

Abstract
This study investigated the actin scavenger function of the vitamin D binding protein (DBP) in vivo using DBP null (−/−) mice. Intravenous injection of G-actin into wild-type (DBP+/+) and DBP−/− mice showed that contrary to expectations, DBP+/+ mice developed more severe acute lung inflammation. Inflammation was restricted to the lung and pathological changes were clearly evident at 1.5 and 4 hours post-injection but were largely resolved by 24 hours. Histology of DBP+/+ lungs revealed noticeably more vascular leakage, hemorrhage and thickening of the alveolar wall. Flow cytometry analysis of whole lung homogenates showed significantly increased neutrophil infiltration into DBP+/+ mouse lungs at 1.5 and 4 hours. Increased amounts of protein and leukocytes were also noted in bronchoalveolar lavage fluid from DBP+/+ mice 4 hours after actin injection. In vitro, purified DBP-actin complexes did not activate complement or neutrophils but induced injury and death of cultured human lung microvascular endothelial cells (HLMVEC) and human umbilical vein endothelial cells (HUVEC). Cells treated with DBP-actin showed a significant reduction in viability at 4 hours, this effect was reversible if cells were cultured in fresh media for another 24 hours. However, a 24-hour treatment with DBP-actin complexes showed a significant increase in cell death (95% for HLMVEC, 45% for HUVEC). The mechanism of endothelial cell death was via both caspase-3 dependent (HUVEC) and independent (HLMVEC) pathways. These results demonstrate that elevated levels and/or prolonged exposure to DBP-actin complexes may induce endothelial cell injury and death, particularly in the lung microvasculature.
Keywords: actin, endothelial cells, inflammation, tissue injury, vitamin D binding protein
INTRODUCTION
Actin is the most abundant and highly conserved protein inside all eukaryotic cells and exists in two forms: monomeric globular actin (G-actin) and polymerized filamentous actin (F-actin) (Rottner and Stradal, 2011). During tissue injury large quantities of actin can be released into extracellular fluids where the ionic conditions and lack of regulators favor spontaneous generation of F-actin filaments (Janmey and Lind, 1987). Circulating F-actin potentially is injurious and previous animal studies have shown that intravascular actin filaments can trigger angiopathic consequences in the microcirculation similar to fibrin (Haddad, et al., 1990, Meier, et al., 2006). Accordingly, higher organisms have evolved a robust extracellular actin scavenger system (EASS) consisting of two plasma proteins: gelsolin that caps and severs F-actin filaments, and the vitamin D binding protein (DBP) that binds G-actin monomers tightly for subsequent clearance from the blood (Meier, et al., 2006). Circulating DBP-actin complexes have been observed in both humans and animals following traumatic injury, and the plasma concentration of actin-free DBP has been shown to be an effective but indirect marker of tissue injury in cases of severe trauma (Antoniades, et al., 2007, Meier, et al., 2006, Schiodt, et al., 2007). Plasma levels of actin-free DBP below 3.5 μM (200 μg/ml) have been shown to significantly correlate with poor prognosis in human cases of sepsis, multiple trauma and acetaminophen-induced liver failure (Antoniades, et al., 2007, Dahl, et al., 2003, Meier, et al., 2006, Schiodt, et al., 2007). Clinical outcome and decreased plasma levels of DBP in trauma have a statistical correlation similar to the APACHE II score, Kings College criteria and the TRISS-like method (Antoniades, et al., 2007, Dahl, et al., 2003, Meier, et al., 2006, Schiodt, et al., 2007). Thus, the capacity to scavenge extracellular actin is a physiologically important role for this multifunctional plasma protein.
DBP, also referred to as Gc-globulin, is an abundant (6–7 μM) 56 kDa plasma protein that is part of the albumin gene family and shares the multiple disulfide linked triple domain structure of albumin (Chun, 2012). As its name implies, it is the primary extracellular transport protein for all vitamin D metabolites. Besides the vitamin D and actin binding functions, DBP can serve as a neutrophil chemotactic cofactor, and a deglycosylated form of DBP acts as a macrophage activating factor (Chun, 2012). There are no known natural deficiencies of DBP in any vertebrate species but a DBP null (−/−) mouse, fully backcrossed on a C57BL/6 background, has been generated. These mice are healthy and develop and reproduce similar to their wild-type counterparts when fed a vitamin D sufficient mouse chow diet (Safadi, et al., 1999, White, et al., 2002). Studies using DBP−/− mice have shown that the primary role of DBP is to maintain circulating vitamin D levels within a physiological range to protect against transient vitamin deficiencies (Zella, et al., 2008). More recently, our lab has shown that DBP−/− mice have significantly reduced (~50%) neutrophil recruitment to the lungs compared to their wild-type DBP+/+ counterparts in three different alveolitis models, two acute and one chronic (Trujillo, et al., 2013). However, the actin scavenger function of DBP has not been investigated in vivo using DBP null mice, and the objective of this study was to characterize how mice with a systemic deficiency of DBP respond to actin in the circulation. Contrary to expectations, results show that DBP−/− mice developed much less acute lung inflammation than their wild-type DBP+/+ counterparts when actin was injected intravenously. Moreover, in vitro studies showed that DBP-actin complexes induce endothelial cell injury and death. These studies suggest that elevated levels and/or prolonged exposure to DBP-actin complexes in vivo may contribute to the sequelae of traumatic tissue injury.
MATERIALS AND METHODS
Reagents
Purified human DBP was obtained from Athens Research & Technology (Athens, GA). The IgG fraction of goat polyclonal anti-human DBP was purchased from DiaSorin (Stillwater, MN) and then affinity-purified in our lab using immobilized DBP. Highly purified rabbit skeletal muscle actin was obtained from Cytoskeleton, Inc. (Denver, CO). Goat polyclonal anti-actin (I-19) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and monoclonal pan anti-actin mAb (ACTN05) was purchased from Thermo Scientific/Lab Vision (Fremont, CA). Polyclonal anti-gelsolin (ab74420) was obtained from Abcam (Cambridge, MA). Sterile, endotoxin-free HBSS, DPBS and RPMI 1640 were purchase from Mediatech, Inc. (Manassas, VA). EDTA solution was purchased from Life Technologies-Gibco (Grand Island, NY). Collagenase and DNase I were purchased from Roche (Indianapolis, IN). Rat anti-mouse monoclonal antibodies for flow cytometry and their corresponding labeled isotype controls, were all purchased from Biolegend (San Diego, CA): PE-labeled anti Gr-1 (RB6-8C5), FITC-labeled anti-F4/80 (BM8). Purified cobra venom factor (CVF) and sheep erythrocytes were obtained from Complement Technology, Inc. (Tyler, TX). Rabbit anti-sheep erythrocyte antibody was purchased from Dako (Accurate Scientific Westbury, NY). Mouse monoclonal anti-human factor B (clone D33/3) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Purified human TNF-α and IL-1β were purchased from R&D Systems (Minneapolis, MN). Bio-Plex 23-mouse cytokine panel was obtained from BioRad (Hercules, CA).
DBP+/+ and −/− mouse colonies
The DBP−/− mouse line has been fully backcrossed on a C57BL6/J background for 11 generations. A detailed description of the DBP−/− and the corresponding DBP+/+ mouse colonies, produced from DBP+/− hemizygotes, has been recently published (Trujillo, et al., 2013). In some experiments, wild type C57BL6/J mice were purchased from Jackson Labs (Bar Harbor, ME). Only mature male age-matched mice (age ranged from 10 weeks to 10 months) were utilized and all animals were housed in a maximum isolation facility at Stony Brook University. Animal experiments were performed using protocols approved by the Institutional Animal Care and Use Committee at Stony Brook University.
Intravenous administration of G-actin
Highly purified rabbit skeletal muscle actin was reconstituted using sterile pyrogen-free DPBS at 1 mg/ml under aseptic conditions. DBP+/+ and −/− mice were injected with 100 μl of G-actin (1 mg/ml) via the tail vein to introduce an actin bolus into the circulation. The control group received 100 μl of DBPS via the tail vein. The mice were observed for signs of respiratory distress. After 1.5, 4 or 24 hours, groups of mice were anesthetized by an i.p. injection (0.3 ml) of ketamine (90 mg/kg) and xylazine (10 mg/kg) solution. Following anesthesia, EDTA plasma was obtained by retro-orbital bleeding and mice were then euthanized by cervical dislocation and secured to a small animal surgery board (Kent Scientific, Torrington, CT). A small midline incision was made in the suprasternal region and the trachea was exposed by blunt dissection. A sterile BD angiocath (18 gauge needle stylet in a 1.3×44 mm long I.V. catheter) was inserted into the anterior portion of the exposed trachea between cartilage rings and the lungs were lavaged once and the BAL fluid was stored on ice until analyzed for total protein and cell count. BAL fluid was centrifuged at 200 × g for 5 min at 4°C to pellet the cells. BAL cells pellets were resuspended in 1 ml of PBS-1% BSA and duplicate counts of total cell number were made using a BioRad TC10 automated cell counter. The protein concentration of the BAL supernatant was measured using a Lowry Protein Assay.
Preparation of organs for histology
In a separate group of mice that were not lavaged, the mouse lungs were inflated with 10% PBS-buffered formalin for 15 min at 25 cm water pressure to ensure the proper degree of lung inflation. The lungs were removed from the thoracic cavity en bloc with the heart and submerged in 10% PBS-buffered formalin for 24 hours at 4°C. The kidneys, and livers were also isolated and submerged into formalin. The fixed organs were transferred to the Stony Brook University Hospital Histology Laboratory for preparation of thin-sectioned slides stained with hematoxylin and eosin (H&E). Digital photos were taken using a Nikon Eclipse Ti inverted microscope with an Insight Spot2 digital camera using 20x and 100x objectives.
Preparation of lung homogenates
Single cell suspensions were prepared from whole lung homogenates at each time point after actin injection. Lungs were isolated and placed into a well in 24-well plate containing 1 ml of complete culture medium (RPMI 1640 + 10% FBS) and thoroughly minced into small pieces using surgical scissors. The minced tissue was transferred to a 15 ml conical tube containing 5 ml of fresh collagenase solution (1 mg/ml type IV collagenase, 25 U/ml DNase I, 5% FBS and the volume adjusted up to 5 ml with RPMI). The samples were then incubated at 37°C for 45 minutes with shaking. After the incubation, the tissue was disaggregated into a cellular suspension by passing it through an 18-gauge needle 20 times. The samples were centrifuged at 500 × g for 5 minutes. The supernatant was aspirated and the pellet was resuspended in RBC lysis buffer and immediately centrifuged (50 × g for 5 minutes). The supernatant was saved and the pellet, containing large tissue aggregates, was discarded. The supernatant was then again centrifuged at 500 × g for 5 min and the cells were resuspended in 500 μl of flow cytometry staining buffer. The samples were counted using an automated cell counter and then stained for Gr-1 and F4/80 and analyzed by flow cytometry.
Nondenaturing PAGE and immunoblot analysis for DBP-actin complexes
EDTA plasma was diluted 1:2 in DPBS and 50 μl of the diluted plasma was loaded to 10% native (no SDS) polyacrylamide gel. After separation proteins were transferred to PVDF membranes (Millipore, MA). The membrane was probed for DBP-actin complexes using 1:1000 dilution of polyclonal goat anti-human DBP (which strongly reacts with mouse DBP) followed by 1:5000 HRP-conjugated donkey anti-goat secondary antibody. The blot was developed with HyGlo Quick spray chemiluminescent HRP antibody detection reagent (Denville Scientific Inc., South Plainfield, NJ) and visualized by autoradiography film.
Endothelial cell culture
Human lung microvascular endothelial cells (HLMVEC) and endothelial cell growth media kits (CC-3156 & CC-4176) were obtained from Lonza (Walkersville, MD). HLMVEC were obtained at passage 3 and immediately thawed and cultured in endothelial growth medium-2 (EGM-2) Bulletkit at 5000 cells/cm2 in a T25 flask. The medium was changed the next day and subsequently every other day until cells were 70 to 80% confluent. HLMVECs were detached using Lonza sub-culturing reagents and the viability was determined by trypan blue assay. Human umbilical cord endothelial cells (HUVECs) were freshly isolated and generously provided by Dr. Martha Furie, Stony Brook University. These cells were grown in the same media as HLMVEC to 70 to 80% confluence.
Cell viability assay
Cell titer blue (CTB) viability assay was obtained from Promega (Madison, WI). Endothelial cells were cultured to 70 to 80% confluence and harvested as described above. 5000 cells in 100 μl of EGM2 complete medium were seeded to 96 well plates and incubated at 37°C, 5% CO2 incubator overnight for the cells to attach. The cells were treated for 4 hours or 24 hours and CTB reagent was added at the end of the treatment. Cells with CTB reagent were incubated for 2 hours and the fluorescent intensity was then measured at 560 nm excitation and 590 nm emission.
Nuclear staining to evaluate cell death
Acridine orange (AO) and ethidium bromide (EB) were purchased from Fisher Scientific (Fairlawn, NJ). Endothelial cells were harvested at 70 to 80% confluence and seeded in 48 well plates at 2 × 105 cells/well and incubated overnight for the cells to attach. AO/EB solution was prepared at 100 μg/ml of each reagent. Cells were then treated for 24 hours with DBP-actin then each sample was stained with 100 μl of AO/EB solution just prior to microscopy and quantification. At least 300 cells were counted for each treatment and the percent of dead (red-orange nucleus) and live (green nucleus) cells was calculated.
Immunoblot for cleaved PARP
Rabbit antibody to the cleaved form of poly ADP ribose polymerase (PARP, #Asp214) was purchased from Cell Signaling Technology (Danvers, MA). Endothelial cells were cultured to 70 – 80% confluence then harvested and seeded into 6 well plates at 106 cells/ml. Cells were incubated overnight to allow attachment and then were treated for 24 hours. After the treatment period cells were washed with DBPS 3 times and then detached. Whole cell lysates were obtained using lysis buffer containing 100 mM Tris pH 7.4, 150 mM NaCl, 1.0 mM EDTA pH 8.0, 1% Triton X-100, 10% glycerol and a complete protease inhibitor cocktail tablet (Millipore). The protein concentration in the cell lysate was determined by a protein assay and 50 μg total protein was loaded on 10% SDS-PAGE. After separation the proteins were transferred to PVDF membranes (Millipore, MA). The PVDF membrane was probed with a 1:500 dilution of the cleaved PARP antibody followed by an HRP-conjugated goat anti-rabbit IgG. The blot was developed with HyGlo Quick spray chemiluminescent HRP antibody detection reagent and visualized by autoradiography film.
Measurement of serum complement activation
The effect of DBP-actin complexes on serum complement was evaluated using a hemolytic assay to test for classical or lectin pathway activation, and an immunoblot for factor B cleavage to assess alternative pathway activation. The hemolytic was performed using antibody-coated sheep erythrocytes (EA) as the target cell for lysis by serum complement. Sheep erythrocytes were obtained from Complement Technology, Inc. (Tyler, TX). Normal human serum with a defined hemolytic value was purchased as lyophilized powder from Sigma-Aldrich (St. Louis, MO). Rabbit anti-sheep erythrocyte antibody was purchased from Dako (Accurate Scientific Westbury, NY). Normal human serum was treated with from 10 nM to 1500 nM of actin or DBP-actin complexes for 1 hour at 37°C. An aliquot (10 μl) of the treated serum was mixed with EA and incubated for 30 min at 37°C. The samples were centrifuged and the red blood cell lysis was measured by reading the absorbance at 412 nm for the release hemoglobin. Water lysed EA were used as a positive control. The data were presented as percentage of hemolysis.
DBP-actin treated serum was also separated using 10% SDS-PAGE and blotted onto a PVDF membrane. The blot was probed with mouse monoclonal anti-human factor B (clone D33/3) primary antibody followed by a goat anti-mouse HRP-conjugated secondary antibody. The blot was developed using HyGlo Quick spray chemiluminescent HRP antibody detection reagent and visualized by autoradiography film. The cleavage of factor B is readily observed by the disappearance of the native 100 kDa factor B band and the appearance of the cleavage product, 33 kDa Ba fragment. The positive control for alternative pathway activation was CVF-treated serum.
Neutrophil chemotaxis assay
Measurement of cell movement of normal human neutrophils and differentiated HL-60 cells was performed using a 48 well microchemotaxis chamber as previously described in detail (Trujillo, et al., 2011). Human neutrophils were isolated from the venous blood of healthy medication-free volunteers who gave informed consent using a standard 3-step purification protocol (Trujillo, et al., 2011). The human promyelocytic cell line HL-60 was also used and differentiated for 4 days using 1.3% DMSO to obtain a neutrophil-like phenotype (Hauert, et al., 2002). In each assay, the migration of 200,000 cells (50 μl of 4×106/ml) was evaluated. Cell movement was quantitated microscopically by measuring the distance in microns (μm) that the leading front of cells had migrated into the filter according to the method described by Zigmond and Hirsch (Zigmond and Hirsch, 1973). In each experiment, five fields per duplicate filter were measured at 400× magnification.
Measurement of reactive oxygen species
Detection of cellular reactive oxygen species (ROS) in neutrophils and endothelial cells was performed using the 2',7'-dichlorofluorescein diacetate (DCFDA) reagent as part of a kit purchased from Abcam (ab112851, Cambridge, MA). Primary human neutrophils were freshly isolated as described above and 4 × 106 cells were used per sample. Endothelial cells were grown to 70 – 80% confluence and 1.5 × 105 cells per well were seeded into 96 well plates and allowed to adhere for 24 hours before treatment. Cells were stained with 20 μM of DCFDA for 30 min at 37°C. Neutrophils were then treated with 50 μM tert-butyl hydrogen peroxide (TBHP, the positive control), 1 μM PMA, 1 μM DBP, 1 μM actin, and 1 μM DBP-actin for 1 hour at 37°C. Endothelial cells were treated with 50 μM TBHP, EGM-2 media, 1 μM DBP, 1 μM actin or 1 μM DBP-actin for 24 hours. The fluorescence intensity was then measured at 485 nm excitation and 535 nm emission.
Statistical analysis
A minimum of 5 mice were used for each experimental treatment, and at least 3 experiments were performed for each in vitro analysis. Statistical differences between the mean values of the treatment groups were evaluated using either an unpaired T-test (comparing two groups) or analysis of variance (ANOVA) followed by a multiple comparisons post-hoc test (comparing three or more groups). Statistical analysis was performed using the software program InStat (GraphPad Software, San Diego, CA).
RESULTS
DBP is the primary scavenger for extracellular actin released from dead or damaged cells, and this function was investigated in vivo using mice with a systemic deficiency of DBP. Both wild-type (DBP+/+) and DBP null (DBP−/−) mice were injected with 100 μg of purified rabbit skeletal actin via the tail vein and then sacrificed after either 1.5, 4 or 24 hours. None of the mice in either group showed signs of overt respiratory distress and displayed normal behavior following actin injection. At the indicated time points, blood, lungs, heart, liver and kidneys were collected to assess the effect of circulating DBP-actin complexes. Plasma samples were analyzed using non-denaturing gels to verify formation of DBP-actin complexes in the blood. Figure 1A demonstrates that, as expected, DBP-actin complexes are observed only in wild-type (DBP+/+) mice. Analysis of the blots by densitometry revealed that the greatest amount of complexes were observed after 1.5 hours (78%) and then declined to about 50% at 4 hours and 30% at 24 hours (Fig. 1B). Furthermore, both DBP+/+ and −/− mice had approximately the same amount of total gelsolin in plasma (Fig. 1C), indicating that DBP−/− mice did not have a compensatory increase in the other plasma EASS protein.
Figure 1.

Plasma DBP-actin complexes and gelsolin levels in DBP+/+ and DBP−/− mice following intravenous actin injection. (A) DBP immunoblot of EDTA plasma samples from three DBP+/+ wild-type (WT) mice and three DBP−/− (KO) mice each obtained 1.5, 4 and 24 hours after actin injection. Plasma aliquots were separated using a 10% native (non-denaturing) gel then blotted for DBP. The electrophoretic separation of unbound DBP and DBP-actin complexes is shown. (B) Densitometry measurement of DBP-actin complexes in panel A. Data is presented as mean ± SEM (n = 3) of DBP-actin as a percent of total DBP (unbound + DBP-actin) in WT plasma. Statistical significance is indicated. (C) Gelsolin immunoblot of plasma separated by SDS-PAGE from three DBP−/− (KO) and two DBP+/+ (WT) mice.
The amount of circulating DBP-actin complexes (Fig. 1A) paralleled the degree of lung inflammation (Fig. 2), where DBP+/+ mice showed extensive interstitial edema and hemorrhage at 1.5 hours that was decreased after 4 hours and largely resolved 24 hours after actin injection. In contrast, DBP−/− mice showed almost no histological signs of inflammation at 1.5 hours, mild inflammation at 4 hours that was noticeably less than the DBP+/+ group (Fig. 2). Histological evidence of lung inflammation in both DBP+/+ and −/− mice was considerably decreased at 24 hours (Fig. 2). The insets in the lower left of each panel are high magnification (1000×) images of the same lung section to show better the degree of cellularity. Interestingly, the lung was the only organ that showed inflammatory injury after intravenous actin injection; the heart, liver and kidneys all appeared essentially identical to the PBS controls (data not shown). The degree of lung inflammation was quantitated by measuring the number of Gr1+ neutrophils by flow cytometry (gating shown in Fig. 3A) in lung homogenates 1.5, 4 or 24 hours after actin injection (Fig. 3B). Both groups showed that largest number of neutrophils after 4 hours that decreased by 24 hours (Fig. 3B). There was also significantly reduced numbers of lung neutrophils in the DBP−/− group at the 1.5 and 4 hour time points (Fig. 3B). In contrast, there was no difference in the number of F4/80+ macrophages in lung homogenates of DBP+/+ versus DBP−/− mice (Fig. 3C).
Figure 2.
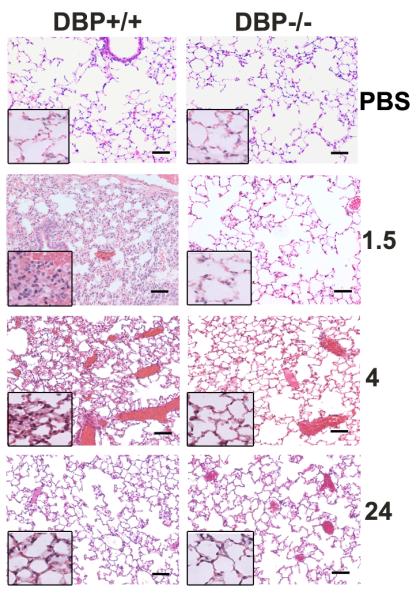
Histology of acute lung inflammation in DBP+/+ and DBP−/− mice 1.5, 4 and 24 hours after intravenous injection of 100 μg purified rabbit skeletal muscle actin in a total volume of 100 μl. Images in all panels are H&E stained sections at 200× magnification, bar = 100 μm. Inset panel is a 1000× magnification of that lung section.
Figure 3.
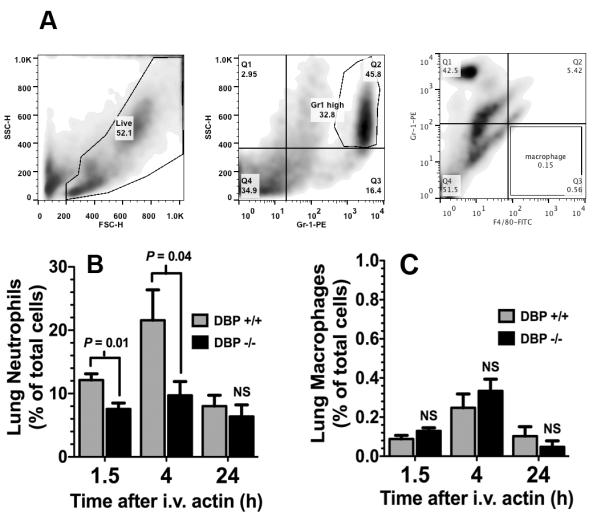
Analysis of total neutrophils and macrophages in whole lung homogenates 1.5, 4 and 24 hours after intravenous injection of purified actin. (A) Flow cytometry gating for analyzing neutrophils and macrophages in whole lung homogenates. A total of 104 cells were collected and the forward scatter (FSC) and side scatter (SSC) gates set to exclude dead cells and debris (left panel). Neutrophils were gated as the Gr-1 PE high, side scatter high cell population (center panel). Macrophages were gated as F4/80-FITC high, Gr-1 PE low population. (B) The percent of Gr-1+ neutrophils in lung homogenates measured by flow cytometry. (C) The percent of F4/80+ macrophages in lung homogenates measured by flow cytometry. Numbers represent mean% ± SEM (n= 5), statistical significance is indicated.
The pulmonary capillary bed is the first microvasculature that an intravenous injected protein complex would encounter. Injury to the capillary endothelium can induce permeability changes and provoke inflammation of the airspaces (alveolitis). To determine if circulating DBP-actin complexes can also induce an alveolitis, bronchoalveolar lavage (BAL) was performed in DBP+/+ and −/− mice at the peak of inflammation, i.e., 4 hours after intravenous injection of actin (Fig. 4). Figure 4A shows that actin injection induced significant protein leakage into the airspaces of DBP+/+ mice that was not observed in DBP−/− animals. Analysis of stained cytospin slides of the BAL fluid showed that DBP+/+ mice also had a significant increase in the number of leukocytes in the alveolar spaces compared to the DBP−/− group (Fig. 4B). The preceding experiments demonstrate that DBP-actin complexes are associated with acute lung inflammation that is largely resolved by 24 hours. Therefore, the possible effects of DBP-actin complexes on triggering immediate inflammatory mechanisms were investigated next. DBP-actin complexes do not activate the complement system, a potent source of early-acting mediators in blood (Supplemental Figure 1). In addition, complexes are not chemotactic for human neutrophils (Supplemental Fig. 2A) and do trigger generation of reactive oxygen species in these cells (Supplemental Fig. 2B). Finally, there were no differences between DBP+/+ and −/− mice in the levels of 23 inflammatory cytokines (Bio-Plex 23 panel mouse cytokine assay) measured in the BAL 4 hours after injection of DBP-actin complexes (data not shown).
Figure 4.
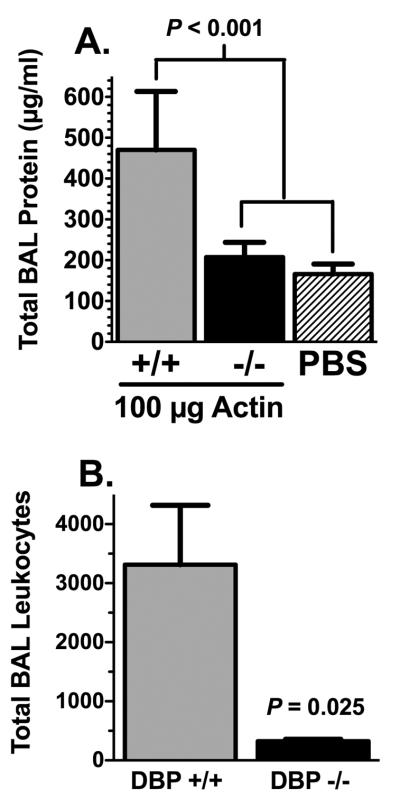
Analysis of total protein and leukocytes in BAL 4 hours after actin injection. (A) Total protein content of cell-free BAL fluid from actin injected DBP+/+ mice (n = 6), actin injected DBP−/− mice (n = 7) and PBS injected mice (n = 5). Statistical significance is indicated. (B) Total BAL leukocytes from actin injected DBP+/+ and DBP−/− mice (n = 5). Numbers represent mean ± SEM, statistical significance is indicated.
The observed lung inflammation presented above (Figs. 2–4) would be consistent with alterations to lung microvascular endothelial cells. Thus, the possibility that DBP-actin complexes have a direct effect on endothelial cells was investigated next in vitro. Primary human endothelial cells from the microvasculature of an adult lung (HLMVEC) and from an umbilical cord (HUVEC) were treated with DBP alone, actin alone or DBP-actin complexes for 4 hours at 37°C and the effect was assessed using the cell-titer blue (CTB) viability assay (Fig. 5). HLMVEC treated with DBP-actin showed a significantly reduced viability compared to untreated cells, DBP treated or actin treated cells (Fig. 5A). The effect of serum starvation is shown as the positive control for reduced cell viability (Fig. 5A). HUVECs are more robust cells and showed increased viability in response to these treatments as compared to HLMVECs, but there was still a significant reduction in viability with DBP-actin treatment (Fig. 5B). A similar reduction in cell viability with DBP-actin treatment was also observed using primary human neonatal dermal endothelial cells (data not shown). The morphology of DBP-actin treated endothelial cells by phase contrast microscopy show that HLMVECs are retracted and partially rounded-up (Supplemental Figure 3), similar results were also noted with HUVECs (data not shown). To determine if damage is reversible, cells were treated with DBP-actin complexes for 4 hours then either washed and placed in fresh media for a 24 hour recovery period or tested immediately using the CTB assay. Figure 6 shows that the injurious effects of a 4 hour DBP-actin treatment were fully reversible in both HLMVECs (Fig. 6A) and HUVECs (Fig. 6B).
Figure 5.

CTB viability assay of HLMVECs and HUVECs treated with DBP-actin complexes. (A) HLMVECs. (B) HUVECs. Endothelial cells were treated for 4 hours with either EGM-2 growth medium (untreated = negative control), EGM-2 base medium without serum (starved = positive control), or 1 μM of the indicated purified proteins in EGM-2 growth medium. CTB reagent was added to the wells at 20 μl/well. Cells were incubated for 1 hour with CTB reagent and the fluorescent intensity was measured at 560 nm excitation and 590 nm emission. Numbers represent mean ± SEM (n = 3), statistical significance is indicated.
Figure 6.

CTB viability assay to test reversible injury in HLMVECs and HUVECs treated with DBP-actin complexes. HLMVECs (A) and HUVECs (B) were treated for 4 hours with either EGM-2 growth medium (untreated = negative control), EGM-2 base medium without serum (starved = positive control), or 1 μM of the indicated purified proteins in EGM-2 growth medium (black bars). A duplicate set of cells were washed after the 4 hour treatment and allowed to recover in EGM-2 growth medium for an additional 24 hours (gray bars). CTB reagent was added to the wells at 20 μl/well. Cells were incubated for 1 hour with CTB reagent and the fluorescent intensity was measured at 560 nm excitation and 590 nm emission. Numbers represent mean ± SEM (n = 3). Triple asterisk indicates that the values of DBP-actin treated cells and serum starved cells were significantly lower (P < 0.001) than all other groups.
The aforementioned cell viability and morphology data would indicate that DBP-actin complexes directly induce endothelial cell injury. To determine if prolonged treatment can cause cell death, primary endothelial cells were treated for 24 hours with DBP-actin complexes and then stained with acridine orange and ethidium bromide to determine the percent live/dead cells. The nucleus of live cells stains green with acridine orange whereas dead cells take up ethidium bromide and display a red or orange nucleus. This assay was quantified by counting the number of dead and live cells in multiple fields. Figure 7A demonstrates that DBP-actin complexes induce the same degree of HLMVEC cell death as the positive control (serum starvation), and significantly less than untreated or single treated (DBP alone, actin alone) cells. Figure 7B shows that HUVECs are more resistant to the injurious effects of DBP-actin complexes or serum starvation, but still display a significant decrease in the number of live cells following a 24 hour treatment.
Figure 7.

Acridine orange and ethidium bromide nuclear staining of HLMVECs (A) and HUVECs (B) after 24 hours of DBP-actin treatment. Cells were treated with EGM-2 growth medium, serum started, 1 μM DBP, 1 μM actin or 1 μM DBP-actin complex for 24 hours. Cells were stained with AO/EB and the percentage of live cells was quantified by counting at least 300 cells per treatment. Statistical significance is indicated.
The kinetics of endothelial cell injury and death following treatment with DBP-actin complexes would be consistent with an apoptotic mechanism. Therefore, potential mechanisms of cell death were next investigated by examining the cleavage of PARP, a key substrate in the caspase-3 dependent apoptosis pathway. Cell lysates of HLMVECs and HUVECs treated with DBP-actin were blotted with a specific antibody that only detects cleaved PARP. HLMVECs showed no PARP cleavage with 24 hour treatment (Fig. 8A), despite the fact that these cells showed clear morphological signs of injury and death (Fig. 7A and Supplemental Fig. 3). In contrast, HUVECs displayed a dramatic increase in the amount of cleaved PARP after a 24 hour treatment with DBP-actin complexes (Fig. 8B) indicating a robust activation of the caspase-3 dependent apoptosis pathway. One mechanism to trigger an apoptotic pathway in endothelial cells is by generation of intracellular reactive oxygen species, however, DBP-actin complexes did not increase the amount of intracellular oxidants in HLMVECs over untreated control cells (Supplemental Figure 4). These results indicate that DBP-actin complexes can directly trigger endothelial cell death by both caspase 3-dependent and independent pathways.
Figure 8.
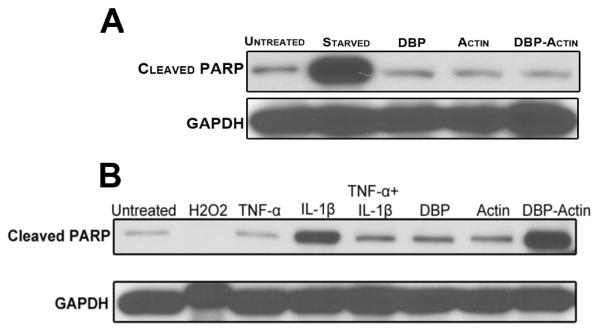
Immunoblot for cleaved PARP in HLMVECs and HUVECs after 24 hours of DBP-actin treatment. (A) HLMVECs were treated with EGM-2 growth medium, serum started, 1 μM DBP, 1 μM actin or 1 μM DBP-actin complex. (B) HUVECs were treated with EGM-2 growth medium, 3% hydrogen peroxide (H2O2), 10 nM TNF-α, 10 nM IL-1β, 10 nM TNF-α+IL-1β, 1 μM DBP, 1 μM Actin, and 1 μM DBP-Actin complex. Cell lysates were separated on 10% SDS-PAGE gel and blotted for cleaved PARP. GAPDH is shown as a loading control.
Finally, because endothelial cells are very sensitive to the effects of lipopolysaccharide (LPS), we considered the possibility that the pure actin and DBP used in the above in vitro assays may be contaminated with LPS. Both DBP and actin were purchased as lyophilized powders that were reconstituted with sterile endotoxin-free reagents using aseptic technique in a laminar flow BSL-2 cabinet. All subsequent experiments were performed with endotoxin-free reagents in a sterile laminar flow hood. In addition, experiments were performed where polymyxin B (that binds and neutralizes LPS) was added to purified DBP and this treatment did not reduce the effect of DBP-actin complexes on endothelial cells. Thus, we conclude that the reagents most likely were not contaminated with LPS and the results were due to the direct effect of DBP-actin complexes on endothelial cells.
DISCUSSION
Massive tissue injury triggered by burns, sepsis or severe trauma will result in the release of large amounts of actin into extracellular fluids. Indeed, previous studies in humans and animals have shown significant quantities of DBP-actin in the plasma following injury (Antoniades, et al., 2007, Dahl, et al., 1998, Dahl, et al., 2003, Harper, et al., 1987, Lind SE, 1986, Meier, et al., 2006, Schiodt, et al., 2007, Young, et al., 1987). The role of DBP-actin complexes as potential mediators of tissue injury has received little attention, presumably because they are cleared rapidly from the circulation largely by the liver (Meier, et al., 2006). In contrast, the role of actin-free DBP has been the focus of several studies in both humans and animals where low plasma DBP levels have been shown to significantly correlate with poor prognosis in human cases of sepsis, multiple trauma and acetaminophen-induced liver failure (Antoniades, et al., 2007, Dahl, et al., 2003, Meier, et al., 2006, Schiodt, et al., 2007). The assumption in these studies is that when a large percentage of the circulating DBP pool is consumed (i.e., bound to G-actin and cleared from the blood) there is very little remaining DBP to neutralize actin released from ongoing injury. Thus, a reduced level of unbound DBP facilitates formation of actin filaments that then induce microvascular damage and tissue ischemia. However, the inverse of this premise is that large amounts of DBP-actin complexes can trigger tissue injury, and this alternative hypothesis has not been investigated. The present study is the first to utilize the DBP null mouse to evaluate the role of DBP-actin complexes in vivo. If the prevailing hypothesis is correct that maintaining a critical concentration of free DBP in blood is essential to protect against the damaging effects of extracellular actin, then a systemic deficiency of DBP should be lethal when actin is injected intravenously. Results presented herein demonstrate that a deficiency of DBP protects the lung against the injurious effects of intravascular actin. Moreover, purified DBP-actin complexes mediate endothelial cell injury and death in vitro.
Remarkably the DBP−/− mice were refractory to lung inflammation induced by intravenous injection of actin. It is highly unlikely that the differences in actin-induced inflammation observed between DBP+/+ and −/− mice are due to strain variations, other than a systemic lack of DBP, since the DBP−/− mice were backcrossed for 11 generations on a C57BL/6J background and both the DBP+/+ and −/− colonies were bred from the same founder hemizygotes (DBP+/−) (Trujillo, et al., 2013). Moreover, the DBP−/− mice do not have an increase in plasma gelsolin, the other EASS protein in blood, to compensate for the lack of DBP (Fig. 1C). Previous studies have shown that plasma gelsolin, much like DBP, becomes depleted during traumatic tissue injury (Dahl, et al., 1999). Nevertheless, plasma gelsolin in DBP−/− mice may help to limit actin filament formation by capping/binding F-actin and possibly weak binding of G-actin monomers (Lee and Galbraith, 1992), thus preventing obstruction of the lung microvasculature. But the EASS role of gelsolin in DBP−/− mice remains to be investigated. The results reported herein also confirm our recent study where we demonstrated that a systemic deficiency of DBP reduces inflammation and neutrophil recruitment to the lungs in acute and chronic models of alveolitis (Trujillo, et al., 2013). Thus, although our current and previous mouse studies would suggest that a lack of DBP is beneficial, a deficiency of DBP may be detrimental in certain infectious and inflammatory conditions. For example, DBP deficiency could enhance lethality in acute bacterial infections where inflammation and a critical tissue concentration of neutrophils are needed for bacterial clearance. However, these models need to be investigated using the DBP−/− mouse.
The mechanism by which DBP-actin complexes trigger lung inflammation in vivo and endothelial injury in vitro is not known. The possibility that complexes could trigger rapid inflammatory mechanisms, such as complement and/or neutrophil activation, was investigated and found to be negative (supplemental figures 1 and 2). Also, DBP-actin complexes did not increase cytokine levels in the BAL 4 hours after actin injection. However, in vivo, DBP-actin complexes induced pulmonary interstitial edema, hemorrhage (Fig. 2) and a permeability leak into the airspaces (Fig. 4A), indicating an effect on the lung microvasculature. In vitro, short-term exposure (4 hours) of endothelial cells to purified DBP-actin complexes induced reversible injury (Figs. 5 and 6) whereas longer exposure (24 hours) resulted in cell death (Fig. 7). These results are consistent with a mechanism that would require either a critical threshold concentration and/or exposure time to induce cell injury. Varying levels of DBP-actin complexes are continually formed in vivo as a result of minor tissue trauma, menstrual cycles, infections or surgery. Therefore, low levels of circulating DBP-actin would be inconsequential. On the other hand, high concentrations of DBP-actin in blood could act as a danger signal of ongoing and significant tissue injury. Indeed, a large percentage (78%) of the total plasma DBP pool was bound to actin 1.5 hours after injection in DBP+/+ mice (Fig. 1A). This percentage is similar to a previous study examining fulminant hepatic necrosis (FHN) in humans where 72% of plasma DBP was complexed with actin in patients who died of the disease whereas only 22% of total DBP was bound to actin in FHN survivors (Goldschmidt-Clermont, et al., 1988).
Although the mechanism by which DBP-actin triggers endothelial cell injury is not known, this study provides potential avenues for further investigation. First, results clearly demonstrate that a caspase-3 dependent apoptotic pathway is activated in HUVECs treated with DBP-actin (Fig. 8B). In contrast, HLMVECs showed no cleaved PARP in response to DBP-actin treatment (Fig. 8A), perhaps indicating that an alternative cell death pathway is triggered in HLMVECs. Furthermore, studies have shown that endothelial cells from different organs can respond differently to the same stimulus. For example, large vessel endothelial cells such as HUVECs are more sensitive to ionophore (A2318) induced calcium toxicity and apoptotic death than microvascular endothelial cells (Kelly, et al., 1998). This could explain the differences in PARP cleavage between HUVECs and HLMVECs. Yet both of these primary endothelial cells are sensitive to injury and death when treated with DBP-actin in vitro. Indeed, a major focus of future studies will be to determine the cell surface ligand for DBP-actin complexes on endothelial cells. Previous reports from our lab and others have shown that unbound DBP binds with low avidity to multiple cell surface macromolecules such as chondroitin sulfate proteoglycans (DiMartino and Kew, 1999), CD44 (McVoy and Kew, 2005), megalin (Nykjaer, et al., 1999), and cubulin (Nykjaer, et al., 2001). However, it is not known if actin alters the binding of DBP to the cell surface and/or its preference of a cellular ligand.
A major limitation of this study is the method of inducing injury in mice, i.e., a single intravenous bolus of purified actin. A more physiological approach would be to employ a model that has continuous injury over a period of hours to days, such as the cardiotoxin-induced muscle necrosis model (Deponti, et al., 2007). In addition, including DBP+/− mice in this study would have provided valuable information on how mice respond to extracellular actin with half-normal levels of circulating DBP. Our previous report showed that DBP+/− mice respond to a C5a-induced alveoltis essentially the same as wild-type DBP+/+ mice (Trujillo, et al., 2013), but how DBP+/− mice would respond to a bolus of intravenous actin is not known. Nevertheless, this report clearly demonstrates that DBP-actin is a mediator of inflammation, and possibly tissue injury, and this knowledge may change the current paradigm that focuses on the concentration of actin-free DBP in plasma as the important prognostic indicator in traumatic injury. The information from this study also may be consequential for the proposed efforts to infuse purified DBP intravenously into trauma patients to augment their actin-free DBP levels as a potential supportive therapy (Pihl, et al., 2010). Results from the mouse experiments reported herein would suggest that approach could be detrimental.
Supplementary Material
ACKNOWLEDGEMENTS
This investigation was supported in part by a grant from the U.S. National Institutes of Health GM063769 to RRK.
Abbreviations used in the paper
- AO/EB
acridine orange/ethidium bromide
- BAL
bronchoalveolar lavage
- CTB
cell-titer blue viability assay
- DBP
vitamin D binding protein
- DCFDA
2',7'–dichlorofluorescein diacetate
- EA
antibody-coated sheep erythrocytes
- EASS
extracellular actin scavenger system
- EGM-2
endothelial growth medium-2
- HLMVEC
human lung microvascular endothelial cell
- HUVEC
human umbilical vein endothelial cell
- PARP
poly ADP ribose polymerase
- ROS
reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Antoniades CG, Berry PA, Bruce M, Cross TJ, Portal AJ, Hussain MJ, Bernal W, Wendon JA, Vergani D. Actin-free Gc globulin: a rapidly assessed biomarker of organ dysfunction in acute liver failure and cirrhosis. Liver transplantation : official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2007;13:1254–1261. doi: 10.1002/lt.21196. [DOI] [PubMed] [Google Scholar]
- Chun RF. New perspectives on the vitamin D binding protein. Cell Biochem. Funct. 2012;30:445–456. doi: 10.1002/cbf.2835. [DOI] [PubMed] [Google Scholar]
- Dahl B, Schiodt FV, Kiaer T, Ott P, Bondesen S, Tygstrup N. Serum Gc-globulin in the early course of multiple trauma. Critical Care Medicine. 1998;26:285–289. doi: 10.1097/00003246-199802000-00027. [DOI] [PubMed] [Google Scholar]
- Dahl B, Schiodt FV, Ott P, Gvozdenovic R, Yin HL, Lee WM. Plasma gelsolin is reduced in trauma patients. Shock. 1999;12:102–104. doi: 10.1097/00024382-199908000-00002. [DOI] [PubMed] [Google Scholar]
- Dahl B, Schiodt FV, Ott P, Wians F, Lee WM, Balko J, O'Keefe GE. Plasma concentration of Gc-globulin is associated with organ dysfunction and sepsis after injury. Crit Care Med. 2003;31:152–156. doi: 10.1097/00003246-200301000-00024. [DOI] [PubMed] [Google Scholar]
- Deponti D, Francois S, Baesso S, Sciorati C, Innocenzi A, Broccoli V, Muscatelli F, Meneveri R, Clementi E, Cossu G, Brunelli S. Necdin mediates skeletal muscle regeneration by promoting myoblast survival and differentiation. J. Cell Biol. 2007;179:305–319. doi: 10.1083/jcb.200701027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMartino SJ, Kew RR. Initial characterization of the vitamin D binding protein (Gcglobulin) binding site on the neutrophil plasma membrane: evidence for a chondroitin sulfate proteoglycan. J. Immunol. 1999;163:2135–2142. [PubMed] [Google Scholar]
- Goldschmidt-Clermont PJ, Lee WM, Galbraith RM. Proportion of circulating Gc (vitamin D-binding protein) in complexed form: relation to clinical outcome in fulminant hepatic necrosis. Gastroenterology. 1988;94:1454–1458. doi: 10.1016/0016-5085(88)90686-5. [DOI] [PubMed] [Google Scholar]
- Haddad JG, Harper KD, Guoth M, Pietra GG, Sanger JW. Angiopathic consequences of saturating the plasma scavenger system for actin. Proceeding of the National Academy of Sciences. 1990;87:1381–1385. doi: 10.1073/pnas.87.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper KD, McLeod JF, Kowalski MA, Haddad JG. Vitamin D binding protein sequesters monomeric actin in the circulation of the rat. J. Clin. Invest. 1987;79:1365. doi: 10.1172/JCI112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauert AB, Martinelli S, Marone C, Niggli V. Differentiated HL-60 cells are a valid model system for the analysis of human neutrophil migration and chemotaxis. The international journal of biochemistry & cell biology. 2002;34:838–854. doi: 10.1016/s1357-2725(02)00010-9. [DOI] [PubMed] [Google Scholar]
- Janmey PA, Lind SE. Capacity of human serum to depolymerize actin filaments. Blood. 1987;70:524–530. [PubMed] [Google Scholar]
- Kelly JJ, Moore TM, Babal P, Diwan AH, Stevens T, Thompson WJ. Pulmonary microvascular and macrovascular endothelial cells: differential regulation of Ca2+ and permeability. The American journal of physiology. 1998;274:L810–819. doi: 10.1152/ajplung.1998.274.5.L810. [DOI] [PubMed] [Google Scholar]
- Lee WM, Galbraith RM. The extracellular actin-scavenger system and actin toxicity. New Engl. J. Med. 1992;326:1335–1341. doi: 10.1056/NEJM199205143262006. [DOI] [PubMed] [Google Scholar]
- Lind SE SD, Janmey PA, Stossel TP. Role of plasma gelosin and the vitamin D binding protein in clearing actin from the circulation. J. Clin. Invest. 1986;79:78. doi: 10.1172/JCI112634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVoy LA, Kew RR. CD44 and annexin A2 mediate the C5a chemotactic cofactor function of the vitamin D binding protein. J. Immunol. 2005;175:4754–4760. doi: 10.4049/jimmunol.175.7.4754. [DOI] [PubMed] [Google Scholar]
- Meier U, Gressner O, Lammert F, Gressner AM. Gc-globulin: roles in response to injury. Clin. Chem. 2006;52:1247–1253. doi: 10.1373/clinchem.2005.065680. [DOI] [PubMed] [Google Scholar]
- Nykjaer A, Dragun D, Walther D, Vorum H, Jacobsen C, Herz J, Melsen F, Christensen EI, Willnow TE. An endocytic pathway essential for renal uptake and activation of the steroid 25-(OH) vitamin D3. Cell. 1999;96:507–515. doi: 10.1016/s0092-8674(00)80655-8. [DOI] [PubMed] [Google Scholar]
- Nykjaer A, Fyfe JC, Kozyraki R, Leheste JR, Jacobsen C, Nielsen MS, Verroust PJ, Aminoff M, de la Chapelle A, Moestrup SK, Ray R, Gliemann J, Willnow TE, Christensen EI. Cubilin dysfunction causes abnormal metabolism of the steroid hormone 25(OH) vitamin D(3) Proc Natl Acad Sci U S A. 2001;98:13895–13900. doi: 10.1073/pnas.241516998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihl TH, Jorgensen CS, Santoni-Rugiu E, Leifsson PS, Hansen EW, Laursen I, Houen G. Safety pharmacology, toxicology and pharmacokinetic assessment of human Gc globulin (vitamin D binding protein) Basic Clin. Pharmacol. Toxicol. 2010;107:853–860. doi: 10.1111/j.1742-7843.2010.00587.x. [DOI] [PubMed] [Google Scholar]
- Rottner K, Stradal TE. Actin dynamics and turnover in cell motility. Curr. Opin. Cell Biol. 2011;23:569–578. doi: 10.1016/j.ceb.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Safadi FF, Thornton P, Magiera H, Hollis BW, Gentile M, Haddad JG, Liebhaber SA, Cooke NE. Osteopathy and resistance to vitamin D toxicity in mice null for vitamin D binding protein. J. Clin. Invest. 1999;103:239–251. doi: 10.1172/JCI5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiodt FV, Bangert K, Shakil AO, McCashland T, Murray N, Hay JE, Lee WM, Acute Liver Failure Study, G. Predictive value of actin-free Gc-globulin in acute liver failure. Liver transplantation : official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2007;13:1324–1329. doi: 10.1002/lt.21236. [DOI] [PubMed] [Google Scholar]
- Trujillo G, Habiel DM, Ge L, Ramadass M, Cooke NE, Kew RR. Neutrophil Recruitment to the Lung in Both C5a- and CXCL1-Induced Alveolitis Is Impaired in Vitamin D-Binding Protein-Deficient Mice. J. Immunol. 2013;191:848–856. doi: 10.4049/jimmunol.1202941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo G, Zhang J, Habiel DM, Ge L, Ramadass M, Ghebrehiwet B, Kew RR. Cofactor regulation of C5a chemotactic activity in physiological fluids. Requirement for the vitamin D binding protein, thrombospondin-1 and its receptors. Mol. Immunol. 2011;49:495–503. doi: 10.1016/j.molimm.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White P, Liebhaber SA, Cooke NE. 129X1/SvJ mouse strain has a novel defect in inflammatory cell recruitment. J. Immunol. 2002;168:869–874. doi: 10.4049/jimmunol.168.2.869. [DOI] [PubMed] [Google Scholar]
- Young WO, Goldschmidt-Clermont PJ, Emerson DL, Lee WM, Jollow DJ, Galbraith RM. Correlation between extent of liver damage in fulminant hepatic necrosis and complexing of circulating group-specific component (vitamin D-binding protein) The Journal of laboratory and clinical medicine. 1987;110:83–90. [PubMed] [Google Scholar]
- Zella LA, Shevde NK, Hollis BW, Cooke NE, Pike JW. Vitamin D-binding protein influences total circulating levels of 1,25-dihydroxyvitamin D3 but does not directly modulate the bioactive levels of the hormone in vivo. Endocrinology. 2008;149:3656–3667. doi: 10.1210/en.2008-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond S, Hirsch J. Leukocyte locomotion and chemotaxis. New methods for evaluation and demonstration of a cell-derived chemotactic factor. J. Exp. Med. 1973;137:387–410. doi: 10.1084/jem.137.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.