

Abstract
Fluorescence provides a mechanism for achieving contrast in biological imaging that enables investigations of molecular structure, dynamics, and function at high spatial and temporal resolution. Small-molecule organic fluorophores have proven essential for such efforts and are widely used in advanced applications such as single-molecule and super-resolution microscopy. Yet, organic fluorophores, like all fluorescent species, exhibit instabilities in their emission characteristics, including blinking and photobleaching that limit their utility and performance. Here, we review the photophysics and photochemistry of organic fluorophores as they pertain to mitigating such instabilities, with a specific focus on the development of stabilized fluorophores through derivatization. Self-healing organic fluorophores, wherein the triplet state is intramolecularly quenched by a covalently attached protective agent, exhibit markedly improved photostabilities. We discuss the potential for further enhancements towards the goal of developing “ultra-stable” fluorophores spanning the visible spectrum and how such fluorophores are likely to impact the future of single-molecule research.
Introduction
Fluorescence imaging, which affords high specificity and imaging contrast,1 has proven to be an indispensable tool for advancing our understanding of biological systems.2 Although biomolecules often contain intrinsic fluorophores, such as aromatic amino acids that can be used to interrogate biological functions, extrinsic fluorophores,3 such as small-molecule organic fluorophores,4, 5 fluorescent proteins,6, 7 and inorganic semiconductor particles (quantum dots),8 have absorbance cross sections and fluorescence quantum yields that dramatically increase image contrast. Extrinsic fluorophores have therefore become essential imaging tools.
Extrinsic fluorophores spanning the visible spectrum are now available that can be specifically attached to almost any biomolecule of interest. These probes can thus serve as versatile messengers of dynamic and functional information in a diverse array of systems that would otherwise be hidden.2 Small-molecule organic fluorophores are the smallest of the known extrinsic fluorophores–only a hundredth to a thousandth the size of fluorescent proteins and quantum dots9, 10(Fig. 1). Correspondingly, organic fluorophores when properly positioned are the least perturbing to the system under interrogation.
Fig. 1.
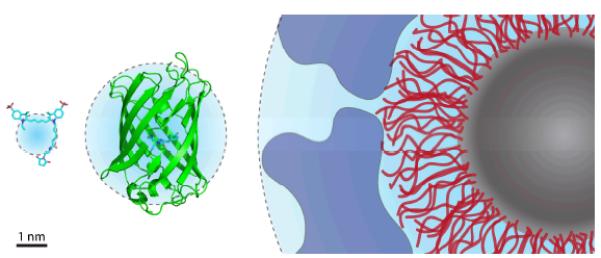
Size comparison of extrinsic fluorophores. From left to right: the organic fluorophore Cy5 (maleimide conjugate), green fluorescent protein, and a quantum dot coated with a passivating polymer layer (red) and a bioconjugating molecule layer (blue). Cyan spheres represent hydrodynamic radii.18, 38, 39
The use of organic fluorophores over the last century has greatly advanced our knowledge and understanding of biological systems.2 They have been used for staining distinct cellular compartments,3 for pH and analyte sensing in vitro and in living cells,11 and for detecting intermolecular interactions via changes in fluorophore excitation, emission, and tumbling properties.2 They have also been used as essential reagents for immunofluorescence, proteomics, as well as a host of medical diagnostic tools.2, 3 In each application, fluorophore choice proves paramount to a successful outcome. The appropriate organic fluorophore is one that maximizes signal and minimizes noise.
While organic fluorophores lack the per se genetic encodability of fluorescent proteins, and can be less photostable than quantum dots, a number of recently developed methods enable the problem of encodability to be overcome12-14 and dramatic improvements in photostability have been achieved (reviewed here) that markedly expand their utility and performance in almost every imaging application. In this review, we focus our discussion on the use and performance of organic fluorophores in investigations using single-molecule fluorescence. Excellent reviews of fluorescent proteins and quantum dots can be found elsewhere.6, 8, 9, 15-19
Since the first single-molecule fluorescence measurements on biological samples at ambient temperatures were made in the 1990s,20, 21 the quest to image biological systems one molecule at a time has grown exponentially.22 A key driving force behind this trend is the emerging understanding that time-dependent fluctuations in the structure and dynamics of molecular systems are essential aspects of function that are lost, or at least obscured, in bulk investigations.20-22 Single-molecule methods also enable the quantification of heterogeneous molecular populations and the tracking of asynchronous events in real time,20-22 information that is inaccessible at the ensemble scale. In vitro analyses of motor protein function,23, 24 RNA folding and catalysis,25 transcription,26 translation,27 DNA recombination,28, 29 splicing,30 telomere maintenance,31, 32 reverse transcription,33 chromatin remodeling,34 and membrane transport35-37 have already lead to unprecedented insights that have advanced our knowledge of molecular structure, dynamics, and function. Single-molecule investigations in living cells,40-49 although still nascent as a field, have further revolutionized our understanding of the transient and stochastic nature of cellular processes and the fundamentally dynamic nature of biological systems.
Yet despite the remarkable progress that has already been achieved, the continued advance of single-molecule research requires new technologies to address the high demands placed on the chemical and physical properties of the fluorophores employed.5 The inherent instabilities of fluorophores –including their propensity to switch between bright and dark states (blinking)50, 51 and permanently terminate fluorescence (photobleaching) –compromises the regularity and duration of photon emission.4, 5 Such phenomena, which stem from fluorophore- and environment-specific photophysical and photochemical reactions,4, 5 limit the spatial and temporal resolution that can be achieved and diminish the information content of the experiment. Therefore, efforts to characterize and to understand each fluorophore’s distinct properties play a critical role in the evolution of fluorophores for distinct applications. Advancements in these areas offer the promise of further broadening the scope and depth of information that can be gained through single-molecule imaging as well as the types of biological that can be interrogated. Here, we discuss our best estimate of the road ahead and the likely obstacles to further progress.
Although blinking can be exploited for reconstructing super-resolution images of cellular structures,52-54 for most single-molecule imaging applications, stochastic blinking, which spans from microsecond to minute timescales, reduces the number of photons emitted per unit time to the detriment of signal quality, resolution and imaging time.55 Blinking can also be misinterpreted as biologically relevant events,56, 57 and significantly hamper efforts to track individual fluorophores in complex environments.58-60 Bright, slow-photobleaching and non-phototoxic61-65 fluorophores with stable fluorescence intensity are correspondingly in great demand.
Below, we discuss key challenges associated with the generation of organic fluorophores spanning the visible spectrum with such performance characteristics and how such properties may be associated with reduced phototoxicity in complex biological settings.61-65 Such species are referred to here as “ultra-stable” organic fluorophores.Progress towards the development of such species is reviewed here in the context of recent advances in the field.
Origins of fluorophore instability
Our discussion of mitigating fluorophore instabilities necessarily begins with a brief review of the photophysics and photochemistry underpinning organic fluorophore performance. (Fig. 2).66 A fluorophore molecule in the ground state (S0 in Fig. 2) that is illuminated with light of appropriate wavelength may absorb a photon to transition into an excited state, where the efficiency of this process is determined by the illumination intensity and the fluorophore’s extinction coefficient. Following rapid (ca. picoseconds), solvent-mediated relaxation, the fluorophore resides in the lowest vibrational level of the first singlet excited state (S1 in Fig. 2). The excitation process can also be described in the framework of molecular orbital theory,66 where an electron within the highest occupied molecular orbital (HO in Fig. 2) transitions to the lowest unoccupied (LU in Fig. 2) molecular orbital.
Fig. 2.
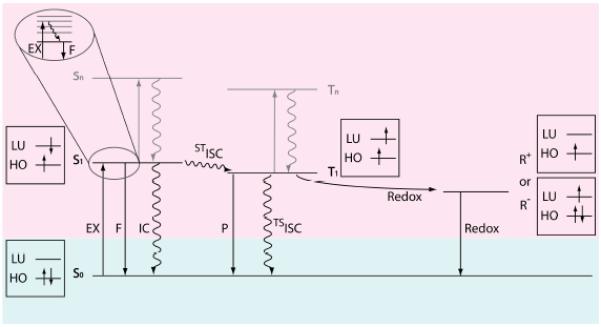
The state energy diagram and the electron spin configuration for fluorophore excitation and deactivation pathways. S0: the ground state of the fluorophore molecule; S1: the first singlet excited state; T1: the first triplet excited state; R+: cationic radical state; R−: anionic radical state; Sn and Tn (n > 1): higher-energy singlet and triplet excited state, respectively; EX: excitation by photon absorption; F: fluorescence; IC: internal conversion; ISC: intersystem crossing; P: phosphorescence; Redox: reduction or oxidation. The boxes show the electron and spin configurations for the corresponding states. HO: highest occupied molecular orbital for the fluorophore molecule; LU: lowest unoccupied molecular orbital.
A fluorophore in S1 can return to the S0 state through either radiative (fluorescence [F] in Fig. 2) or a non-radiative (internal conversion [IC] in Fig. 2) relaxation pathways, at timescales on the order of 10−10 – 10−9 seconds (for fluorophores typically used in single-molecule imaging).4, 67, 68 Due to rapid vibrational relaxation following excitation, the energy of the photon emitted from S1 is lower than the excitation photon, resulting in an increase in wavelength ranging from 5-50 nm (Stokes shift).66
While an ideal fluorophore rapidly cycles between S1 and S0, resulting in regular photon emission, deviations from this simple two-state model feature prominently in the experimentally observed behaviours of organic fluorophores. For instance, a fluorophore in S1 can also undergo intersystem crossing (ISC in Fig. 2) to a non-fluorescent triplet excited state (T1 in Fig. 2). Although ISC to T1 is typically a rare event for organic fluorophores used in single-molecule imaging (< 0.01),69-72 its high energy and long lifetime (typically 10−6 – 10−4 seconds)70, 73-76 make it a key determinant of fluorophore performance. Excursions to the triplet state attenuate the observed photon emission rate (blinking) and open chemical pathways to irreversible damage (photobleaching). For example, fluorophores in T1 are particularly active in electron transfer reactions66 that result in the formation of non-fluorescent radical species (R+ and R− in Fig. 2) and subsequent degradation of the fluorophore. Here, oxidation or reduction of the fluorophore can be mediated by a solvent impurity (e.g. metal ions), molecular oxygen (O2), components of the biological molecule to which it is attached or another fluorophore.
Molecular oxygen, present at a concentration of approximately 0.3 mM in aqueous solutions at ambient pressure,77 is a ubiquitous and reactive participant in reactions with organic fluorophores. Electron transfer from a triplet fluorophore to molecular oxygen produces a superoxide radical (O2−) and a non-fluorescent, cationic state (R+) of the fluorophore. Energy transfer from a triplet fluorophore to molecular oxygen produces excited singlet oxygen (1O2), an oxidizing agent stronger than ground state molecular oxygen. Superoxide radicals and singlet oxygen, along with other downstream reactive oxygen species (ROS), including HO·, HO2·, and H2O2, can cause photobleaching by reacting with the fluorophore78-80 and phototoxicity by reacting with nearby biomolecules.65, 81, 82
The aforementioned discussion provides a simplified framework for understanding the experimentally observed instability of organic fluorophores (Fig. 3). While the ideal fluorophore cycles exclusively between the S0 and S1 states, leading to a non-blinking and long-lasting fluorescent signal, such behavior is never achieved in practice because the rate of intersystem crossing to the triplet state is non-negligible. A simple calculation illustrates this point. Approximately 100 detected photons per time point are needed to achieve a sufficient signal-to-noise ratio (SNR) for a single-molecule measurement.5 Assuming that the quantum yield of triplet state formation (ΦISC) is 0.001, the quantum yield of fluorescence (Φfl) is 0.5, and the efficiency of photon detection is 10%,5 a fluorophore will make about two transitions to T1 during each integration period.
Fig. 3.
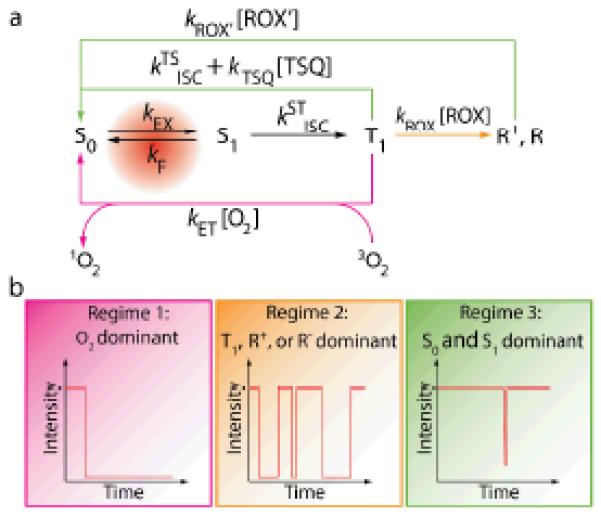
(a) A framework for understanding the nature of fluorophore instabilities. (b) Kinetic regimes that lead to different behaviors of a fluorophore. TSQ: triplet state quencher; ROX and ROX’: reducing or oxidizing agents. texp: the exposure time for each frame of the measurement. Regime 1 occurs when
ket[O2] >> ktsisc+ktsq[TSQ], krox[ROX],
and the fluorophore photobleaches quickly. Regime 2 occurs when
krox[ROX] >> ket[O2], ktsisc +ktsq[TSQ];
krox’[ROX’] ≤ 1/ texp,
and the fluorophore blinks frequently. Regime 3 occurs when
ktsisc+ ktsq[TSQ] >> ket[O2], krox[ROX];
or krox[ROX] >> ket[O2], ktsisc +ktsq[TSQ];
krox’[ROX’] >> 1/ texp,
and the fluorophore lasts long and rarely blinks.
In air-saturated solutions, reactions between molecular oxygen and T1 are rapid (on the order of 106 s−1),83 leading to substantial ROS generation and rapid photobleaching (Regime 1 in Fig. 3). In the absence of molecular oxygen, radical states of the fluorophore may be rapidly formed through electron transfer with its surroundings (Regime 2 in Fig. 3). As R+ and R− radical states of the fluorophore are non-emissive and can be long lived, pronounced blinking and photobleaching occur. As will be discussed in detail below, triplet state quenchers (TSQ in Fig. 3) and reducing and oxidizing agents (ROX in Fig. 3) can quench T1 and radical states74, 76, 84 to recover the ground state. When such quenching occurs rapidly, triplet and radical states are shortened resulting in a non-blinking and long-lasting fluorescent signal (Regime 3 in Fig. 3).
Reactions independent of the triplet state may also contribute to fluorophore instability. The following two examples are relevant to many applications. First, excited states higher than S1 and T1 can be produced by the absorption of one or multiple photons (eg. S0→Sn, S0→S1→Sn, and S0→T1→Tn) (Fig. 2). However, since Sn and Tn generally relax to S1 or T1 (ca. femtoseconds to picoseconds) faster than they undergo other transitions (Kasha’s rule),66 higher excited states are usually not explicitly discussed in the context of fluorophore photophysics. Nevertheless, an excess of photobleaching is often observed in single-molecule Förster Resonance Energy Transfer (smFRET) and in multi-color excitation studies, which typically demand intense illumination, suggesting an involvement of Sn or Tn states.83, 85-87 Second, the polymethine chain in cyanine fluorophores in the S1 excited state can undergo cis-trans isomerization to produce poorly fluorescent cis isoforms,88-90 leading to attenuations in brightness, including microsecond timescale fluctuations and blinking.4, 91, 92
Selecting fluorophores for single-molecule imaging
For single-molecule fluorescence imaging, it is imperative to maximize the signal detected from each individual fluorophore molecule. Selection of a fluorophore with robust emission properties is a critical first step. An appropriate fluorophore should be bright enough to provide the desired spatial and temporal resolution and photostable enough to ensure a sufficiently long observation window. A gamut of commercially available fluorophores meet such requirements, including blue- (Cy2,93 Atto 488,93 Alexa 48893), yellow- (Cy3,94 Cy3B,4 Cy3.5,95 Atto 550,94 Alexa 55594), red- (Cy5,94 Cy5.5,96 Atto 647N,94 Atto 655,97 Alexa 64794), and near-infrared- (Cy744, 95) emitting molecules of the cyanine (Cy2, Cy3, Cy3B, Cy3.5, Cy5, Alexa 647, Cy5.5, Cy7), rhodamine (Alexa 488, Atto 488, Alexa 555), oxazine (Atto 655), and carbopyronine (Atto 647N) classes.
In certain super-resolution imaging modalities, fluorophore blinking and photobleaching properties are key determinants of spatial and temporal resolution.48, 52-54 A systematic evaluation by Zhuang and colleagues demonstrated that Atto 488, Cy3B, Cy5, Cy7 and Cy7 derivatives (Alexa 750, DyLight 750), are particularly amenable to such applications.98 In ratiometric fluorescence methods like smFRET imaging, the chosen fluorophores should not exhibit intrinsic spectral shifts or intensity fluctuations. While such effects may be less relevant in applications such as particle tracking,58, 60 they confound smFRET measurements seeking to quantify functional conformational events.56, 57 For example, while Alexa 488, Atto 488, and Atto 647N possess high extinction coefficients, quantum yields and stability, their tendency to exhibit spectral shifts generally makes them poor choices for quantitative smFRET measurements.5, 93 Cis-trans isomerization, particularly relevant to the cyanine fluorophore class,88 can also contribute to fluctuations in fluorescence quantum yield, and hence brightness.89, 90 Such phenomena render the observed fluorescence of Cy3 strongly dependent on the local environment.91, 92 However, in our experience, fluctuations of this kind have only a limited impact on the observed signal in the context of FRET as variations in Cy3 intensity tend to propagate to the acceptor fluorophore equally, resulting in correlated reductions in intensity with negligible impact on the apparent FRET efficiency.
In the following sections, we focus our discussion on the sulfonated derivatives of the cyanine fluorophores99 (Fig. 4) as they remain the most widely employed in single-molecule research. Despite their aforementioned non-idealities, cyanine-class fluorophores exhibit properties that make them highly amenable to imaging, including high aqueous solubility, high brightness and photostability as well as low spectral shift propensities and controllable blinking behaviors. They are also particularly amenable to systematic chemical and photophysical investigations as they are closely related in structure and can be chemically synthesized in high yields.
Fig. 4.
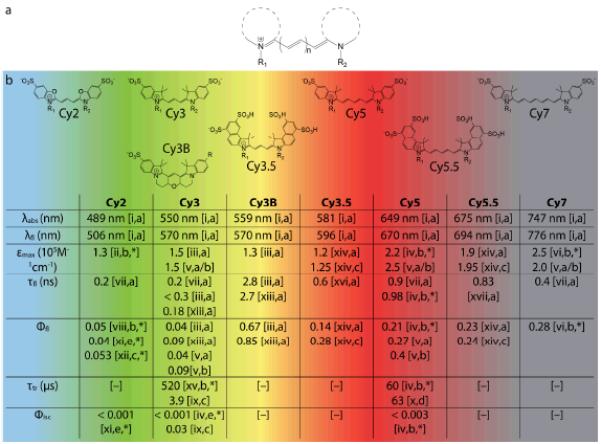
(a) Generic structure of cyanine fluorophores. (b) Structures of commercially available cyanine fluorophores along with important spectroscopic properties. Note that the values cited here are for free dyes in solution and may change significantly upon conjugation to biomolecules. λabs, λem – wavelengths of absorption, emission maximum; εmax – extinction coefficient; τfl, τtr – lifetimes of fluorescence and triplet state; Φfl, Φisc – quantum yields of fluorescence and intersystem crossing. The R groups represent the various linkers available for bioconjugation of the fluorophores.
Source: [i] Dempsey et al. 2011;98 [ii] Kassab 2002;65 [iii] Cooper et al. 2004;100 [iv] Chibisov et al. 1996;70 [v] Mujumdar et al. 1993;99 [vi] Rurack and Spieles 2011;101 [vii] unpublished data; [viii] Ponterini and Caselli 1992;102 [ix] Jia et al. 2007;75 [x] Zheng et al. 2012;76 [xi] Chibisov 1977;103 [xii] Roth and Craig 1974;104 [xiii] Sanborn et al. 2007;105 [xiv] Mujumdar et al. 1996;106 [xv] Chibisov et al. 1995;107 [xvi] Gu et al. 2013;108 [xvii] Buschmann et al. 2003.67 Solvent: [a] Water; [b] Ethanol; [c] Methanol; [d] Acetonitrile; [e] Butanol. [*] non-sulfonated form; [–] no data available.
Fluorophore stabilization through oxygen depletion
Given that the rate constant for quenching of fluorophore triplet states by molecular oxygen is on the order of 109 M−1s−1,66 the effective rate of triplet state quenching in air-saturated solutions approaches 106 s−1. This rate is substantially faster than the intrinsic decay of triplet states for most organic fluorophore species (ca.104 - 106 s−1). In the absence of high concentrations (e.g. > 1 mM) of oxidants or reductants, the rate of triplet state quenching by molecular oxygen is also faster than the formation of radical states. Thus, in the presence of molecular oxygen, where the triplet state lifetime is on the order of 1 μs, fluorophore blinking stemming from the formation of radical states is typically negligible. While this may be a preferred regime for live cell single-molecule imaging and some in vitro systems, the generation of ROS can lead to rapid fluorophore photobleaching (Regime 1 in Fig. 3; Fig. 4a) and unwanted phototoxicity.61, 64, 65
Molecular oxygen’s recognized role in the photobleaching of organic fluorophores has motivated extensive investigations into practical means of removing it from solution. Dissolved oxygen can be efficiently removed by degassing techniques and saturating the solution with oxygen-free gases.109 However, such methods are prone to variability and reverse rapidly. For this reason, enzymatic oxygen scavenging systems have become the method of choice for many fluorescence applications.110-113 This is particularly true in the case of single-molecule imaging, where the demand for fluorophore performance is greatest. The GOD:CAT system, comprised of glucose, glucose oxidase and catalase, and the PCA/PCD system, comprised of protocatechuic acid (PCA) and protocatechuate-3,4-dioxygenase (PCD), are the most widely employed. Alternative methods include the usage of pyranose oxidase, d-glucose, and catalase,112 or methylene blue and thiol,113 but such systems are less common and have yet to be fully characterized.
In an air sealed container, the PCA/PCD system can reduce the molecular oxygen concentration to approximately 3 μM when operating properly.111 In doing so, the collision frequency between the fluorophore and molecular oxygen is lowered by roughly two orders of magnitude (from ~1 μs−1 to ~0.01 μs−1). Although the removal of molecular oxygen from solution can reduce fluorophore photobleaching rates by an order of magnitude or more (Fig. 5a and b), doing so accentuates the redox characteristics of the fluorophore’s triplet state. In solution conditions and biological settings, this typically results in severe blinking (Fig. 5b) due to the formation of radical states lasting anywhere from several milliseconds to hours (Regime 2 in Fig. 3).114 As we discuss below, improved fluorophore performance can be realized through the addition of exogenous chemical additives as well as fluorophore engineering.
Fig. 5.
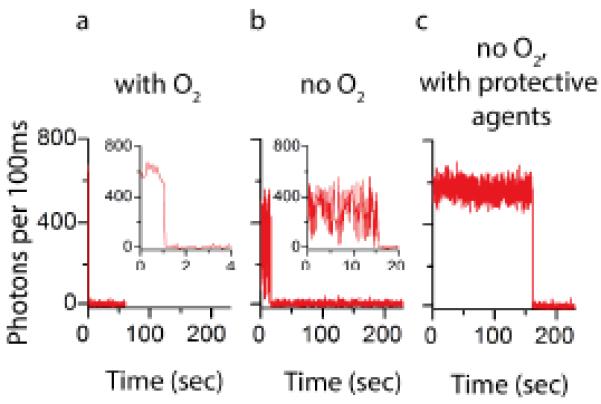
Representative single-molecule fluorescence traces for Cy5 in (a) air saturated buffer, (b) deoxygenated buffer, and (c) deoxygenated buffer plus 1mM COT, NBA, and Trolox.
Fluorophore stabilization using solution additives
A small but growing number of specific chemical additives –collectively referred to here and elsewhere83 as “protective agents” –have been identified that afford significant improvements in fluorophore performance. Protective agents may operate through a wide range of mechanisms. However, they are generally characterized by their capacity to reduce fluorophore photobleaching rates, to increase the mean fluorescence intensity, to reduce variances in fluorescence intensity, and to reduce blinking frequency.55, 83, 84, 115
The reducing agent, β-mercaptoethanol, was one of the first protective agents to be employed for fluorophore stabilization.116 Consistent with the idea that the reactive oxygen species contribute to poor fluorophore performance, the antioxidants cysteamine,62 N-propyl gallate,117 ascorbic acid,118p-phenylenediamine,119 and 1,4-diazabicyclo[2.2.2]octane (DABCO),120 have since been found to reduce the apparent blinking and photobleaching rates in bulk and single-molecule imaging.83 Some of these compounds constitute the active ingredients in commercially available anti-fading agents employed for fixed cell imaging applications.121
Chemicals such as 1,3,5,7 cyclooctatetraene (COT),27, 83, 115, 122 4-nitrobenzylalchohol (NBA),27, 115, 122 and Trolox55, 83, 115, 123, 124 are on the shortlist of preferred compounds for fluorophore stabilization. The combined use of COT, NBA and Trolox under oxygen scavenging conditions can drastically increase the mean fluorescence intensity as well as the duration of photon emission (Fig. 5c).115 Although these reagents were understood to operate through a collision-based mechanism and to exhibit the greatest benefits when used in combination, the precise mechanisms of these protective agents were not known at the time that they were initially employed.55, 83, 115, 123 The utility of solution-based protective agents as a strategy for fluorophore stabilization is highlighted by the fact that protective agents have been used for the vast majority of single-molecule fluorescence applications over the past decade.
A significant advance in the use of protective agents for fluorophore photostabilization has been the development of reducing and oxidizing systems (ROXS).84 These systems elegantly address the tendency of organic fluorophores to enter dark states via the formation of radical fluorophore species that are directly on path to photobleaching (Fig. 2). The proper balance of reducing agents, such as ascorbic acid and n-propyl gallate, and oxidizing agents, such as methylviologen, can reduce the lifetime of the triplet state by forcing the formation of fluorophore radicals that can then be quickly returned to the ground state by providing a readily available source of redox agents (Regime 3 in Fig. 3). ROXS can also reduce the lifetime of spontaneously formed radical states through similar means. Trolox and two commercially available anti-fading agents (Vectashield and Ibidi-MM) have also been shown to function through ROXS mechanisms.121, 125 Importantly, ROXS has proven effective for controlling the duration of bright and dark states for a variety of commercially available fluorophores by adjusting the concentration of reducing and oxidizing agents used. Sub-millimolar ROXS concentrations generally leads to reversible blinking, which can be exploited for super-resolution applications based on stochastic blinking,126 while at millimolar concentrations and above, redox agents collide with the fluorophore on the microsecond timescale, resulting in much shorter triplet and radical state lifetimes.84 Under such conditions, ROXS can significantly increase fluorophore stability by effectively short-circuiting photobleaching pathways. However, ROXS performance is strongly dependent on fluorophore type and experimental demands. It also functions by enforcing the entry into dark states and thus only appears to eliminate blinking when imaging at integration times substantially slower than the blinking frequency.
Research in the dye laser field dating back to the 1960s demonstrated that certain agents can increase fluorophore brightness and photostability by operating through a triplet-triplet energy transfer mechanism.127 For this mechanism to be efficient, the triplet energy of the fluorophore (donor) must be higher than the triplet energy of the triplet state quencher (acceptor). In the presence of molecular oxygen, the triplet energy of the quencher should also be lower than the triplet-singlet energy gap of molecular oxygen (~94 kJ/mol) to prevent singlet oxygen generation. COT, which has a low-energy triplet state (~92 kJ/mol),128, 129 fits this description and has recently been shown to greatly improve fluorophore stability in single-molecule imaging in aqueous environments.35, 115, 130
Despite the remarkable advancements afforded by solution-based protective agents, such approaches face severe constraints. First, the benefit to fluorophore performance by exogenous protective agents depends on the collision frequency.115 Thus, millimolar concentrations are typically required for stabilizing effects. This concentration regime is at, or near, the solubility limit for many of these compounds. Correspondingly, protective agents may lead to a non-specific inhibition of biological activities and their effects must be carefully examined for each system under investigation. Second, the hydrophobic nature and redox properties of protective agents pose serious limitations for investigations of biological systems at the membrane or in living cells. Indeed, it has now been shown that COT, NBA and Trolox interact with biological membranes to quantitatively alter the function of integral membrane proteins.131 Third, the effects of protective agents may depend on the fluorophore and the labeling context.84, 115 Such considerations pose significant challenges for ROXS-based stabilization strategies in particular, as the redox properties of organic fluorophores are dependent on fluorophore type, solution conditions, and biological context. These issues suggest that a universal, fluorophore-independent solution for optimizing photostability using this approach may not exist.
Self-healing fluorophores
To address the aqueous insolubilities and potential toxicities exhibited by protective agents, we have recently turned to a strategy of chemically engineering organic fluorophores to improve their performance.76, 95, 132 Efforts along these lines have been successfully employed previously to develop fluorophores for the dye laser field,133 to improve the aqueous solubility,134 and to develop fluorescence-based biosensors.11 As indicated above, we have focused our recent endeavors on the cyanine fluorophores (Fig. 4), although we believe the mechanistic insights from these investigations are likely applicable to other fluorophore classes.
Building on the observation that the protective agents COT, NBA and Trolox operate through a concentration-dependent mechanism,115 we set out to covalently conjugate them to the fluorogenic center to achieve the highest effective concentration possible. In our initial efforts,132 we focused on the commercially available cyanine fluorophore, Cy5, one of the most widely employed organic fluorophores in fluorescence and FRET imaging. Cy5 serves an important role in cellular imaging and FRET due to its red-shifted emission properties but is prone to frequent blinking and rapid photobleaching in the absence of protective agents. In this work, we chose a generalizable synthetic strategy in which a bis-N-hydroxysuccinimide activated Cy5 fluorophore was coupled to amine-activated COT, NBA and Trolox molecules to create fluorophores linked to these protective agents through a flexible 13-atom linker (termed Cy5-COT(13), Cy5-NBA(11), and Cy5-Trolox(11), respectively; Scheme 1).
Scheme 1.
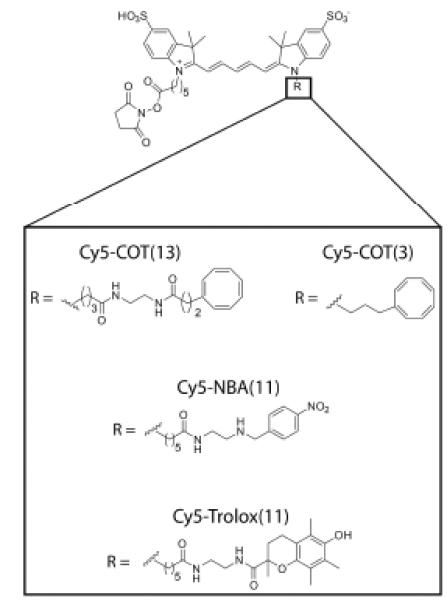
The structures of Cy5-COT(13), Cy5-COT(3), Cy5-NBA(11), and Cy5-Trolox(11).
Remarkably, these fluorophores showed little blinking and reduced photobleaching rates compared to the parent Cy5 fluorophore in distinct biological contexts (linked to a DNA molecule in vitro, and to the Dopamine D2 receptor on the surface of living cells). Consistent with the hypothesis that covalent attachment of the protective agent would increase the effective local concentration, each of the conjugates performed better than Cy5 with the respective protective agent in solution at near-saturating (1 mM) concentration. Improvements in photostability were also found in oxygenated buffers, where protective agents in solution had little or no effect.132 These findings showed that a single, proximally linked protective agent could mediate photostabilization of the Cy5 fluorophore. Coined “self-healing fluorophores”,135 Tinnefeld and Cordes speculated that the observed photostabilization could be mediated by ping-pong redox chemistry via the conjugated protective agent, thereby mitigating the formation of triplet and radical states directly.135
In a later report,95 this approach was extended to cyanine fluorophores spanning the visible and near-infrared spectrum (Cy2, Cy3, Cy3.5, Cy5.5, and Cy7), to find that each cyanine fluorophore responds uniquely to the attached protective agent, where the difference in photon count varied by several orders of magnitude. Pursuing the observation in this report that COT-linked fluorophores tended to exhibit the broadest and most substantial enhancements in fluorophore performance, including increased brightness, photon count, and signal-to-noise ratio (Fig. 6a), a mechanistic investigation was undertaken to explore whether further improvements could be achieved.76 In this study, we demonstrated that the COT moiety in Cy5-COT(13) improved Cy5 performance by directly reducing the lifetime of the triplet state through triplet-triplet energy transfer (see discussion in the previous section). In acetonitrile, we observed a 4.8-fold reduction in the Cy5 triplet state lifetime (from ~63 μsec to ~13 μsec), while no effect on the triplet state was observed for Cy5-Trolox(11) or Cy5-NBA(11). This finding demonstrated that COT operates through a distinct mechanism from NBA and Trolox. It also agreed with the notion that beneficial effects of NBA and Trolox on Cy5 performance operated through a charge transfer mechanism,135 which was efficiently suppressed in the organic solvent used.
Fig. 6.
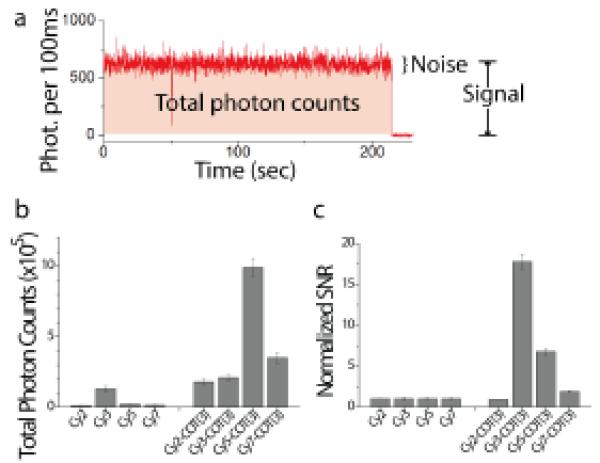
(a) A representative trace of Cy5-COT(3). The signal is defined as the average intensity before photobleaching. The noise is the standard deviation of the intensity. (b) Average number of photons detected before the fluorophore photobleaches, and (c) the signal-to-noise ratio (SNR) observed for Cy2, Cy3, Cy5, Cy7, and their COT-linked derivatives.
As triplet-triplet energy transfer is a collision-based process, these experiments also led us to test the hypothesis that further improvements in Cy5 performance could be achieved by reducing the length of the linker between COT and the fluorogenic center. This led to the development of synthetic strategies enabling us to bring COT to within 3 atoms of the Cy5 fluorophore (Scheme 1), where we observed an additional 12-fold reduction in triplet state lifetime (1.1 μsec vs. 13 μsec for Cy5-COT(13)). By quantifying the number of photons detected for each ensemble of single molecules, we showed that the number of detected photons before blinking or photobleaching for Cy5, Cy5-COT(13), and Cy5-COT(3) (2×104, 4×105, and 1×106, respectively) was inversely correlated with the triplet state lifetime, demonstrating that the triplet state plays a key role in fluorophore instability (Fig. 3). Following analogous strategies to synthesize Cy2-COT(3), Cy3-COT(3) and Cy7-COT(3), we show here that each of these fluorophores also exhibits marked enhancements in photon emission rates, total photon counts and signal-to-noise ratios (Fig. 6).
Experiments enabled by self-healing fluorophores
Organic fluorophores with improved stability are anticipated to enable new types of single-molecule experiments that were not previously feasible. For instance, the enhanced brightness, signal-to-noise ratio, and total photon counts should facilitate imaging at faster temporal resolution. In the field of particle tracking, sub-millisecond resolution using large scattering targets has enabled important findings in membrane diffusion and organization.136 The combination of self-healing fluorophores with feedback-driven tracking instrumentation137, 138 and new high-speed sCMOS cameras offers the promise of similar success using fluorescence.139, 140 Self-healing fluorophores are also anticipated to vastly expand the range of systems that can be investigated using single-molecule imaging methods. Today, fluorophore performance places highly restrictive, practical limitations on imaging time scales. In practice, single-molecule fluorescence studies are generally restricted to a temporal resolution of 10 ms or longer, where adequate photon counts (ca. >100 photons per time step) can be achieved over time periods that are functionally relevant to biological systems (ca. seconds to minutes). While sub-millisecond temporal resolution has been reported for fluorescence detected in a confocal geometry, the duration of fluorescence under these conditions is restricted to very short (ca < 1 ms) bursts.141
To assess the feasibility of high-temporal resolution imaging using self-healing fluorophores, we compared the single-molecule fluorescence signals detected from Cy5 and Cy5-COT(3) – both conjugated to DNA oligonucleotides – over a wide range of excitation intensities using a wide-field TIRF configuration. In these experiments molecular oxygen was depleted using the PCA/PCD system and samples were imaged in a simplified Tris-buffer solution (pH = 7.5) containing 50 mM potassium chloride and 5 mM β-mercaptoethanol. The data obtained show that individual Cy5-COT(3) fluorophores exhibit a linear increase in photon emission rate over the full range of excitation intensities tested, reaching up to 150 detected photons ms−1 at 1KW/cm2. In sharp contrast, fluorescence detected from the Cy5 fluorophore saturated at an emission rate of 15 photons ms−1 at much lower powers (Fig. 7). These findings reveal that the Cy5-COT(3) fluorophore has enough stability for 1 millisecond time scale imaging and suggest that additional head room may be available to image even faster. Further experiments are needed to verify this possibility.
Fig. 7.
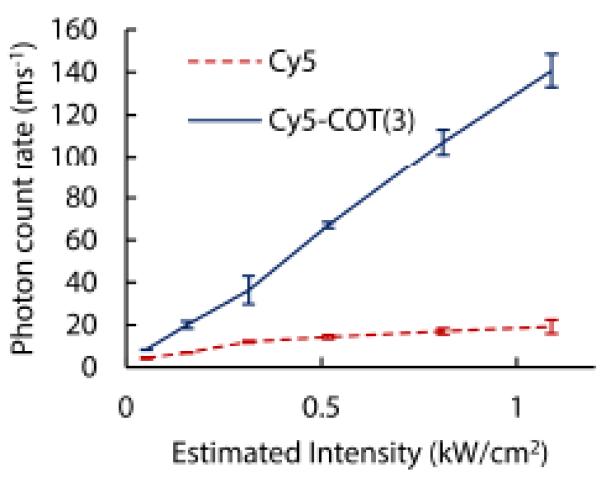
Photon emission rate as a function of excitation intensity (640 nm) for Cy5 (red, dashed line) and Cy5-COT (blue line) in the presence of the PCA/PCD oxygen scavenging system.
To demonstrate that self-healing fluorophores enable millisecond-timescale smFRET imaging, we prepared a labeled ribosome complex as previously described,142 in which donor (either Cy3 or Cy3-COT(3)) and acceptor (either Cy5 or Cy5-COT(3)) fluorophores were site-specifically conjugated to ribosomal proteins L1 and S13 within the large and small subunit, respectively (Fig. 8a). This system gives rise to FRET changes that report on spontaneous conformational processes in the ribosome that facilitate rotation of the large and small subunits with respect to each other (inter-subunit rotation).142, 143 The reversible process of inter-subunit rotation gives rise to fluctuations between low- and high-FRET states in individual molecules as a function of time. In the absence of molecular oxygen, ribosomes bearing self-healing donor and acceptor fluorophores yielded robust FRET recordings over extended periods (ca. 1 second) (Fig. 8b). Here, an equivalent of roughly ~150 photons could be detected per millisecond where the total fluorescence intensity observed showed remarkably little variance. Under the same conditions, FRET recordings could not be obtained using the Cy3 and Cy5 fluorophores as photobleaching occurred in less than one frame (data not shown).
Fig. 8.
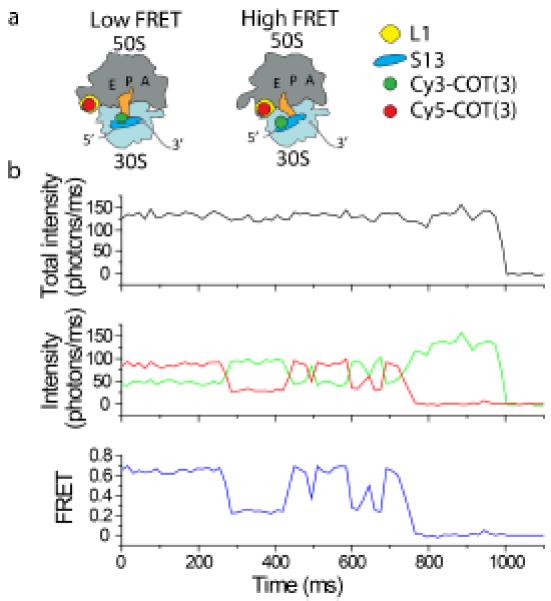
Self-healing fluorophores enable robust smFRET recordings at emission rates compatible with millisecond time scale imaging. Data were collected in Tris-polymix buffer using a wide-field TIRF illumination system as previously described142 at a frame rate of 66 Hz. (a) Cartoon illustrating the labeling strategy designed to probe inter-subunit rotation of the bacterial ribosome using smFRET. Low FRET reports on an unrotated ribosome conformation; high FRET reports on a rotated ribosome conformation. (b) A long-lived trace with emission rate of >100 photons per millisecond enabled by imaging with self-healing dyes. Molecular oxygen was depleted using the PCA/PCD system.
Although single-molecule studies in complex biological and cellular contexts have been reported for more than a decade,42, 144, 145 the signals and timescales achieved thus far have been severely limited by rapid fluorophore photobleaching and low signal-to-noise ratios. Traditional methods to improve photostability, including oxygen scavenging systems and solution additives, may be incompatible in complex biological settings or have little or no benefit.42 Protective agents that exhibit redox properties and/or hydrophobicity may also interfere with the system under investigation.131 Self-healing fluorophores bypass these limitations and may be enabling for future endeavors aiming to perform single-molecule measurements in living cells. Self-healing fluorophores will be particularly important for multicolor fluorescence studies, where different fluorophores –or the same fluorophore in unique environments –respond differently, or even negatively, to protective agents such that optimal conditions for multiple fluorophores cannot be achieved using solution-based approaches.
Future perspectives and the development of next-generation ultra-stable organic fluorophores
While substantial progress has been made in developing photo-protection methods for organic fluorophores and in gaining a deeper phenomenological and quantitative understanding of the associated mechanisms, many open questions and significant challenges remain. Ultimately, advancements in the breadth and scope of systems that can be investigated using single-molecule imaging require even greater improvements in organic fluorophore photostability and brightness than what has already been achieved. Biological systems that exhibit dynamics on the 1-100 μs timescale and interact with ligands in the micromolar to millimolar regime146 are beyond the reach of the photon emission rates and detection efficiencies that can be presently achieved. A diverse range of multistep and complex biological processes (e.g. translation of a whole protein or changes in gene expression) also lay outside the range of timescales that can be reliably tracked under continuous illumination. As a consequence, the single-molecule field has been largely restricted to investigations of biological systems that exhibit dynamics within a fairly narrow imaging regime (Fig. 9). Although self-healing fluorophores will doubtless enable faster imaging timescales and longer observation windows for many systems, the ultimate goals of tracking minute-long reactions at timescales approaching those achieved through molecular dynamics simulations are still beyond reach. Efforts towards this goal are essential to bridging the knowledge gap that currently exists between single-molecule imaging and complete atomistic descriptions of molecular systems and biological functions.77, 99, 147, 148
Fig. 9.
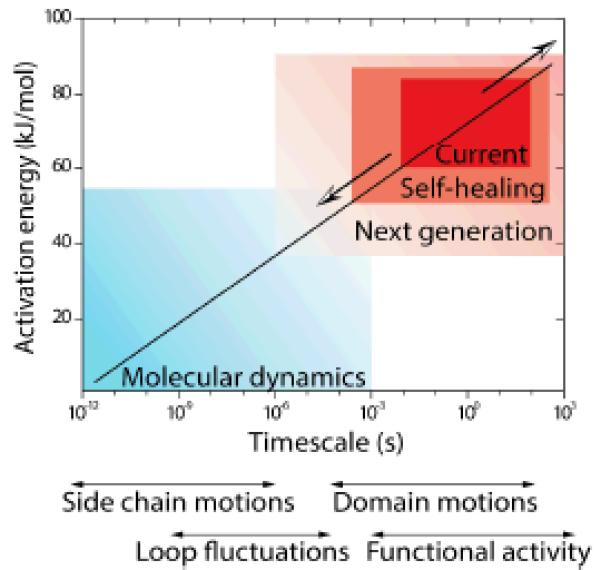
Ultra-stable fluorophores will enable the observation of previously unexplored kinetic regimes and may close remaining gaps between experimental and computational methods. The plot shows approximate timescales of biomolecular dynamic processes along with the associated activation energies.
These goals call for further efforts towards the development of ultra-stable fluorophores, where triplet state lifetimes are engineered to be significantly shorter than the temporal resolution of interest and the rate of molecular oxygen’s collision with the fluorophore (ca. <100 ns). Given the rate constants of diffusion-controlled quenching mechanisms (109-1010 M−1s−1), the physical limits of solubility, and issues surrounding biological toxicity, the “self-healing” strategy, rather than solution additive approach, is likely to be the preferred route to meeting this demand. While our achievements in this area point to this potential, today we are more than an order of magnitude away from this quenching rate using a single protective agent. One immediate goal is to explore the possibility that multiple protective agents may be conjugated to the fluorogenic center to achieve additive or even synergistic shortening of the triplet state lifetime. Yet, distinct strategies may also be warranted. Eliminating the formation of triplet and radical states through enhancing the fluorescence decay rate (kF in Fig. 3) and intersystem crossing rate (kISC and k’ISC in Fig. 3) may be considered equally attractive.149 While proof-of-concept studies in this direction have been published recently,150, 151 biologically amenable strategies of this nature have yet to be realized.
Our working framework (Fig. 3) also falls short of explaining experimental observations showing that red-emitting fluorophores photobleach much faster when illuminated with short wavelength light than with direct excitation.86, 87 Such complications, which may involve photoreactions from higher-energy excited states,85 are particularly challenging for FRET-based measurements, where short-wavelength light must be used to excite a donor fluorophore. Although the violation of Kasha’s rule66 has not yet been shown unambiguously, experimental observations of this kind suggest that investigations into the formation and features of higher excited states are greatly needed.
In parallel, the field of single-molecule imaging will also be greatly advanced by continued efforts to facilitate the goal of making organic fluorophores genetically encodable. A growing number of approaches – including non-natural amino acid chemistry, expressed protein ligation and enzymatic peptide targeting strategies – already enable organic fluorophores to be site-specifically attached to a biological molecule of interest in increasingly complex milieus.12 Further improvements in the specificity of these methods, and the means to induce fluorogenicity upon fluorophores attachment to its target13, 14 are advents that are likely to propel single-molecule research far beyond its current scope into realms unimaginable just a few years ago.
Acknowledgments
This work was carried out in memory of Dr. Nicholas J. Turro through the support of NIH grants GM098859 and GM079238 to S.C.B. and a National Science Foundation grant (NSF-CHE-11-11398) to S.J. We are grateful to members of the Blanchard lab for their critical comments and discussions during the writing of this manuscript. In particular, we would like to thank Dr. Daniel S. Terry for critical discussions and review of the written text.
References
- 1.Cheng PC. In: Handbook of Biological Confocal Microscopy. Pawley JB, editor. 2006. pp. 162–206. [Google Scholar]
- 2.Lakowicz J. Principles of Fluorescence Spectroscopy. Third Springer Science + Business Media, LLC; New York, NY: 2006. [Google Scholar]
- 3.Giepmans BN, Adams SR, Ellisman MH, Tsien RY. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 4.Levitus M, Ranjit S. Q Rev Biophys. 2011;44:123–151. doi: 10.1017/S0033583510000247. [DOI] [PubMed] [Google Scholar]
- 5.Ha T, Tinnefeld P. Annual Review of Physical Chemistry. 2012;63:595–617. doi: 10.1146/annurev-physchem-032210-103340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsien RY. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 7.Shaner NC, Steinbach PA, Tsien RY. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 8.Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS, Weiss S. Science. 2005;307:538–544. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Resch-Genger U, Grabolle M, Cavaliere-Jaricot S, Nitschke R, Nann T. Nat Meth. 2008;5:763–775. doi: 10.1038/nmeth.1248. [DOI] [PubMed] [Google Scholar]
- 10.Yang F, Moss LG, Phillips GN. Nat Biotech. 1996;14:1246–1251. doi: 10.1038/nbt1096-1246. [DOI] [PubMed] [Google Scholar]
- 11.Ueno T, Nagano T. Nat Meth. 2011;8:642–645. doi: 10.1038/nmeth.1663. [DOI] [PubMed] [Google Scholar]
- 12.Chen I, Ting AY. Curr Opin Biotechnol. 2005;16:35–40. doi: 10.1016/j.copbio.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Szent-Gyorgyi C, Schmidt BF, Creeger Y, Fisher GW, Zakel KL, Adler S, Fitzpatrick JAJ, Woolford CA, Yan Q, Vasilev KV, Berget PB, Bruchez MP, Jarvik JW, Waggoner A. Nat Biotech. 2008;26:235–240. doi: 10.1038/nbt1368. [DOI] [PubMed] [Google Scholar]
- 14.Paige JS, Wu KY, Jaffrey SR. Science. 2011;333:642–646. doi: 10.1126/science.1207339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandez-Suarez M, Ting AY. Nat Rev Mol Cell Biol. 2008;9:929–943. doi: 10.1038/nrm2531. [DOI] [PubMed] [Google Scholar]
- 16.Lippincott-Schwartz J, Patterson GH. Science. 2003;300:87–91. doi: 10.1126/science.1082520. [DOI] [PubMed] [Google Scholar]
- 17.Fu A, Gu W, Larabell C, Alivisatos AP. Current Opinion in Neurobiology. 2005;15:568–575. doi: 10.1016/j.conb.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 18.Bruchez MP. Current Opinion in Chemical Biology. 2011;15:775–780. doi: 10.1016/j.cbpa.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dedecker P, De Schryver FC, Hofkens J. Journal of the American Chemical Society. 2013;135:2387–2402. doi: 10.1021/ja309768d. [DOI] [PubMed] [Google Scholar]
- 20.Weiss S. Science. 1999;283:1676–1683. doi: 10.1126/science.283.5408.1676. [DOI] [PubMed] [Google Scholar]
- 21.Moerner WE. The Journal of Physical Chemistry B. 2002;106:910–927. [Google Scholar]
- 22.Joo C, Balci H, Ishitsuka Y, Buranachai C, Ha T. Annual Review of Biochemistry. 2008;77:51–76. doi: 10.1146/annurev.biochem.77.070606.101543. [DOI] [PubMed] [Google Scholar]
- 23.Vale RD, Funatsu T, Pierce DW, Romberg L, Harada Y, Yanagida T. Nature. 1996;380:451–453. doi: 10.1038/380451a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR. Science. 2003;300:2061–2065. doi: 10.1126/science.1084398. [DOI] [PubMed] [Google Scholar]
- 25.Zhuang X, Bartley LE, Babcock HP, Russell R, Ha T, Herschlag D, Chu S. Science. 2000;288:2048–2051. doi: 10.1126/science.288.5473.2048. [DOI] [PubMed] [Google Scholar]
- 26.Kapanidis AN, Margeat E, Ho SO, Kortkhonjia E, Weiss S, Ebright RH. Science. 2006;314:1144–1147. doi: 10.1126/science.1131399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blanchard SC, Gonzalez RL, Kim HD, Chu S, Puglisi JD. Nat Struct Mol Biol. 2004;11:1008–1014. doi: 10.1038/nsmb831. [DOI] [PubMed] [Google Scholar]
- 28.McKinney SA, Declais A-C, Lilley DMJ, Ha T. Nat Struct Mol Biol. 2003;10:93–97. doi: 10.1038/nsb883. [DOI] [PubMed] [Google Scholar]
- 29.Galletto R, Amitani I, Baskin RJ, Kowalczykowski SC. Nature. 2006;443:875–878. doi: 10.1038/nature05197. [DOI] [PubMed] [Google Scholar]
- 30.Abelson J, Blanco M, Ditzler MA, Fuller F, Aravamudhan P, Wood M, Villa T, Ryan DE, Pleiss JA, Maeder C, Guthrie C, Walter NG. Nat Struct Mol Biol. 2010;17:504–512. doi: 10.1038/nsmb.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stone MD, Mihalusova M, O'Connor C M, Prathapam R, Collins K, Zhuang X. Nature. 2007;446:458–461. doi: 10.1038/nature05600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoskins AA, Friedman LJ, Gallagher SS, Crawford DJ, Anderson EG, Wombacher R, Ramirez N, Cornish VW, Gelles J, Moore MJ. Science. 2011;331:1289–1295. doi: 10.1126/science.1198830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abbondanzieri EA, Bokinsky G, Rausch JW, Zhang JX, Le Grice SFJ, Zhuang X. Nature. 2008;453:184–189. doi: 10.1038/nature06941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blosser TR, Yang JG, Stone MD, Narlikar GJ, Zhuang X. Nature. 2009;462:1022–1027. doi: 10.1038/nature08627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao Y, Terry D, Shi L, Weinstein H, Blanchard SC, Javitch JA. Nature. 2010;465:188–193. doi: 10.1038/nature09057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao Y, Terry DS, Shi L, Quick M, Weinstein H, Blanchard SC, Javitch JA. Nature. 2011;474:109–113. doi: 10.1038/nature09971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akyuz N, Altman RB, Blanchard SC, Boudker O. Nature. 2013;502:114–118. doi: 10.1038/nature12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loman A, Dertinger T, Koberling F, Enderlein J. Chemical Physics Letters. 2008;459:18–21. [Google Scholar]
- 39.Hink MA, Griep RA, Borst JW, Hoek A. v., Eppink MHM, Schots A, Visser AJWG. Journal of Biological Chemistry. 2000;275:17556–17560. doi: 10.1074/jbc.M001348200. [DOI] [PubMed] [Google Scholar]
- 40.Sako Y, Minoghchi S, Yanagida T. Nature cell biology. 2000;2:168–172. doi: 10.1038/35004044. [DOI] [PubMed] [Google Scholar]
- 41.Wieser S, Schütz GJ. Methods. 2008;46:131–140. doi: 10.1016/j.ymeth.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 42.Sakon JJ, Weninger KR. Nat Meth. 2010;7:203–205. doi: 10.1038/nmeth.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Babcock HP, Chen C, Zhuang X. Biophys J. 2004;87:2749–2758. doi: 10.1529/biophysj.104.042234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shav-Tal Y, Darzacq X, Shenoy SM, Fusco D, Janicki SM, Spector DL, Singer RH. Science. 2004;304:1797–1800. doi: 10.1126/science.1099754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Biteen JS, Thompson MA, Tselentis NK, Bowman GR, Shapiro L, Moerner WE. Nature Methods. 2008;5:947–949. doi: 10.1038/NMETH.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manley S, Gillette JM, Patterson GH, Shroff H, Hess HF, Betzig E, Lippincott-Schwartz J. Nature Methods. 2008;5:155–157. doi: 10.1038/nmeth.1176. [DOI] [PubMed] [Google Scholar]
- 47.Xie XS, Choi PJ, Li G-W, Lee and G. Lia NK. Annual Review of Biophysics. 2008;37:417–444. doi: 10.1146/annurev.biophys.37.092607.174640. [DOI] [PubMed] [Google Scholar]
- 48.Toomre DK, Bewersdorf J. Annual Review of Cell and Developmental Biology. 2010;26:285–314. doi: 10.1146/annurev-cellbio-100109-104048. [DOI] [PubMed] [Google Scholar]
- 49.Persson F, Barkefors I, Elf J. Current Opinion in Biotechnology. 2013;24:1–8. doi: 10.1016/j.copbio.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 50.Trautman JK, Macklin JJ. Chemical Physics. 1996;205:221–229. [Google Scholar]
- 51.Orrit M. Photochemical & Photobiological Sciences. 2010;9:637–642. doi: 10.1039/b9pp00192a. [DOI] [PubMed] [Google Scholar]
- 52.Rust MJ, Bates M, Zhuang X. Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 54.Hess ST, Girirajan TPK, Mason MD. Biophysical Journal. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rasnik I, McKinney SA, Ha T. Nat Meth. 2006;3:891–893. doi: 10.1038/nmeth934. [DOI] [PubMed] [Google Scholar]
- 56.Koopmans WJ, Brehm A, Logie C, Schmidt T, van Noort J. J Fluoresc. 2007;17:785–795. doi: 10.1007/s10895-007-0218-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tomschik M, Zheng H, Holde K. v., Zlatanova J, Leuba SH. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:3278–3283. doi: 10.1073/pnas.0500189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saxton MJ, Jacobson K. Annual Review of Biophysics and Biomolecular Structure. 1997;26:373–399. doi: 10.1146/annurev.biophys.26.1.373. [DOI] [PubMed] [Google Scholar]
- 59.Kusumi A, Nakada C, Ritchie K, Murase K, Suzuki K, Murakoshi H, Kasai RS, Kondo J, Fujiwara T. Annual Review of Biophysics and Biomolecular Structure. 2005;34:351–378. doi: 10.1146/annurev.biophys.34.040204.144637. [DOI] [PubMed] [Google Scholar]
- 60.Cang H, Shan Xu C, Yang H. Chemical Physics Letters. 2008;457:285–291. [Google Scholar]
- 61.Dixit R, Cyr R. The Plant Journal. 2003;36:280–290. doi: 10.1046/j.1365-313x.2003.01868.x. [DOI] [PubMed] [Google Scholar]
- 62.Sheetz MP, Koppel DE. Proceedings of the National Academy of Sciences. 1979;76:3314–3317. doi: 10.1073/pnas.76.7.3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vigers GP, Coue M, McIntosh JR. The Journal of Cell Biology. 1988;107:1011–1024. doi: 10.1083/jcb.107.3.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dobrucki JW, Feret D, Noatynska A. Biophysical Journal. 2007;93:1778–1786. doi: 10.1529/biophysj.106.096636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kassab K. Journal of Photochemistry and Photobiology B: Biology. 2002;68:15–22. doi: 10.1016/s1011-1344(02)00325-1. [DOI] [PubMed] [Google Scholar]
- 66.Turro NJ, Ramamurthy V, Scaiano JC. Modern Molecular Photochemistry of Organic Molecules. University Science Books; 2010. [Google Scholar]
- 67.Buschmann V, Weston KD, Sauer M. Bioconjugate Chem. 2003;14:195–204. doi: 10.1021/bc025600x. [DOI] [PubMed] [Google Scholar]
- 68.Properties of ATTO-dyes. http://www.atto-tec.com/fileadmin/user_upload/Katalog_Flyer_Support/Dye_Properties_01.pdf.
- 69.Webb JP, McColgin WC, Peterson OG, Stockman DL, Eberly JH. The Journal of Chemical Physics. 1970;53:4227–4229. [Google Scholar]
- 70.Chibisov AK, Zakharova GV, Gorner H. Journal of the Chemical Society, Faraday Transactions. 1996;92:4917–4925. [Google Scholar]
- 71.Menzel R, Thiel E. Chemical Physics Letters. 1998;291:237–243. [Google Scholar]
- 72.Kuzmin VA, Darmanyan AP. Chemical Physics Letters. 1978;54:159–163. [Google Scholar]
- 73.Tian H, Chen K. Dyes and Pigments. 1994;26:167–174. [Google Scholar]
- 74.Widengren J, Chmyrov A, Eggeling C, Lofdahl PA, Seidel CA. The journal of physical chemistry. A. 2007;111:429–440. doi: 10.1021/jp0646325. [DOI] [PubMed] [Google Scholar]
- 75.Jia K, Wan Y, Xia A, Li S, Gong F, Yang G. The Journal of Physical Chemistry A. 2007;111:1593–1597. doi: 10.1021/jp067843i. [DOI] [PubMed] [Google Scholar]
- 76.Zheng Q, Jockusch S, Zhou Z, Altman RB, Warren JD, Turro NJ, Blanchard SC. The Journal of Physical Chemistry Letters. 2012;3:2200–2203. doi: 10.1021/jz300670p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Montalti M, Credi A, Prodi L, Gandolfi MT. Handbook of photochemistry. 3rd CRC/Taylor & Francis; Boca Raton: 2006. [Google Scholar]
- 78.Byers GW, Gross S, Henrichs PM. Photochemistry and Photobiology. 1976;23:37–43. doi: 10.1111/j.1751-1097.1976.tb06768.x. [DOI] [PubMed] [Google Scholar]
- 79.Kuramoto N, Kitao T. Dyes and Pigments. 1982;3:49–58. [Google Scholar]
- 80.Bonnett R, Martínez G. Tetrahedron. 2001;57:9513–9547. [Google Scholar]
- 81.Sies H, Menck CFM. Mutation Research/DNAging. 1992;275:367–375. doi: 10.1016/0921-8734(92)90039-r. [DOI] [PubMed] [Google Scholar]
- 82.Davies MJ. Photochemical & Photobiological Sciences. 2004;3:17–25. doi: 10.1039/b307576c. [DOI] [PubMed] [Google Scholar]
- 83.Eggeling C, Widengren J, Rigler R, Seidel CAM. Applied Fluorescence in Chemistry, Biology and Medicine. Springer Berlin Heidelberg; 1999. pp. 193–240. [Google Scholar]
- 84.Vogelsang J, Kasper R, Steinhauer C, Person B, Heilemann M, Sauer M, Tinnefeld P. Angew Chem Int Ed Engl. 2008;47:5465–5469. doi: 10.1002/anie.200801518. [DOI] [PubMed] [Google Scholar]
- 85.Eggeling C, Widengren J, Rigler R, Seidel CAM. Analytical Chemistry. 1998;70:2651–2659. doi: 10.1021/ac980027p. [DOI] [PubMed] [Google Scholar]
- 86.Kong X, Nir E, Hamadani K, Weiss S. J Am Chem Soc. 2007;129:4643–4654. doi: 10.1021/ja068002s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eggeling C, Widengren J, Brand L, Schaffer J, Felekyan S, Seidel CAM. The Journal of Physical Chemistry A. 2006;110:2979–2995. doi: 10.1021/jp054581w. [DOI] [PubMed] [Google Scholar]
- 88.Zechmeister L, Pinckard JH. Experientia. 1953;9:16–17. doi: 10.1007/BF02147696. [DOI] [PubMed] [Google Scholar]
- 89.Dempster DN, Morrow T, Rankin R, Thompson GF. Journal of the Chemical Society, Faraday Transactions 2: Molecular and Chemical Physics. 1972;68:1479–1496. [Google Scholar]
- 90.Di Paolo RE, Scaffardi LB, Duchowicz R, Bilmes GM. The Journal of Physical Chemistry. 1995;99:13796–13799. [Google Scholar]
- 91.Luo G, Wang M, Konigsberg WH, Xie XS. Proceedings of the National Academy of Sciences. 2007;104:12610–12615. doi: 10.1073/pnas.0700920104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hwang H, Kim H, Myong S. Proceedings of the National Academy of Sciences. 2011;108:7414–7418. doi: 10.1073/pnas.1017672108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee J, Lee S, Ragunathan K, Joo C, Ha T, Hohng S. Angewandte Chemie International Edition. 2010;49:9922–9925. doi: 10.1002/anie.201005402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Roy R, Hohng S, Ha T. Nat Methods. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Altman RB, Zheng Q, Zhou Z, Terry DS, Warren JD, Blanchard SC. Nature Methods. 2012;9:428–429. doi: 10.1038/nmeth.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hohng S, Joo C, Ha T. Biophys J. 2004;87:1328–1337. doi: 10.1529/biophysj.104.043935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vogelsang J, Cordes T, Forthmann C, Steinhauer C, Tinnefeld P. Proc Natl Acad Sci U S A. 2009;106:8107–8112. doi: 10.1073/pnas.0811875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dempsey GT, Vaughan JC, Chen KH, Bates M, Zhuang X. Nature Methods. 2011;8:1027–1036. doi: 10.1038/nmeth.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mujumdar RB, Ernst LA, Mujumdar SR, Lewis CJ, Waggoner AS. Bioconjugate Chem. 1993;4:105–111. doi: 10.1021/bc00020a001. [DOI] [PubMed] [Google Scholar]
- 100.Cooper M, Ebner A, Briggs M, Burrows M, Gardner N, Richardson R, West R. Journal of Fluorescence. 2004;14:145–150. doi: 10.1023/b:jofl.0000016286.62641.59. [DOI] [PubMed] [Google Scholar]
- 101.Rurack K, Spieles M. Analytical Chemistry. 2011;83:1232–1242. doi: 10.1021/ac101329h. [DOI] [PubMed] [Google Scholar]
- 102.Ponterini G, Caselli M. Berichte der Bunsengesellschaft für physikalische Chemie. 1992;96:564–573. [Google Scholar]
- 103.Chibisov AK. Journal of Photochemistry. 1976;6:199–214. [Google Scholar]
- 104.Roth NJL, Craig AC. The Journal of Physical Chemistry. 1974;78:1154–1155. [Google Scholar]
- 105.Sanborn ME, Connolly BK, Gurunathan K, Levitus M. The Journal of Physical Chemistry B. 2007;111:11064–11074. doi: 10.1021/jp072912u. [DOI] [PubMed] [Google Scholar]
- 106.Mujumdar SR, Mujumdar RB, Grant CM, Waggoner AS. Bioconjugate Chem. 1996;7:356–362. doi: 10.1021/bc960021b. [DOI] [PubMed] [Google Scholar]
- 107.Chibisov AK, Zakharova GV, Goerner H, Sogulyaev YA, Mushkalo IL, Tolmachev AI. The Journal of Physical Chemistry. 1995;99:886–893. [Google Scholar]
- 108.Gu L, Hall DJ, Qin Z, Anglin E, Joo J, Mooney DJ, Howell SB, Sailor MJ. Nat Commun. 2013:4. doi: 10.1038/ncomms3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lemke EA, Gambin Y, Vandelinder V, Brustad EM, Liu H-W, Schultz PG, Groisman A, Deniz AA. Journal of the American Chemical Society. 2009;131:13610–13612. doi: 10.1021/ja9027023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Benesch RE, Benesch R. Science. 1953;118:447–448. doi: 10.1126/science.118.3068.447. [DOI] [PubMed] [Google Scholar]
- 111.Aitken CE, Marshall RA, Puglisi JD. Biophys J. 2008;94:1826–1835. doi: 10.1529/biophysj.107.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Swoboda M, Henig J, Cheng H-M, Brugger D, Haltrich D, Plumeré N, Schlierf M. ACS Nano. 2012;6:6364–6369. doi: 10.1021/nn301895c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schäfer P, van de Linde S, Lehmann J, Sauer M, Doose S. Analytical Chemistry. 2013;85:3393–3400. doi: 10.1021/ac400035k. [DOI] [PubMed] [Google Scholar]
- 114.van de Linde S, Krstic I, Prisner T, Doose S, Heilemann M, Sauer M. Photochemical & Photobiological Sciences. 2011;10:499–506. doi: 10.1039/c0pp00317d. [DOI] [PubMed] [Google Scholar]
- 115.Dave R, Terry DS, Munro JB, Blanchard SC. Biophys J. 2009;96:2371–2381. doi: 10.1016/j.bpj.2008.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yanagida T, Nakase M, Nishiyama K, Oosawa F. Nature. 1984;307:58–60. doi: 10.1038/307058a0. [DOI] [PubMed] [Google Scholar]
- 117.Giloh H, Sedat JW. Science. 1982;217:1252–1255. doi: 10.1126/science.7112126. [DOI] [PubMed] [Google Scholar]
- 118.Leslie RJ, Saxton WM, Mitchison TJ, Neighbors B, Salmon ED, McIntosh JR. The Journal of Cell Biology. 1984;99:2146–2156. doi: 10.1083/jcb.99.6.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Johnson GD, de GM, Nogueira Araujo C. Journal of Immunological Methods. 1981;43:349–350. doi: 10.1016/0022-1759(81)90183-6. [DOI] [PubMed] [Google Scholar]
- 120.Von Trebra R, Koch TH. Chemical Physics Letters. 1982;93:315–317. [Google Scholar]
- 121.Cordes T, Maiser A, Steinhauer C, Schermelleh L, Tinnefeld P. Physical Chemistry Chemical Physics. 2011;13:6699–6709. doi: 10.1039/c0cp01919d. [DOI] [PubMed] [Google Scholar]
- 122.Blanchard SC, Kim HD, Gonzalez RL, Jr., Puglisi JD, Chu S. Proc Natl Acad Sci USA. 2004;101:12893–12898. doi: 10.1073/pnas.0403884101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Grunwell JR, Glass JL, Lacoste TD, Deniz AA, Chemla DS, Schultz PG. J Am Chem Soc. 2001;123:4295–4303. doi: 10.1021/ja0027620. [DOI] [PubMed] [Google Scholar]
- 124.Glazer AN. The FASEB Journal. 1988;2:2487–2491. doi: 10.1096/fasebj.2.9.3371593. [DOI] [PubMed] [Google Scholar]
- 125.Cordes T, Vogelsang J, Tinnefeld P. J Am Chem Soc. 2009;131:5018–5019. doi: 10.1021/ja809117z. [DOI] [PubMed] [Google Scholar]
- 126.Vogelsang J, Steinhauer C, Forthmann C, Stein IH, Person-Skegro B, Cordes T, Tinnefeld P. ChemPhysChem. 2010;11:2475–2490. doi: 10.1002/cphc.201000189. [DOI] [PubMed] [Google Scholar]
- 127.Marling JB, Gregg DW, Wood L. Appl. Phys. Lett. 1970;17:527–530. [Google Scholar]
- 128.Wenthold PG, Hrovat DA, Borden WT, Lineberger WC. Science. 1996;272:1456–1459. doi: 10.1126/science.272.5267.1456. [DOI] [PubMed] [Google Scholar]
- 129.Frutos LM, Castano O, Andres JL, Merchan M, Acuna AU. The Journal of Chemical Physics. 2004;120:1208–1216. doi: 10.1063/1.1631418. [DOI] [PubMed] [Google Scholar]
- 130.Weninger K, Bowen ME, Chu S, Brunger AT. Proc Natl Acad Sci U S A. 2003;100:14800–14805. doi: 10.1073/pnas.2036428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Alejo Jose L., Blanchard Scott C., Andersen Olaf S. Biophysical Journal. 2013;104:2410–2418. doi: 10.1016/j.bpj.2013.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Altman RB, Terry DS, Zhou Z, Zheng Q, Geggier P, Kolster RA, Zhao Y, Javitch JA, Warren JD, Blanchard SC. Nature Methods. 2012;9:68–71. doi: 10.1038/nmeth.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liphardt B, Liphardt B, Lüttke W. Optics Communications. 1981;38:207–210. [Google Scholar]
- 134.Ernst LA, Gupta RK, Mujumdar RB, Waggoner AS. Cytometry. 1989;10:3–10. doi: 10.1002/cyto.990100103. [DOI] [PubMed] [Google Scholar]
- 135.Tinnefeld P, Cordes T. Nat Meth. 2012;9:426–427. doi: 10.1038/nmeth.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Murase K, Fujiwara T, Umemura Y, Suzuki K, Iino R, Yamashita H, Saito M, Murakoshi H, Ritchie K, Kusumi A. Biophysical journal. 2004;86:4075–4093. doi: 10.1529/biophysj.103.035717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Juette MF, Rivera-Molina FE, Toomre DK, Bewersdorf J. Appl. Phys. Lett. 2013:102. [Google Scholar]
- 138.Dupont A, Lamb DC. Nanoscale. 2011;3:4532–4541. doi: 10.1039/c1nr10989h. [DOI] [PubMed] [Google Scholar]
- 139.Huang F, Hartwich TMP, Rivera-Molina FE, Lin Y, Duim WC, Long JJ, Uchil PD, Myers JR, Baird MA, Mothes W, Davidson MW, Toomre D, Bewersdorf J. Nat Meth. 2013;10:653–658. doi: 10.1038/nmeth.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Saurabh S, Maji S, Bruchez MP. Optics Express. 2012;20:7338–7349. doi: 10.1364/OE.20.007338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Campos LA, Liu J, Wang X, Ramanathan R, English DS, Munoz V. Nat Meth. 2011;8:143–146. doi: 10.1038/nmeth.1553. [DOI] [PubMed] [Google Scholar]
- 142.Wang L, Pulk A, Wasserman MR, Feldman MB, Altman RB, Doudna Cate JH, Blanchard SC. Nat Struct Mol Biol. 2012;19:957–963. doi: 10.1038/nsmb.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dunkle JA, Wang L, Feldman MB, Pulk A, Chen VB, Kapral GJ, Noeske J, Richardson JS, Blanchard SC, Cate JH. Science. 2011;332:981–984. doi: 10.1126/science.1202692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Murakoshi H, Iino R, Kobayashi T, Fujiwara T, Ohshima C, Yoshimura A, Kusumi A. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:7317–7322. doi: 10.1073/pnas.0401354101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Reyes-Lamothe R, Possoz C, Danilova O, Sherratt DJ. Cell. 2008;133:90–102. doi: 10.1016/j.cell.2008.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Henzler-Wildman K, Kern D. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 147.Geggier P, Dave R, Feldman MB, Terry DS, Altman RB, Munro JB, Blanchard SC. J Mol Biol. 2010;399:576–595. doi: 10.1016/j.jmb.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schweitzer C, Schmidt R. Chemical Reviews. 2003;103:1685–1758. doi: 10.1021/cr010371d. [DOI] [PubMed] [Google Scholar]
- 149.Geddes C, Lakowicz J. Journal of Fluorescence. 2002;12:121–129. doi: 10.1023/A:1021341305229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kinkhabwala A, Yu Z, Fan S, Avlasevich Y, Mullen K, Moerner WE. Nat Photon. 2009;3:654–657. [Google Scholar]
- 151.Acuna GP, Möller FM, Holzmeister P, Beater S, Lalkens B, Tinnefeld P. Science. 2012;338:506–510. doi: 10.1126/science.1228638. [DOI] [PubMed] [Google Scholar]