. Curr Opin Chem Biol. 2006;10:115. doi: 10.1016/j.cbpa.2006.02.012. [DOI] [PubMed] [Google Scholar]; f Punniyamurthy T, Rout L. Coord Chem Rev. 2008;252:134. [Google Scholar]; g Himes RA, Karlin KD. Curr Opin Chem Biol. 2009;13:119. doi: 10.1016/j.cbpa.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Gunay A, Theopold KH. Chem Rev. 2010;110:1060. doi: 10.1021/cr900269x. [DOI] [PubMed] [Google Scholar]; i Wendlandt AE, Suess AM, Stahl SS. Angew Chem Int Ed. 2011;50:11062. doi: 10.1002/anie.201103945. [DOI] [PubMed] [Google Scholar]; j Garcia-Bosch I, Ribas X, Costas M. Eur J Inorg Chem. 2012;179 [Google Scholar]; k van Eldik R, Reedijk J, editors. Advances in Inorganic Chemistry: Homogeneous Biomimetic Oxidation Catalysis. Vol. 58. Elsevier; London: 2006. [Google Scholar]; l Itoh S, Fukuzumi S. Acc Chem Res. 2007;40:592. doi: 10.1021/ar6000395. [DOI] [PubMed] [Google Scholar]
- 11.Kraatz HB, Metzler-Nolte N. Concepts and Models in Bioinorganic Chemistry. Wiley-VCH; Weinheim: 2006. [Google Scholar]
- 12.Karlin KD, Itoh S. Copper-Oxygen Chemistry. Wiley; Hoboken, N.J: 2011. [Google Scholar]
- 13.Tolman WB. Acc Chem Res. 1997;30:227. [Google Scholar]
- 14.Meyer F, Limberg C, editors. Organometallic Oxidation Catalysis. Springer; New York: 2007. [Google Scholar]
- 15.Stack TDP. Dalton Trans. 2003;1881 [Google Scholar]
- 16.a Messerschmidt A, Huber R, Poulos T, Wieghardt K. Handbook of Metalloproteins. Wiley; Chichester: 2001. [Google Scholar]; b Solomon EI, Chen P, Metz M, Lee SK, Palmer AE. Angew Chem Int Ed. 2001;40:4570. doi: 10.1002/1521-3773(20011217)40:24<4570::aid-anie4570>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]; c Pascaly M, Jolk I, Krebs B. Chem Unserer Zeit. 1999;33:334. [Google Scholar]; d Kaim W, Rall J. Angew Chem Int Ed. 1996;35:43. [Google Scholar]; e Solomon EI, Sundaram UM, Machonkin TE. Chem Rev. 1996;96:2563. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- 17.a Henson MJ, Mukherjee P, Root DE, Stack TDP, Solomon EI. J Am Chem Soc. 1999;121:10332. [Google Scholar]; b Spuhler P, Holthausen MC. Angew Chem Int Ed. 2003;42:5961. doi: 10.1002/anie.200352231. [DOI] [PubMed] [Google Scholar]; c Cramer CJ, Smith BA, Tolman WB. J Am Chem Soc. 1996;118:11283. [Google Scholar]
- 18.a Liang HC, Zhang CX, Henson MJ, Sommer RD, Hatwell KR, Kaderli S, Zuberbühler AD, Rheingold AL, Solomon EI, Karlin KD. J Am Chem Soc. 2002;124:4170. doi: 10.1021/ja0125265. [DOI] [PubMed] [Google Scholar]; b Park GY, Qayyum MF, Woertink J, Hodgson KO, Hedman B, Narducci Sarjeant AA, Solomon EI, Karlin KD. J Am Chem Soc. 2012;134:8513. doi: 10.1021/ja300674m. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cahoy J, Holland PL, Tolman WB. Inorg Chem. 1999;38:2161. doi: 10.1021/ic990095+. [DOI] [PubMed] [Google Scholar]; d Henson MJ, Vance MA, Zhang CX, Liang HC, Karlin KD, Solomon EI. J Am Chem Soc. 2003;125:5186. doi: 10.1021/ja0276366. [DOI] [PubMed] [Google Scholar]; e Maiti D, Woertink JS, Narducci Sarjeant AA, Solomon EI, Karlin KD. Inorg Chem. 2008;47:3787. doi: 10.1021/ic702437c. [DOI] [PubMed] [Google Scholar]
- 19.Mahadevan V, Henson MJ, Solomon EI, Stack TDP. J Am Chem Soc. 2000;122:10249. [Google Scholar]
- 20.Mahapatra S, Kaderli S, Llobet A, Neuhold YM, Palanché T, Halfen JA, Young VG, Kaden TA, Que L, Zuberbühler AD, Tolman WB. Inorg Chem. 1997;36:6343. [Google Scholar]
- 21.a Halfen JA, Mahapatra S, Wilkinson EC, Kaderli S, Young VG, Que L, Zuberbuhler AD, Tolman WB. Science. 1996;271:1397. doi: 10.1126/science.271.5254.1397. [DOI] [PubMed] [Google Scholar]; b Mahapatra S, Halfen JA, Wilkinson EC, Pan G, Wang X, Young VG, Cramer CJ, Que L, Tolman WB. J Am Chem Soc. 1996;118:11555. [Google Scholar]
- 22.Ottenwaelder X, Rudd DJ, Corbett MC, Hodgson KO, Hedman B, Stack TDP. J Am Chem Soc. 2006;128:9268. doi: 10.1021/ja061132g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki M. Acc Chem Res. 2007;40:609. doi: 10.1021/ar600048g. [DOI] [PubMed] [Google Scholar]
- 24.Himes RA, Karlin KD. Proc Natl Acad Sci USA. 2009;106:18877. doi: 10.1073/pnas.0911413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Himes RA, Barnese K, Karlin KD. Angew Chem Int Ed. 2010;49:6714. doi: 10.1002/anie.201003403. [DOI] [PubMed] [Google Scholar]
- 26.Vanelderen P, Hadt RG, Smeets PJ, Solomon EI, Schoonheydt RA, Sels BF. J Catal. 2011;284:157. doi: 10.1016/j.jcat.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karlin KD, Gultneh Y, Hayes JC, Zubieta J. Inorg Chem. 1984;23:519. [Google Scholar]
- 28.Sanyal I, Mahroof-Tahir M, Nasir MS, Ghosh P, Cohen BI, Gultneh Y, Cruse RW, Farooq A, Karlin KD. Inorg Chem. 1992;31:4322. [Google Scholar]
- 29.Obias HV, Lin Y, Murthy NN, Pidcock E, Solomon EI, Ralle M, Blackburn NJ, Neuhold YM, Zuberbühler AD, Karlin KD. J Am Chem Soc. 1998;120:12960. [Google Scholar]
- 30.Kitajima N, Koda T, Moro-oka Y. Chem Lett. 1988:347. [Google Scholar]
- 31.Kitajima N, Koda T, Iwata Y, Moro-oka Y. J Am Chem Soc. 1990;112:8833. [Google Scholar]
- 32.Kitajima N, Koda T, Hashimoto S, Kitagawa T, Moro-oka Y. J Am Chem Soc. 1991;113:5664. [Google Scholar]
- 33.Sorrell TN. Tetrahedron. 1989;45:3. [Google Scholar]
- 34.a Churchill MR, Davies G, El-Sayed MA, El-Shazly MF, Hutchinson JP, Rupich MW. Inorg Chem. 1980;19:201. [Google Scholar]; b Churchill MR, Davies G, El-Sayed MA, Hutchinson JP, Rupich MW. Inorg Chem. 1982;21:995. [Google Scholar]; c El-Sayed MA, Davies G, Kasem TS. Inorg Chem. 1990;29:4730. [Google Scholar]
- 35.a Reglier M, Jorand C, Waegell B. J Chem Soc Chem Commun. 1990;1752 [Google Scholar]; b Lapinte C, Riviere H, Roselli A, Fabre C. J Chem Soc Chem Commun. 1981;1109 [Google Scholar]
- 36.Paul PP, Tyeklar Z, Jacobson RR, Karlin KD. J Am Chem Soc. 1991;113:5322. [Google Scholar]
- 37.Groothaert MH, Smeets PJ, Sels BF, Jacobs PA, Schoonheydt RA. J Am Chem Soc. 2005;127:1394. doi: 10.1021/ja047158u. [DOI] [PubMed] [Google Scholar]
- 38.Woertink JS, Smeets PJ, Groothaert MH, Vance MA, Sels BF, Schoonheydt RA, Solomon EI. Proc Natl Acad Sci USA. 2009;106:18908. doi: 10.1073/pnas.0910461106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smeets PJ, Hadt RG, Woertink JS, Vanelderen P, Schoonheydt RA, Sels BF, Solomon EI. J Am Chem Soc. 2010;132:14736. doi: 10.1021/ja106283u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lieberman RL, Rosenzweig AC. Nature. 2005;434:177. doi: 10.1038/nature03311. [DOI] [PubMed] [Google Scholar]
- 41.Balasubramanian R, Smith SM, Rawat S, Yatsunyk LA, Stemmler TL, Rosenzweig AC. Nature. 2010;465:115. doi: 10.1038/nature08992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Culpepper MA, Cutsail GE, Hoffman BM, Rosenzweig AC. J Am Chem Soc. 2012;134:7640. doi: 10.1021/ja302195p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hakemian AS, Kondapalli KC, Telser J, Hoffman BM, Stemmler TL, Rosenzweig AC. Biochemistry. 2008;47:6793. doi: 10.1021/bi800598h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith SM, Rawat S, Telser J, Hoffman BM, Stemmler TL, Rosenzweig AC. Biochemistry. 2011;50:10231. doi: 10.1021/bi200801z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Solomon EI, Ginsbach JW, Heppner DE, Kieber-Emmons MT, Kjaergaard CH, Smeets PJ, Tian L, Woertink JS. Faraday Discuss. 2010;148:11. doi: 10.1039/c005500j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.a Shilov AE, Shteinman AA. Russ Chem Rev. 2012;81:291. [Google Scholar]; b Vanelderen P, Vancauwenbergh J, Sels BF, Schoonheydt RA. Coord Chem Rev. 2013;257:483. [Google Scholar]
- 47.Haack P, Limberg C, Ray K, Braun B, Kuhlmann U, Hildebrandt P, Herwig C. Inorg Chem. 2011;50:2133. doi: 10.1021/ic101249k. [DOI] [PubMed] [Google Scholar]
- 48.Pilz MF, Limberg C, Ziemer B. J Org Chem. 2006;71:4559. doi: 10.1021/jo060394y. [DOI] [PubMed] [Google Scholar]
- 49.a Kadish K, Smith K, Guilard R. Handbook of Porphyrin Science. Vol. 11. World Scientific; 2010. [Google Scholar]; b Loh ZH, Miller SE, Chang CJ, Carpenter SD, Nocera DG. J Phys Chem A. 2002;106:11700. [Google Scholar]
- 50.a Hlavinka ML, Hagadorn JR. Chem Commun. 2003:2686. doi: 10.1039/b309732c. [DOI] [PubMed] [Google Scholar]; b Kwong FY, Klapars A, Buchwald SL. Org Lett. 2002;4:581. doi: 10.1021/ol0171867. [DOI] [PubMed] [Google Scholar]
- 51.Janiak C. J Chem Soc Dalton Trans. 2000;3885 [Google Scholar]
- 52.a Pasquali M, Marini G, Floriani C, Gaetani-Manfredotti A. J Chem Soc Chem Commun. 1979;937 [Google Scholar]; b Eckenhoff WT, Pintauer T. Inorg Chem. 2007;46:5844. doi: 10.1021/ic700908m. [DOI] [PubMed] [Google Scholar]
- 53.Fulmer GR, Miller AJM, Sherden NH, Gottlieb HE, Nudelman A, Stoltz BM, Bercaw JE, Goldberg KI. Organometallics. 2010;29:2176. [Google Scholar]
- 54.Kau LS, Spira-Solomon DJ, Penner-Hahn JE, Hodgson KO, Solomon EI. J Am Chem Soc. 1987;109:6433. [Google Scholar]
- 55.a Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]; b Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 56.a Schultz NE, Zhao Y, Truhlar DG. J Phys Chem A. 2005;109:11127. doi: 10.1021/jp0539223. [DOI] [PubMed] [Google Scholar]; b Sousa SF, Fernandes PA, Ramos MJ. J Phys Chem A. 2007;111:10439. doi: 10.1021/jp0734474. [DOI] [PubMed] [Google Scholar]
- 57.a Weigend F, Furche F, Ahlrichs R. J Chem Phys. 2003;119:12753. [Google Scholar]; b Weigend F, Ahlrichs R. Phys Chem Chem Phys. 2005;7:3297. doi: 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- 58.a Noodleman L, Norman JG. J Chem Phys. 1979;70:4903. [Google Scholar]; b Noodleman L. J Chem Phys. 1981;74:5737. [Google Scholar]
- 59.a Quintal MM, Karton A, Iron MA, Boese AD, Martin JML. J Phys Chem A. 2006;110:709. doi: 10.1021/jp054449w. [DOI] [PubMed] [Google Scholar]; b Sousa SF, Fernandes PA, Ramos MJ. J Phys Chem A. 2007;111:10439. doi: 10.1021/jp0734474. [DOI] [PubMed] [Google Scholar]
- 60.a Becke AD. Phys Rev A. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]; b Perdew J. Phys Rev B. 1986;33:8822. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 61.a Greco C, Bruschi M, Fantucci P, Gioia Lde. Eur J Inorg Chem. 2007:1835. [Google Scholar]; b Greco C. Dalton Trans. 2013 [Google Scholar]; c Schneider CJ, Zampella G, Greco C, Pecoraro VL, Gioia Lde. Eur J Inorg Chem. 2007:515. [Google Scholar]
- 62.Yanai T, Tew DP, Handy NC. Chem Phys Lett. 2004;393:51. [Google Scholar]
- 63.Mikolajczyk MM, Zaleśny R, Czyznikowska Z, Toman P, Leszczynski J, Bartkowiak W. J Mol Model. 2011;17:2143. doi: 10.1007/s00894-010-0865-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Casida ME, Salahub DR. J Chem Phys. 2000;113:8918. [Google Scholar]
- 65.a Gillard RD, Laurie SH, Stephens FS. J Chem Soc A. 1968;2588 [Google Scholar]; b Zurowska B, Ochocki J, Mroziński J, Ciunik Z, Reedijk J. Inorg Chim Acta. 2004;357:755. [Google Scholar]; c Massoud SS, Louka FR, Mikuriya M, Ishida H, Mautner FA. Inorg Chem Commun. 2009;12:420. [Google Scholar]; d Ran J, Li X, Zhao Q, Qu Z, Li H, Shi Y, Chen G. Inorg Chem Commun. 2010;13:526. [Google Scholar]
- 66.a Zurowska B, Mrozinski J, Ciunik Z. Polyhedron. 2007;26:1251. [Google Scholar]; b Kleinstein A, Webb G. J Inorg Nucl Chem. 1971;33:405. [Google Scholar]
- 67.Vad MS, Nielsen A, Lennartson A, Bond AD, McGrady JE, McKenzie CJ. Dalton Trans. 2011:10698. doi: 10.1039/c1dt10594a. [DOI] [PubMed] [Google Scholar]
- 68.a Mahapatra S, Halfen JA, Tolman WB. J Am Chem Soc. 1996;118:11575. [Google Scholar]; b Cramer CJ, Pak Y. Theor Chem Acc. 2001;105:477. [Google Scholar]; c Mahapatra S, Halfen JA, Wilkinson EC, Que L, Tolman WB. J Am Chem Soc. 1994;116:9785. [Google Scholar]; d Taki WB, Teramae S, Nagatomo S, Tachi Y, Kitagawa T, Itoh S, Fukuzumi S. J Am Chem Soc. 2002;124:6367. doi: 10.1021/ja026047x. [DOI] [PubMed] [Google Scholar]; e Shearer J, Zhang CX, Hatcher LQ, Karlin KD. J Am Chem Soc. 2003;125:12670. doi: 10.1021/ja0359409. [DOI] [PubMed] [Google Scholar]; f Shearer J, Zhang CX, Zakharov LN, Rheingold AL, Karlin KD. J Am Chem Soc. 2005;127:5469. doi: 10.1021/ja045191a. [DOI] [PubMed] [Google Scholar]
- 69.Müller E, Mayer R, Narr B, Rieker A, Scheffler K. Justus Liebigs Ann Chem. 1961;645:25. [Google Scholar]
- 70.Meier H, Schneider HP, Rieker A, Hitchcock PB. Angew Chem Int Ed. 1978;17:121. [Google Scholar]
- 71.a Bowman DF, Hewgill FR. J Chem Soc C. 1971;1777 [Google Scholar]; b Becker HD, Gustafsson K. Tetrahedron Lett. 1976;17:4883. [Google Scholar]
- 72.a Cole AP, Root DE, Mukherjee P, Solomon EI, Stack TDP. Science. 1996;273:1848. doi: 10.1126/science.273.5283.1848. [DOI] [PubMed] [Google Scholar]; b Herres-Pawlis S, Ver-ma P, Haase R, Kang P, Lyons CT, Wasinger EC, Flörke U, Henkel G, Stack TDP. J Am Chem Soc. 2009;131:1154. doi: 10.1021/ja807809x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gupta R, Mukherjee R. Tetrahedron Lett. 2000;41:7763. [Google Scholar]; d Wünnemann S, Fröhlich R, Hoppe D. Eur J Org Chem. 2008;2008:684. [Google Scholar]; e Itoh S, Kumei H, Taki M, Nagatomo S, Kitagawa T, Fukuzumi S. J Am Chem Soc. 2001;123:6708. doi: 10.1021/ja015702i. [DOI] [PubMed] [Google Scholar]; f Halfen JA, Young VG, Tolman WB. Inorg Chem. 1998;37:2102. doi: 10.1021/ic971216d. [DOI] [PubMed] [Google Scholar]; g Yuan DR, Yan JM, Yu CZ, Xie RG. Chinese Chem Lett. 2005;16:147. [Google Scholar]; h Cole AP, Mahadevan V, Mirica LM, Ottenwaelder X, Stack TDP. Inorg Chem. 2005;44:7345. doi: 10.1021/ic050331i. [DOI] [PubMed] [Google Scholar]; i Alexakis A, Polet D, Rosset S, March S. J Org Chem. 2004;69:5660. doi: 10.1021/jo049359m. [DOI] [PubMed] [Google Scholar]; j Buisman GJH, Kamer PCJ, van Leeuwen PWNM. Tetrahedron: Asymmetry. 1993;4:1625. [Google Scholar]
- 73.Kushioka K. J Org Chem. 1983;48:4948. [Google Scholar]
- 74.Kushioka K. J Org Chem. 1984;49:4456. [Google Scholar]
- 75.a Kushioka K, Tanimoto I, Maruyama K. J Chem Soc Perkin Trans. 1989;2:1303. [Google Scholar]; b Kushioka K, Tanimoto I, Maruyama K. Bull Chem Soc Jpn. 1989;62:1147. [Google Scholar]; c Allen SE, Walvoord RR, Padilla-Salinas R, Kozlowski MC. Chem Rev. 2013;113:6234. doi: 10.1021/cr300527g. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.