

Abstract
As a result of their inherent planarity, DNA base radicals generated by one electron oxidation/reduction or bond cleavage form π- or σ-radicals. While most DNA base systems form π-radicals there are a number of nucleobase analogs such as one-electron oxidized 6-azauraci1, 6-azacytosine, and 2-thiothymine or one-electron reduced 5-bromouracil that form more reactive σ-radicals. Elucidating the availability of these states within DNA, base radical electronic structure is important to the understanding of the reactivity of DNA base radicals in different environments. In this work, we address this question by the calculation of the relative energies of π- and σ-radical states in DNA/RNA bases and their analogs. We used density functional theory B3LYP/6-31++G** method to optimize the geometries of π- and σ-radicals in Cs symmetry (i.e., planar) in the gas phase and in solution using the polarized continuum model (PCM). The calculations predict that σ- and π-radical states in one electron oxidized bases of thymine, T(N3-H)•, and uracil, U(N3-H)• are very close in energy, i.e., the π-radical is only ca. 4 kcal/mol more stable than the σ-radical. For the one electron oxidized radicals of cytosine, C•+, C(N4-H)•, adenine, A•+, A(N6-H)•, and guanine, G•+, G(N2-H)•, G(N1-H)• the π-radicals are ca. 16 to 41 kcal/mol more stable than their corresponding σ-radicals. Inclusion of solvent (PCM) is found to stabilize the π- over σ-radical of each of the systems. U(N3-H)• with three discrete water molecules in the gas phase, is found to form a three-electron σ bond between N3 atom of uracil and O atom of a water molecule but on inclusion of full solvation and discrete hydration the π-radical remains most stable..
Keywords: DNA base radical, sigma-radical, pi-radical, radical of DNA bases, DFT, cation radical of bases
Introduction
DNA/RNA base radicals produced by ionizing radiation, UV-radiation, OH• and chemical attack have been identified by electron spin resonance (ESR) and pulse radiolysis experiments.1–10 The electronic nature of these radicals (σ- or π-type) determine the mechanisms of the development of further DNA damage. The initial cation or anion radicals of the DNA bases are usually π-radicals in nature. However, this is not always the case. For γ-irradiated samples of pyridine and several N-heteroaromatic hydrocarbons σ-cation radicals were found by Kato and Shida using ESR at 77 K.11,12 Sevilla and Swarts13 found the σ-radicals of one-electron oxidized 6-azauracil (6-azaU) and 6-azacytosine (6-azaC) employing ESR spectroscopy at 77 K. One-electron addition to 5-halouracils produces π-radicals in 5-bromo-uracil and 5-chloro-uracil that form σ-σ*-bonded radicals as the C-X (X= Cl, Br) bond lengthens and on bond rupture the reactive uracilyl σ-radical is formed.14,15 ESR spectroscopic results show that γ-irradiated single crystals of 2-thiothymine form σ-radicals while 2-thiouracil and 2-thiocytosine were found to produce π-radicals, suggesting σ- and π-radical states are close in energy for all of these species.16 The succinimidyl radical with the same C(O)-N-C(O) functional group as found in thymine and uracil has been recognized to produce near degenerate σ- and π-radicals with distinct chemical properties. The σ-radical is found to be much more reactive and undergo C-C bond cleavage while the π-radical does not.17
Though, the σ-radicals for a number of substituted one-electron oxidized pyrimidines were recognized experimentally,11–17 σ-cation radicals of the one electron oxidized DNA/RNA bases (guanine (G), adenine (A), thymine (T), cytosine (C) and uracil (U)) have not been found as yet. The breaking of a bond such as the C-Br bound in 5-bromouracil results in a σ-radical while the deprotonation of a DNA base π-cation radical from a nitrogen site produces the same π-radical species as formed by N-H bond fragmentation. An important question in this context is how close in energy are the σ- and π-radical states and under what conditions will the π-cation radical, on deprotonation, become a σ-radical. Thus, in this present work we employ density functional theory to calculate the relative stabilities of σ- and π-radicals of one-electron oxidized DNA bases and their N-H deprotonated neutral radicals and show that for pyrimidine DNA bases the σ- and π-radicals are close in energy and there are conditions for which the σ-radicals become more stable than π-radicals.
Method of Calculations
The present work employs the B3LYP method which has been found to be an excellent and cost-effective choice for calculation of various properties of DNA bases and base-pairs in their neutral and radical state.3,15,18–26 The B3LYP functional utilizes Becke’s exchange functional with the Lee-Yang-Parr nonlocal correlation functional (LYP).27 The basis set used in the calculation is 6-31++G** and the unrestricted open shell method was employed in the calculation. The geometries of all the species in their radical cation states or nitrogen deprotonated state in the gas phase were optimized in the Cs symmetry (planar) and “guess=alter” keyword was used to obtain optimized σ- and π-radical states, respectively. The stable states were then fully optimized in C1 symmetry to observe the effect of structural distortion (nonplanarity) on the σ- and π-nature and energy of these species. To incorporate the effect of bulk aqueous solvent, we used the self-consistent reaction field and the integral equation formalism polarized continuum model (IEFPCM) with a dielectric constant ε = 78.4 as implemented in the Gaussian09 program. The IEFPCM model was employed to optimize the geometries of few representative cases with outcomes presented in the result and discussion section. All the calculations were carried out using the Gaussian 09 program28, spin density distribution of molecule was plotted (0.004 electron/bohr3) using the GaussView program29 and JMOL was used to draw structures.30
From earlier works,22,25, 26 it is well established that use of a small basis set (6-31G*) with the B3LYP method is most suitable to reproduce the experimental HFCCs. In the present study we use the 6-31++G** basis set to calculate optimal total energies of π- and σ-radicals but also find this basis set gives good estimates of HFCCs.
Results and Discussion
The molecular structures of all the molecules used in the present study to explore their π- and σ-radical formation in the gas phase and in solution are presented in Figure 1. The spin density distribution and relative stabilities of the π- and σ-radicals in the gas phase and in solution are presented in Tables 1 – 4 and Figures 2, respectively. The anisotropic hyperfine couplings (HFCs) calculated using the B3LYP/6-31++G** method, for all the radicals presented in Tables 1 – 4, along with the available experimental values are given in Table S1 in the supporting information. In Table S1, we provide only the couplings of atoms with the largest spin densities. We also note that radical designations such as T(N3-H)•, indicates that the thymine radical is deprotonated from N3; whereas the designation π(C5) for T(N3-H)• indicates this radical has a π radical state with high spin density at atom C5. A single radical such as T(N3-H)•, can have several π- or σ-radical states. The relative stability of π- and σ-radical states of a radical is of considerable interest to the radical chemistry of these intermediates.11–17
Figure 1.

Structures of molecules used for the calculation of their π- and σ-radicals.
Table 1.
The B3LYP/6-31++G** calculated spin density distribution of π- and σ-radicals of succinimidyl• and 2-thiothymine(N3-H)•. The relative stabilities of the radicals in the gas phase and (aqueous solution) in kcal/mol are given at the bottom of each figure. The molecular symmetry is given in parentheses.
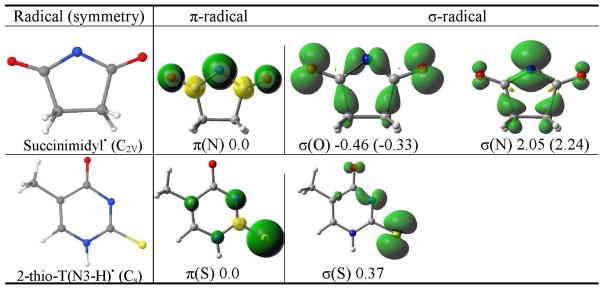
|
Table 4.
The B3LYP/6-31++G** calculated spin density distribution of π- and σ-radicals of adenine and guanine. The relative stabilities of the radicals in the gas phase (solution) in kcal/mol are given at the bottom of each figure. The molecular symmetry is given in parentheses.b
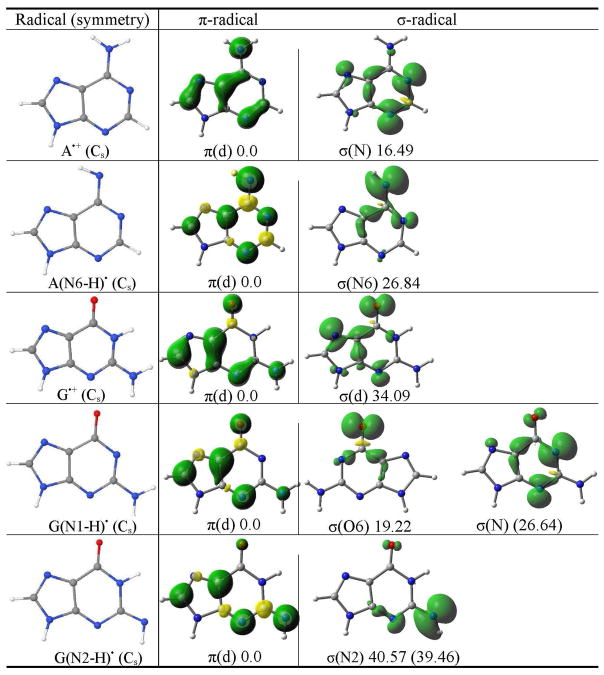
|
π(d) designates the delocalized nature of the radical.
Figure 2.
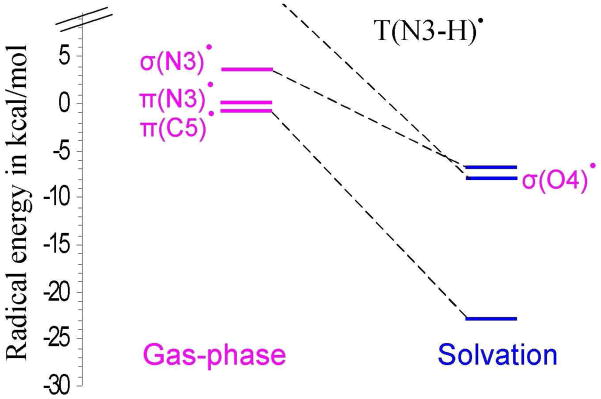
Relative energies of σ- and π-radicals of T(N3-H)• in the gas phase and in aqueous solution using PCM.
(i) Succinimidyl•
The succinimidyl radical has been shown to have both π- and σ-radical forms. These two succinimidyl radical types were first identified by Skell et al.17 in a thermal chain reaction and shown to have distinct chemical reactivities. The geometry of succinimidyl• is optimized by the B3LYP/6-31++G** method specifying C2V symmetry. The π-radical state is 2B1 while the two σ-radical states are 2B2 (oxygen(O)-centered) and 2A1 (N-centered), respectively. The O-centered σ-radical state is found to be the most stable in the gas phase and in solution using the PCM model. The σ (O)-radical in the gas phase (solution) is only 0.5 (0.33) kcal/mol lower in energy than the π-radical whereas the σ (N)-centered radical is 2.05 (2.24) kcal/mol higher in energy than the π-radical (Table 1). Using the B3LYP method and different basis sets Gainsforth et al.31 also predicted the O-centered σ-radical to be the most stable whereas an ab initio calculation including electron correlation predicted the π-radical to be more stable than the σ (N)-radical by ca. 5 kcal/mol.32
From Table 1, it is evident that the spin density distribution in the p-radical is delocalized on the N atom and CO groups, while in σ (O)-radical, the spin is localized in the molecular plane at both the CO groups and N atom and in σ (N)-radical the spin is localized mostly on the N atom in the molecular plane. The B3LYP method predicts the presence of all three types of the radicals within ca. 3 kcal/mol. The succinimide radical state would therefore be very sensitive to environment and factors such as hydrogen bonding at specific sites, stacking, and solvation would be expected to stabilize different states. In fact the succinimidyl π-radical is reported in an ESR study of X-irradiated succinimide single crystals at 26 K where the crystal structure provides stacking and specific hydrogen bonds that apparently stabilize the π-radical over the σ-states.33
The principle values of the anisotropic hyperfine couplings for the N atom observed experimentally by ESR10 is 31, ca. 0, ca. 0 G and characterized succinimidyl radical as a π-radical. The theoretically calculated anisotropic HFCs of the N atom of π-radical are 28.3, 0.1, 0.4 G, respectively, which is in excellent agreement with the experimental values. The calculated anisotropic HFCs of the N atom in σ (N)-radical is (8.8 G, 40.2 G, 7.4 G) is in poor agreement with the experiment.
(ii) 2-Thiothymine(N3-H)•
Sulfur substituted nucleic acid bases are known photosensitive probes. For example, 6-thioguanine has unusual optical properties. It absorbs ultraviolet A (UVA) (340 nm) while canonical DNA bases absorbs UVB (< 300 nm).34 2-Thiothymine is also used for specific targeting of selected sites in nucleic acids.35 ESR experiments on γ-irradiated single crystal of 2-thiothymine find the sulfur centered σ-radical deprotonated from N3 at 77 K.36 It was also observed that σ-radical formed by one electron oxidation converts to the allylic 7-yl carbon centered π-radical on warming the crystal to room temperature.36
The structure of one electron oxidized N3-deprotonated 2-thiothymine (2-thioT(N3-H)•) is shown in Figure 1, and the relative stability of its sulfur-centered π- and σ-radicals along with spin density distributions are presented in Table 1. The structure was optimized in the Cs symmetry by the B3LYP/6-31++G** method. The present calculation predicted π- and σ-radicals isoenergetic having energy difference ca. 0.4 kcal/mol between them. This energy difference (0.4 kcal/mol) is very small and shows both the radicals would be present at room temperature. For the π-radical the spin density is largely localized on the sulfur atom perpendicular to the molecular plane while in the σ-radical the spin density is localized mainly on the sulfur atom in the molecular plane, see Table 1.
For 2-thioT(N3-H)•, the 14N couplings for the sulfur-centered σ-radical were reported by experiment10 and the principal coupling values are 46.6, 55.5 and 45.2 G, respectively. The calculated total N3 atom couplings for the sulfur-centered σ-radical are 13.4 G, 19.2 G and 13.6 G which are far smaller than the reported experimental values. The theoretically calculated total 33S couplings for the σ-radical are reported in Table S1 in the supporting information. In the experiment S couplings were not observed. For π-radical (Tables 1 and S1 in the supporting information) the theoretically calculated HFCCs for N3 atom are −0.1 G, −0.2 G and 6.0 G. This shows that both the σ and π-radical N3 couplings are far smaller than the corresponding couplings for the σ-radical.
(iii) Uracil radicals
Uracil can be one electron oxidized by various chemical species such as Cl2•−, HPO4•−, H2PO4• and SO4•− and ionizing radiation.1,37 The uracil radical cation (U•+) produced by one electron oxidation is expected to be far more acidic than the parent neutral uracil. For example, the pKa’s of the thymine radical cation toward deprotonation at N1 and N3 sites are ca. 3.2 and 3.5, respectively.1 It is likely that U•+ is slightly more acidic than the T•+ and thus has pKa’s which are slightly lower than those of T•+. When U•+ deprotonates either from N1 or N3 sites in aqueous solution it becomes a neutral radical (U(N1-H)• or U(N3-H)•).1,37 The structure of U(N3-H)• is shown in Figure 1 and the spin density distribution of the π-, σ (O)- and σ (N)-radicals are shown in Table 2. The radical structures are optimized in the Cs symmetry and π- and σ-radicals have 2A″ and 2A′ symmetry. The present calculation shows that like succinimidyl radical, U(N3-H)• can also form σ (O)- and σ (N)-radicals. The π-radical is found to be the more stable than the σ-radicals and the relative stability in the gas phase in kcal/mol is π(0.0) > σ (O)(3.39) > σ (N)(3.58), respectively. Under the influence of the aqueous solution the σ (O)- and σ (N)-radicals are further destabilized so that they are 8.11 kcal/mol and 9.54 kcal/mol less stable than the π-radical, see Table 2. It is also found by our calculations that in going from the gas phase to solution the π-spin density which is largely localized on the N3 atom in the gas phase becomes largely localized on the C5 atom in solution, i.e., thus the π(N3)-radical in the gas phase becomes a π(C5)-radical in solution. These two π-radicals are in near degenerate states, thus the environment determines the most stable state. We note that experiments in solution find the C5 π-radical as predicted in this work.10 The energy difference between the two π-radicals and the σ-radical of uracil are not large (< 4(10) kcal/mol in gas phase (solution)) and the σ-radical states are clearly thermally accessible.
Table 2.
The B3LYP/6-31++G** calculated spin density distribution of π- and σ-radicals of uracil, thymine and cytosine. The relative stabilities of the radicals in the gas phase (aqueous solution) in kcal/mol are given at the bottom of each figure. The molecular symmetry is given in parentheses.a
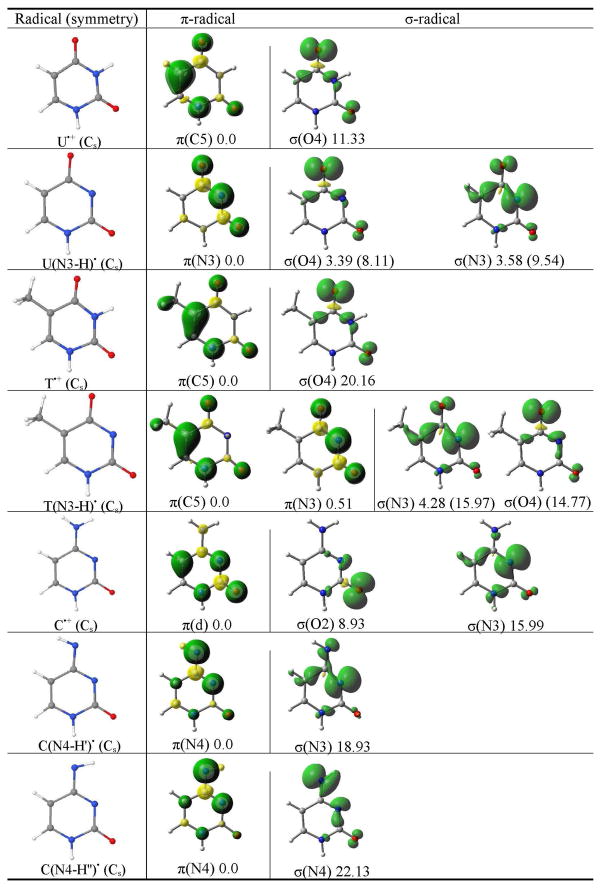
|
π(d) designates the delocalized nature of the radical.
The spin density distribution of π- and σ-radicals of U•+ (one-electron oxidized uracil) are shown in Table 2. The spin density in the π-radical is mainly localized on the C5 atom followed by a smaller spin distributions on N1, O4 and O2 atoms, however, for the σ-radical, the spin is localized on the O4 and O2 atoms in the molecular plane. The π-radical is found to be more stabilized than the σ-radical by ca. 11 kcal/mol. On deprotonation of U•+ from N3 site, the σ-radical becomes close in energy to the π-radical as discussed above, see Table 2.
π cation of uracil deprotonated at N1 was reported by ESR10. The α-proton at C5 gave principal H5 anisotropic couplings of −8, −24, −15 G (−16 G isotropic coupling). The calculated H5 anisotropic HFCs are −6.7, −23 G, −16.5 G (−15.4 G isotropic coupling) of the uracil cation radical, U•+, which match the experimental values well especially considering the difference in protonations state, see Table S1 in the supporting information. The σ (O4)-radical of U•+ shows no appreciable H5 couplings.
(iv) Thymine radicals
Geimer and Beckert generated thymine cation radical (T•+) in solution from photoexcited anthraquinone-2,6-disulfonate with thymine and its methyl derivatives through electron transfer reaction and characterized T•+ by Fourier transform EPR (electron paramagnetic resonance).38,39 The T•+ deprotonates at N1 and when N1 site was blocked by a methyl group the deprotonation takes place from the N3 site.38,39 The pKa for N1-deprotonation of T•+ was determined to be 3.240 and the pKa for N3-deprotonation thymidine and 1-methylthymine cation radicals are 3.6 and 3.8, respectively.41 The N3-centered radical was also produced by photoionization of thymidine 5′-monophosphate, thymidine, and 1-substituted thymine in basic 8 M NaC1O4-D2O glasses at 77 K by Sevilla and were investigated by ESR spectroscopy.42 From these results we expect that the T(N3-H) radical will be the form produced after ionization and deprotonation in DNA. For T(N3-H)• we find in this work two π- and two σ-radicals just as obtained for uracil. In the gas phase the π-radical localized at C5, i.e., π(C5) radical, is found to be nearly equal in energy but slightly more stable than π-radical at N3 by 0.5 kcal/mol, see Table 2. The π(C5) radical is also found to be more stable than the σ (N3)-radical by 4.23 kcal/mol. When solvated the π(C5) radical was ca. 23 kcal/mol more stable than in the gas phase and substantially more stable by ca. 16 kcal/mol and 15 kcal/mol over the σ (N3)- and σ (O4)-radicals, respectively, see Figure 2. For uracil π(N3)-radical is found to be the most stable species in the gas phase and on solvation π(C5)-radical is found to be the most stable species.
In solution, as modeled by PCM for waters high dielectric (ε = 78.4) the charge distribution within T(N3-H)• rearranges significantly compared to the charge distribution in the gas phase. This is most easily seen from the calculated dipole moments of T(N3-H)• in the gas phase and in solution. In the gas phase T(N3-H)• has a dipole moments of 6.67 Debye for π(N3) radical and 9.85 Debye for slightly more stable π(C5) radical. However, in water (ε = 78.4) the π(C5) radical becomes by far the most stable and its dipole moment increases to 13.62 Debye. Thus, in water the most polar state of the T(N3-H)• is stabilized and it becomes even more polar with N3 gaining charge density. We note that in agreement, experiments on T(N3-H)• in nucleosides or in DNA in aqueous systems at low temperatures only the C5 π-radical is found.10,20
For one-electron oxidized thymine (T•+), the most stable state is the C5 π-radical which is ca. 20 kcal/mol more stable than the σ (O)-radical. The spin density in the π-radical is localized on the C5 atom followed by N1, O4 and O2 atoms and in σ (O)-radical, spin is localized on O4 and O2 atoms as found for U•+, see Table 2.
The experimental N1 and average methyl hydrogen couplings of the π-cation radical of thymine are obtained by ESR experiments.42 The experimental N1 principal anisotropic couplings and the average isotropic coupling of the three hydrogens of CH3 for both radical are in good agreement with the calculated HFCCs of the thymine π-cation radical (see Table S1 in the supporting information). The calculated N1 HFCCs of σ (O4)-radical are very small and can not account for the observed couplings.
Our calculated couplings of π(C5)-radical of T(N3-H)• are in good agreement with the reported experimental values, see Table S1 in the supporting information. The calculated N3 couplings of π(N3)- and σ (N3)-radicals of T(N3-H)• are far too large to explain the experimental results.
(v) Cytosine radicals
The one-electron oxidation of cytosine has been shown to occur by reaction with SO4•−, Cl2•− or directly by ionizing radiation.1 The one-electron oxidized cytosine radical cation (C•+) has pKa 4.0 for its deprotonation from N4 (NH2) site.43–45 The pKa of deoxycytidine itself is > 13, thus C•+ is about 9 orders of magnitude more acidic than neutral cytosine. Using the Fourier-transform EPR study, Geimer et al.46 characterized the deprotonation of C•+ from the N1 site and deprotonation of 1-methylcytosine radical cation from the N4 site. Pulse radiolysis experiment with conductance detection estimated the life time of deoxycytidine radical cation to be approx. 200 nanoseconds at pH 5.2.44
The structures of C•+ and two of its deprotonated forms (C(N4-H′)• and C(N4-H″)•) are presented in Figure 1 and Table 2, respectively. The structures of C•+, C(N4-H′)• and C(N4-H″)• were optimized by the B3LYP/6-31++G** method in the Cs symmetry. For C•+, the calculation predicted the π-radical to be the more stable than the σ (O)- and σ (N)-radicals. The relative stabilities of these radicals in kcal/mol are 0.0 (π), 8.93 σ (O) and 15.99 σ (N), respectively, see Table 2. In the π-radical, spin density is largely localized on the C5 atom. In the σ (O)- and σ (N)-radicals, spin densities are largely localized on the O and N3 atoms in the molecular plane.
For C(N4-H′)• (see Table 2), the π-spin density is mainly localized on the N4 atom in the π-orbital system perpendicular to the molecular plane. Interestingly, for σ (N)-radical of C(N4-H′)•, the spin density is mainly localized on the N3 atom in the molecular plane. The energy difference between π-radical and less stable σ-radical was predicted to be ca. 19 kcal/mol, see Table 2.
In C(N4-H″)•, shown in Table 2, the spin densities in the π- and σ-radicals are both mainly localized on N4 atom. Thus, in σ-radical the location of spin density distribution is affected by the site of deprotonation of the NH2 group. The energy difference between π- and σ-radicals of C(N4-H″)• was predicted to be ca. 22 kcal/mol which is slightly larger than the energy difference between π- and σ-radicals of C(N4-H′)•, see Table 2.
The calculated HFCCs of C•+ (Table S1) are also close to the experimental couplings determined for N1-deprotonated cytosine π-cation radical by the ESR.10 The calculated σ (O2)- and σ (N3)-radicals couplings eliminate them as candidates to explain the experimental values.
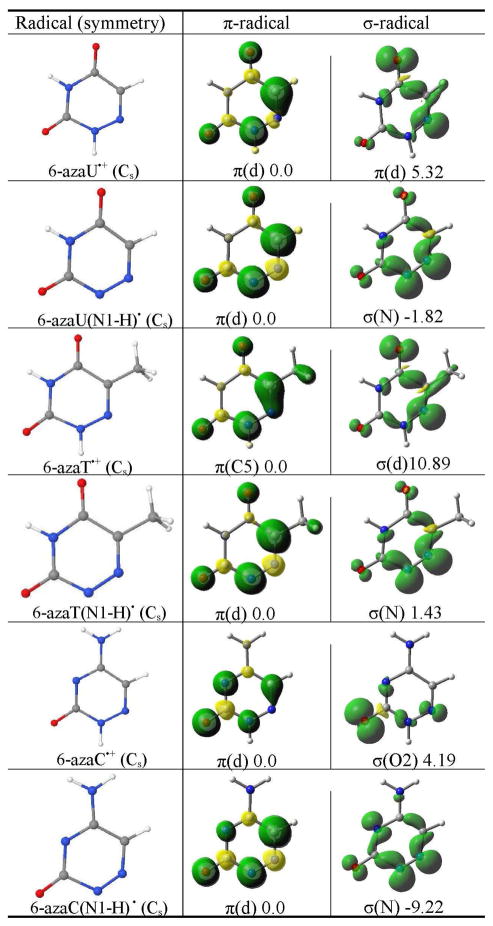
(vi) 6-Azauracil radicals
One-electron oxidized 6-azauracil deprotonated at N1 (6-azaU(N1-H)•) was produced by Cl2− attack in basic 12 M LiCl glasses and investigated by ESR spectroscopy at low temperatures by Sevilla and Swarts.13 The 6-azaU(N1-H)• was found to be a σ- radical whose ESR spectrum shows large couplings (ca. 30 Gauss) to the N1 and N6 atoms of 6-azauracil.
In this work we find that the structure of 6-azauracil cation radical, 6-azaU•+, (Figure 1) optimized in the Cs symmetry has the π-radical as most stable by ca. 5 kcal/mol relative to the σ-radical, see Table 3. The spin density on the π-radical is delocalized over the 6-azauracil ring largely localizing on C5 atom, while in the σ (N)-radical the spin is on N6 and O4 atoms, see Table 3.
Table 3.
The B3LYP/6-31++G** calculated spin density distribution of π- and σ-radicals of 6-azauracil, 6-azathymine, and 6-azacytosine. The relative stabilities of the radicals in the gas phase in kcal/mol are given at the bottom of each figure. The molecular symmetry is given in parentheses.a
|
π(d) designates the delocalized nature of the radical.
However, for the N1-deprotonated 6-azauracil radical (6-azaU(N1-H)•) we find that the N1-deprotonated σ-radical is more stable than its π-radical by ca. 2 kcal/mol (Table 3) as observed experimentally.13 The nature of the spin density distribution in the π-radicals of 6-azauracil(N1-H)• and 6-azauracil•+ are almost identical, however, the spin density distribution in the σ-radicals of 6-azaU(N1-H)• and 6-azaU•+ are quite different, see Table 3. In the σ-radical of 6-azaU(N1-H)•, the spin density is mainly localized on N1 and N6 atoms.
The N1 and N6 couplings of σ-radical of 6-azaU(N1-H)• are measured by the ESR experiment13 and 30.2 G (isotropic) and (26 G, 38.5 G, 26 G) anisotropic couplings were measured for both N1 and N6 atoms. The calculated N1 and N6 couplings of 6-azaU(N1-H)• are overestimated by ca. 7 – 16 G, see Table S1 in the supporting information.
(vii) 6-Azathymine radicals
One-electron oxidized 6-azathymine was produced by Cl2− attack in basic 12 M LiCl glasses and studied by ESR spectroscopy at low temperatures.13 The 6-azathymine radical was also generated by photoionization in 8 M NaClO4.13 These ESR experiments confirmed the formation of only the π-radical of 6-azathymine.13 In our study, we consider 6-azathymine•+ (6-azaT•+) and N1-deprotonated 6-azathymine radical (6-azaT(N1-H)•) and optimized their structures in the Cs symmetry. For 6-azatT•+, we find the π-radical to be more stable than its σ-radical by ca. 11 kcal/mol. The spin density in the π-radical is delocalized largely on C5 atom and the spin density in the σ (N)-radical is localized on N6 and O4 atoms as found for 6-azauracil, see Table 3.
From the calculations, the π- and σ-radicals of 6-azaT(N1-H)• are found to be nearly isoenergetic, with the π-radical more stable than the σ-radical by ca. 2 kcal/mol. The spin density distribution in π- and σ-radicals is also similar to those found for 6-azaU(N1-H)•.
The ESR experiment13 predicted 6-azaT(N1-H)• as a π-radical and the measured isotropic couplings of CH3 is 14.7 G, couplings of N1 atom are 8 – 10 G (isotropic) and (< 3 G, < 3 G, 23 G) anisotropic and unresolved < 4 G couplings for the N6 atom. The calculated HFCCs are in excellent agreement with those measured experimentally, see Table S1 in the supporting information.
(viii) 6-Azacytosine radicals
Using ESR spectroscopy at 77 K Sevilla and Swarts characterized the formation of the σ-radical of one electron oxidized 6-azacytosine.13 In this case, we optimized the structures of 6-azacytosine cation radical, 6-azaC•+ and the N1 deprotonated one electron oxidized 6-azacytosine radical, 6-azaC(N1-H)• in the Cs symmetry and determined the relative stability of π- and σ-radicals. From Table 3, it is evident that π-radical of 6-azaC•+ is more stable than the σ-radical by ca. 4 kcal/mol. To the best of our knowledge 6-azaC•+ has not been observed experimentally. The spin density in the π-radical is delocalized on the 6-azacytosine ring, while in the σ-radical spin is localized on the O2 atom in the molecular plane.
The σ-radical of 6-azaC(N1-H)• is found to more stable than its π-radical by ca. 9 kcal/mol. The spin density distribution in the π-radical of 6-azaC(N1-H)• is similar to the spin density distribution in the π-radical of 6-azaC•+. The spin density in the σ-radical of 6-azaC(N1-H)• is largely localized on the N1 and N6 atoms as found for 6-azauracil and 6-azathymine. Thus, the present calculations predict the formation of ground state σ-radical of 6-azaC(N1-H)• as observed by ESR experiments.13
For 6-azacC(N1-H)• the theoretically calculated couplings of N1, N3 and N6 atoms of σ-radical are in close agreement with those measured experimentally by ESR 13, see Table S1 in the supporting information.
(ix) Adenine radicals
Adenine cation radical (A•+) is produced by one-electron oxidation of adenine and its 2′-deoxynucleosides by SO4•−, photolysis and γ-irradiation and was extensively studied using, (i) pulse radiolysis and flash photolysis in aqueous solution,1,4,44 (ii) ESR in γ-irradiated aqueous (D2O) glassy systems at low temperature,47,48 and (iii) X-ray irradiated single crystals.49–51 A number of theoretical treatments have also been reported.52–54 One electron oxidized adenine (A•+) becomes very acidic and has pKa in the range ca. 1 – 4.4,44,45,48,53–55 The pKa of neutral adenine is ≥ 14, thus, the acidity of A•+ is increased by 10 – 13 orders of magnitude over the parent molecule.4,44 Although Steenken reported the pKa of A•+ deprotonated from NH2 group as ≤ 1 from experiment44 other more recent work suggests pKa values near 4.45,55 The high acidity of A•+ (with a pKa of 4 or less) produces a strong driving force for deprotonation from it’s NH2 group. This deprotonation is found in X-irradiated single crystals of adenosine at 10 K.49 Thus, we calculated the π- and σ-radicals of A•+ as well as the expected A(N6-H)• in the Cs symmetry using the B3LYP/6-31++G** method. The π-radical of A•+ is ca. 17 kcal/mol more stable than its σ-radical, see Table 4. From the nature of the spin density distribution, it is evident that in π-radical spin is delocalized on the adenine ring while in the σ-radical spin is mainly localized on the N1, N3 and N7 atoms in the molecular plane.
The spin density distributions of the π- and σ-radicals of A(N6-H)• are shown in Table 4. The spin density distribution in the π-radical of A(N6-H)• shows more localization at N6 than found for A•+, see Table 4. The spin density distribution in the σ-radical of A(N6-H)• is quite different than the corresponding σ-radical of A•+ and spin is largely localized on the N6 atom, see Table 4. We also found that the π-radical of A(N6-H)• is highly stabilized relative to its σ-radical by ca. 27 kcal/mol.
(x) Guanine radicals
The one-electron oxidized guanine (G•+) and its nucleosides have been extensively studied using theory and experiment.2–9,18,21,22,56–58 The pKa of neutral deoxyguanosine is 9.5 and it can be oxidized by several oxidants, for example, SO4•−, Cl2•−, Br2•−, Ti(II), photoionization and γ-irradiation to produce G•+.1,2,4,5,56,58 Candeias and Steenken estimated two pKas of G•+ (i) 3.9 for its deprotonation from the N1 site in deoxyguanosine and guanosine, and (ii) 4.7 for its deprotonation from NH2 site in 1-methylguanosine by time-resolved optical and conductance techniques.56 In single crystal G•+ was found to be present as neutral radical (G•) at ca. 10 K59,60 and the ESR experiments find G•+ and G(N1 H)• in aqueous glasses at low temperatures.22 Using ESR and thermal annealing of one-electron oxidized guanine in dsDNA at 155K, G•+ was found to exist mainly as G(N1-H)•:C(N3+H), however, upon annealing the sample to 175 K, deprotonation to the solvent occurs and an equilibrium mixture of G(N1 H)•:C and G(N2 H)•:C is found.57,58 This experimental finding was also supported by theory.18 Thus, in the present study, we considered the formation of π- and σ-radicals of G(N1-H)• and G(N2-H)•, respectively. The structures are optimized in the Cs symmetry in the gas phase and in solution and the nature of their spin density distribution is presented in Table 4. For G(N1-H)• we found that its π-radical is more stable than its σ (O)- and σ (N)-radicals by ca. 19 kcal/mol and ca. 27 kcal/mol, respectively, see Table 4. In the gas phase only σ (O)-radical was obtained by the theory while in solution only σ (N)-radical was obtained. The spin density distribution in the π-radical is delocalized on the guanine ring and in σ-radicals, spin densities are localized on the O6 atom in σ (O)-radical and on N3 and N1 atoms in σ (N)-radical, respectively.
In G(N2-H)•, only π- and σ (N)-radicals were obtained by the calculation in the gas phase and in solution and π-radical was found to be more stabilized than its σ-radical by ca. 40 kcal/mol in the gas phase as well as in solution. The spin density distribution in the π-radical is similar to those obtained for G(N1-H)• and for σ (N)-radical the spin density is mainly localized on the N2 atom.
The π- and σ-radicals of guanine radical cation (G•+) is shown in Table 4. In σ-radical of G•+ the spin is mainly localized on N7, O6 and N3 atoms in the molecular plane while in the π-radical, the spin is delocalized on the purine ring, see Table 4. The π-radical is found to be more highly stabilized than the corresponding σ-radical by ca. 34 kcal/mol.
The π-cation radical of 2′-deoxyguanosine in D2O was studied using ESR at 77 K.22 The measured isotropic couplings of C8-H is 7 G and the measured anisotropic couplings are (−3.5 G, −10.5 G, −7.5 G), respectively. The calculated couplings of C8-H are −8.9 G (isotropic) and (−3.8 G, −13.4 G, −9.5 G) (anisotropic) are in good agreement the experimental values. The calculated couplings of σ (N7) and σ (O6) atoms of G•+ are far too large to explain experimental values as calculated for other σ (N)- and σ (O)-radicals.
(xi) Uracil(N3-H)•-3H2O
As a model for gas phase clusters, the stabilization of π- and σ-radicals of N3-deprotonated uracil radical (U(N3-H)•) is considered in the presence of three water molecules. We arranged three water molecules near the C(O)-N3-C(O) region of the uracil in the molecular plane in a hydrogen-bonding fashion in Cs symmetry and optimized the Uracil(N3-H)•-3H2O structure by the B3LYP/6-31++G** method. The spin density distribution is shown in Figure 3. From Figure 3, it is evident that the nature of the spin density distribution in the π- and σ-radicals are similar to those found for the corresponding radicals in the gas phase for U(N3-H)•, see Table 2. The π-radical of U(N3-H)•-3H2O is more stable than its σ-radical by ca. 5 kcal/mol as obtained in the gas phase.
Figure 3.
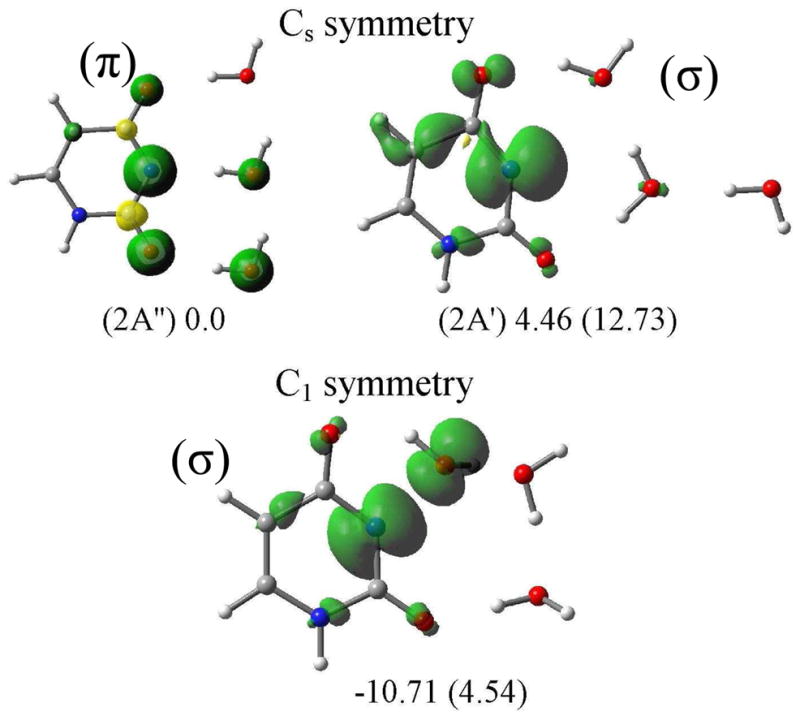
Spin density plots of σ- and π-radicals of U(N3-H)• in the presence of three water molecules in Cs and C1 symmetries. The relative stabilities in the gas phase (solution) are given in kcal/mol at the bottom of each figure. In C1 symmetry the uracil σ-radical is stabilized by a three-electron bond with one of the water molecules. All the structures are optimized by the B3LYP/6-31++G** method.
Instead of Cs symmetry, we optimized the σ-radical of U(N3-H)•-3H2O in C1 symmetry which allows the water molecules to become nonplanar with the uracil. Here we found that the σ-radical in C1 symmetry is more stable than the π-radical in Cs symmetry by ca. 11 kcal/mol. In this particular arrangement, the σ-radical is stabilized by making a three-electron bond with one of the water molecules. The σ-spin density localized on the N3 atom of uracil forms the bond to the O atom of a water molecule, see Figure 3. The Uracil(N3-H)•-3H2O structures were further optimized considering the effect of full solvent through PCM. With full solvation we find that the σ (N)-radical in the Cs symmetry (planarity forced) is less stable by ca. 13 kcal/mol than the corresponding π-radical with spin at N3 (Figure 3) while the σ (N)-radical in the C1 symmetry (nonplanar) was less stable by ca. 5 kcal/mol than the π(C5)-radical. Thus our calculations show that on full solvation (PCM) the π-radical with high spin density at C5 was found instead of the π-radical with high spin at N3. The stabilization of the σ (N)-radical is confined to gas phase clusters.”
(xii) Fully optimized radicals and mixed states
In the work above the structures of all the one-electron oxidized DNA/RNA bases and their radicals considered in this study were optimized with the symmetry constraint of Cs symmetry (planar) to emphasize their σ- and π-radical nature. To assess the affect of nonplanarity on the energies, we also optimized all the ground state radicals both σ and π without symmetry constraints (i.e., in C1 symmetry). For all the DNA π-radicals considered in this study we found the energies in the C1 symmetry did not change significantly (ΔE < 0.6 kcal/mol) from the energies in Cs symmetry and all these structures in the C1 symmetry maintained their near planar π-radical nature. For the DNA analog structures two cases showed little change. The energy of the σ-radicals of 6-azaC(N1-H)• in C1 and Cs symmetries were found to be essentially identical (within 0.004 kcal/mol) with no mixing of σ and π states. The optimized σ- and π-radicals in C1 and Cs symmetries of 2-thiothymine(N3-H)• were also found to be isoenergetic having energy differences of 0.01 kcal/mol and 0.004 kcal/mol, for σ- and π-radicals, respectively. Although the energy changes were not large, the spin density distribution in non-planar radicals (C1 symmetry) of 6-azaU(N1-H)•, and 6-azaT(N1-H)• shows a clear mixing of π and σ states. The energy of the mixed state (mainly σ-radical) of 6-azaU(N1-H)• in C1 symmetry was ca. 1.7 kcal/mol lower than the pure σ-radical in Cs symmetry, whereas the mixed state of the still mainly π-radical of 6-azaT(N1-H)• in C1 symmetry was ca. 2.2 kcal/mol lower than the pure π-radical in the Cs symmetry. In general, we find that as the energy between σ- and π-states separated little or no mixing of states is found.
(xiii) Distinction between σ- and π-radicals by HFCs
From the above discussion and Table S1, it is evident that the B3LYP/6-31++G** calculated HFCs for both π- and σ-radicals of a molecule when compared to their experimental HFCs one can often easily distinguish the π- from σ-radicals. For example, a comparison of the calculated and the experimental HFCs of 6-azaU(N1-H)• and 6-azaT(N1-H)• allow for easy identification of σ- and π-radicals in each case. The π-radical of 6-azaT(N1-H)• yields calculated HFCs for the C5 methyl group (15.8G isotropic) and a single N1 HFC of 0.8, 1.0, 22.8G whereas the σ-radical gives two large nitrogen couplines from N1 and N6 and no significant methyl coupling, see Table S1. ESR experiment13 gives a C5 methyl group (14.7G isotropic) and a single N1 HFC of <3, <3, 23G which provide very clear evidence for a π-radical. For 6-azaU(N1-H)• which differs from 6-azaT(N1-H)• by only the 5-methyl group, experiment shows two large N1, N6 couplings of 26, 39, 26 G each. Theory predicts for the π-radical of 6-azaU(N1-H)•, a single N1 anisotropic coupling of 0.9, 1.1, and 24.7 G and C5 proton couplings of −6.1, −20.4, 14.3 G while the σ-radical gives two large N1, N6 couplings of 36, 54 and 38 G (avg) and no significant C5 proton coupling, see Table S1. Clearly, the comparison of experimental and calculated hyperfine couplings show that 6-azaU(N1-H)• is a σ-radical and that 6-azaT(N1-H)• is a π-radical. The methyl substitution at C5 was sufficient to interchange the energies of the two states so that the σ-radical in 6-azaU(N1-H)• becomes a π-radical in 6-azaT(N1-H)• and this was predicted by theory (Table 3 and Figure 4).
Figure 4.

Lowest σ-radical energy relative to the most stable π-radical energy for gas phase pyrimidine and purine systems considered in the present study (in kcal/mol).
Conclusions
In this work the DNA/RNA radicals were optimized as planar structures (Cs symmetry). In addition, the σ or π stable states were also fully optimized without symmetry restrictions (in C1 symmetry). The energies and structures did not change significantly (ΔE < 0.6 kcal/mol) except for several cases where near degeneracy in the σ and π states lead to mix states which induced some nonplanarity. In Figure 4 we present a summary of the energies of σ–radicals relative to their respective π-radicals for all the radicals considered in the present study under Cs symmetry. The results summarized in the figure clearly show that on going from pyrimidines to purines the energy difference between their π- and σ-radicals states increases with the purine σ-radicals being the most unstable. It is especially interesting to note that for the pyrimidine radicals, U(N3-H)•, T(N3-H)•, and 2-thiothymine(N3-H)•, the σ- and π-radicals are within 4 kcal/mol of each other. These radicals are similar in nature to succinimidyl radical whose σ- and π-radicals are within a few kcal/mol and are known to react to form different molecular products, see Tables 1 and 2. Our work clearly indicates that environments with low polarity tend to stabilize the σ-radicals vs the π-radicals and thus can alter reaction mechanisms. Thus for a radicals such as, T(N3-H)•, in a low polarity stacked base DNA system specific interactions within the local environment may tend to favor one state over another. The point of interest is that under specific conditions both σ- and π-states are thermally accessible and are likely to contribute to the chemistry of the radical intermediates.
The deprotonation state of the radical is a major factor in the stabilization of the σ-DNA base radicals. For the cation radicals, U•+, T•+ and G•+ σ-radicals are found which lie ca. 11 kcal/mol, 20 kcal/mol and 34 kcal/mol above their π-radicals. After deprotonation from N3 site, the σ (N)-radical of U(N3-H)• and T(N3-H)• are found to lie only ca. 4 kcal/mol above their π-radicals. The π-radicals of 6-azauracil•+, 6-azathymine•+ and 6-azacytosine•+ are predicted to be more stable than their σ-radicals by ca. 5 to 11 kcal/mol; however, on deprotonation from the N1 site, the σ-radicals of 6-azauracil(N1-H)• and 6-azacytosine(N1-H)• were found to be more stable than their π-radicals in agreement with ESR experiments.13 For G•+, σ (N)-radicals resulting from deprotonation at N1 or N2 sites are still highly unfavorable energetically relative to their respective lowest energy π-radicals, see Table 4.
As indicated above, PCM results show that a polar solvent tends to stabilize the π state in these systems. However, paradoxically inclusion of specific waters of hydration are found to behave differently than inclusion of a continuous dielectric in the PCM model. For example, we found the σ-radical of U(N3-H)• complexed with three waters of hydration in the gas phase (Figure 3) in C1 symmetry is lower by 11 kcal than the π-radical. The spin distribution and structure show the formation of a three-electron σ bond between the N3 atom of uracil and the O atom of one of the water molecules. When PCM is included the σ-radical becomes ca. 5 kcal/mol less stable than the π-radical; in this regard, we note that since the dielectric is low in the stacked DNA system61,62 σ bond formation to water becomes possible. The extent of this behavior in contributing to the chemistry of the DNA base radicals is not fully understood and is clearly an area for future work.
Supplementary Material
Acknowledgments
The authors thank the NCI of the NIH for support under Grant No. R01CA045424. The authors thank Prof. D. Becker for his helpful suggestions and critically reviewing the manuscript.
Footnotes
Calculated (B3LYP/6-31++G**) and experimentally observed hyperfine couplings as well as spin density distributions for all radicals considered in the present study are given as supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.von Sonntag C. Free-Radical-Induced DNA Damage and Its Repair: A Chemical Perspective. Springer-Verlag; Berlin Heidelberg: 2006. [Google Scholar]
- 2.Kumar A, Sevilla MD. Proton-Coupled Electron Transfer in DNA on Formation of Radiation-Produced Ion Radicals. Chem Rev. 2010;110:7002–7023. doi: 10.1021/cr100023g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X, Sevilla MD. DFT Treatment of Radiation Produced Radicals in DNA Model Systems. Adv Quantum Chem. 2007;52:59–87. [Google Scholar]
- 4.Steenken S. Purine bases, nucleosides, and nucleotides: aqueous solution redox chemistry and transformation reactions of their radical cations and e- and OH adducts. Chem Rev. 1989;89:503. [Google Scholar]
- 5.Becker D, Adhikary A, Sevilla MD. In: In Charge Migration in DNA. Chakraborty T, editor. Springer-Verlag; Berlin, Germany: 2007. pp. 139–175. [Google Scholar]
- 6.Kumar A, Sevilla MD. In Radiation Induced Molecular Phenomena in Nucleic Acids. In: Shukla MK, Leszczynski J, editors. Challenges and Advances in Computational Chemistry and Physics. Vol. 5. Springer Science; Dordrecht, The Netherlands: 2008. pp. 577–617. [Google Scholar]
- 7.Kumar A, Sevilla MD. In: In Radical and Radical Ion Reactivity in Nucleic Acid Chemistry. Greenberg M, editor. John Wiley & Sons, Inc; Hoboken, NJ: 2009. pp. 1–40. [Google Scholar]
- 8.Sevilla MD, Becker D, Yan M, Summerfield SR. Relative Abundances of Primary Ion Radicals in γ-Irradiated DNA: Cytosine vs Thymine Anions and Guanine vs Adenine Cations. J Phys Chem. 1991;95:3409–3415. [Google Scholar]
- 9.Close DM. In Radiation Induced Molecular Phenomena in Nucleic Acids. In: Shukla MK, Leszczynski J, editors. Challenges and Advances in Computational Chemistry and Physics. Vol. 5. Springer Science; Dordrecht, The Netherlands: 2008. pp. 493–529. [Google Scholar]
- 10.Sevilla MD, Suryanarayana D, Morehouse KM. ESR Study of DNA Base Cation Radicals Produced by Attack of Oxidizing Radicals. J Phys Chem. 1981;85:1027–1031. [Google Scholar]
- 11.Kato T, Shida T. Electronic Structures of Ion Radicals of N-Heteroaromatic Hydrocarbons as Studied by ESR and Optical Spectroscopy. J Am Chem Soc. 1979;101:6869–6876. [Google Scholar]
- 12.Shida T, Kato T. ESR and optical studies on the cation-radical of pyridine in a γ-irradiated rigid matrix at low temperatures. Chem Phys Lett. 1979;68:106–110. [Google Scholar]
- 13.Sevilla MD, Swarts S. Electron Spin Resonance Study of Radicals Produced by One-Electron Loss from 6-Azauraci1, 6-Azathymine, and 6-Azacytosine. Evidence for both σ and π Radicals. J Phys Chem. 1982;86:1751–1755. [Google Scholar]
- 14.Riederer H, Hüttermann J, Symons MCR. σ*-Electron addition to 5-halogenouracils in neutral glasses. Chem Commun. 1978:313–314. [Google Scholar]
- 15.Li Xi, Sevilla MD, Sanche L. DFT Investigation of Dehalogenation of Adenine-Halouracil Base Pairs upon Low-Energy Electron Attachment. J Am Chem Soc. 2003;125:8916–8920. doi: 10.1021/ja034286u. [DOI] [PubMed] [Google Scholar]
- 16.Bešić E, Sanković K, Gomzi V, Herak JN. Sigma radicals in gamma-irradiated single crystals of 2-thiothymine. Phys Chem Chem Phys. 2001;3:2723–2725. [Google Scholar]
- 17.Skell PS, Day JC, Slangs JP. More about π- and σ-Succinimidyl Radicals: Ring Opening Reactions. Angew Chem Int Ed Engl. 1978;17:515–516. [Google Scholar]
- 18.Kumar A, Sevilla MD. Influence of Hydration on Proton Transfer in the Guanine–Cytosine Radical Cation (G•+–C) Base Pair: A Density Functional Theory Study. J Phys Chem B. 2009;113:11359–11361. doi: 10.1021/jp903403d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar A, Sevilla MD. Low-Energy Electron Attachment to 5′-Thymidine Monophosphate: Modeling Single Strand Breaks Through Dissociative Electron Attachment. J Phys Chem B. 2007;111:5464–5474. doi: 10.1021/jp070800x. [DOI] [PubMed] [Google Scholar]
- 20.Adhikary A, Kumar A, Heizer AN, Palmer BJ, Pottiboyina V, Liang Y, Wnuk SF, Sevilla MD. Hydroxyl Ion Addition to One-Electron Oxidized Thymine: Unimolecular Interconversion of C5 to C6 OH-Adducts. J Am Chem Soc. 2013;135:3121–3135. doi: 10.1021/ja310650n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar A, Sevilla MD, Suhai S. Microhydration of the Guanine–Cytosine (GC) Base Pair in the Neutral and Anionic Radical States:–A Density Functional Study. J Phys Chem B. 2008;112:5189–5198. doi: 10.1021/jp710957p. [DOI] [PubMed] [Google Scholar]
- 22.Adhikary A, Kumar A, Becker D, Sevilla MD. The Guanine Cation Radical: Investigation of Deprotonation States by ESR and DFT. J Phys Chem B. 2006;110:24171–24180. doi: 10.1021/jp064361y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jena NR, Mishra PC. Mechanisms of Formation of 8-Oxoguanine Due To Reactions of One and Two OH• Radicals and the H2O2 Molecule with Guanine: A Quantum Computational Study. J Phys Chem B. 2005;109:14205–14218. doi: 10.1021/jp050646j. [DOI] [PubMed] [Google Scholar]
- 24.Sreeruttun RK, Ramasami P, Wannere CS, Simmonett AC, Schaefer HF. π and σ-Phenylethynyl Radicals and Their Isomers o-, m-, and p-Ethynylphenyl: Structures, Energetics, and Electron Affinities. J Phys Chem A. 2008;112:2838–2845. doi: 10.1021/jp0763329. [DOI] [PubMed] [Google Scholar]
- 25.Raiti MJ, Sevilla MD. Density Functional Theory Investigation of the Electronic Structure and Spin Density Distribution in Peroxyl Radicals. J Phys Chem A. 1999;103:1619–1626. [Google Scholar]
- 26.Hermosilla L, Calle P, García A, de la Vega JM, Sieiro C. Density Functional Theory Study of 14N Isotropic Hyperfine Coupling Constants of Organic Radicals. J Phys Chem A. 2006;110:13600–13608. doi: 10.1021/jp064900z. and references therein. [DOI] [PubMed] [Google Scholar]
- 27.Cohen AJ, Mori-Sánchez P, Yang W. Challenges for Density Functional Theory. Chem Rev. 2012;112:289–320. doi: 10.1021/cr200107z. [DOI] [PubMed] [Google Scholar]
- 28.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, et al. Gaussian 09: revision B01. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 29.GaussView. Gaussian, Inc; Pittsburgh, PA: 2003. [Google Scholar]
- 30.Jmol. An Open-Source Java Viewer for Chemical Structures in 3D. 2004 see http://jmol.sourceforge.net.
- 31.Gainsforth JL, Klobukowski M, Tanner DD. Structure and Reactions of the Succinimidyl Radical: A Density Functional Study. J Am Chem Soc. 1997;119:3339–3346. [Google Scholar]
- 32.Field MJ, Hiller H, Guest MF. Accurate calculations of the structure and energy of the π- and σ-succinimidyl radicals: an ab initio study including electron correlation. J Chem Soc Perkin Trans. 1987;2:1311–1316. [Google Scholar]
- 33.Lund A, Samskog PO, Eberson L, Lunell S. ESR single-crystal study of the succinimidyl π-electron radical in x-irradiated succinimide at 26 K. J Phys Chem. 1982;86:2458–2462. [Google Scholar]
- 34.Martínez-Fernández L, Gonzálezb L, Corral I. An ab initio mechanism for efficient population of triplet states in cytotoxic sulfur substituted DNA bases: the case of 6-thioguanine. Chem Commun. 2012;48:2134–2136. doi: 10.1039/c2cc15775f. [DOI] [PubMed] [Google Scholar]
- 35.Kutyavin IV, Rhinehart RL, Lukhtanov EA, Gorn VV, Meyer RB, Gamper HB. Oligonucleotides Containing 2-Aminoadenine and 2-Thiothymine Act as Selectively Binding Complementary Agents. Biochemistry. 1996;35:11170–11176. doi: 10.1021/bi960626v. [DOI] [PubMed] [Google Scholar]
- 36.Besšić E, Sanković K, Gomzi V, Herak JN. Sigma radicals in gamma-irradiated single crystals of 2-thiothymine. Phys Chem Chem Phys. 2001;3:2723–2725. [Google Scholar]
- 37.Luke TL, Mohan H, Manoj VM, Manoj P, Mittal JP, Aravindakumar CT. Reaction of sulphate radical anion (SO4•−) with hydroxy-and methyl-substituted pyrimidines: a pulse radiolysis study. Res Chem Intermed. 2003;29:379–391. [Google Scholar]
- 38.Geimer J, Brede O, Beckert D. Fourier Transform EPR study of N-centered pyrimidine radicals in the nanosecond time scale. Chem Phys Lett. 1997;276:411–417. [Google Scholar]
- 39.Geimer J, Beckert D. Direct Evidence for 1-Methylthymine Radical Cations and Their Transformation to Successor Radicals by Fourier Transform Electron Paramagnetic Resonance in the Nanosecond Time Scale. J Phys Chem A. 1999;103:3991–3998. [Google Scholar]
- 40.Geimer J, Beckert D. Study of radical pairs generated by photoreduction of anthraquinone-2,6-disulfonic acid with thymine by Fourier transform electron paramagnetic resonance. Chem Phys Lett. 1998;288:449–458. [Google Scholar]
- 41.Deeble DJ, Schuchmann MN, Steenken S, von Sonntag C. Direct Evidence for the Formation of Thymine Radical Cations from the Reaction of SO4•− with Thymine Derivatives: A Pulse Radiolysis Study with Optical and Conductance Detection. J Phys Chem. 1990;94:8186–8192. [Google Scholar]
- 42.Sevilla MD. Electron spin resonance study of N1-substituted thymine p-cation radicals. J Phys Chem. 1976;80:1898–1901. [Google Scholar]
- 43.Ts’o POP. Bases, Nucleosides, and Nucleotides. In: Ts’o POP, editor. Basic Principles in Nucleic Acid Chemistry. Vol. 1. Academic Press; New York: 1974. pp. 454–584. [Google Scholar]
- 44.Steenken S. Electron-transfer-induced acidity/basicity and reactivity changes of purine and pyrimidine bases. Consequences of redox processes for DNA base pairs. Free Radical Res Commun. 1992;16:349–379. doi: 10.3109/10715769209049187. [DOI] [PubMed] [Google Scholar]
- 45.Close DM. Calculated pKa’s of the DNA Base Radical Ions. J Phys Chem A. 2013;117:473–480. doi: 10.1021/jp310049b. and references therein. [DOI] [PubMed] [Google Scholar]
- 46.Geimer J, Hildenbrand K, Naumov S, Beckert D. Radicals formed by electron transfer from cytosine and 1-methylcytosine to the triplet state of anthraquinone-2,6-disulfonic acid. A Fourier-transform EPR study. Phys Chem Chem Phys. 2000;2:4199–4205. [Google Scholar]
- 47.Adhikary A, Khanduri D, Kumar A, Sevilla MD. Photoexcitation of Adenine Cation Radical [A•+] in the near UV-vis Region Produces Sugar Radicals in Adenosine and in Its Nucleotides. J Phys Chem B. 2008;112:15844–15855. doi: 10.1021/jp808139e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adhikary A, Kumar A, Khanduri D, Sevilla MD. Effect of Base Stacking on the Acid-Base Properties of the Adenine Cation Radical [A•+] in Solution: ESR and DFT Studies. J Am Chem Soc. 2008;130:10282–10292. doi: 10.1021/ja802122s. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Close DM, Nelson WH. ESR and ENDOR Study of Adenosine Single Crystals X-Irradiated at 10 K. Radiat Res. 1989;117:367–378. [PubMed] [Google Scholar]
- 50.Close DM, Nelson WH, Sagstuen E, Hole EO. ESR and ENDOR Study of Single Crystals of Deoxyadenosine Monohydrate X-Irradiated at 10 K. Radiat Res. 1994;137:300–309. [PubMed] [Google Scholar]
- 51.Kar L, Bernhard WA. Electron Gain and Electron Loss Radicals Stabilized on the Purine and Pyrimidine of a Cocrystal Exhibiting Base-Base Interstacking: ESR-ENDOR of X-Irradiated Adenosine:5-Bromouracil. Radiat Res. 1983;93:232–253. [Google Scholar]
- 52.Wetmore SD, Boyd RJ, Eriksson LA. Theoretical Investigation of Adenine Radicals Generated in Irradiated DNA Components. J Phys Chem B. 1998;102:10602–10614. [Google Scholar]
- 53.Baik MH, Silverman JS, Yang IV, Ropp PA, Szalai VS, Thorp HH. Using Density Functional Theory To Design DNA Base Analogues with Low Oxidation Potentials. J Phys Chem B. 2001;105:6437–6444. [Google Scholar]
- 54.Chen X, Syrstad EA, Nguyen MT, Gerbaux P, Turebek F. Distonic Isomers and Tautomers of the Adenine Cation Radical in the Gas Phase and Aqueous Solution. J Phys Chem A. 2004;108:9283–9293. [Google Scholar]
- 55.Kobayashi K. Evidence of Formation of Adenine Dimer Cation Radical in DNA: The Importance of Adenine Base Stacking. J Phys Chem. 2010;114:5600–5604. doi: 10.1021/jp100589w. [DOI] [PubMed] [Google Scholar]
- 56.Candeias LP, Steenken S. Structure and Acid-Base Properties of One-Electron-Oxidized Deoxyguanosine, Guanosine, and 1–Methylguanosine. J Am Chem Soc. 1989;111:1094–1099. [Google Scholar]
- 57.Adhikary A, Kumar A, Munafo SA, Khanduri D, Sevilla MD. Prototropic equilibria in DNA containing one-electron oxidized GC:intra-duplex vs. duplex to solvent deprotonation. Phys Chem Chem Phys. 2010;12:5353–5368. doi: 10.1039/b925496j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adhikary A, Khanduri D, Sevilla MD. Direct Observation of the Hole Protonation State and Hole Localization Site in DNA-Oligomers. J Am Chem Soc. 2009;131:8614–8619. doi: 10.1021/ja9014869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hole EO, Nelson WH, Sagstuen E, Close DM. Free Radical Formation in Single Crystals of 2′-Deoxyguanosine 5′-Monophosphate Tetrahydrate Disodium Salt: An EPR/ENDOR Study. Radiat Res. 1992;129:119–138. [PubMed] [Google Scholar]
- 60.Hole EO, Nelson WH, Close DM, Sagstuen E. ESR and ENDOR study of the guanine cation: Secondary product in 5′-dGMP. J Chem Phys. 1987;86:5218–5219. [Google Scholar]
- 61.Schwalb NK, Temps F. Ultrafast electronic relaxation in guanosine is promoted by hydrogen bonding with cytidine. J Am Chem Soc. 2007;129:9272–9273. doi: 10.1021/ja073448+. [DOI] [PubMed] [Google Scholar]
- 62.Kumar A, Sevilla MD. Excited state proton-coupled electron transfer in 8-oxoG–C and 8-oxoG–A base pairs: a time dependent density functional theory (TD-DFT) study. Photochem Photobiol Sci. 2013;12:1328–1340. doi: 10.1039/c3pp25430e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.