

Abstract
Accumulation of globotriaosylceramide (Gb3) and other neutral glycosphingolipids with galactosyl residues is the hallmark of Fabry disease, a lysosomal storage disorder caused by deficiency of the enzyme alpha-galactosidase A (α-gal A). These lipids are incorporated into the plasma membrane and intracellular membranes, with a preference for lipid rafts. Disruption of raft mediated cell processes is implicated in the pathogenesis of several human diseases, but little is known about the effects of the accumulation of glycosphingolipids on raft dynamics in the context of Fabry disease. Using siRNA technology, we have generated a polarized renal epithelial cell model of Fabry disease in Madin-Darby canine kidney cells. These cells present increased levels of Gb3 and enlarged lysosomes, and progressively accumulate zebra bodies. The polarized delivery of both raft-associated and raft-independent proteins was unaffected by α-gal A knockdown, suggesting that accumulation of Gb3 does not disrupt biosynthetic trafficking pathways. To assess the effect of α-gal A silencing on lipid raft dynamics, we employed number and brightness (N&B) analysis to measure the oligomeric status and mobility of the model glycosylphosphatidylinositol (GPI)-anchored protein GFP-GPI. We observed a significant increase in the oligomeric size of antibody-induced clusters of GFP-GPI at the plasma membrane of α-gal A silenced cells compared with control cells. Our results suggest that the interaction of GFP-GPI with lipid rafts may be altered in the presence of accumulated Gb3. The implications of our results with respect to the pathogenesis of Fabry disease are discussed.
Keywords: Fabry disease, lipid rafts, GPI-anchored proteins, number and brightness analysis, kidney, polarized sorting
Introduction
Fabry disease is a lysosomal storage disorder (LSD) caused by deficient activity of the lysosomal hydrolase α-galactosidase A (α-gal A) (EC 3.2.1.22), encoded by the gene GLA [1, 2]. The presence of dysfunctional α-gal A or its absence leads to progressive accumulation of neutral glycosphingolipids with α-D-galactosyl residues, mainly globotriaosylceramide (Gb3), in a variety of cell types, and results in multi-system disease [3].
The kidney is one of the most affected organs in Fabry disease and end-stage renal disease is a significant cause of morbidity in these patients [4]. While advanced stages of chronic renal disease usually develop in middle adulthood in males, the first histological signs of kidney involvement are seen as early as in utero [5, 6]. Additionally, pediatric patients may present early signs of nephropathy such as microalbuminuria, overt proteinuria, and hyperfiltration [7]. Histopathologic analyses of kidney biopsies of Fabry disease patients show Gb3 inclusions in most renal segments and cell types [8]. As Fabry nephropathy progresses, mesangial expansion, interstitial fibrosis, tubular atrophy, and glomerulosclerosis are often observed [9].
Recombinant Enzyme Replacement Therapy (ERT) for Fabry disease has been clinically available since 2001 and its administration improves overall clinical status and quality of life of Fabry patients [10–12]. However, ERT regimens only slightly retard the progression of chronic kidney disease, and a steady decline in glomerular filtration rate is still observed in Fabry disease patients receiving long-term ERT [13–15]. While our understanding of the molecular mechanisms and clinical progression of the disease has exponentially increased over the past several years, the pathogenic link between glycosphingolipid accumulation and renal cellular dysfunction that culminates in kidney failure remains unclear. A better appreciation of how these are connected may contribute to identification of novel drug targets for optimized therapy for the disease.
After its synthesis from lactosylceramide in the Golgi apparatus, Gb3 reaches the outer leaflet of the plasma membrane via vesicle-mediated transport [16, 17]. Like other glycosphingolipids, Gb3 resides preferentially in specialized membrane domains termed lipid rafts [18]. Lipid rafts constitute tightly packed dynamic assemblies of the plasma membrane and of biosynthetic and endocytic compartments that are enriched in sphingolipids and/or cholesterol [19]. Differential protein partitioning into these rafts can be conferred by lipid modifications such as glycosylphosphatidylinositol (GPI) lipid anchors, acylation, and palmitoylation, or by binding of N-glycans to raft-associated lectins [20, 21] Raft domains orchestrate the distribution and diffusion of a variety of proteins and lipids to enable or prevent lipid-lipid, protein-lipid, and protein-protein interactions [22].
Lipid rafts play important roles in post-Golgi membrane trafficking, intra- and inter-cellular signaling, and cell adhesion [23–25]. A primary function of these domains is to serve as scaffolds that enable the formation of higher order protein associations required for proper sorting and signal transduction. In polarized epithelial cells, a subset of newly synthesized proteins requires association with lipid rafts in the trans-Golgi network for efficient delivery to the apical plasma membrane [26]. Clustering of these raft-associated proteins into high molecular weight complexes is essential for their proper polarized delivery [27–29]. Similarly, oligomerization and higher order clustering of proteins is also important for the formation of signaling synapses in membrane microdomains in response to physiological stimuli [30].
Perturbations in lipid raft composition or dynamics contribute to the pathogenesis of several human diseases including atherosclerosis [31] and Alzheimer’s disease [32]. Furthermore, changes in raft composition have been described for some lysosomal storage disorders such as Niemann-Pick type C [33], Gaucher disease type I [34], Sandhoff disease [35], Sanfilippo disease [36], neuronal ceroid lypofuscinosis [37], and Krabbe disease [38]. Whether lipid raft structure is altered in Fabry disease is not known, however recent studies have suggested that trafficking of the glycosphingolipid lactosylceramide and of the apical glycoprotein dipeptidylpeptidase IV are perturbed in fibroblasts of Fabry disease patients compared to control fibroblasts [39, 40].
Changes in the lipid composition of lipid rafts may alter the stoichiometry of their protein components, with possible functional effects [41]. A recent advance in our ability to study spatial and temporal dynamics of proteins in living cells is the establishment of fluorescence fluctuation technique of Number and Brightness (N&B) analysis. In this method, quantitation of temporal fluctuations in the emission intensity of fluorescent molecules is used to calculate the oligomerization of fluorescently-tagged proteins expressed at low levels [42–45].
Here we have established and characterized a polarized cell model of Fabry disease in the canine kidney cell line MDCK (for Madin-Darby canine kidney). Cells silenced for α-gal A had increased levels of Gb3 and developed ultrastructural changes consistent with those observed in histopathological analyses of Fabry disease patient samples. Accumulation of Gb3 in intracellular compartments and at the cell surface did not alter the targeted delivery of either raft-associated or raft-independent proteins to the apical and basolateral membranes. To investigate lipid raft dynamics, we examined the effect of α-gal A depletion on the clustering of a model raft-associated GPI-anchored protein (GFP-GPI). N&B analysis in living cells revealed an increased oligomeric size of GFP-GPI at the plasma membrane of α-gal A silenced cells upon antibody-induced clustering compared with control cells. We speculate that accumulation of Gb3 in Fabry disease may lead to changes in protein-protein and protein-lipid interactions in lipid rafts.
Materials and Methods
Cell culture
MDCK cells were cultured in MEM with 10% heat-inactivated fetal bovine serum. Growth media was replaced every 48 h. Cells at 70–90% confluence were detached using TrypLE select (Life Technologies), transfected as described below, and seeded on 0.4 μm Transwell® permeable supports (Corning Life Sciences) for up to six days prior to experiments.
siRNA-mediated knockdown
An siRNA oligonucleotide targeting the following sequence 5′-GATAGATCTGCTGAAATT-3′ was designed for transient silencing of the canine α-gal A using Dharmacon siDESIGN center (http://www.thermoscientificbio.com/design-center/). SiRNA against firefly luciferase (Dharmacon) targeting the sequence 5′-GAATATTGTTGCACGATTT-3′ was used as a control. MDCK cells were transfected with either α-gal A or control siRNA using Lipofectamine™ 2000 transfection reagent (Life Technologies) and Opti-MEM® I Reduced Serum Media (Life Technologies). Briefly, 260 pmol of either α-gal A or control siRNA were incubated with 10 μL of lipofectamine and 500 μL of Opti-MEM for 30 min at ambient temperature. An aliquot (125 μL) of the transfection mix and 5 × 105 subconfluent MDCK cells suspended in 333 μL of MEM with serum were seeded in 12-well plates or on Transwell® permeable supports. Growth media was replaced within 8 h of transfection and every other day. Experiments were typically performed after 3 to 4 days. For longer knockdown periods, siRNA transfections were repeated every 3–4 days.
PCR analysis of knockdown efficiency
Isolation of total RNA was performed using the RNAqueous® kit (Ambion) according to the manufacturer’s recommendations. Purified total RNA underwent reverse transcription by incubation with Moloney murine leukemia virus reverse transcriptase (Ambion) at 42°C for 1 h. PCR of canine alpha-galactosidase was performed using the Phusion® High-Fidelity PCR kit (New England BioLabs, Inc.), with the following sense and antisense primers: 5′-TGTGCAACGTTGACTGCCAAGAAG-3′ and 5′-TCCTGCAGGTTTACCATAGCCACA-3′. As a control, canine β-actin was also amplified (sense and antisense primers 5′-CTGCTGGAAGGTGGACAG-3′; and 5′-ACCTTCAACTCCATCATGAAG-3′). Reactions were run under the following parameters: 30 sec at 98°C, 5 cycles at [98°C for 10 sec, 72°C for 30 sec, 65°C for 30 sec (reducing temperature by 1°C each cycle), 72°C for 30 sec], followed by 27 cycles at [98°C for 10 sec, 56°C for 30 sec, 72°C for 30 sec], and a final cycle of 72°C for 10 min. For real-time quantitative reverse transcription-PCR (qRT-PCR), 40 ng of RNA was loaded per experimental well. The following primers targeting canine mRNA products were used: α-gal A sense and antisense: 5′-ACCGGCTTAGAAAGGAAGACA-3′ and 5′-TTTACCATAGCCACAGCCCA-3′; β-actin sense and antisense: 5′-GATCAAGATCATCGCACCCC-3′ and 5′-ACAGTCCGCCTAGAAGCATT-3. Six-point standard curves were generated for each primer using progressive 1:2 dilutions of cDNA. Maxima SYBR (Thermo Scientific) was added to each experimental well containing sample and primers. qRT-PCR was performed on the 7300 Real Time System (Applied Biosystems). Reactions were run using the following parameters: 2 min at 50°C, 10 min at 95°C, and 40 cycles at 95°C for 15 sec followed by 60°C for 1 min. All samples were run in triplicate and normalized to β-actin controls.
Recombinant adenoviruses and adenoviral infection
Generation of replication-defective recombinant adenovirus encoding influenza hemagglutinin (HA) has been previously described [46]. Adenovirus encoding GFP-GPI was generated as described in [46]. Adenovirus stocks encoding dipeptidylpeptidase IV (DPPIV), and p75-GFP were generous gifts from A. Musch (Albert Einstein College of Medicine, New York, NY) and E. Rodriguez-Boulan (Weill Cornell Medical College, New York, NY), respectively. Sixteen to eighteen hours prior to experiments, MDCK cells grown on permeable supports were rinsed in Ca2+-free PBS containing 1 mM MgCl2 (PBS-Mg) and the indicated replication-defective adenoviruses (encoding HA, DPPIV, GFP-GPI, or p75-GFP) and the tetracycline transactivator (necessary for expression of some constructs) were added in PBS-Mg to the apical chamber at a multiplicity of infection of 25 to 75. PBS-Mg (500 μL) was added to the basolateral chamber and cells were incubated at 37°C with 5% CO2 for 1 h. Cells were then rinsed with PBS-Mg and incubated overnight in growth media.
Indirect Immunofluorescence and confocal microscopy
MDCK cells cultured on permeable supports were fixed for 15 min in 4% paraformaldehyde, 100 mM sodium cacodylate, pH 7.4 warmed to 37°C. Cells were then quenched in PBS containing 20 mM glycine and 75 mM NH4Cl for 5 min and permeabilized with 0.1% TritonX-100 in quench solution for 8 min with gentle shaking. After blocking in 1% BSA and 0.1% saponin in PBS for 30 min at ambient temperature, cells were incubated in primary antibody for 1 h. Rat IgM Anti-CD77 mAb (clone 38-13, Abcam), which recognizes Gb3, was used at a dilution of 1:20. Rabbit anti-giantin (from A Linstedt, Carnegie Mellon University, Pittsburgh, PA) was diluted at 1:400. Mouse mAb to LAMP-2 (AC17; from E. Rodriguez-Boulan) was diluted at 1:2,000. Mouse mAb against the early endosomal antigen EEA1 (Affinity Bioreagents) was used at 1:200. Rat mAb against HA (clone 3F10; Roche Applied Biosystems) was used at 1:500. Rat anti-ZO-I (hybridoma R40.76 supernatant) was used neat. Monoclonal anti-DPPIV antibody was a gift from Ann Hubbard (Johns Hopkins School of Medicine, Baltimore, MD) and was used at 1:500. Rabbit polyclonal Ab against caveolin-1 (Cell Signaling Technology) was used at 1:300. Mouse mAb against Na+/K+ ATPase (Abcam) was used at 1:400. Mouse mAb against E-cadherin (BD Biosciences) was used at 1:400. After incubation with primary antibodies of interest, cells were washed four times for 5 min each in PBS containing 0.5% BSA and 0.05% saponin and incubated in secondary antibody for 30 min. Alexa-Fluor (Invitrogen Molecular Probes) and Cy3 (Jackson ImmunoResearch Inc.) secondary antibodies were used at a dilution of 1:500 and 1:200, respectively. Primary and secondary antibodies were diluted in PBS containing 0.5% BSA and 0.05% saponin. Filters were washed four times for 5 min each and mounted using ProLong antifade gold with DAPI (Life Technologies). For surface staining, MDCK cells seeded on permeable supports were washed five times with HEPES/MES-buffered MEM, pH 7.0 (binding medium) for 3 min on ice. After blocking with HEPES-buffered MEM, 0.6% BSA, pH 7.4 for 15 min on ice, cells were incubated with rat IgM anti-CD77 mAb (diluted at 1:20) in binding medium on ice for 10 min. Cy3 goat anti-rat IgM secondary antibody was used at concentration of 1:200 diluted in binding medium on ice in anti-CD77 treated cells. Cells were washed five times with binding medium following incubation with both primary and secondary antibodies and then fixed and quenched as described above before, mounting using ProLong antifade gold with DAPI. Where stated, SYTOX Green Nucleic Acid Stain was used to visualize nuclei. Images were captured on an Olympus Fluoview FV1000 laser-scanning confocal microscope and processed using ImageJ (National Institutes of Health, Bethesda, MD), Metamorph imaging system (Universal Imaging Corp, Brandywine, PA) and Adobe Photoshop® CS6. Statistical background subtraction of the images was performed using Metamorph Imaging system (Universal Imaging Corp, Brandywine, PA).
Electron microscopy
Monolayers of MDCK cells seeded on permeable supports were fixed in solution of 2.5% glutaraldehyde and 4% paraformaldehyde in PBS for 30 min and post-fixed in 1% Osmium tetroxide overnight. Samples were dehydrated in graded series of ethanol solutions and embedded in EPON according to standard procedures. Ultra-thin sections were stained with uranyl acetate and lead citrate and examined using a JEOL electron microscope.
N&B measurements
Cells transfected with control or α-gal A siRNA for three to seven days were transfected with 0.2 μg of DNA plasmid encoding GFP fused to the GPI-anchor of 5′ nucleotidase (GFP-GPI; provided by G. Ihrke; Uniformed Services University of the Health Sciences, Bethesda, MD) using Lipofectamine 2000 for 16–20 h. The oligomeric status of both GFP-tagged proteins in the absence or presence of 50 μg/mL rabbit anti-GFP antibody (Invitrogen) was determined by number and brightness (N&B) analysis as previously described [47]. Briefly, a set of 100 confocal images for control or α-gal A silenced cells were acquired on a Leica TCS system equipped with a 63X/1.2 NA water objective with 488-nm laser line and 500–570 nm emission filter. Images were collected at 200 Hz using a hybrid detector set in the photon counting mode. Measurements were performed at 37°C and the average molecular brightness (B) and number of particles (N) per pixel were calculated using the SimFCS software ([42]; Laboratory of Fluorescence Dynamics, University of California, Irvine). N and B are defined as:
| (Eq. 1) |
| (Eq. 2) |
The variable k represents the signal intensity and σ is the variance of the signal. The molecular brightness of either control or α-gal A silenced cells was divided by the average brightness of monomeric GFP measured in the cytosol to obtain the normalized brightness.
To measure expression levels of heterologously expressed proteins in the cells analyzed by N&B analysis, the average intensity of fluorescence of a solution containing 30 nM of purified EGFP (Biovision) as well as the average intensity of fluorescence of cells expressing GFP-GPI were measured under the same experimental settings and quantitated using SimFCS. Approximate concentration of GFP-GPI in each cell was determined by dividing the measured value of intensity of fluorescence for GFP-GPI in that cell divided by the intensity of fluorescence for the EGFP solution multiplied by 30 nM. For calculation purposes, we considered autofluorescence to be negligible under the experimental conditions assessed.
Results
SiRNA mediated silencing of α-gal A efficiently reduces α-gal A mRNA in kidney epithelial cells
Despite their lack of α-gal A activity, knockout α-gal A mice do not fully reproduce the human clinical phenotype of Fabry disease [48]. As alternative approaches, cell models have been useful tools for understanding disease cellular pathogenesis, for identifying biomarkers of disease progression, and for developing and testing novel diagnostic and therapeutic avenues [49–52]. To generate a renal epithelial cell model of Fabry disease, we transfected MDCK cells with siRNA oligonucleotides targeting α-gal A or a control oligonucleotide. Because commercially available antibodies against human α-gal A failed to recognize the canine enzyme, we measured knockdown efficiency using RT-PCR. As shown in figure 1A, we observed a significant reduction in α-gal A mRNA in MDCK cells treated with α-gal A siRNA compared to control siRNA, without significant differences in β-actin mRNA levels. We could maintain this level of α-gal A knockdown for at least four days with a single transfection and up to six weeks by repeating the transfection every three to four days (data not shown). Efficient knockdown was further confirmed by quantitative RT-PCR, which showed an average of 88% reduction in α-gal A mRNA in α-gal A siRNA transfected cells (figure 1B).
Figure 1. Efficient siRNA-mediated silencing of α-gal A in MDCK cells.
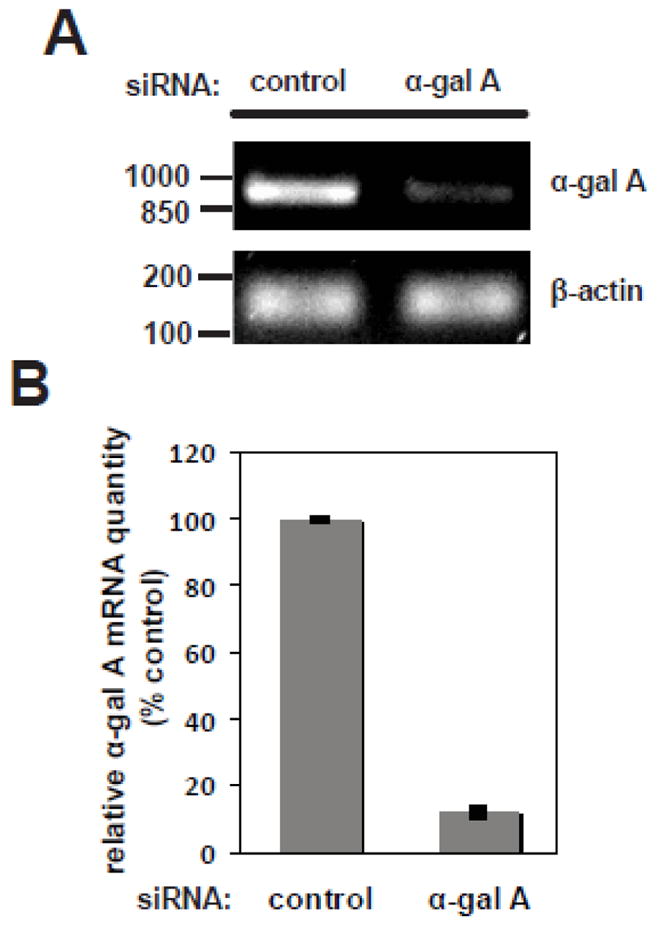
MDCK cells were transfected with control siRNA or siRNA targeted against α-gal A. After three days or seven days with repeated transfection, cells were solubilized and mRNA was extracted. (A) The efficiency of knockdown was quantified by RT-PCR, and a representative gel is shown. The migration of DNA ladder standards is shown on the left. The predicted PCR product for α-gal A is 898 bp. β-actin was used as a PCR control (expected product size 160 bp). (B) Quantitation of knockdown efficiency by qRT-PCR, performed as described in Methods. The mean +/− range of two independent experiments is plotted.
Accumulation of Gb3 in MDCK cells silenced for α-gal A
Gb3 is the main substrate of α-gal A and accumulates in a variety of cell types in Fabry disease patients [53]. To investigate the consequences of α-gal A silencing on cellular Gb3 levels, we compared the intensity and pattern of Gb3 staining in polarized MDCK cells three days after transfection with α-gal A or control siRNA. Cells were fixed and processed for indirect immunofluorescence using a monoclonal antibody against Gb3 (Anti-CD77). As shown in figure 2, we observed a marked increase in anti-CD77 staining in MDCK cells transfected for three days with α-gal A siRNA compared with control siRNA. The majority of the staining was present in intracellular compartments likely to correspond to lysosomes. Increased levels of Gb3 were also observed at the plasma membrane of non-permeabilized MDCK cells silenced for α-gal A (figure 2, bottom). Cell surface Gb3 is also known to be elevated in Fabry disease and measurement of this pool has been recently proposed as a means to monitor the response to ERT [50].
Figure 2. α-gal A siRNA silencing increases cellular levels of Gb3.

(Top) Filter-grown MDCK cells transfected with control (left panel) and α-gal A siRNA (right panel) for seven days were processed for indirect immunofluorescence using anti-CD77 antibody to detect Gb3 Gb3 and imaged by epifluorescence microscopy. SYTOX Green Nucleic Acid Stain was used to visualize nuclei. Scale bar: 10 μm (Bottom) Surface staining of CD77/Gb3 in non-permeabilized MDCK control cells (left) and α-gal A siRNA silenced cells (right). Maximum projections of five optical sections taken at the apical surface are shown. Scale bar: 5 μm.
Ultrastructural changes upon α-gal A silencing in MDCK cells
Fabry disease patient fibroblasts have enlarged lysosomes that decrease in size upon disease-targeted therapy [54]. We therefore examined whether α-gal A silencing altered lysosome size in MDCK cells. We performed confocal microscopy of MDCK cells transfected with control or α-gal A siRNA and immunostained for the lysosome marker LAMP-2. By visual inspection there appeared to be an increase in the size of LAMP-2 positive compartments in α-gal A silenced cells compared with cells treated with control siRNA (figure 3A). To quantify this difference, the average area of individual LAMP-2 positive compartments in confocal image stacks was determined using ImageJ software. While there was variability in individual lysosome sizes, on average, the lysosomal area in α-gal A silenced cells was 233 % greater per field than that measured in control cells (figure 3B). In contrast, the morphology of early endosomes and the Golgi complex were unaffected (supplementary figure 1).
Figure 3. α-gal A siRNA silencing causes accumulation of zebra bodies and increased lysosome size.

(A) Indirect immunofluorescence of the late endosome/lysosome marker LAMP-2 in control (left panel) and α-gal A silenced (right panel) MDCK cells. SYTOX Green Nucleic Acid Stain was used to visualize nuclei. Scale bar: 10 μm (B) The average area of individual LAMP-2 positive compartments in control and α-gal A silenced cells was quantified using ImageJ. The graph shows data from 20 fields in three independent experiments. *t-test p<0.001. (C) Transmission electron micrographs of MDCK cells treated with α-gal A siRNA for six days (left and middle panels) or six weeks (right panel). Transversely-stacked, osmiophilic myelin-like membranes also known as “zebra bodies” (arrow heads) are evident within six days of transfection and are more prevalent after six weeks of repeated transfections. Scale bar: left: 2 μm, middle and right: 500 nm.
Light and electron microscopy of biopsied tissues of patients with Fabry disease reveals cell-type dependent ultra-structural changes that are characteristic of lipid deposition seen in LSDs [9]. In cell types with a greater degree of lipid deposition, the accumulation of intracellular inclusions formed of concentric or parallel arrays of lipid aggregates, also called “zebra bodies”, is evident by electron microscopy [55]. To investigate renal epithelial cell ultrastructure upon α-gal A silencing, we fixed MDCK cells six days after siRNA transfection and processed the samples for transmission electron microscopy (figure 3C). Osmiophilic myelin-like structures similar to the zebra bodies observed in affected tissues of patients with Fabry disease were observed in α-gal A silenced cells and absent in cells transfected with control siRNA. These structures increased in number after six weeks of repeated α-gal A siRNA transfection (figure 3C). These results confirm that siRNA-mediated silencing of α-gal A MDCK cells reproduces the Gb3 deposition pattern and ultrastructural changes observed in Fabry disease patient cells. Therefore, this model can be used to study cellular processes that when perturbed could potentially contribute to the disease mechanism in α-gal A deficiency.
Polarized sorting of raft-associated and raft-independent proteins is unaffected by α-gal A silencing
Association with lipid rafts is essential for efficient biosynthetic delivery of a subset of newly synthesized apically-destined proteins [56]. To test whether accumulation of Gb3 alters apical delivery pathways, we examined the steady state distribution of apical cargoes in α-gal A silenced polarized MDCK cells grown on permeable supports. As shown in figure 4A, staining of the tight junction protein ZO-1 was unaffected by α-gal A knockdown. Moreover, we found no difference in the steady state distribution of the raft-associated protein HA (figure 4B) or the raft-independent protein p75-GFP (figure 4C) in α-gal A silenced cells. A previous study reported changes in the subcellular localization of DPPIV in fibroblasts cultured from Fabry patients [39]. However, we found no effect of α-gal A silencing on DPPIV distribution (figure 4D). Additionally, we observed no differences in the steady state distribution of the basolaterally targeted proteins E-cadherin and Na+/K+ ATPase, as well as the raft-associated protein caveolin-1 in MDCK cells silenced for α-gal A and in control cells (supplementary figure 2). Thus, Gb3 accumulation in α-gal A-depleted cells is apparently insufficient to alter apical sorting in our model system.
Figure 4. Steady-state distribution of raft-associated and raft-independent apical cargoes is not affected by α-gal A siRNA silencing. (A).
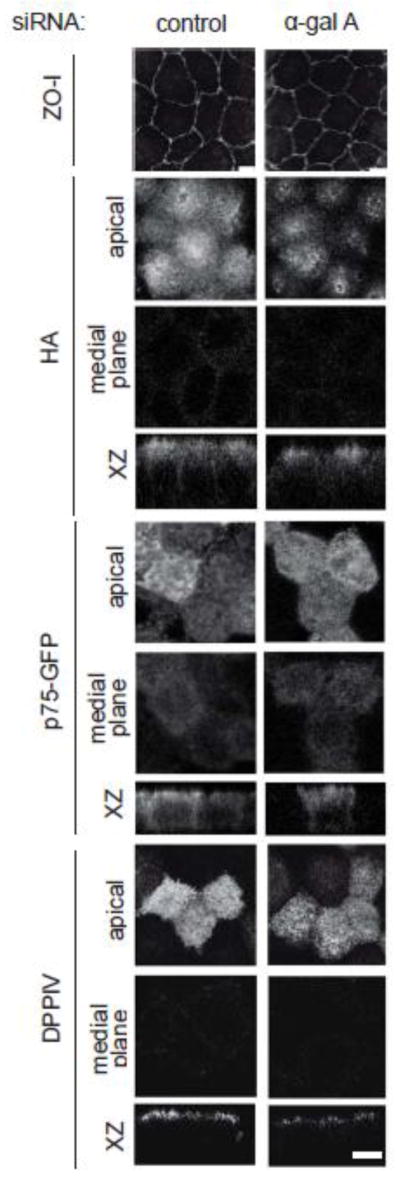
Indirect immunofluorescence staining of ZO-I (tight junction marker) in control and α-gal A silenced MDCK cells confirms that tight junctions are intact. Control and α-gal A siRNA treated polarized MDCK cells were infected with replication-defective adenoviruses encoding (B) the raft-associated protein HA; (C) the raft-independent neurotrophin receptor p75 (tagged with GFP); or (D) the glycoprotein dipeptidylpeptidase IV (DPPIV). Cells were fixed, processed for immunofluorescence, and imaged by confocal microscopy. Images taken at the level of the apical surface and at a medial plane are shown for each protein, and xz reconstructions are below. Scale bar: 5 μm.
Dynamics of a model raft-associated protein upon α-gal A silencing in MDCK cells
Recent studies have demonstrated that the steady-state cellular distribution of lactosylceramide differs in fibroblasts of Fabry disease patients when compared to control fibroblasts, suggesting perturbed lipid raft trafficking in Fabry disease [40]. However, the effects of Gb3 accumulation on lipid raft dynamics and any consequences to the pathogenesis of Fabry disease remain unknown. We postulated that α-gal A silencing and subsequent accumulation of neutral glycosphingolipids at the plasma membrane would impact how proteins interact with lipid rafts and potentially induce changes in oligomerization of raft-associated proteins. As a proof of principle we chose to study the oligomeric status of GFP-GPI in our MDCK model of Fabry disease. GPI-anchored proteins are well-characterized raft-associated proteins and serve as models to study cell processes that occur in lipid rafts [57]. GFP-GPI is efficiently recruited to lipid rafts, and alteration of raft composition is known to change the oligomeric state of GFP-GPI, thus making it a useful probe for monitoring lipid raft microdomains [58].
Using N&B analysis, we measured the self-assembly of GFP-GPI at the plasma membrane of MDCK cells transfected with α-gal A and control siRNA. In N&B analysis, a series of confocal images of cells expressing the fluorescent protein of interest are acquired and the average intensity and the variance of the fluorescence signal per pixel are measured. The molecular brightness is then calculated as the ratio of the signal variance to intensity (see schematic in figure 5A). For example, considering samples with the same average in intensity of fluorescence, tetramers would have a four-fold greater brightness compared to monomers since the signal variance of the many dim monomers is much smaller compared to the variance of several bright oligomers as they move in and out of the confocal volume. In both control and α-gal A siRNA treated cells, the majority of the GFP-GPI fluorescence was uniformly distributed at the cell surface, with some intracellular staining as has been previously reported [59] (figure 5B). The expression level of GFP-GPI in our experiments ranged from 0.2 to 1.4 μM, which is similar to the endogenous expression range observed for membrane tyrosine kinase receptors and associated downstream signaling molecules [60]. The brightness measured under these conditions corresponded to the total values across the area of interest because no identifiable puncta could be gated for analysis (see supplementary figure 3). The average molecular brightness for GFP-GPI at the plasma membrane was 2.21 and 2.24 for control and α-gal A silenced cells, respectively (figure 5C, dashed line). These data are in good agreement with previous studies showing that under non-stimulated conditions GPI-anchored proteins are highly mobile and form transient homodimers at the plasma membrane [61]. Upon activation or ligand binding, GPI-anchored proteins form stable homodimers and higher order clusters composed of GPI-anchored proteins and other raft-associated proteins [62, 63]. Experimentally, clustering of GPI-anchored proteins can be induced by addition of antibodies, and leads to activation of downstream signaling pathways [64]. Addition of anti-GFP antibody for ten minutes caused rapid and visible coalescence of GFP-GPI into large patches at the cell surface (figure 5B). Quantitation of the average brightness in these patches revealed an increase in clustering of GFP-GPI that was significantly greater in α-gal A silenced cells (5.08 ± 0.45; median ± SEM) compared to control cells (2.74 ± 0.24; median ± SEM; p<0.005; figure 5C). Inclusion of brightness values across the region of interest (i.e., not limited to the patches) in these samples did not change the observation that α-gal A depletion leads to larger cluster size but would lead to an underestimation of the brightness in the patches (supplementary figure 3).
Figure 5. α-gal A silencing alters antibody induced-clustering of a lipid raft associated protein.

Schematic of the theory of number and brightness (N&B) analysis. Left: depiction of cell surface expression of GFP-GPI protein in the absence (−Ab), or presence (+Ab) of anti-GFP antibody (red). The magenta circle in the region of interest (roi) represents the focal volume of the microscope. The magenta hourglass is an expanded view of the focal volume with a representation of GFP-GPI clusters (membrane not shown for clarity) diffusing in the −Ab or +Ab conditions. Center: In this example, the total concentration in the roi before and after Ab addition is similar, giving rise to the same average fluorescence intensity (k). When antibody is added to induce clustering, the same average fluorescence intensity is now redistributed into a few oligomeric species within the focal volume, which causes a dramatic increase in the signal variance (σ) as the large oligomers diffuse through the focal volume. The ratio of variance to intensity is termed molecular brightness (brightness) and provides information on the oligomeric status of a protein when compared to the monomeric fluorescent probe. Right: Depiction of brightness map for the roi in the −Ab and +Ab conditions with cool and warm colors representing less and more clustering, respectively. (B) Control or α-gal A silenced (KD) cells expressing GFP-GPI in the absence or presence of anti-GFP antibody. Left panels: Fluorescence images showing control and or α-gal A silenced cells before (−Ab), or 10 min after addition of anti-GFP antibody (+Ab); scale bar: 20 μm. Middle panels: higher magnification of the boxed region where N&B measurements were acquired; scale bar: 2 μm. Right panels: map of molecular brightness for the inset regions shown in the fluorescence images; heat map scale bar = normalized molecular brightness per pixel (e.g. 2 = 2 GFP molecules per cluster). (C) Scatter plot of normalized molecular brightness values for control and or α-gal A silenced cells (n=13 and n=15, respectively, from two experiments). The dotted line represents the average normalized molecular brightness (~2.2) for control and α-gal A silenced cells in the absence of antibody. *p < 0.005 by unpaired t-test. (D) Control or α-gal A silenced (KD) cells expressing GFP-endolyn in the absence or presence of anti-GFP antibody. Image layouts same as in panel B. (E) Scatter plot of normalized molecular brightness values for GFP-endolyn in control or α-gal A silenced cells (n=16 and n=14, respectively, from two experiments). The fine dotted line represents the average normalized molecular brightness for control and the coarse dotted line for α-gal A silenced cells in the absence of antibody (1.65 and 1.27, respectively).
In order to determine if the change in oligomeric stability seen for GFP-GPI extended to non-raft membrane proteins, we next measured the molecular brightness of a lumenally GFP-tagged version of endolyn (GFP-endolyn), an apically destined sialomucin that traffics in a raft independent matter. [65] The average molecular brightnesses for GFP-endolyn at the plasma membrane were 1.65 and 1.27 in control and α-gal A silenced cells, respectively (figure 5E, dashed lines). Similar to GFP-GPI, addition of anti-GFP antibody caused the rapid accumulation of clusters of protein at the cell surface (figure 5D). In control cells the GFP-endolyn clusters ranged in size from ~2–4 protein molecules per cluster with a median cluster size of 2.88. While GFP-endolyn in α-gal A silenced cells exhibited a much broader range of cluster sizes (~2–8 molecules, median 2.96; figure 5E), cluster size in control and knockdown cells was not statistically significantly different. Together, our results suggest that there is greater stabilization of GFP-GPI oligomers in rafts of α-gal A silenced cells upon antibody-induced clustering and that this effect does not extend globally to all plasma membrane proteins.
Discussion
The cellular phenotype of Fabry disease is not well understood. In particular, how the accumulation of the raft-associated lipid Gb3 might alter the dynamics and efficiency of lipid raft dependent processes has never been examined. As lipid raft formation and composition have been widely studied in MDCK cells for several years [25], we decided to generate a cell model to study raft-dependent processes using this well-characterized kidney tubular epithelial cell line. We effectively knocked down α-gal A mRNA levels by nearly 90% using an siRNA approach. MDCK cells silenced for α-gal A had elevated levels of Gb3, enlarged lysosomes, and progressively accumulated zebra bodies, consistent with the cellular phenotype of Fabry disease in humans.
In nonpolarized cells, lipid rafts are estimated to cover 30–40% of the plasma membrane, and are even more prevalent in the apical membrane of polarized cells [66]. Oligomerization of raft-associated proteins either by ligand binding or antibody crosslinking is known to increase their affinity for lipid rafts and their stabilization in these microdomains [67]. Additionally, oligomerization of raft components causes lipid rafts to coalesce and form larger domains [27]. Our results indicate that the alterations in lipid raft composition observed in Fabry disease induce changes in lipid raft dynamics that may occur upon activation/oligomerization of raft-associated proteins. We speculate that this could impact raft-mediated signal transduction in Fabry disease.
Using N&B analysis, we spatially mapped the dynamics of a GPI-anchored protein in our cell model of Fabry disease. This imaging approach allows us to measure the size of oligomeric complexes in live intact cells at physiologically relevant expression levels. Strikingly, we found that the average oligomeric size for the ligated GPI-anchored protein was ~two-fold higher in α-gal A silenced cells compared to cells transfected with control siRNA. We observed ~2.5–7 molecules of GFP-GPI per oligomer upon antibody addition. This range is consistent with single molecule studies of the GPI-anchored receptor CD59 that identified clustering of 3–9 molecules of CD59 per oligomer upon gold-induced crosslinking and activation of the receptor [59]. In contrast, the clustering of a lipid raft independent protein was not significantly affected in α-gal A silenced cells suggesting that the observed change in oligomeric stability was specific to raft proteins. Measurements in live cells are critical given recent studies demonstrating that remodeling of G protein coupled receptors and GPI-anchored protein complexes can occur on the millisecond to second timescale [68–70].
What are the implications of our results with respect to Fabry disease? High-resolution studies of CD59 have identified transient stalling, or immobilization, as a key requirement for the recruitment of downstream signaling effectors, such as Lyn kinase and PLCγ2, to activate intracellular calcium signaling [59, 71]. The increased oligomer size we observed of a model GPI-anchored protein upon α-gal A depletion might represent “stalled” oligomers due to alteration in lipid-lipid interactions or protein-lipid interactions. Thus, we predict that signaling from raft-associated complexes may be altered in Fabry disease. In this regard, one signaling pathway that will require future exploration in the context of α-gal A silencing is that mediated by TGF-β. TGF-β signals through three different receptor isoforms that traffic via both raft-dependent and raft-independent pathways. [72] The binding ratio of monomeric TGF-β to heterotetrameric complexes of these receptors differs upon membrane compartmentalization, resulting in activation of different downstream effectors [73]. Human podocytes loaded with lyso-Gb3, the deacylated form of Gb3, showed increased expression of TGF-β1, which mediates an increase in the expression of the extracellular matrix components fibronectin and collagen type IV [74]. This dysregulation of TGF-β signaling was proposed as a contributor to development of glomerular injury in Fabry disease. The cell model that we have developed combined with N&B analysis will be a powerful tool to dissect the alterations that occur in lipid raft and cellular signaling in Fabry disease.
Supplementary Material
Highlights.
We have established a kidney cell model for Fabry disease that replicates key features of the cellular pathology, including elevated levels of the raft associated lipid Gb3
Apical sorting of raft-associated proteins is not disrupted in our polarized cell model
Antibody induced clustering of a model raft associated protein is enhanced in our alpha-galactosidase A-silenced cells
Our results have potential implications for raft-dependent signal transduction in Fabry disease.
Acknowledgments
This work was supported by National Institutes of Health grant R01-DK54407 (to OAW), an American Heart Association Scientist Development Grant (AHA 12SDG8960000; to RTY) and a Brazilian National Council for Scientific and Technological Development (CNPq) predoctoral grant (CsF 238084/2012-4 to AL). AL and SJHM were supported in part by Shire HGT. We are grateful to the Kidney Imaging and Physiology Cores of the P30 Pittsburgh Center for Kidney Research (National Institutes of Health DK079307) for technical support. We also thank Drs. Adam Linstedt, Anne Müsch, Enrique Rodriguez-Boulan, Ann Hubbard, and Gudrun Ihrke for their generous gifts of reagents. We acknowledge Youssef Rbaibi, Di Mo, Venkatesan Raghavan, and Christina M Szalinski for their advice and technical assistance.
Abbreviations
- α-gal A
α-galactosidase A
- DPPIV
dipeptidylpeptidase IV
- EEA1
early endosomal antigen 1
- ERT
Enzyme replacement therapy
- Gb3
globotriaosylceramide
- GPI
glycosylphosphatidylinositol
- HA
hemagglutinin
- LAMP-2
lysosomal membrane associated protein 2
- LSD
lysosomal storage disorder
- MDCK
Madin-Darby canine kidney
- N&B
number and brightness
- RT-PCR
reverse transcription polymerase chain reaction
- qRT-PCR
real-time quantitative reverse-transcription polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yoshimitsu M, et al. Identification of novel mutations in the alpha-galactosidase A gene in patients with Fabry disease: pitfalls of mutation analyses in patients with low alpha-galactosidase A activity. J Cardiol. 2011;57(3):345–53. doi: 10.1016/j.jjcc.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. doi: 10.1186/1750-1172-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Desnick RJ, Ioannou YA, Eng CM. alpha-galactosidase A deficiency: Fabry disease. In: Scriver CR, et al., editors. The Metabolic and Molecular Bases of Inherited Disease. 8. McGraw-Hill; New York: 2001. pp. 3733–3774. [Google Scholar]
- 4.Eng CM, et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis. 2007;30(2):184–92. doi: 10.1007/s10545-007-0521-2. [DOI] [PubMed] [Google Scholar]
- 5.Ortiz A, et al. End-stage renal disease in patients with Fabry disease: natural history data from the Fabry Registry. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2010;25(3):769–75. doi: 10.1093/ndt/gfp554. [DOI] [PubMed] [Google Scholar]
- 6.Pintos-Morell G, Beck M. Fabry disease in children and the effects of enzyme replacement treatment. Eur J Pediatr. 2009;168(11):1355–63. doi: 10.1007/s00431-009-0937-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hopkin RJ, et al. Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. Pediatr Res. 2008;64(5):550–5. doi: 10.1203/PDR.0b013e318183f132. [DOI] [PubMed] [Google Scholar]
- 8.Tondel C, et al. Agalsidase benefits renal histology in young patients with Fabry disease. Journal of the American Society of Nephrology: JASN. 2013;24(1):137–48. doi: 10.1681/ASN.2012030316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fogo AB, et al. Scoring system for renal pathology in Fabry disease: report of the International Study Group of Fabry Nephropathy (ISGFN) Nephrol Dial Transplant. 2010;25(7):2168–77. doi: 10.1093/ndt/gfp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watt T, et al. Agalsidase beta treatment is associated with improved quality of life in patients with Fabry disease: findings from the Fabry Registry. Genet Med. 2010;12(11):703–12. doi: 10.1097/GIM.0b013e3181f13a4a. [DOI] [PubMed] [Google Scholar]
- 11.Wilcox WR, et al. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet. 2004;75(1):65–74. doi: 10.1086/422366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughes DA, et al. A randomised, double-blind, placebo-controlled, crossover study to assess the efficacy and safety of three dosing schedules of agalsidase alfa enzyme replacement therapy for Fabry disease. Molecular genetics and metabolism. 2013 doi: 10.1016/j.ymgme.2013.04.015. [DOI] [PubMed] [Google Scholar]
- 13.Weidemann F, et al. Long-term outcome of enzyme-replacement therapy in advanced Fabry disease: evidence for disease progression towards serious complications. Journal of internal medicine. 2013 doi: 10.1111/joim.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feriozzi S, et al. The Effectiveness of Long-Term Agalsidase Alfa Therapy in the Treatment of Fabry Nephropathy. Clin J Am Soc Nephrol. 2012;7(1):60–69. doi: 10.2215/CJN.03130411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warnock DG, et al. Renal outcomes of agalsidase beta treatment for Fabry disease: role of proteinuria and timing of treatment initiation. Nephrol Dial Transplant. 2011 doi: 10.1093/ndt/gfr420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lannert H, et al. Functional organization of the Golgi apparatus in glycosphingolipid biosynthesis. Lactosylceramide and subsequent glycosphingolipids are formed in the lumen of the late Golgi. The Journal of biological chemistry. 1998;273(5):2939–46. doi: 10.1074/jbc.273.5.2939. [DOI] [PubMed] [Google Scholar]
- 17.Keusch JJ, et al. Cloning of Gb3 synthase, the key enzyme in globo-series glycosphingolipid synthesis, predicts a family of alpha 1, 4-glycosyltransferases conserved in plants, insects, and mammals. J Biol Chem. 2000;275(33):25315–21. doi: 10.1074/jbc.M002630200. [DOI] [PubMed] [Google Scholar]
- 18.Nutikka A, Lingwood C. Generation of receptor-active, globotriaosyl ceramide/cholesterol lipid ‘rafts’ in vitro: A new assay to define factors affecting glycosphingolipid receptor activity. Glycoconjugate journal. 2004;20(1):33–8. doi: 10.1023/B:GLYC.0000016740.69726.fb. [DOI] [PubMed] [Google Scholar]
- 19.Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327(5961):46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 20.Melkonian KA, et al. Role of lipid modifications in targeting proteins to detergent-resistant membrane rafts. Many raft proteins are acylated, while few are prenylated. The Journal of biological chemistry. 1999;274(6):3910–7. doi: 10.1074/jbc.274.6.3910. [DOI] [PubMed] [Google Scholar]
- 21.Delacour D, et al. Galectin-4 and sulfatides in apical membrane trafficking in enterocyte-like cells. The Journal of cell biology. 2005;169(3):491–501. doi: 10.1083/jcb.200407073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Owen DM, et al. The lipid raft hypothesis revisited--new insights on raft composition and function from super-resolution fluorescence microscopy. BioEssays: news and reviews in molecular, cellular and developmental biology. 2012;34(9):739–47. doi: 10.1002/bies.201200044. [DOI] [PubMed] [Google Scholar]
- 23.Wang R, et al. Lipid raft regulates the initial spreading of melanoma A375 cells by modulating beta1 integrin clustering. The international journal of biochemistry & cell biology. 2013 doi: 10.1016/j.biocel.2013.04.031. [DOI] [PubMed] [Google Scholar]
- 24.Simons K, Toomre D. Lipid rafts and signal transduction. Nature reviews Molecular cell biology. 2000;1(1):31–9. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 25.Fullekrug J, Simons K. Lipid rafts and apical membrane traffic. Annals of the New York Academy of Sciences. 2004;1014:164–9. doi: 10.1196/annals.1294.017. [DOI] [PubMed] [Google Scholar]
- 26.Weisz OA, Rodriguez-Boulan E. Apical trafficking in epithelial cells: signals, clusters and motors. J Cell Sci. 2009;122(Pt 23):4253–66. doi: 10.1242/jcs.032615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuck S, Simons K. Polarized sorting in epithelial cells: raft clustering and the biogenesis of the apical membrane. Journal of cell science. 2004;117(Pt 25):5955–64. doi: 10.1242/jcs.01596. [DOI] [PubMed] [Google Scholar]
- 28.Paladino S, et al. Protein oligomerization modulates raft partitioning and apical sorting of GPI-anchored proteins. The Journal of cell biology. 2004;167(4):699–709. doi: 10.1083/jcb.200407094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galmes R, et al. Oligomerization is required for normal endocytosis/transcytosis of a GPI-anchored protein in polarized hepatic cells. Journal of cell science. 2013 doi: 10.1242/jcs.126250. [DOI] [PubMed] [Google Scholar]
- 30.Shi D, et al. Smoothened oligomerization/higher order clustering in lipid rafts is essential for high hedgehog activity transduction. The Journal of biological chemistry. 2013;288(18):12605–14. doi: 10.1074/jbc.M112.399477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lemaire-Ewing S, Lagrost L, Neel D. Lipid rafts: a signalling platform linking lipoprotein metabolism to atherogenesis. Atherosclerosis. 2012;221(2):303–10. doi: 10.1016/j.atherosclerosis.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 32.Ehehalt R, et al. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. The Journal of cell biology. 2003;160(1):113–23. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kosicek M, et al. Cholesterol accumulation in Niemann Pick type C (NPC) model cells causes a shift in APP localization to lipid rafts. Biochemical and biophysical research communications. 2010;393(3):404–9. doi: 10.1016/j.bbrc.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langeveld M, et al. Type I Gaucher disease, a glycosphingolipid storage disorder, is associated with insulin resistance. The Journal of clinical endocrinology and metabolism. 2008;93(3):845–51. doi: 10.1210/jc.2007-1702. [DOI] [PubMed] [Google Scholar]
- 35.te Vruchte D, et al. Glycosphingolipid storage leads to the enhanced degradation of the B cell receptor in Sandhoff disease mice. J Inherit Metab Dis. 2010;33(3):261–70. doi: 10.1007/s10545-010-9109-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dawson G, et al. Abnormal gangliosides are localized in lipid rafts in Sanfilippo (MPS3a) mouse brain. Neurochemical research. 2012;37(6):1372–80. doi: 10.1007/s11064-012-0761-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rusyn E, et al. CLN3p impacts galactosylceramide transport, raft morphology, and lipid content. Pediatric research. 2008;63(6):625–31. doi: 10.1203/PDR.0b013e31816fdc17. [DOI] [PubMed] [Google Scholar]
- 38.White AB, et al. Persistence of psychosine in brain lipid rafts is a limiting factor in the therapeutic recovery of a mouse model for Krabbe disease. J Neurosci Res. 2011;89(3):352–64. doi: 10.1002/jnr.22564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maalouf K, et al. A modified lipid composition in Fabry disease leads to an intracellular block of the detergent-resistant membrane-associated dipeptidyl peptidase IV. J Inherit Metab Dis. 2010;33(4):445–9. doi: 10.1007/s10545-010-9114-6. [DOI] [PubMed] [Google Scholar]
- 40.Chen CS, et al. Broad screening test for sphingolipid-storage diseases. Lancet. 1999;354(9182):901–5. doi: 10.1016/S0140-6736(98)10034-X. [DOI] [PubMed] [Google Scholar]
- 41.Simons K, Vaz WL. Model systems, lipid rafts, and cell membranes. Annual review of biophysics and biomolecular structure. 2004;33:269–95. doi: 10.1146/annurev.biophys.32.110601.141803. [DOI] [PubMed] [Google Scholar]
- 42.Digman MA, et al. Mapping the number of molecules and brightness in the laser scanning microscope. Biophysical journal. 2008;94(6):2320–32. doi: 10.1529/biophysj.107.114645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crosby KC, et al. Quantitative analysis of self-association and mobility of annexin a4 at the plasma membrane. Biophysical journal. 2013;104(9):1875–85. doi: 10.1016/j.bpj.2013.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hellriegel C, et al. Number and brightness image analysis reveals ATF-induced dimerization kinetics of uPAR in the cell membrane. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2011;25(9):2883–97. doi: 10.1096/fj.11-181537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herrick-Davis K, et al. Oligomer size of the serotonin 5-hydroxytryptamine 2C (5-HT2C) receptor revealed by fluorescence correlation spectroscopy with photon counting histogram analysis: evidence for homodimers without monomers or tetramers. The Journal of biological chemistry. 2012;287(28):23604–14. doi: 10.1074/jbc.M112.350249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Henkel JR, et al. Selective perturbation of apical membrane traffic by expression of influenza M2, an acid-activated ion channel, in polarized madin-darby canine kidney cells. Mol Biol Cell. 1998;9(9):2477–90. doi: 10.1091/mbc.9.9.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Youker RT, et al. Multiple motifs regulate apical sorting of p75 via a mechanism that involves dimerization and higher-order oligomerization. Molecular biology of the cell. 2013;24(12):1996–2007. doi: 10.1091/mbc.E13-02-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ohshima T, et al. alpha-Galactosidase A deficient mice: a model of Fabry disease. Proc Natl Acad Sci U S A. 1997;94(6):2540–4. doi: 10.1073/pnas.94.6.2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liebau MC, et al. Dysregulated Autophagy Contributes to Podocyte Damage in Fabry’s Disease. PloS one. 2013;8(5):e63506. doi: 10.1371/journal.pone.0063506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thomaidis T, et al. Downregulation of alpha-galactosidase A upregulates CD77: functional impact for Fabry nephropathy. Kidney Int. 2009;75(4):399–407. doi: 10.1038/ki.2008.576. [DOI] [PubMed] [Google Scholar]
- 51.Namdar M, et al. Globotriaosylsphingosine accumulation and not alpha-galactosidase-A deficiency causes endothelial dysfunction in Fabry disease. PloS one. 2012;7(4):e36373. doi: 10.1371/journal.pone.0036373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Medin JA, et al. Correction in trans for Fabry disease: expression, secretion and uptake of alpha-galactosidase A in patient-derived cells driven by a high-titer recombinant retroviral vector. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(15):7917–22. doi: 10.1073/pnas.93.15.7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Askari H, et al. Cellular and tissue localization of globotriaosylceramide in Fabry disease. Virchows Arch. 2007;451(4):823–34. doi: 10.1007/s00428-007-0468-6. [DOI] [PubMed] [Google Scholar]
- 54.Yam GH, et al. Pharmacological chaperone corrects lysosomal storage in Fabry disease caused by trafficking-incompetent variants. American journal of physiology Cell physiology. 2006;290(4):C1076–82. doi: 10.1152/ajpcell.00426.2005. [DOI] [PubMed] [Google Scholar]
- 55.Alroy J, Sabnis S, Kopp JB. Renal pathology in Fabry disease. J Am Soc Nephrol. 2002;13(Suppl 2):S134–8. [PubMed] [Google Scholar]
- 56.Ikonen E. Roles of lipid rafts in membrane transport. Current opinion in cell biology. 2001;13(4):470–7. doi: 10.1016/s0955-0674(00)00238-6. [DOI] [PubMed] [Google Scholar]
- 57.Glebov OO, Nichols BJ. Distribution of lipid raft markers in live cells. Biochemical Society transactions. 2004;32(Pt 5):673–5. doi: 10.1042/BST0320673. [DOI] [PubMed] [Google Scholar]
- 58.Nohe A, et al. Dynamics of GPI-anchored proteins on the surface of living cells. Nanomedicine: nanotechnology, biology, and medicine. 2006;2(1):1–7. doi: 10.1016/j.nano.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 59.Suzuki KG, et al. GPI-anchored receptor clusters transiently recruit Lyn and G alpha for temporary cluster immobilization and Lyn activation: single-molecule tracking study 1. The Journal of cell biology. 2007;177(4):717–30. doi: 10.1083/jcb.200609174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Legewie S, et al. Recurrent design patterns in the feedback regulation of the mammalian signalling network. Molecular systems biology. 2008;4:190. doi: 10.1038/msb.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suzuki KG, et al. Transient GPI-anchored protein homodimers are units for raft organization and function. Nature chemical biology. 2012;8(9):774–83. doi: 10.1038/nchembio.1028. [DOI] [PubMed] [Google Scholar]
- 62.Friedrichson T, Kurzchalia TV. Microdomains of GPI-anchored proteins in living cells revealed by crosslinking. Nature. 1998;394(6695):802–5. doi: 10.1038/29570. [DOI] [PubMed] [Google Scholar]
- 63.Hofman EG, et al. EGF induces coalescence of different lipid rafts. Journal of cell science. 2008;121(Pt 15):2519–28. doi: 10.1242/jcs.028753. [DOI] [PubMed] [Google Scholar]
- 64.Horejsi V, et al. Signal transduction in leucocytes via GPI-anchored proteins: an experimental artefact or an aspect of immunoreceptor function? Immunology letters. 1998;63(2):63–73. doi: 10.1016/s0165-2478(98)00054-6. [DOI] [PubMed] [Google Scholar]
- 65.Potter BA, et al. Specific N-glycans direct apical delivery of transmembrane, but not soluble or glycosylphosphatidylinositol-anchored forms of endolyn in Madin-Darby canine kidney cells. Mol Biol Cell. 2004;15(3):1407–16. doi: 10.1091/mbc.E03-08-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. The Journal of clinical investigation. 2002;110(5):597–603. doi: 10.1172/JCI16390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheng PC, et al. Translocation of the B cell antigen receptor into lipid rafts reveals a novel step in signaling. Journal of immunology. 2001;166(6):3693–701. doi: 10.4049/jimmunol.166.6.3693. [DOI] [PubMed] [Google Scholar]
- 68.Hern JA, et al. Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(6):2693–8. doi: 10.1073/pnas.0907915107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kasai RS, et al. Full characterization of GPCR monomer-dimer dynamic equilibrium by single molecule imaging. The Journal of cell biology. 2011;192(3):463–80. doi: 10.1083/jcb.201009128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Calebiro D, et al. Single-molecule analysis of fluorescently labeled G-protein-coupled receptors reveals complexes with distinct dynamics and organization. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(2):743–8. doi: 10.1073/pnas.1205798110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Suzuki KG, et al. Dynamic recruitment of phospholipase C gamma at transiently immobilized GPI-anchored receptor clusters induces IP3-Ca2+ signaling: single-molecule tracking study 2. The Journal of cell biology. 2007;177(4):731–42. doi: 10.1083/jcb.200609175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Finger EC, et al. Endocytosis of the type III transforming growth factor-beta (TGF-beta) receptor through the clathrin-independent/lipid raft pathway regulates TGF-beta signaling and receptor down-regulation. The Journal of biological chemistry. 2008;283(50):34808–18. doi: 10.1074/jbc.M804741200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Di Guglielmo GM, et al. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nature cell biology. 2003;5(5):410–21. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- 74.Sanchez-Nino MD, et al. Globotriaosylsphingosine actions on human glomerular podocytes: implications for Fabry nephropathy. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2011;26(6):1797–802. doi: 10.1093/ndt/gfq306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.