

Abstract
CD8 T cell activation and differentiation is tightly controlled, and dependent on the context in which naïve T cells encounter antigen, can either result in functional memory or T cell dysfunction, including exhaustion, tolerance, anergy, or senescence. With the identification of phenotypic and functional traits shared in different settings of T cell dysfunction, distinctions between such dysfunctional `states' have become blurred. Here, we discuss distinct states of CD8 T cell dysfunction, with emphasis on (i) T cell tolerance to self-antigens (self-tolerance), (ii) T cell exhaustion during chronic infections, and (iii) tumor-induced T cell dysfunction. We highlight recent findings on cellular and molecular characteristics defining these states, cell-intrinsic regulatory mechanisms that induce and maintain them, and strategies that can lead to their reversal.
Keywords: CD8 T cell differentiation, CD8 T cell dysfunction, self-tolerance, exhaustion, chronic infection, tumors
T cell activation and differentiation can result in functional memory or T cell dysfunction
When naïve CD8 T cells encounter (foreign) antigen in a stimulatory and inflammatory context (e.g. acute infection), a cell-intrinsic program is initiated that drives responding CD8 T cells to greatly expand and differentiate into cytotoxic effector cells that control and eventually clear the pathogen/antigen (expansion phase). Effector T cells secrete high amounts of effector cytokines (e.g. interferon-γ (IFNγ) and tumor necrosis factor (TNFα)) and produce cytolytic molecules (e.g. granzymes and perforin). After the peak of the response, if the pathogen/antigen has been eliminated, most effector T cells undergo apoptosis (contraction phase), but a fraction survive and differentiate into central memory and effector memory T cells (memory phase) (Figure 1). Genome-wide molecular profiling has revealed that naïve, effector and memory T cell differentiation states each have unique gene signatures that dictate their functional and phenotypic properties [1,2].
Figure 1.
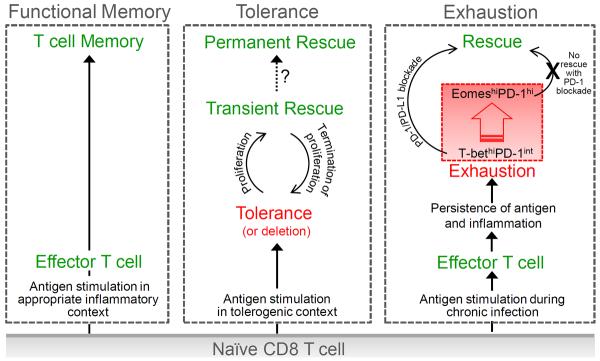
T cell differentiation of naïve CD8 T cells results in either functional T cell memory or T cell dysfunction as reflected by self-tolerance or exhaustion in chronic infections. Functional memory: When naïve CD8 T cells encounter (foreign) antigen in a stimulatory and inflammatory context (e.g. acute infection), T cells differentiate into effector and eventually into memory T cells. Tolerance: Peripheral self-reactive CD8 T cells that encounter self-antigen in a tolerogenic context acquire a program of functional unresponsiveness. Tolerant T cells can be transiently rescued by inducing cell proliferation, e.g. by cytokines (IL-2, IL-15) or lymphopenia. However, once proliferation stops, rescued self-reactive T cells are re-tolerized. If self-tolerant T cells can be permanently reprogrammed and rescued remains to be determined. Exhaustion: Virus-specific T cells initially acquire some effector functions early during chronic infections, but, due to persistence of viral antigen and inflammation, T cells become progressively exhausted. Exhausted T cells represent a heterogeneous T cell population containing T-bethiPD-1int and EomeshiPD-1hi subpopulations (see text). T-bethiPD-1int but not EomeshiPD-1hi exhausted T cells can be functionally rescued by PD-1 blockade.
CD8 T cell differentiation is tightly controlled, and changes in the nature, context and duration of antigen encounter can cause substantial alterations in the T cell activation and differentiation process, potentially leading to T cell dysfunction, unresponsiveness and/or even death. Various states of T cell dysfunction have been described as a consequence of altered activation and differentiation processes, and, depending on the experimental or clinical settings and phenotypic and functional features of the T cells, terms such as exhaustion, tolerance, anergy, senescence, and even ignorance have been used to describe the dysfunctional state (Table 1).
Table 1.
Terms associated with hyporesponsive, dysfunctional, or unresponsive T cells
| I | Ignorance | Peripheral (self-) reactive T cells are `unaware' or `ignorant' of (self-) antigen due to physical sequestration or low level expression of antigen. (Self-) ignorant T cells are antigen-inexperienced, but potentially functional. |
|
| II | Tolerance | a) Central tolerance | Deletion (negative selection) of self-reactive thymocytes expressing T cell receptors (TCR) with too high affinity for self antigen/MHC complexes. |
| b) Peripheral (self-) tolerance | To prevent autoimmunity, self-reactive T cells that escape negative selection are inactivated in the periphery by:
Self-tolerant T cells have encountered self-antigen and are antigen-experienced. |
||
| III | Anergy | a) `In vitro anergy' | Dysfunction of T cells from stimulation in vitro in the absence of co-stimulatory signals. |
| b) `In vivo anergy' or `adaptive tolerance | Initially described as the absence of delayed skin test hypersensitivity responses to recall antigens in patients. Also now used to describe dysfunction of T cells induced by sub-optimal in vivo stimulation. | ||
| IV | Exhaustion | Persistent antigen and inflammation during chronic infection induces progressive loss of effector function in virus-specific T cells. Exhaustion represents a state of functional hyporesponsiveness.
|
|
| V | Senescence | Irreversible, permanent cell-cycle arrest commonly reflected by telomere shortening (Hayflick limit).
|
A large number of inhibitory receptors associated with dysfunction have been identified, with most characterized and functionally assessed in a mouse model of T cell exhaustion during chronic viral infection [3,4]. Subsequently, most of these receptors have also been detected on T cells in different experimental and clinical settings of T cell dysfunction, including tumor-reactive T cells in cancers, self-tolerant T cells, and exhausted T cells in the context of other mouse and human chronic infections [5–9]. With the identification of phenotypic traits shared in different settings of T cell dysfunction, distinctions between such `states' have become blurred, resulting in confused use in the literature of the words exhaustion, tolerance, anergy, and ignorance. Clear definitions for such terms based on their functional traits and molecular choreography are needed to facilitate interpretation of basic and clinical research findings and selection of strategies to modulate T cell dysfunction in different settings.
Here we discuss the various states of T cell dysfunction, focusing on two well characterized and defined settings: peripheral CD8 T cell tolerance to self-antigens (self-tolerance) and CD8 T cell exhaustion during chronic infections -- disparate settings that have in common the persistence of the inciting antigen. We will highlight recent findings on the cellular and molecular characteristics that define these two states, the cell-intrinsic regulatory mechanisms that induce, mediate and maintain them, and strategies and factors that can lead to their reversal. As tumor-reactive CD8 T cells in the context of established cancers can feature similar characteristics as exhausted virus-specific CD8 T cells during chronic infection, aspects of tumor-induced T cell dysfunction are also discussed.
Induction and characteristics of self-tolerance
Tolerance in self-antigen specific T cells is a dysfunctional state required to prevent autoimmunity (self-tolerance). Unresponsiveness to `self' results from both central and peripheral immune tolerance mechanisms (Table 1). Central tolerance is established during T cell development in the thymus, with thymocytes expressing T cell receptors (TCR) of too high affinity for self-antigen/MHC complexes eliminated (negative selection) [10]. However, central tolerance is incomplete, in part because not all peripheral self-antigens are adequately presented in the thymus; self-reactive T cells that escape negative selection must be inactivated in the periphery by a series of tolerizing mechanisms that can include deletion [11–13], suppression by regulatory CD4 T cells [14], and/or induction of cell-intrinsic programs that force self-reactive T cells into a state of functional unresponsiveness [9,15,16]. T cell fate following peripheral encounter with self-antigen is partly dictated by the activation state of the antigen-presenting cell (APC) [17,18]: T cells encountering self-antigen presented by non-activated or non-professional APCs receive incomplete priming signals, and either undergo programmed cell death or become functionally tolerant, exhibiting an antigen-experienced CD44hi phenotype. Such peripheral tolerance is manifested in the inability of tolerant T cells to proliferate and expand in number in response to antigen stimulation, but may not necessarily completely disrupt effector functions such as cytolytic activity and effector cytokine production (split tolerance) [19]. In some settings maintenance of tolerance requires continual exposure of T cells to the self-antigen [20–22], whereas in others the impairment of self-reactive T cells is more profound and even withdrawal of antigen is not adequate to reverse the unresponsive state [9], likely reflecting differences in antigen level, the nature and site of exposure, and T cell avidity.
Self-tolerance versus self-ignorance
Self-reactive T cells can fail to provoke autoimmune disease due to ignorance (Table 1): when anatomical barriers sequester antigen from immune surveillance (immune privileged site), or when self-antigen is expressed and/or cross-presented at concentrations too low to stimulate T cells, peripheral self-reactive T cells can simply remain `unaware' or `ignorant' of self-antigen [23–27]. Thus, `self-ignorant' T cells, in contrast to self-tolerant T cells, are not rendered dysfunctional from self-antigen encounter, but are antigen-inexperienced and persist as naïve, potentially functional T cells in the periphery. If self-ignorant T cells become activated by external stimuli (e.g. by infection [24,28], inflammatory stimuli [29,30] or cytokines [31]), ignorance can be easily overcome, potentially inducing autoimmunity.
Self-tolerance -- a unique state of T cell differentiation with a “tolerance-specific” gene program
Genome-wide transcriptional analysis has revealed self-tolerant CD8 T cells to harbor a “tolerance-specific” gene program, with hundreds of genes differentially expressed compared to their naïve or memory counterparts [9]. Self-tolerance thus represents a distinct state of T cell differentiation. Functional impairment of self-reactive T cells was shown to be associated with: (i) lack of expression of genes encoding effector molecules (Infγ, Prf1, Gzmm, Grn), and altered expression of master transcription factors (T-bet, Eomes, Gata3, Egr1 and Egr2) and chemokine and cytokine receptors (CXCR3, CCR5,IL-12Rβ), (ii) expression of genes associated with reduced immune function and `exhaustion' (e.g., Lag-3), and (iii) expression of genes not previously associated with T cell unresponsiveness (cell cycle, cell division, nucleosome and spindle assembly, DNA-replication).
Tolerance versus anergy
Although first described as the absence of delayed skin test hypersensitivity responses to recall antigens in cancer patients [32,33], anergy is now commonly used to describe the dysfunctional state of T cells stimulated in vitro in the absence of co-stimulatory signals [34], which likely reflects a different mechanism. A functional characteristic of anergic T cells induced in vitro is the inability to produce IL-2 or proliferate in response to later antigen stimulation under optimal conditions. Numerous studies have also described T cell unresponsiveness in vivo with `anergy-like' characteristics as a result of sub-optimal stimulation, referred to as `in vivo anergy', or `adaptive tolerance' [35,36] (Table 1). However, functional, phenotypic and molecular analyses revealed that, despite some overlapping functional and phenotypic traits, many of the in vitro and in vivo induced states called `anergy' are regulated and maintained by distinct cell-intrinsic molecular and cell-extrinsic factors, and require different strategies to restore cell function [35]. As self-tolerance, similar to anergy, is thought to result from stimulation by self-antigen without co-stimulatory and/or inflammatory signals, and is associated with the inability to proliferate in response to antigen, the terms `tolerance' and `anergy' have often been used interchangeably. However, despite some functional and molecular similarities between `anergic' and `tolerant' states, including the expression of the transcriptional regulator early growth response gene 2 (Egr2) [9,37], these are not equivalent terms, given the existence of significant differences in functional characteristics and underlying molecular programs [9,38,39].
Fate commitment and plasticity of self-tolerant T cells
In tolerance settings in which removal from the tolerizing environment is not sufficient to restore cell function, rescue can still be achieved by (i) inducing proliferation by exogenous cytokines such as IL-15 in vitro [40], (ii) inducing proliferation of tolerant dual-receptor T cells in vivo through a second TCR not reactive with “self” [41], or (iii) inducing homeostatic proliferation by lymphopenia [9] (Figure 1). The fact that tolerance can be broken through the induction of proliferation by alternative non-TCR mediated signaling pathways provides important mechanistic insights into how self-tolerance might be regulated. T cells generally exist in a cellular state of quiescence - a reversible non-proliferative state, and cognate antigen stimulation can trigger naïve or memory T cells to exit the quiescent state, enter cell cycle, and undergo clonal expansion. TCR signaling in tolerant T cells is disengaged from cell cycle reentry control mechanisms, but alternative intact signaling pathways, e.g. through cytokine receptors, enable tolerant T cells to undergo proliferation independently of cognate antigen encounter creating a “rescued state” during which tolerant T cells become capable of responding to antigen. However, once the proliferative stimulus ends and rescued, self-tolerant T cells exit the cell cycle, tolerance is re-established and self-tolerant T cells largely restore their tolerance-associated molecular profile [9]. Re-tolerization not only occurs in a tolerizing environment, but can occur even in the absence of self-antigen, suggesting that self-tolerant T cells “remember” the tolerance program established during the initial encounter(s) with self-antigen in the periphery. How precisely fate commitment of self-tolerant T cells is encoded, and how tolerance-associated “memory” can be erased to mediate long-term functionality of self-antigen specific T cells, remains to be determined (Figure 1).
Proliferation-induced rescue and enhancement of T cell function by cytokine stimulation (IL-2, IL-15) or lymphopenia have been described not only for self-tolerant T cells, but also for other dysfunctional states, including ignorance [31,42–44] and anergy [45,46]. As lymphopenia is associated with the induction and exacerbation of autoimmune diseases, including graft-versus-host disease after autologous stem cell transplant (auto-GVHD)[47–51], proliferation-mediated re-programming of self-reactive tolerant or anergic T cells and activation of self-ignorant T cells could be the underlying cell-intrinsic mechanism(s) of lymphopenia-associated autoimmunity that operates in concert with extrinsic factors such as decreased numbers of regulatory T cells in lymphopenic hosts. Whether lymphopenia mediates transient or permanent rescue of anergic T cells and whether and how epigenetic changes mediate the cell-intrinsic programming associated with functional rescue, is currently not known.
Imprinted, epigenetic “memory” of T cell dysfunction
Conrad Waddington's concept of epigenetic landscapes was the first postulation that cellular states are not only regulated by gene sequence but also sequence-independent regulatory mechanisms [52]. The different gene expression patterns and phenotypic traits observed in genetically identical cells in the absence of changes in Watson-Crick DNA base-pairing reflect distinct epigenetic states. Such imprinting is encoded at multiple levels, including methylation of DNA, modifications of histones, organization of nucleosomes, and expression of non-coding RNAs [53]. Naïve, effector and memory CD8 T cell differentiation states are associated with specific epigenetic or chromatin states [54–57]: as T cells differentiate from naïve to effector T cells, e.g. during acute infection, epigenetic marks are laid down in promoter regions and regulatory elements of relevant gene loci that define and mediate functional and phenotypic properties [55,58–60]. These epigenetic marks persist through cell division and are maintained throughout memory development, allowing memory T cells to “remember” and retain an active chromatin state at specific sites (`gene poising') -- the molecular basis for the robust and rapid execution of effector functions seen in memory T cells upon antigen re-encounter [54,55,60–62].
The demonstration that self-tolerant CD8 T cells rescued by cell division in a non-tolerogenic host eventually re-tolerize suggests that, similar to the epigenetic memory imprinted in long-lived memory T cells, heritable and imprinted epigenetic states are established during acquisition of self-tolerance, which function independent of external cues. Persistence of functional and phenotypic traits associated with unresponsiveness in the absence of antigen has not only been observed in CD8 T cell self-tolerance. In a transgenic mouse model of inducible antigen presentation, CD4 T cells retain at least parts of their dysfunctional state long after antigen removal [63], and, as discussed below, exhausted CD8 T cells during chronic LCMV infection maintain some phenotypic and functional properties associated with exhaustion after transfer into antigen-free hosts [64–66]. Thus, maintenance of phenotypic and/or functional features associated with T cell unresponsiveness can be mediated by distinct and sometimes concurrent regulatory mechanisms, one which becomes cell-intrinsic and independent of external cues, and one which is dependent on continuous, external signaling (instructional model).
T cell exhaustion in chronic viral infections
Unlike acute infections, where the pathogen is rapidly cleared, in chronic infections pathogens are not quickly eliminated but rather persist, leading to chronic antigen stimulation and persistent inflammation, and potentially to `exhaustion' and/or clonal deletion of pathogen-specific CD8 T cells [7,67–69]. T cell exhaustion is not limited to chronic viral infection, but also includes bacterial and parasitic infections; however, here we will focus on the more extensively defined characteristics of exhaustion in viral infections associated with high viral replication, including lymphocytic choriomeningitis virus (LCMV) clone 13, hepatitis C virus (HCV), hepatitis B virus (HBV), human immunodeficiency virus (HIV), and simian immunodeficiency virus (SIV).
Infection of mice with LCMV clone 13 recapitulates many aspects of chronic infection in humans, with virus-specific CD8 T cells initially expanding and acquiring some effector functions, but, as the infection progresses, gradually losing effector functions in a hierarchical manner. Proliferative capacity and production of IL-2 are lost first, followed by the ability to produce TNFα, and ultimately to produce IFNγ [7,68]. Thus, in contrast to anergy, which is induced rapidly after stimulation, exhaustion is progressive over a period of weeks or months depending on the chronic stimulus. Loss of function generally coincides with expression of inhibitory surface receptors, including PD-1, LAG-3, CD160, 2B4, TIM-3, BTLA, and CTLA-4 [3]. High antigen load, long duration of the infection, and absence or loss of CD4 T cell help amplify the stage and severity of exhaustion, reflected in increasingly more profound functional impairment, distinct patterns of co-expression of inhibitory receptors, and higher expression levels of individual receptors per cell [3,7,69].
PD-1 has emerged as a major inhibitory receptor associated with T cell exhaustion [70–73]. Although PD-1 is transiently expressed during CD8 T cell activation [74], presumably to prevent hyper-activation and/or autoimmunity [74,75], persistent antigen stimulation in the context of chronic infection causes epigenetic alterations in the Pdcd1 locus, resulting in maintenance of high, long-term PD-1 expression on virus-specific T cells [76,77]. However, high PD-1 expression is also found on functional effector memory T cells in healthy adult humans [78], suggesting that PD-1 should not be considered as a definitive marker for T cell exhaustion and dysfunction.
The PD-1/PD-L1 signaling pathway can regulate exhaustion by directly attenuating functional and proliferative capabilities (e.g. by repressing TCR signaling [74] and inducing genes that impair T cell function such as BATF [79]). Furthermore, PD-1 has recently been shown to promote immune exhaustion by inducing paralysis of T cell motility, thereby preventing T cells from performing effector functions and target cell killing and preventing immunopathology in persistently infected tissues [80]. PD-1 and other inhibitory receptors such as LAG-3, 2B4 and TIM-3 act, at least in part, synergistically, contributing via non-redundant signaling pathways to establishment of T cell exhaustion [3]. Thus, inhibitory-receptor mediated exhaustion is “tuned” by the availability of ligands in the environment. Furthermore, suppressive cytokines such as IL-10 and transforming growth factor (TGF-β), immune cells including myeloid suppressor cells [81], Foxp3+ CD4+ regulatory T cells [7], and prolonged production of type I interferons (IFN-α/β) that paradoxically up-regulate expression and production of inhibitory molecules, contribute to the maintenance of persistent infections and/or to the functional state of T cell exhaustion during chronic infections [82,83].
Exhaustion - a distinct differentiation state associated with functional hyporesponsiveness
Exhausted CD8 T cells, similar to self-tolerant CD8 T cells, harbor a unique molecular signature markedly distinct from naïve, effector or memory T cells, with alterations in TCR and cytokine signaling pathways, migration, metabolism, as well as expression of transcription factors (Blimp-1, BATF, NFAT, T-bet, Eomes) [7,79,84–90]. However, exhaustion is neither a fixed, irreversible, terminal differentiation state, nor an unresponsive T cell state. Instead, exhaustion represents an adaptive state of hyporesponsiveness that, although insufficient to completely clear the pathogen, can provide the host with the ability to control the infection without causing detrimental immunological pathology. During chronic infections, the emergence of immunologic escape mutants and the rapid and dramatic increase in viremia after CD8 T cell depletion, point to an effective state of hyporesponsiveness of exhausted T cells rather than unresponsiveness or senescence (Table 1) [91–93]. Such hyporesponsiveness may be important for the host, as fatal disease can be induced in some settings if anti-viral immune responses become fully unleashed and exhausted T cells mediate unrestrained effector functions [80,94]. Thus, during chronic infections a `host-pathogen stalemate' is maintained by establishment of an exhausted state in virus-specific T cells. However, this stalemate is dynamic, and interference with immune function or escape from the existing immune response can give the pathogen the edge.
Recent studies are shedding light on the underlying mechanism(s) and factors mediating immune control or subsequent loss of immune control during chronic infections. Previously defined `exhausted' T cells represent a heterogeneous T cell population, including at least two distinct virus-specific CD8 T cell subpopulations: a progenitor and a more mature, terminally differentiated T cell pool, with both required for immune control of chronic infections [89] (Figure 1). T-bethiPD-1int CD8 T cells represent the progenitor T cell subset, which proliferate in response to persisting antigen, and ultimately give rise to EomeshiPD-1hi CD8 T cells, the terminal progeny. EomeshiPD-1hi T cells display higher levels of other inhibitory receptors (LAG-3, CD160, 2B4, TIM-3) and do not replicate, but exhibit high levels of cytotoxic activity. Over time, persisting antigen results in progressive differentiation and loss of T-bethiPD-1int progenitor T cells and accumulation of EomeshiPD-1hi T cells. This loss of the progenitor pool and dramatic imbalance of progenitor-terminally differentiated T cells is thought to be one reason for the loss of immune control during chronic infections. Consistent with these observations in the LCMV mouse model, in patients with chronic HCV infection exhaustion is associated with few T-bethi precursors and accumulation of an Eomeshi terminally differentiated population in the liver [89]. Interestingly, during the early phase of chronic infection, exhausted virus-specific CD8 T cells can continue their differentiation process and form functional T cell memory if transferred into infection-/antigen-free mice, however, exhausted T cells from established chronic infections are unable to differentiate into memory T cells when removed from antigen, do not restore effector functions and retain traits of their exhausted state [64,66]. In fact, exhausted T cells that persist long-term during a chronic infection ultimately require tonic antigen stimulation for their continued survival, a feature referred to as antigen addiction [64]. This is in contrast to functional memory CD8 T cells, which persist and very intermittently proliferate in the absence of antigen as a result of IL-7 and IL-15-mediated homeostatic proliferation, or self-tolerant T cells, which persist in antigen-free hosts, and, dependent on their history, may restore cell function in that setting. Together, these observations demonstrate that the acquisition of a terminally differentiated exhausted state is a progressive process and that the survival of terminally differentiated, exhausted T cells is actively maintained by TCR-signaling from antigen encounter, but that some characteristics associated with exhaustion might be imprinted and operate independent of external cues. However, a recent study reported that exhausted T cells after transfer into naive mice can exhibit memory-like properties including the ability to robustly proliferate and control a subsequent infection, but at the same time maintain an `exhausted' phenotype including the expression of PD-1 [65]. The differences between these studies likely reflect differences in the severity of the infection and consequently composition of the exhausted T cell pool (e.g. different fractions of progenitors versus terminally differentiated exhausted T cells). Thus, some phenotypic traits such as PD-1 expression can be imprinted early during chronic infection, but establishing the exhausted T cell state requires further imprinting and acquisition over time of additional functional and phenotypic changes.
Rescue of exhausted T cells during chronic infection
Antibody blockade of inhibitory receptors has become a promising therapeutic intervention for chronic viral infections. Blocking the PD-1 pathway has demonstrated therapeutic benefits in vivo in both LCMV-infected mice and SIV-infected macaques by increasing T cell function and viral clearance [72,95]. However, effectiveness of PD-1 blockade depends on the nature of the exhausted T cell pool, as PD-1 blockade reverses `exhaustion' of PD-1int T cells but not terminally differentiated PD-1hi T cells [96]. Combinatorial approaches, including blockade of multiple inhibitory receptors and/or activation of co-stimulatory receptor pathways, might be necessary to promote functional rescue of more terminally differentiated PD-1hi T cells. Combined blockade of Tim-3 and PD-1 [97] or of LAG-3 and PD-1 [3], PD-1 blockade and IL-2 therapy [98], or PD-1 blockade and anti-4-1BB agonistic antibody therapy [99] synergistically improve CD8 T cell responses and viral control in chronically infected mice. Evaluating (i) if such combinatorial strategies actually reverse the dysfunction of terminally differentiated PD-1hi T cells or again only effectively target the less differentiated PD-1int T cells, and (ii) if T cells forced to continue responding with sustained use of checkpoint regulators such as prolonged PD-1/PD-L1 blockade will ultimately be driven to senescence and reach their “Hayflick limit” [100], should provide insights into the mechanism(s) underlying maintenance and/or rescue of T cell exhaustion.
T cells in tumors exhibit an exhausted profile
It has been commonly assumed that T cells in the context of established progressing cancers exhibit an `exhausted' state similar to chronic infection due to high tumor-antigen load and immunosuppressive factors in the tumor microenvironment. This hypothesis is largely based on the observation that T cells isolated from human tumors as well as experimental tumor models share many phenotypic and functional characteristics of exhausted T cells in chronic infections: tumor-infiltrating CD8 T lymphocytes (TIL) are impaired in production of effector cytokines, express inhibitory receptors including PD-1, LAG-3, 2B4, TIM-3, CTLA-4, and display alterations in signaling pathways described for exhausted T cells (Figure 2) [6,101–105]. Furthermore, genome-wide transcriptome analysis of Melan-A/Mart-1-specific CD8 T cells isolated from metastases of melanoma patients revealed an exhaustion profile very similar to murine exhausted T cells during chronic LCMV clone 13 infection [106].
Figure 2.
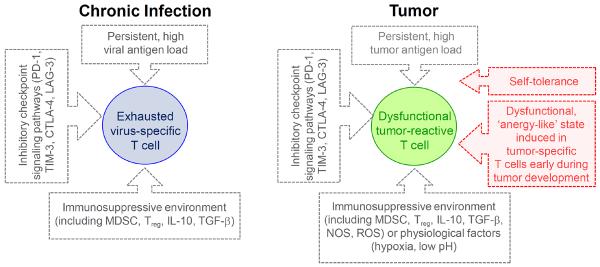
Factors mediating exhaustion in chronic infections and tumor-induced T cell dysfunction. Exhausted virus-specific CD8 T cells in chronic infections (left) and dysfunctional tumor-infiltrating CD8 T cells (TIL, right) exhibit an `exhausted' state induced by high viral or tumor antigen load and immunosuppressive factors. `Exhausted' T cells in both settings share phenotypic and functional characteristics, including the expression of inhibitory receptors such as PD-1, LAG-3, 2B4, TIM-3, CTLA-4. However, TIL can represent a heterogeneous cell population with distinct individual `states' of dysfunction. Such dysfunctional states can be mediated by cell-intrinsic programs, including a tolerance program imprinted in self/tumor antigen-specific T cells, or programs induced in tumor-specific T cells that encounter tumor antigen early during a pre-malignant non-inflammatory phase of tumor development (red). Consequently, depending on the nature of the T cells and the microenvironment in which they must function, TIL might require different immunotherapeutic strategies to restore cell function. Abbreviations: MDSC = myeloid-derived suppressor cells; Treg = regulatory CD4 T cells; TGF-β = transforming growth factor β; NOS = Nitric Oxide Synthase; ROS = reactive oxygen species.
In spite of these overlapping functional and phenotypic traits, a clear picture of the `state' of tumor-induced T cell dysfunction is lacking because it has not been possible to dissect the extent to which functional unresponsiveness results from (i) cell-intrinsic self-tolerance programs imprinted in self/tumor antigen-specific T cells [9], (ii) cell-intrinsic programs induced in tumor-specific T cells that encountered tumor antigen during the early or pre-malignant, non-inflammatory phase of tumor development [107,108], (iii) cell-intrinsic and extrinsic immune-suppressive factors in the tumor microenvironment including chronic tumor-antigen encounter, expression of inhibitory receptors and availability of their ligands, myeloid derived suppressor cells, regulatory Foxp3+ CD4 T cells, cytokines such as IL-10 and TGF-ß, and compounds such as nitric oxide synthase (NOS), reactive oxygen species (ROS), nitrogen species and indoleamine-2,3 dioxygenase (IDO), and/or (iv) physiological changes within tumors including hypoxia, low nutrient levels, low pH, and/or high interstitial fluid pressure (Figure 2). All these factors, as well as antigen-specificity, TCR affinity, level of tumor antigen, and T cell differentiation state, ultimately contribute to the state of unresponsiveness of tumor-specific, tumor-infiltrating T cells. Moreover, T cell exhaustion due to persistent viral antigen stimulation during chronic infection might be the initial underlying cause of T cell dysfunction in some virus-induced cancers (e.g. hepatitis B virus- and hepatitis C virus–associated hepatocellular carcinoma [109,110], or human T-cell leukemia virus-1-asssociated adult T-cell leukemia/lymphoma [111]). Thus, TIL represent heterogeneous cell populations with distinct individual `states' of dysfunction that likely will require different strategies to restore cell function, which potentially explains why at least some therapeutic interventions have been unpredictably effective in apparently similar as well as disparate clinical settings (see below). Decoding the various molecular programs mediating T cell dysfunction in tumors, and understanding which programs are actively maintained by external cues or which are epigenetically imprinted, will be important for the design of effective immunotherapies.
Overcoming tumor-induced T cell dysfunction
Cancer immunotherapies aim to stimulate and enhance T cell function as well as target immune-suppressive and tumor-promoting pathways mediated by the tumor microenvironment. Similar to chronic infections, blockade of negative checkpoint receptors has emerged as a promising approach for treatment of cancers. Ipilimumab, a CTLA-4-blocking monoclonal antibody was the first FDA approved cancer immunotherapy for treatment of melanoma, and is currently being tested in other malignancies [112]. Blockade of PD-1/PD-L1, another inhibitory checkpoint signaling pathway, has demonstrated clinical efficacy in some types of cancers including melanoma, non-small-cell lung cancer and renal cell cancer [113,114]. However, similar to the setting of chronic infections, reversing T cell hyporesponsiveness by PD-1 (or CTLA-4) blockade comes at a cost: adverse immune-related toxicities, including some with fatal outcomes, have been observed in a fraction of patients, although initial results with anti-PD-1 suggest it may cause less severe side effects and autoimmune toxicities than anti-CTLA-4 blockade [115].
Animal studies and ex vivo studies with human tumor-reactive T cells have demonstrated that combination checkpoint blockades, e.g. PD-1+CTLA-4 [116], PD-1+LAG-3 [103], PD-1+TIM-3 [104,117], or PD-1+LAG-3+CTLA-4 [118] may be required in some settings for rescue of T cell function and/or effective T cell-mediated cancer regression. Importantly, phase 1 clinical trials recently reported that combinatorial PD-1+CTLA-4 blockade results in improved treatment outcomes in melanoma patients, although with higher incidence of adverse toxicities compared to monotherapy [119,120].
In addition to blockade of inhibitory receptors, targeting co-stimulatory receptors by agonistic antibodies including anti-CD137, anti-CD40, anti-OX40, and anti-GITR are being investigated in clinical trials as a means to overcome hypo-responsiveness to tumors (reviewed in [121]). Identifying which T cell population(s) and/or state(s) are rendered functional by these various immunomodulatory strategies, as well as how and in what settings the particular reagents are effective, will be essential for designing predictably effective cancer immunotherapies.
Concluding remarks
Tolerance to a self-antigen and exhaustion resulting from a chronic infection represent two states of T cell dysfunction associated with unique molecular programs. Neither tolerance nor exhaustion is a fixed or irreversible differentiation state, but instead represents a flexible and plastic cell state that, depending on external conditions, can allow transient re-programming and functional rescue. Recent studies now suggest that tolerance and exhaustion are associated with distinct epigenetic landscapes and that early during the induction and establishment of such T cell dysfunction, a heritable imprinted epigenetic `memory' is formed that ultimately mediates and maintains the phenotypic and functional traits of self-tolerance or exhaustion. While the imprinted cell-intrinsic program can function independent of external cues, the cell can remain susceptible to external signals from the environment, and in settings of chronic infection the cell-intrinsic program acts in concert with cell-extrinsic regulatory mechanism(s) resulting in a progressively more severe `exhausted' state over the course of infection.
Precisely how molecular programs of T cell dysfunction are epigenetically encoded, and whether self-tolerant and/or exhausted T cells can be permanently re-programmed to differentiate into long-term functional memory T cells remains unanswered. Elucidating the genetic and epigenetic regulatory mechanism(s) mediating and maintaining or required to break T cell unresponsiveness in settings of self-tolerance, exhaustion or dysfunction in tumors may identify broader principles of T cell dysfunction and reveal clinical opportunities for intervention to treat chronic infections and cancers, as well as expose potential strategies for mitigating the dangers of autoimmunity and transplant rejection.
Question Box
Phenotypic and functional characteristics (e.g. the expression of inhibitory receptors and loss of effector functions) are often shared in different settings of T cell dysfunction. Are there common regulatory pathways and transcription factors that are shared between these different states of dysfunction? Is there a common, “master” transcriptional regulator necessary and sufficient to mediate T cell dysfunction?
States of T cell dysfunction, including exhaustion during chronic infections and self-tolerance, represent distinct T cell differentiation states associated with specific molecular programs. Do these molecular programs become epigenetically encoded, and if yes, how and when does that occur?
Exhaustion during chronic infection is a progressive process ultimately leading to the generation of terminally differentiated T cells. What is the molecular and epigenetic basis for T cell exhaustion early during a persistent infection, and how does this change at later stages of chronic infections? Is tumor-induced T cell dysfunction a progressive process, similarly to exhaustion during chronic infections?
What are the molecular and epigenetic regulatory mechanism(s) that determine whether a dysfunctional T cell can or cannot be rescued? For dysfunctional cells that can be rescued, what determines whether this is permanent or only transient? Can dysfunctional self-tolerant and/or exhausted T cells be permanently re-programmed to differentiate into long-term functional memory T cells?
Will T cells of patients that receive prolonged PD-1/PD-L1 blockade (or other immunomodulatory therapies over long periods of time) to overcome dysfunction in the presence of persistent antigen ultimately be driven to apoptosis or reach their “Hayflick limit” and become senescent?
Highlights
Exhaustion and tolerance represent distinct CD8 T cell differentiation states.
These distinct states are associated with unique molecular programs.
Exhaustion and tolerance are not fixed, but flexible and plastic cell states.
Certain characteristics are epigenetically imprinted and independent from external cues.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kaech SM, et al. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 2002;111:837–51. doi: 10.1016/s0092-8674(02)01139-x. [DOI] [PubMed] [Google Scholar]
- 2.Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12:749–61. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blackburn SD, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Odorizzi PM, Wherry EJ. Inhibitory receptors on lymphocytes: insights from infections. J Immunol. 2012;188:2957–65. doi: 10.4049/jimmunol.1100038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baitsch L, et al. The three main stumbling blocks for anticancer T cells. Trends Immunol. 2012;33:364–72. doi: 10.1016/j.it.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 6.Kim PS, Ahmed R. Features of responding T cells in cancer and chronic infection. Curr Opin Immunol. 2010;22:223–30. doi: 10.1016/j.coi.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–9. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 8.Grosso JF, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest. 2007;117:3383–92. doi: 10.1172/JCI31184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schietinger A, et al. Rescued tolerant CD8 T cells are preprogrammed to reestablish the tolerant state. Science. 2012;335:723–7. doi: 10.1126/science.1214277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hogquist KA, et al. Central tolerance: learning self-control in the thymus. Nat Rev Immunol. 2005;5:772–82. doi: 10.1038/nri1707. [DOI] [PubMed] [Google Scholar]
- 11.Hernandez J, et al. Phenotypic and functional analysis of CD8(+) T cells undergoing peripheral deletion in response to cross-presentation of self-antigen. J Exp Med. 2001;194:707–17. doi: 10.1084/jem.194.6.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Redmond WL, et al. Distinct requirements for deletion versus anergy during CD8 T cell peripheral tolerance in vivo. J Immunol. 2005;174:2046–53. doi: 10.4049/jimmunol.174.4.2046. [DOI] [PubMed] [Google Scholar]
- 13.Kurts C, et al. Class I-restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8(+) T cells. J Exp Med. 1997;186:239–45. doi: 10.1084/jem.186.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. 2010;11:7–13. doi: 10.1038/ni.1818. [DOI] [PubMed] [Google Scholar]
- 15.Redmond WL, Sherman LA. Peripheral tolerance of CD8 T lymphocytes. Immunity. 2005;22:275–84. doi: 10.1016/j.immuni.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 16.Mueller DL. Mechanisms maintaining peripheral tolerance. Nat Immunol. 2010;11:21–7. doi: 10.1038/ni.1817. [DOI] [PubMed] [Google Scholar]
- 17.Steinman RM, et al. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 18.Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol. 2001;19:47–64. doi: 10.1146/annurev.immunol.19.1.47. [DOI] [PubMed] [Google Scholar]
- 19.Ohlen C, et al. CD8(+) T cell tolerance to a tumor-associated antigen is maintained at the level of expansion rather than effector function. J Exp Med. 2002;195:1407–18. doi: 10.1084/jem.20011063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rocha B, et al. Clonal anergy blocks in vivo growth of mature T cells and can be reversed in the absence of antigen. J Exp Med. 1993;177:1517–21. doi: 10.1084/jem.177.5.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramsdell F, Fowlkes BJ. Maintenance of in vivo tolerance by persistence of antigen. Science. 1992;257:1130–4. doi: 10.1126/science.257.5073.1130. [DOI] [PubMed] [Google Scholar]
- 22.Calbo S, et al. Functional tolerance of CD8+ T cells induced by muscle-specific antigen expression. J Immunol. 2008;181:408–17. doi: 10.4049/jimmunol.181.1.408. [DOI] [PubMed] [Google Scholar]
- 23.Pittet MJ, et al. High frequencies of naive Melan-A/MART-1-specific CD8(+) T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J Exp Med. 1999;190:705–15. doi: 10.1084/jem.190.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohashi PS, et al. Ablation of "tolerance" and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 1991;65:305–17. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- 25.Overwijk WW, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–80. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parish IA, Heath WR. Too dangerous to ignore: self-tolerance and the control of ignorant autoreactive T cells. Immunol Cell Biol. 2008;86:146–52. doi: 10.1038/sj.icb.7100161. [DOI] [PubMed] [Google Scholar]
- 27.Kurts C, et al. CD8 T cell ignorance or tolerance to islet antigens depends on antigen dose. Proc Natl Acad Sci U S A. 1999;96:12703–7. doi: 10.1073/pnas.96.22.12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oldstone MB, et al. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: role of anti-self (virus) immune response. Cell. 1991;65:319–31. doi: 10.1016/0092-8674(91)90165-u. [DOI] [PubMed] [Google Scholar]
- 29.Lang KS, et al. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat Med. 2005;11:138–45. doi: 10.1038/nm1176. [DOI] [PubMed] [Google Scholar]
- 30.Millar DG, et al. Hsp70 promotes antigen-presenting cell function and converts T-cell tolerance to autoimmunity in vivo. Nat Med. 2003;9:1469–76. doi: 10.1038/nm962. [DOI] [PubMed] [Google Scholar]
- 31.Ramanathan S, et al. Exposure to IL-15 and IL-21 enables autoreactive CD8 T cells to respond to weak antigens and cause disease in a mouse model of autoimmune diabetes. J Immunol. 2011;186:5131–41. doi: 10.4049/jimmunol.1001221. [DOI] [PubMed] [Google Scholar]
- 32.Schier WW. Cutaneous anergy and Hodgkin's disease. N Engl J Med. 1954;250:353–61. doi: 10.1056/NEJM195403042500902. [DOI] [PubMed] [Google Scholar]
- 33.Ashikawa K, et al. Immune response in tumor-bearing patients and animals. II. Incidence of tuberculin anergy in cancer patients. Gann. 1967;58:565–73. [PubMed] [Google Scholar]
- 34.Schwartz RH. T cell anergy. Annu Rev Immunol. 2003;21:305–34. doi: 10.1146/annurev.immunol.21.120601.141110. [DOI] [PubMed] [Google Scholar]
- 35.Choi S, Schwartz RH. Molecular mechanisms for adaptive tolerance and other T cell anergy models. Semin Immunol. 2007;19:140–52. doi: 10.1016/j.smim.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med. 1987;165:302–19. doi: 10.1084/jem.165.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng Y, et al. Transcriptional regulator early growth response gene 2 (Egr2) is required for T cell anergy in vitro and in vivo. J Exp Med. 2012;209:2157–63. doi: 10.1084/jem.20120342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Villarino AV, et al. Posttranscriptional silencing of effector cytokine mRNA underlies the anergic phenotype of self-reactive T cells. Immunity. 2011;34:50–60. doi: 10.1016/j.immuni.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Macian F, et al. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–31. doi: 10.1016/s0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- 40.Teague RM, et al. Interleukin-15 rescues tolerant CD8+ T cells for use in adoptive immunotherapy of established tumors. Nat Med. 2006;12:335–41. doi: 10.1038/nm1359. [DOI] [PubMed] [Google Scholar]
- 41.Teague RM, et al. Peripheral CD8+ T cell tolerance to self-proteins is regulated proximally at the T cell receptor. Immunity. 2008;28:662–74. doi: 10.1016/j.immuni.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oelert T, et al. Irradiation and IL-15 promote loss of CD8 T-cell tolerance in response to lymphopenia. Blood. 2010;115:2196–202. doi: 10.1182/blood-2009-06-227298. [DOI] [PubMed] [Google Scholar]
- 43.Gattinoni L, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–12. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson LD, Jameson SC. Self-Specific CD8+ T Cells Maintain a Semi-naive State Following Lymphopenia-Induced Proliferation. J Immunol. 2010 doi: 10.4049/jimmunol.1000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeSilva DR, et al. Clonal anergy is induced in vitro by T cell receptor occupancy in the absence of proliferation. J Immunol. 1991;147:3261–7. [PubMed] [Google Scholar]
- 46.Brown IE, et al. Homeostatic proliferation as an isolated variable reverses CD8+ T cell anergy and promotes tumor rejection. J Immunol. 2006;177:4521–9. doi: 10.4049/jimmunol.177.7.4521. [DOI] [PubMed] [Google Scholar]
- 47.Krupica T, Jr., et al. Autoimmunity during lymphopenia: a two-hit model. Clin Immunol. 2006;120:121–8. doi: 10.1016/j.clim.2006.04.569. [DOI] [PubMed] [Google Scholar]
- 48.King C, et al. Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell. 2004;117:265–77. doi: 10.1016/s0092-8674(04)00335-6. [DOI] [PubMed] [Google Scholar]
- 49.Le Campion A, et al. Lymphopenia-induced spontaneous T-cell proliferation as a cofactor for autoimmune disease development. Blood. 2009;114:1784–93. doi: 10.1182/blood-2008-12-192120. [DOI] [PubMed] [Google Scholar]
- 50.Datta S, Sarvetnick N. Lymphocyte proliferation in immune-mediated diseases. Trends Immunol. 2009;30:430–8. doi: 10.1016/j.it.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 51.Jones RJ, et al. Induction of graft-versus-host disease after autologous bone marrow transplantation. Lancet. 1989;1:754–7. doi: 10.1016/s0140-6736(89)92575-0. [DOI] [PubMed] [Google Scholar]
- 52.Goldberg AD, et al. Epigenetics: a landscape takes shape. Cell. 2007;128:635–8. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 53.Guil S, Esteller M. DNA methylomes, histone codes and miRNAs: tying it all together. Int J Biochem Cell Biol. 2009;41:87–95. doi: 10.1016/j.biocel.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 54.Cuddapah S, et al. Epigenomics of T cell activation, differentiation, and memory. Curr Opin Immunol. 2010;22:341–7. doi: 10.1016/j.coi.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zediak VP, et al. The contribution of epigenetic memory to immunologic memory. Curr Opin Genet Dev. 2011;21:154–9. doi: 10.1016/j.gde.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 56.Youngblood B, et al. T-cell memory differentiation: insights from transcriptional signatures and epigenetics. Immunology. 2013;139:277–84. doi: 10.1111/imm.12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Denton AE, et al. Differentiation-dependent functional and epigenetic landscapes for cytokine genes in virus-specific CD8+ T cells. Proc Natl Acad Sci U S A. 2011;108:15306–11. doi: 10.1073/pnas.1112520108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schmidl C, et al. Lineage-specific DNA methylation in T cells correlates with histone methylation and enhancer activity. Genome Res. 2009;19:1165–74. doi: 10.1101/gr.091470.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Araki Y, et al. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity. 2009;30:912–25. doi: 10.1016/j.immuni.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weng NP, et al. The molecular basis of the memory T cell response: differential gene expression and its epigenetic regulation. Nat Rev Immunol. 2012;12:306–15. doi: 10.1038/nri3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Youngblood B, et al. Making memories that last a lifetime: heritable functions of self-renewing memory CD8 T cells. Int Immunol. 2010;22:797–803. doi: 10.1093/intimm/dxq437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rothenberg EV, Zhang JA. T-cell identity and epigenetic memory. Curr Top Microbiol Immunol. 2012;356:117–43. doi: 10.1007/82_2011_168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Han S, et al. Role of antigen persistence and dose for CD4+ T-cell exhaustion and recovery. Proc Natl Acad Sci U S A. 2010;107:20453–8. doi: 10.1073/pnas.1008437107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wherry EJ, et al. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc Natl Acad Sci U S A. 2004;101:16004–9. doi: 10.1073/pnas.0407192101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Utzschneider DT, et al. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat Immunol. 2013;14:603–10. doi: 10.1038/ni.2606. [DOI] [PubMed] [Google Scholar]
- 66.Angelosanto JM, et al. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J Virol. 2012;86:8161–70. doi: 10.1128/JVI.00889-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moskophidis D, et al. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. 1993;362:758–61. doi: 10.1038/362758a0. [DOI] [PubMed] [Google Scholar]
- 68.Wherry EJ, et al. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–27. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. 2004;78:5535–45. doi: 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Trautmann L, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 71.Day CL, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–4. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 72.Barber DL, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 73.Petrovas C, et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med. 2006;203:2281–92. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Keir ME, et al. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nishimura H, et al. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–51. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 76.Youngblood B, et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity. 2011;35:400–12. doi: 10.1016/j.immuni.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Youngblood B, et al. Cutting Edge: Prolonged Exposure to HIV Reinforces a Poised Epigenetic Program for PD-1 Expression in Virus-Specific CD8 T Cells. J Immunol. 2013;191:540–4. doi: 10.4049/jimmunol.1203161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Duraiswamy J, et al. Phenotype, function, and gene expression profiles of programmed death-1(hi) CD8 T cells in healthy human adults. J Immunol. 2011;186:4200–12. doi: 10.4049/jimmunol.1001783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Quigley M, et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat Med. 2010;16:1147–51. doi: 10.1038/nm.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zinselmeyer BH, et al. PD-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. J Exp Med. 2013;210:757–74. doi: 10.1084/jem.20121416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Norris BA, et al. Chronic but not acute virus infection induces sustained expansion of myeloid suppressor cell numbers that inhibit viral-specific T cell immunity. Immunity. 2013;38:309–21. doi: 10.1016/j.immuni.2012.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Teijaro JR, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science. 2013;340:207–11. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wilson EB, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013;340:202–7. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Doering TA, et al. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37:1130–44. doi: 10.1016/j.immuni.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wherry EJ, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–84. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 86.Wang C, et al. Loss of the signaling adaptor TRAF1 causes CD8+ T cell dysregulation during human and murine chronic infection. J Exp Med. 2012;209:77–91. doi: 10.1084/jem.20110675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rangachari M, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3-mediated cell death and exhaustion. Nat Med. 2012;18:1394–400. doi: 10.1038/nm.2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shin H, et al. A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity. 2009;31:309–20. doi: 10.1016/j.immuni.2009.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Paley MA, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. 2012;338:1220–5. doi: 10.1126/science.1229620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Larsson M, et al. Molecular signatures of T-cell inhibition in HIV-1 infection. Retrovirology. 2013;10:31. doi: 10.1186/1742-4690-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jin X, et al. Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J Exp Med. 1999;189:991–8. doi: 10.1084/jem.189.6.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schmitz JE, et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283:857–60. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- 93.Pircher H, et al. Viral escape by selection of cytotoxic T cell-resistant virus variants in vivo. Nature. 1990;346:629–33. doi: 10.1038/346629a0. [DOI] [PubMed] [Google Scholar]
- 94.Frebel H, et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J Exp Med. 2012;209:2485–99. doi: 10.1084/jem.20121015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Velu V, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458:206–10. doi: 10.1038/nature07662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Blackburn SD, et al. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci U S A. 2008;105:15016–21. doi: 10.1073/pnas.0801497105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jin HT, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 2010;107:14733–8. doi: 10.1073/pnas.1009731107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.West EE, et al. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J Clin Invest. 2013;123:2604–15. doi: 10.1172/JCI67008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vezys V, et al. 4-1BB signaling synergizes with programmed death ligand 1 blockade to augment CD8 T cell responses during chronic viral infection. J Immunol. 2011;187:1634–42. doi: 10.4049/jimmunol.1100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 101.Zippelius A, et al. Effector function of human tumor-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res. 2004;64:2865–73. doi: 10.1158/0008-5472.can-03-3066. [DOI] [PubMed] [Google Scholar]
- 102.Fourcade J, et al. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. 2012;72:887–96. doi: 10.1158/0008-5472.CAN-11-2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Woo SR, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72:917–27. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fourcade J, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207:2175–86. doi: 10.1084/jem.20100637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Matsuzaki J, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A. 2010;107:7875–80. doi: 10.1073/pnas.1003345107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Baitsch L, et al. Exhaustion of tumor-specific CD8 T cells in metastases from melanoma patients. J Clin Invest. 2011;121:2350–60. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. 2005;437:141–6. doi: 10.1038/nature03954. [DOI] [PubMed] [Google Scholar]
- 108.Willimsky G, et al. Immunogenicity of premalignant lesions is the primary cause of general cytotoxic T lymphocyte unresponsiveness. J Exp Med. 2008;205:1687–700. doi: 10.1084/jem.20072016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Castello G, et al. HCV-related hepatocellular carcinoma: From chronic inflammation to cancer. Clin Immunol. 2010;134:237–50. doi: 10.1016/j.clim.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 110.Willimsky G, et al. Virus-induced hepatocellular carcinomas cause antigen-specific local tolerance. J Clin Invest. 2013;123:1032–43. doi: 10.1172/JCI64742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kozako T, et al. PD-1/PD-L1 expression in human T-cell leukemia virus type 1 carriers and adult T-cell leukemia/lymphoma patients. Leukemia. 2009;23:375–82. doi: 10.1038/leu.2008.272. [DOI] [PubMed] [Google Scholar]
- 112.Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Brahmer JR, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Callahan MK, Wolchok JD. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J Leukoc Biol. 2013;94:41–53. doi: 10.1189/jlb.1212631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Duraiswamy J, et al. Dual Blockade of PD-1 and CTLA-4 Combined with Tumor Vaccine Effectively Restores T-Cell Rejection Function in Tumors. Cancer Res. 2013;73:3591–603. doi: 10.1158/0008-5472.CAN-12-4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sakuishi K, et al. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207:2187–94. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Berrien-Elliott MM, et al. Durable adoptive immunotherapy for leukemia produced by manipulation of multiple regulatory pathways of CD8+ T-cell tolerance. Cancer Res. 2013;73:605–16. doi: 10.1158/0008-5472.CAN-12-2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wolchok JD, et al. Nivolumab plus Ipilimumab in Advanced Melanoma. N Engl J Med. 2013 doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hamid O, et al. Safety and Tumor Responses with Lambrolizumab (Anti-PD-1) in Melanoma. N Engl J Med. 2013 doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yao S, et al. Advances in targeting cell surface signalling molecules for immune modulation. Nat Rev Drug Discov. 2013;12:130–46. doi: 10.1038/nrd3877. [DOI] [PMC free article] [PubMed] [Google Scholar]