

Abstract
Sphingosine-1-phosphate (S1P), a pleiotropic bioactive lipid mediator, and the kinase that produces it have now emerged as key regulators of numerous cellular processes involved in inflammation and cancer. Here, we review the importance of S1P in colitis and colitis-associated cancer (CAC) and discuss our recent work demonstrating that S1P produced by upregulation of SphK1 during colitis and associated cancer is essential for production of the multifunctional NF-kB-regulated cytokine IL-6, persistent activation of the transcription factor Stat3, and consequent upregulation of the S1P receptor, S1PR1. The effectiveness of the pro-drug FTY720 (known as fingolimod), approved for the treatment of multiple sclerosis, has become the gold standard for S1P-centric drugs, and will be used to illustrate the therapeutic value of modulating SphK1 and S1P receptor functions. We will discuss our recent results showing that FTY720/fingolimod administration interferes with the SphK1/S1P/S1PR1 axis and suppresses the NF-kB/IL-6/Stat3 malicious amplification loop and CAC. These preclinical studies suggest that FTY720/fingolimod may be useful in treating colon cancer in individuals with ulcerative colitis.
Keywords: sphingosine-1-phosphate, sphingosine kinase, colitis, colitis associated cancer, inflammation, FTY720
Introduction
It is now more than two decades since it was discovered that S1P is a signaling molecule that regulates cell growth and the suggestion that it may play a role in cancer (Olivera et al., 1993; Zhang et al., 1991). A large subsequent body of work has demonstrated that S1P regulates many processes important for inflammation and cancer, including cell proliferation, survival, migration, invasion, cytokine and chemokine production, angiogenesis and lymphangiogenesis (Nagahashi et al., 2012; Nagahashi et al., 2010; Pyne et al., 2010; Spiegel et al., 2011). S1P is generated intracellularly by two sphingosine kinase isoenzymes, SphK1 and SphK2. Numerous agonists and stimuli, such as growth factors, hormones, and pro-inflammatory cytokines activate SphK1 by inducing its phosphorylation and translocation from the cytosol to the plasma membrane where its substrate sphingosine is localized (Alvarez et al., 2007; Maceyka et al., 2012). This translocation enables localized production of S1P that in turn activates a family of five G protein coupled receptors (S1PR1-5), a process known as “inside-out signaling” by S1P (Takabe et al., 2008). S1P is exported out of the cells by ATP binding cassette transporters, ABCA1 (Sato et al., 2007), ABCC1, ABCG2 (Mitra et al., 2006; Takabe et al., 2010) and Spns2 (Hisano et al., 2011; Kawahara et al., 2009; Nagahashi et al., 2012). In contrast, much less is known about the roles of S1P produced by SphK2, which is present in several subcellular compartments including mitochondria and nucleus (Hait et al., 2009; Strub et al., 2011).
Intracellular S1P levels are tightly maintained by the balance between synthesis and degradation. S1P can be dephosphorylated by the action of two specific endoplasmic reticulum localized S1P phosphatases back to sphingosine, which is reutilized for synthesis of ceramide and complex sphingolipids. As the end product of metabolism of all cellular sphingolipids, S1P is also irreversibly degraded by S1P lyase to hexadecenal and phosphoethanolamine (Fyrst et al., 2010). By degrading tissue S1P, S1P lyase plays a major role in generation of a S1P concentration gradient between the circulation and tissues (Cyster et al., 2012) - S1P levels in blood and lymph are high, whereas levels are orders of magnitude lower in tissues and interstitial fluids (Cyster and Schwab, 2012). Immune cells, such as lymphocytes, hematopoietic progenitor cells, and dendritic cells utilize this S1P gradient to regulate their trafficking. Interestingly, the successful development of FTY720/fingolimod for treatment of multiple sclerosis (MS) and the elucidation of its mechanism of action has provided insight into the importance of the S1P/S1PR1 axis for the immune system.
In addition to the canonical extracellular actions of S1P that are now well known, it has long been suspected that S1P also has receptor independent intracellular actions that remained mysterious until the last few years. This situation has changed with discovery of several intracellular targets of S1P (Alvarez et al., 2010; Hait et al., 2009). The first example is TRAF2, an adaptor protein containing a RING domain that is implicated in the regulatory ubiquitination of RIP1, a critical event in activation of NF-κB in response to TNF-α. We showed that S1P produced by SphK1 binds to TRAF2 and is a cofactor required for its E3 ligase activity and consequently, Lys-63-linked polyubiquitination of RIP1 and NF-κB activation (Alvarez et al., 2010), explaining the importance of SphK1 and S1P in cytoprotection, inflammation, and immune responses (Spiegel and Milstien, 2011). Intriguingly, S1P, produced in the nucleus by SphK2, is an endogenous inhibitor of histone deacetylase (HDAC) (Hait et al., 2009). Nuclear SphK2 was shown to be a component of HDAC containing repressor complexes that are present at the promoters of specific genes, producing S1P that binds to and inhibits both HDAC1 and HDAC2, and regulates gene transcription, including the cyclin dependent kinase inhibitor p21. These results link sphingolipid metabolism in the nucleus to epigenetic regulation (Hait et al., 2009).
FTY720, the basic properties
FTY720 was first synthesized in 1992 in an attempt to simplify the complex structure of myriocin (ISP-1), a fungal metabolite with immunosuppressive properties isolated from Isaria sinclairii culture broth. A phenylene moiety was introduced in the side chain and this compound, designated FTY720 (2-amino-2-[2-(4octylphenyl)ethyl]propane-1,3-diol) was found to have even more potent immunosuppressive activity than myriocin (Chiba et al., 1998). Although it was initially thought that FTY720 itself was biologically active, it was later noted that FTY720 is structurally homologous to sphingosine, and then shown that it is phosphorylated in vivo to FTY720-P, a mimetic of S1P and an agonist all of the S1P receptors except S1PR2 (Brinkmann et al., 2002; Mandala et al., 2002). Subsequent studies demonstrated that FTY720 is primarily phosphorylated by SphK2 (Kharel et al., 2005a; Paugh et al., 2003; Zemann et al., 2006) and to a lesser extent by SphK1 (Liang et al., 2013).
Since FTY720 inhibits lymphocyte emigration from lymphoid organs and FTY720-P binds and activates S1P receptors, it was expected that activation of S1P receptors might play a role in immunosuppression. Indeed, S1PR1-deficient and T cell specific conditional knockout mice demonstrated conclusively that lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1PR1 (Allende et al., 2004; Matloubian et al., 2004). Although it is an S1PR1 agonist, FTY720-P is a functional antagonist because it induces internalization and degradation of S1PR1 and prolonged receptor downregulation, resulting in T cells that do not respond to S1P exit signal from lymph nodes. Hence, the immunosuppressive actions of FTY720 are due to the induction of lymphopenia.
Inflammation and cancer
Associations between inflammation and development of cancer have long been observed. As early as 1863, Rudolf Virchow, who is referred to as “the father of modern pathology”, noted leukocytes in neoplastic tissues and suggested that the “lymphoreticular infiltrate” reflected the origin of cancer at sites of chronic inflammation (Balkwill et al., 2001). Evidence has only been obtained in the past decade demonstrating that inflammation has a critical role in turmorigenesis and the molecular mechanisms involved have begun to be unraveled (Grivennikov et al., 2010; Karin et al., 2006). Epidemiological as well as clinical studies have revealed that chronic inflammation contributes to different stages of tumor development, including initiation, progression, invasion, and metastasis. Inflammation can be caused by various environmental and genetic factors such as cigarette smoking, Helicobactor pylori, hepatitis B and C, human papilloma virus, solar irradiation, asbestos, pancreatitis, autoimmunity, diet/obesity and diabetes mellitus (Grivennikov et al., 2010). Inflammation also affects immune surveillance and susceptibility to cancer therapy. Immune cells that infiltrate tumors have extensive and dynamic interactions with tumor cells that can enhance or suppress tumor growth and metastasis (Grivennikov et al., 2010). Understanding of the relationship between inflammation and cancer is growing and approaches are being developed to target not only cancer but also associated inflammation for cancer prevention and therapy.
Roles of NF-kB and Stat3 in colorectal cancer
Inflammatory bowel diseases (IBD) including ulcerative colitis (UC) and Crohn’s disease are a salient example of the link between chronic inflammation and cancer. An unfortunate consequence of persistent inflammation of the colon or UC is an increased risk factor for developing colorectal cancer (Ullman et al., 2011). Animal models that recapitulate many aspects of the human disease have provided several clues to the critical roles of inflammatory mediators and molecular events leading to development of colitis-associated cancer (CAC) (Saleh et al., 2011). Particular emphasis has been given to the roles of the master transcription factors NF-κB and signal transducer and activator of transcription-3 (Stat3) and their downstream pro-inflammatory cytokines TNF-α and IL-6 in the development of colitis-associated cancer (CAC) (Bollrath et al., 2009; Greten et al., 2004; Grivennikov et al., 2009).
Activation of NF-kB in intestinal epithelial cells (IECs) enhances survival pathways that are needed for formation of tumors (Greten et al., 2004). Activation of NF-κB in myeloid-derived inflammatory cells enhances inflammation in the tumor microenvironment, mainly by increasing expression of pro-inflammatory cytokines, such as TNF-α and IL-6, that in turn stimulate proliferation of tumor and stromal cells to further fuel and promote CAC (Greten et al., 2004; Popivanova et al., 2008). IL-6 then enhances both initiation and progression of CAC and stimulates Stat3 activation, which is necessary for intestinal mucosal regeneration after injury and for the development of CAC (Bollrath et al., 2009; Greten et al., 2004; Grivennikov et al., 2009). NF-κB and Stat3 can also act in positive feedback loops to enhance production of cytokines and chemokines that recruit additional immune cells that maintain tumor-associated inflammation (Bollrath et al., 2009; Greten et al., 2004; Grivennikov et al., 2009). Clinical studies have revealed that expression of TNF-α and IL-6 and activation of NF-κB and Stat3 are increased in patients with active UC and particularly in those who progressed to colorectal cancer (Li et al., 2010; Popivanova et al., 2008).
Sphingosine-1-phosphate and colon cancer
As mentioned above, there is abundant evidence that S1P is involved in inflammation and cancer (Pyne and Pyne, 2010; Spiegel and Milstien, 2011). S1P and SphK1 have long been implicated in the actions of pro-inflammatory cytokines, such as TNF-α (Billich et al., 2005; Pettus et al., 2003; Xia et al., 2002). TNF-α activates and translocates SphK1 to the plasma membrane (Pitson et al., 2003; Xia et al., 2002) and “inside-out signaling” by S1P promotes certain TNF-α functions (De Palma et al., 2006; Scherer et al., 2010). Nevertheless, it is also possible that SphK1 and intracellular S1P, by stimulating the E3 ubiquitin ligase activity of TRAF2, play a direct role in TNF-α signaling and the canonical NF-κB activation pathway important in inflammatory, anti-apoptotic, and immune processes (Alvarez et al., 2010).
Investigations of clinical samples demonstrated that expression of SphK1 is elevated in the colons of patients with UC (Snider et al., 2009) or colon cancer (Kawamori et al., 2006). Previous studies demonstrated that Sphk1 is expressed and is required for small intestinal tumor cell proliferation in ApcMin/+ mice (Kohno et al., 2006). Adenoma size and epithelial cell proliferation in the polyps but not incidence were dramatically reduced in Apc Min/+ Sphk1−/− mice, suggesting that Sphk1 regulates adenoma progression. Although S1p1rR, S1pr2, and S1pr3 were expressed, polyp incidence or size were unchanged in ApcMin/+ mice with deletions of these receptors suggesting that extracellular S1P signaling via its receptors is not involved in adenoma cell proliferation. Increased SphK1 and elevated levels of S1P in colon cancers and in the circulation were also observed in mice treated with the colonotropic mutagen azoxymethane (AOM) followed by chronic intestinal inflammation triggered by administration of the luminal toxin dextran sodium sulfate (DSS) (Kawamori et al., 2008). Furthermore, deletion of Sphk1 reduced DSS-induced colitis (Snider et al., 2009) and also the aberrant crypt formation and colon cancer development in this CAC model (Kawamori et al., 2008). However, the molecular mechanisms by which S1P produced by SphK1 promotes early and late stages of CAC development and the role of SphK2 have not been elucidated and this was the focus of our recent studies.
SphK1/S1P/S1PR1 axis in chronic intestinal inflammation and cancer
Surprisingly, we found that in contrast to the protective effects of Sphk1 deletion (Kawamori et al., 2008), knockout of Sphk2 markedly increased the severity of DSS-induced colitis and associated tumorigenesis (Liang et al., 2013). Consistent with the profound effect on tumor size and large increase in adenomas in SphK2 knockout mice, there was increased expression of cyclooxygenase 2, which plays a causal role in colon carcinogenesis (Oshima et al., 1996). Moreover, we found in agreement with others (Snider et al., 2009) that SphK1 and S1P in the colon and in the circulation were increased during acute colitis. Surprisingly, however, SphK1 expression in the colon was greatly elevated in SphK2 null mice with a corresponding increase in S1P levels and both were exacerbated during colitis. Interestingly, in agreement with the key role of SphK1/S1P in the NF-kB pathway, in the SphK2 null colon, NF-κB was constitutively activated, and was stimulated further during colitis development. This led to enormous production of the NF-κB regulated proinflammatory cytokines TNF-α and IL-6, which are key mediators in colitis and development of CAC (Bollrath et al., 2009; Grivennikov et al., 2009), that stimulates Stat3 and as also shown by others (Lee et al., 2010; Yu et al., 2009), upregulates expression of S1pr1.
Although it has been revealed that NF-κB and Stat3 play a role in chronic inflammation and cancer development, negative feedback loops usually tightly limit the duration of activation of these factors (Lee et al., 2010), but it was previously not clear how their persistent activation is maintained during colitis and the progression of CAC. Our work revealed that one of the missing links between inflammation and cancer is the SphK1/S1P/S1PR1 axis that contributes to the NF-kB/IL-6/Stat3 amplification loop. Similar to SphK1, we found that S1PR1 is also upregulated in murine models of colitis and CAC. We suggested that upregulation of SphK1 in intestinal epithelial cells (IECs) and increased formation of S1P during intestinal inflammation, and more so in CAC, leads to activation of S1PR1 and downstream activation of Src or JAK, which can phosphorylate and activate Stat3 (Yu et al., 1995). Stat3 then regulates expression of many genes involved in both cell survival and proliferation, including S1pr1 (Yu et al., 2009). Moreover, SphK1 and intracellular, or extracellular S1P by inside-out signaling, are also involved in activation of NF-κB by TNF-α (Figure 1). This leads to recruitment of myeloid cells that produce IL-6 and TNF-α, pro-inflammatory cytokines, that are important for CAC growth (Bollrath et al., 2009; Grivennikov et al., 2009; Popivanova et al., 2008). Moreover, NF-kB also induces expression of genes that regulate survival of IECs and anti-apoptotic genes during early tumor promotion that increase tumor incidence (Greten et al., 2004). Therefore, NF-κB can regulate Stat3 activation in IEC in a three-fold manner: i. by recruiting myeloid cells, such as lamina propria macrophages, dendritic cells, and tumor-associated macrophages that secrete Stat3 activating cytokines; ii. by regulating the transcription of these cytokines in myeloid cells; and iii., these cytokines, including TNF-α and IL-1, stimulate SphK1 and upregulate its expression (Spiegel and Milstien, 2011), leading to amplification of the activation of both Stat3 (via S1PR1) and NF-κB (by intracellular and extracellular S1P) (Figure 1). Interestingly, although TNF-α activates NF-κB in IECs, ablation of IKKβ in myeloid cells, which prevents TNF-α production, does not affect NF-κB or the survival of premalignant IECs (Greten et al., 2004). It is tempting to speculate that the robust upregulation of SphK1 in IECs is responsible at least in part for NF-κB activation in IECs. Of importance, SphK1 and S1P increase during chronic intestinal inflammation. In CAC, altered epithelial and neoplastic cells and recruited immune cells continue to produce S1P and pro-inflammatory cytokines such as IL-6 (Bollrath et al., 2009; Grivennikov et al., 2009) that activate oncogenic Stat3 to sustain tumor-associated inflammation and to fuel tumor initiation, promotion, and progression. Taken together our work showed that upregulation of SphK1 expression, formation S1P, and subsequent activation of its S1PR1 receptor play an essential role in maintaining persistent activation of the critical transcription factors NF-kB and Stat3 in a malicious feed-forward amplification loop that leads to chronic inflammation and CAC development.
Figure 1. Role of the SphK1/S1P/S1PR1 axis in activation of NF-kB and Stat3 linking inflammation and cancer.
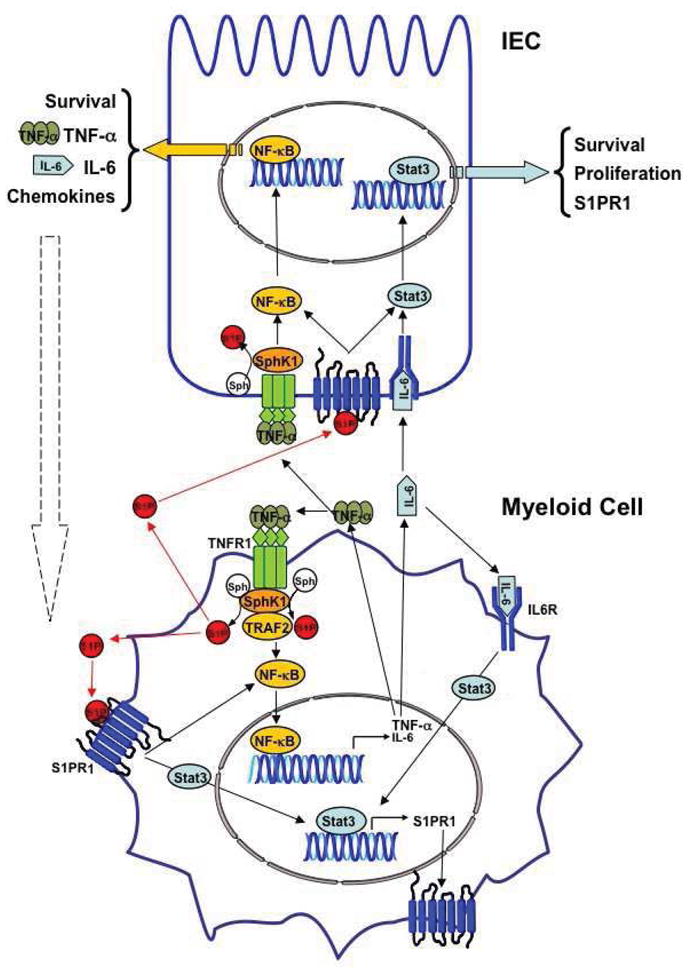
SphK1 is upregulated in colitis and CAC, increasing S1P that activates its receptor S1PR1, leading to Stat3 activation. Reciprocally, Stat3 enhances transcription of its target genes, including S1PR1. Intracellular and/or extracellular S1P also induces activation of NF-κB. NF-kB induces transcription of the proinflammatory cytokines TNF-α and IL-6, which induces activation of Stat3. TNF-α stimulates SphK1 to sustain NF-kB activation. These feed-forward amplification loops in cancer cells (IECs) and in the stromal myeloid cells are important for malignant progression. Note: NF-κB and Stat3 may also be activated independently of the SphK1/S1P axis in CAC.
Potential therapy of colitis and CAC with FTY720/fingolimod
As mentioned above, FTY720 is phosphorylated in vivo to a S1P mimetic that modulates the immune system by acting as a functional antagonist of S1PR1, inducing its internalization and degradation (Brinkmann et al., 2010). Therefore it was of interest to examine the effects of interfering with S1PR1 functions and this feed-forward amplification loop with FTY720. We observed that daily treatment with FTY720 suppressed colitis. Surprisingly, FTY720 was also effective in SphK2 null mice, in contrast to previous studies indicating that SphK2 was the sole enzyme responsible for its phosphorylation in vivo (Kharel et al., 2005b; Sensken et al., 2009). However, this conclusion was based on the inability of a single dose of FTY720 to induce lymphopenia in SphK2 null mice. Consistent with previous results showing that FTY720 was also a weak substrate for SphK1 (Billich et al., 2003; Paugh et al., 2003), our study indicates that upregulation of SphK1 can compensate for the lack of SphK2. FTY720 also reduced expression of SphK1 in colitis and CAC and consequently prevented activation of NF-kB, elevation of IL-6 and S1PR1, and subsequently reduced persistent activation of Stat3. Daily treatment of mice with FTY720 during CAC induction drastically reduced tumor multiplicity and load probably due to decreased infiltration of inflammatory cells. Remarkably, even when FTY720 was administered later after tumors were initiated, it reduced tumor growth and development and prevented the amplification loop involving SphK1, S1P, and S1PR1 leading to persistent phosphorylation of Stat3.
In agreement with our findings and their immunosuppressive actions, FTY720 and other S1PR1 modulators are efficacious in several different models of colitis (Daniel et al., 2007; Deguchi et al., 2006; Fujii et al., 2008; Sanada et al., 2011; Song et al., 2008). Although targeting the inflammatory microenvironment may be beneficial since immune cells are not subject to the same mechanisms of drug resistance as cancer cells, anti-inflammatory therapies do not kill cancer cells. Treatment with FTY720, however, blocked CAC progression even when only administered after tumors were initiated, and thus has cytostatic effects on cancer cells that are independent of immunosuppression. In agreement, FTY720 inhibits proliferation and enhances apoptosis of several types of cancer cells in vitro and suppresses xenograft tumor growth and metastasis in immunodeficient mice (reviewed in (Pyne and Pyne, 2010)). Our work shows that FTY720 by interfering with the upregulation of SphK1 and S1PR1 curtails the S1P-SphK1-S1PR1 feedforward amplification loop that leads to NF-κB and persistent Stat3 activation critical for CAC. Hence, targeting the upstream machinery leading to NF-κB and Stat3 activation is a feasible alternative approach to inhibit this signaling pathway. Our work has broad implications for pathogenesis and treatment of colitis and associated colorectal cancer and suggests that FTY720 may be useful in treating colon cancer patients with inflammatory bowel disease.
Conclusions
Targeted chemotherapy is a promising therapeutic modality; however, it also has some limitations as do other forms of cancer therapy. One of the most serious limitations is the development of resistance to a drug that limits its efficacy. In some cases, another targeted therapy might overcome this resistance. Therefore, targeted therapies are expected to work best in combination, or with conventional cytotoxic therapies. Since FTY720 has unique mechanisms to suppress cancer progression by targeting S1P/SphK1/S1PR1 axis, combination therapies with FTY720 may provide more therapeutic options for colon cancer patients. FTY720/fingolimod already has several advantages for implementation as an anti-cancer drug: it is an orally bio-available drug with good pharmacokinetics and a long half-life; and it has already been approved for human use for the treatment of multiple sclerosis with few and manageable side effects. It is hoped that the studies reviewed here will serve as the basis for new clinical trials for treatment of IBD and colorectal cancer.
Acknowledgments
This work was supported by NIH grants R37GM043880 and R01CA61774 (to S.S.) and R01CA160688 and the Susan G. Komen for the Cure Research Foundation grant (to K.T) in part by NIH grant P30CA16059 to the Massey Cancer Center. M.N. is a Japan Society for the Promotion of Science Postdoctoral Fellow.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allende ML, Dreier JL, Mandala S, Proia RL. Expression of the sphingosine-1-phosphate receptor, S1P1, on T-cells controls thymic emigration. J Biol Chem. 2004;279:15396–401. doi: 10.1074/jbc.M314291200. [DOI] [PubMed] [Google Scholar]
- Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465:1084–8. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez SE, Milstien S, Spiegel S. Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol Metab. 2007;18:300–7. doi: 10.1016/j.tem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–45. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- Billich A, Bornancin F, Devay P, Mechtcheriakova D, Urtz N, Baumruker T. Phosphorylation of the imunomodulatory drug FTY720 by sphingosine kinases. J Biol Chem. 2003;278:47408–15. doi: 10.1074/jbc.M307687200. [DOI] [PubMed] [Google Scholar]
- Billich A, Bornancin F, Mechtcheriakova D, Natt F, Huesken D, Baumruker T. Basal and induced sphingosine kinase 1 activity in A549 carcinoma cells: function in cell survival and IL-1beta and TNF-alpha induced production of inflammatory mediators. Cell Signal. 2005;17:1203–17. doi: 10.1016/j.cellsig.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9:883–97. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, et al. The immune modulator, FTY720, targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277:21453–7. doi: 10.1074/jbc.C200176200. [DOI] [PubMed] [Google Scholar]
- Chiba K, Yanagawa Y, Masubuchi Y, Kataoka H, Kawaguchi T, Ohtsuki M, et al. FTY720, a novel immunosuppressant, induces sequestration of circulating mature lymphocytes by acceleration of lymphocyte homing in rats. I. FTY720 selectively decreases the number of circulating mature lymphocytes by acceleration of lymphocyte homing. J Immunol. 1998;160:5037–44. [PubMed] [Google Scholar]
- Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. 2012;30:69–94. doi: 10.1146/annurev-immunol-020711-075011. [DOI] [PubMed] [Google Scholar]
- Daniel C, Sartory N, Zahn N, Geisslinger G, Radeke HH, Stein JM. FTY720 ameliorates Th1-mediated colitis in mice by directly affecting the functional activity of CD4+CD25+ regulatory T cells. J Immunol. 2007;178:2458–68. doi: 10.4049/jimmunol.178.4.2458. [DOI] [PubMed] [Google Scholar]
- De Palma C, Meacci E, Perrotta C, Bruni P, Clementi E. Endothelial nitric oxide synthase activation by tumor necrosis factor alpha through neutral sphingomyelinase 2, sphingosine kinase 1, and sphingosine 1 phosphate receptors: a novel pathway relevant to the pathophysiology of endothelium. Arterioscler Thromb Vasc Biol. 2006;26:99–105. doi: 10.1161/01.ATV.0000194074.59584.42. [DOI] [PubMed] [Google Scholar]
- Deguchi Y, Andoh A, Yagi Y, Bamba S, Inatomi O, Tsujikawa T, et al. The S1P receptor modulator FTY720 prevents the development of experimental colitis in mice. Oncol Rep. 2006;16:699–703. [PubMed] [Google Scholar]
- Fujii T, Tomita T, Kanai T, Nemoto Y, Totsuka T, Sakamoto N, et al. FTY720 suppresses the development of colitis in lymphoid-null mice by modulating the trafficking of colitogenic CD4+ T cells in bone marrow. Eur J Immunol. 2008;38:3290–303. doi: 10.1002/eji.200838359. [DOI] [PubMed] [Google Scholar]
- Fyrst H, Saba JD. An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat Chem Biol. 2010;6:489–97. doi: 10.1038/nchembio.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–96. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–13. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325:1254–7. doi: 10.1126/science.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisano Y, Kobayashi N, Kawahara A, Yamaguchi A, Nishi T. The sphingosine 1-phosphate transporter, SPNS2, functions as a transporter of the phosphorylated form of the immunomodulating agent FTY720. J Biol Chem. 2011;286:1758–66. doi: 10.1074/jbc.M110.171116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–35. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- Kawahara A, Nishi T, Hisano Y, Fukui H, Yamaguchi A, Mochizuki N. The sphingolipid transporter spns2 functions in migration of zebrafish myocardial precursors. Science. 2009;323:524–7. doi: 10.1126/science.1167449. [DOI] [PubMed] [Google Scholar]
- Kawamori T, Kaneshiro T, Okumura M, Maalouf S, Uflacker A, Bielawski J, et al. Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J. 2008;23:405–14. doi: 10.1096/fj.08-117572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamori T, Osta W, Johnson KR, Pettus BJ, Bielawski J, Tanaka T, et al. Sphingosine kinase 1 is up-regulated in colon carcinogenesis. FASEB J. 2006;20:386–8. doi: 10.1096/fj.05-4331fje. [DOI] [PubMed] [Google Scholar]
- Kharel Y, Lee S, Snyder AH, Sheasley-O’neill SL, Morris MA, Setiady Y, et al. Sphingosine kinase 2 is required for modulation of lymphocyte traffic by FTY720. J Biol Chem. 2005a;280:36865–72. doi: 10.1074/jbc.M506293200. [DOI] [PubMed] [Google Scholar]
- Kharel Y, Lee S, Snyder AH, Sheasley-O’neill SL, Morris MA, Setiady Y, et al. Sphingosine kinase 2 is required for modulation of lymphocyte traffic by FTY720. J Biol Chem. 2005b;280:36865–72. doi: 10.1074/jbc.M506293200. [DOI] [PubMed] [Google Scholar]
- Kohno M, Momoi M, Oo ML, Paik JH, Lee YM, Venkataraman K, et al. Intracellular role for sphingosine kinase 1 in intestinal adenoma cell proliferation. Mol Cell Biol. 2006;26:7211–23. doi: 10.1128/MCB.02341-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Deng J, Kujawski M, Yang C, Liu Y, Herrmann A, et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med. 2010;16:1421–8. doi: 10.1038/nm.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, de Haar C, Chen M, Deuring J, Gerrits MM, Smits R, et al. Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut. 2010;59:227–35. doi: 10.1136/gut.2009.184176. [DOI] [PubMed] [Google Scholar]
- Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang W-C, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. 2013;23:107–20. doi: 10.1016/j.ccr.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012;22:50–60. doi: 10.1016/j.tcb.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296:346–9. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–60. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- Mitra P, Oskeritzian CA, Payne SG, Beaven MA, Milstien S, Spiegel S. Role of ABCC1 in export of sphingosine-1-phosphate from mast cells. Proc Natl Acad Sci USA. 2006;103:16394–9. doi: 10.1073/pnas.0603734103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahashi M, Ramachandran S, Kim EY, Allegood JC, Rashid OM, Milstien S, et al. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by tumor-induced angiogenesis and lymphangiogenesis. Cancer Res. 2012;72:726–35. doi: 10.1158/0008-5472.CAN-11-2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahashi M, Ramachandran S, Rashid OM, Takabe K. Lymphangiogenesis: A new player in cancer progression. World J Gastroenterology. 2010;16:4003–12. doi: 10.3748/wjg.v16.i32.4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera A, Spiegel S. Sphingosine-1-phosphate as a second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature. 1993;365:557–60. doi: 10.1038/365557a0. [DOI] [PubMed] [Google Scholar]
- Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, et al. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–9. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- Paugh SW, Payne SG, Barbour SE, Milstien S, Spiegel S. The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2. FEBS Lett. 2003;554:189–93. doi: 10.1016/s0014-5793(03)01168-2. [DOI] [PubMed] [Google Scholar]
- Pettus BJ, Bielawski J, Porcelli AM, Reames DL, Johnson KR, Morrow J, et al. The sphingosine kinase 1/sphingosine-1-phosphate pathway mediates COX-2 induction and PGE2 production in response to TNF-alpha. FASEB J. 2003;17:1411–21. doi: 10.1096/fj.02-1038com. [DOI] [PubMed] [Google Scholar]
- Pitson SM, Moretti PA, Zebol JR, Lynn HE, Xia P, Vadas MA, et al. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 2003;22:5491–500. doi: 10.1093/emboj/cdg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008;118:560–70. doi: 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- Saleh M, Trinchieri G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat Rev Immunol. 2011;11:9–20. doi: 10.1038/nri2891. [DOI] [PubMed] [Google Scholar]
- Sanada Y, Mizushima T, Kai Y, Nishimura J, Hagiya H, Kurata H, et al. Therapeutic effects of novel sphingosine-1-phosphate receptor agonist W-061 in murine DSS colitis. PLoS One. 2011;6:e23933. doi: 10.1371/journal.pone.0023933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Malchinkhuu E, Horiuchi Y, Mogi C, Tomura H, Tosaka M, et al. Critical role of ABCA1 transporter in sphingosine 1-phosphate release from astrocytes. J Neurochem. 2007;103:2610–9. doi: 10.1111/j.1471-4159.2007.04958.x. [DOI] [PubMed] [Google Scholar]
- Scherer EQ, Yang J, Canis M, Reimann K, Ivanov K, Diehl CD, et al. Tumor necrosis factor-alpha enhances microvascular tone and reduces blood flow in the cochlea via enhanced sphingosine-1-phosphate signaling. Stroke. 2010;41:2618–24. doi: 10.1161/STROKEAHA.110.593327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensken SC, Bode C, Graler MH. Accumulation of fingolimod (FTY720) in lymphoid tissues contributes to prolonged efficacy. J Pharmacol Exp Ther. 2009;328:963–9. doi: 10.1124/jpet.108.148163. [DOI] [PubMed] [Google Scholar]
- Snider AJ, Kawamori T, Bradshaw SG, Orr KA, Gilkeson GS, Hannun YA, et al. A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. FASEB J. 2009;23:143–52. doi: 10.1096/fj.08-118109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Matsuda C, Kai Y, Nishida T, Nakajima K, Mizushima T, et al. A novel sphingosine 1-phosphate receptor agonist, 2-amino-2-propanediol hydrochloride (KRP-203), regulates chronic colitis in interleukin-10 gene-deficient mice. J Pharmacol Exp Ther. 2008;324:276–83. doi: 10.1124/jpet.106.119172. [DOI] [PubMed] [Google Scholar]
- Spiegel S, Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat Rev Immunol. 2011;11:403–15. doi: 10.1038/nri2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strub GM, Paillard M, Liang J, Gomez L, Allegood JC, Hait NC, et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011;25:600–12. doi: 10.1096/fj.10-167502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takabe K, Kim RH, Allegood JC, Mitra P, Ramachandran S, Nagahashi M, et al. Estradiol induces export of sphingosine 1-phosphate from breast cancer cells via ABCC1 and ABCG2. J Biol Chem. 2010;285:10477–86. doi: 10.1074/jbc.M109.064162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takabe K, Paugh SW, Milstien S, Spiegel S. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev. 2008;60:181–95. doi: 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology. 2011;140:1807–16. doi: 10.1053/j.gastro.2011.01.057. [DOI] [PubMed] [Google Scholar]
- Xia P, Wang L, Moretti PA, Albanese N, Chai F, Pitson SM, et al. Sphingosine kinase interacts with TRAF2 and dissects tumor necrosis factor-alpha signaling. J Biol Chem. 2002;277:7996–8003. doi: 10.1074/jbc.M111423200. [DOI] [PubMed] [Google Scholar]
- Yu CL, Meyer DJ, Campbell GS, Larner AC, Carter-Su C, Schwartz J, et al. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science. 1995;269:81–3. doi: 10.1126/science.7541555. [DOI] [PubMed] [Google Scholar]
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemann B, Kinzel B, Muller M, Reuschel R, Mechtcheriakova D, Urtz N, et al. Sphingosine kinase type 2 is essential for lymphopenia induced by the immunomodulatory drug FTY720. Blood. 2006;107:1454–8. doi: 10.1182/blood-2005-07-2628. [DOI] [PubMed] [Google Scholar]
- Zhang H, Desai NN, Olivera A, Seki T, Brooker G, Spiegel S. Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J Cell Biol. 1991;114:155–67. doi: 10.1083/jcb.114.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]