

Abstract
Secreted and cell surface-localized members of the immunoglobulin superfamily (IgSF) play central roles in regulating adaptive and innate immune responses, and are prime targets for the development of protein-based therapeutics. An essential activity of the ectodomains of these proteins is the specific recognition of cognate ligands, which are often other members of the IgSF. In this work we provide functional insight for this important class of proteins through the development of a clustering algorithm that groups together extracellular domains of the IgSF with similar binding preferences. Information from hidden Markov model-based sequence profiles and domain structure is calibrated against manually curated protein interaction data to define functional families of IgSF proteins. The method is able to assign 82% of the 477 extracellular IgSF protein to a functional family, while the rest are either single proteins with unique function or proteins that could not be assigned with the current technology. The functional clustering of IgSF proteins generates hypotheses regarding the identification of new cognate receptor:ligand pairs and reduces the pool of possible interacting partners to a manageable level for experimental validation.
Keywords: Immunoglobulin superfamily, protein-protein interaction, functional prediction
Introduction
The immunoglobulin superfamily is one of the largest domain families in the human proteome, encompassing over 700 cell surface and soluble proteins1. Members of the IgSF contain at least one immunoglobulin (Ig) domain, which is a 70–110 residue long β-sandwich fold, many of which contain a conserved disulfide bond connecting its B and F strands. The ancestral function of IgSF proteins is believed to be the mediation of homotypic cell-cell adhesion2. In vertebrates, IgSF proteins have evolved to play key roles in cell recognition and adhesion, developmental and morphogenetic processes, and innate and adaptive immune responses3. In addition to antibodies and T-cell receptors (TCRs), the human IgSF contains 477 cell-surface or secreted proteins (hereon referred to as ‘extracellular IgSF’, see Supplementary Table 1). Many of these extracellular IgSF proteins contribute to the immune response through specific cell-to-cell (trans) receptor:ligand interactions. While some of these IgSF members have been extensively studied, many remain uncharacterized with regard to interacting ligand(s) and specific function. There is strong biomedical motivation for defining the receptor:ligand relationships of these molecules. For example, CD80 and CD86 on antigen presenting cells bind to CD28 or Cytotoxic T-lymphocyte protein 4 (CTLA-4) on T-cells, triggering co-stimulatory or inhibitory T-cell responses, respectively4. Orencia™, a soluble version the CTLA-4 ectodomain, engages the CD80/CD86 ligands on antigen-presenting cells, interferes with their interaction with CD28 and results in blockade of the CD28-based co-stimulation of T-cells5. This behavior results in global suppression of T cell immunity, making Orencia a leading therapy for autoimmune diseases such as rheumatoid arthritis. A high affinity variant, Belatacept, has received FDA approval for the prevention of kidney transplant rejection6. Furthermore, Ipilimumab, a function blocking mAb targeting CTLA4, results in systemic immune activation and is the most recently developed therapeutic for the treatment of late stage melanoma7.
With the long-term goal of systematically defining the entire ensemble of receptor:ligand interactions involving the extracellular IgSF proteins, we present a computational method to cluster these proteins into functional families. As the primary function of the IgSF ectodomains is binding, we define a functional family as a group of IgSF proteins that have similar binding properties, i.e., the recognition of the same extracellular partner in a similar fashion (binding site and pose). This clustering generates binding partner predictions that reduce the number of potential candidate interactions requiring experimental verification. In addition, the resulting functional families can be analyzed to identify family-specific physico-chemical features, such as conserved side chain patterns that enable members of the same family to bind related ligands in a similar manner. This clustering can also reveal unique structural features that contribute to novel function. These considerations are of the upmost importance for large-scale structural genomics efforts, as they provide powerful criteria for identifying and prioritizing those functional families that lack any experimental characterization, as well as unclustered singletons that have no significant similarity to other IgSF proteins8. These are the targets that are most likely to benefit from structural analysis.
Functional clustering using sequence or structural similarity is a well-established approach, but has many subtleties. One complication is that unrelated proteins can have the same function due to convergent evolution. While this is a rather rare situation, there are several such documented cases9; 10. Even between proteins with common ancestries, it is difficult to assert common or related functionalities using clustering methods that are based on pairwise sequence identity (e.g. BlastClust11 and CD-HIT12). This difficulty arises because most functionally-related proteins diverged so far over time that their global pairwise sequence identities are indistinguishable from those of otherwise unrelated proteins13. For instance, in case of IgSF proteins, the ectodomains of CD80 and the functionally related CD86 share only 27% sequence identity, whereas CD80 shares greater than 27% sequence identity with other, functionally unrelated IgSF proteins such as immunoglobulin superfamily DCC subclass member 4 (IGDC4)14 and neural cell adhesion molecule L1 (L1CAM)15. Thus, consideration of pairwise sequence comparisons alone does not allow for robust prediction of functional relatedness.
Agglomerative clustering methods have been more successful in subfamily identification than direct sequence comparisons. SCI-PHY16 uses Dirichlet mixture densities to construct profiles for subtrees, and relative entropy as a distance function for merging subtrees. Similarly, GEMMA17 uses profile-profile comparison to cluster Gene3D superfamilies into functional subfamilies (available as ‘FunFams’18 from the CATH-Gene3D database). BAR+19, ProtoNet20, and TRIBE-MCL21 are large-scale databases that generate protein clusters based on sequence similarities from BLAST for all proteins in the Uniprot database.
A central challenge to subfamily clustering is the selection of an appropriate cutoff for the established phylogenetic tree of functional relatedness such that the granularity of the resulting families is appropriate for the research question at hand. SCI-PHY uses a minimum-encoding-cost criterion to automatically determine the cutoff. FunFams offers two subfamily granularities: a coarse FunFams layer that exploits available GO annotation data to select a protein-family specific cutoff, and a fine FunFams layer using a fixed cutoff that is based on a training set of six families from the enzyme Structure– Function Linkage Database22. TRIBE-MCL21 uses Markov cluster (MCL) method to detect natural clusters based on the distribution of edges. In contrast to these generic purpose clustering approaches, Rubinstein et. al. used a sequence-based method, termed the Brotherhood algorithm, that utilizes intermediate sequence information to cluster IgSF proteins into functionally related families23. Brotherhood method was calibrated empirically using a set of 14 hand-curated IgSF functional families, but without paying attention if these really share common ligands.
A related field to functional clustering is functional annotation, where a sequence of unknown function is explored using information of amino acid sequence, phylogeny, genomic context, protein-protein interaction networks, protein structure, microarray expression data or a combination of these data types. Clustering is one of the approaches that guides transfer of functional annotation (PANNZER24, BAR+19). Conversely functional annotation can be used to guide functional clustering (FunFams). A recent large scale critical assessment of protein function annotation (CAFA)24 evaluated 54 functional annotation algorithms, using Gene Ontology (GO) terms as performance benchmarks. A phylogenomics-based method, SIFTER25, shows promise by taking a reconciled phylogeny for a protein family and using a statistical model of function evolution that accounts for lineage-specific rate variation to incorporate annotations throughout an evolutionary tree. However, the current algorithm transfers rather generic GO annotations, which are usually not specific enough for identifying trans-cellular binding partners. For instance, the most specific GO molecular function annotation currently available for CD80 and CD86 is ‘coreceptor activity’, which does not provide ligand information to generate experimental hypothesis and would be indistinguishable from many of the hundreds of secreted, cell surface IgSF proteins.
We present a method for functional clustering that differs from the usual sequence similarity methods. First, we assessed the sequence similarity between two proteins by comparing their respective sequence profile-based hidden Markov models (HMMs)26. This amplifies signals from the conserved (and often functionally more important) portions of the sequences and downplays the role of less conserved segments. Second, and most importantly, we introduced a novel approach to calibrate profile similarity among functionally related proteins using the available experimental protein-protein interaction data. We compiled a training set of IgSF pairs that share the same experimentally verified ligands from the STRING protein interaction database27, and used it to guide the optimal grouping of candidate proteins. Furthermore, in our similarity measure of IgSF proteins, we can directly include other empirical information about the specific functions of these proteins. For instance, previous studies of IgSF domain activity in cell adhesion have identified the N-terminal IgSF domain as the domain most frequently involved in binding interactions28; 29; 30. We have included this information as a criterion in our similarity measure, so that IgSF pairs that share sequence homology at the N-terminus are considered more functionally similar than those that share homology over disparate segments of their sequences. Finally, we reduce erroneous linkages in our clustering by hierarchically clustering our IgSF pair distances into a graph tree31.
The method, Protein Interactions Calibrated hidden Markov model Tree (PICTree) was applied to the subproteome of 477 extracellular human IgSF proteins, resulting in the assignment of 390 to respective functional families. The resulting functional groups can serve as a starting platform to form hypothesis about possible new receptor-ligand interactions. We discuss one such case for the VSIG8 and the cortical thymocyte marker in Xenopus (CTX) family of proteins. The method can be readily adapted to handle other classes of proteins, and can be easily updated to include additional empirical information about the binding modes of proteins.
Results and Discussion
Functional clustering of all known 477 human IgSF proteins
Positive and negative training sets for the calibration profile similarity were prepared from the STRING database27, a web resource for protein-protein interactions that integrates meta information from experiments, computational methods, and text-mining. The positive training set contained 55 manually curated non-redundant IgSF pairs, each binding at least one common trans-binding ligand according to STRING (Table 1). As an example, high-quality protein-protein interaction records in STRING identified both CD80 and CD86 as binding partners to CD28 and CTLA-4. These interactions were further manually verified to be cell-to-cell, trans-binding interactions. CD80/CD86 [common ligand: CD28 and CTLA-4] and CD28/CTLA-4 [common ligand: CD80 and CD86] hence constitute two common ligand IgSF pairs in our positive training set.
Table 1.
Positive training set of non-redundant common-ligand binding IgSF pairs, with and corresponding node-to-node heights (h). Bold: outlying IgSF pairs with h greater than cutoff; Ig-only: pairs where both proteins have only Ig domain(s) in their extracellular segments (this subset was used for benchmarking with FunFams)
| IgSF1 | IgSF2 | Common Extracellular Interactors | h | Ig-only |
|---|---|---|---|---|
| CD226 | PVRL3 | PVR PVRL2 | 2.925 | X |
| CD226 | TACT | PVR | 0.410 | X |
| CD28 | CTLA4 | CD80 CD86 | 0.071 | X |
| CD80 | CD86 | CD28 CTLA4 | 0.100 | X |
| CNTN2 | L1CAM | NCAN | 0.117 | |
| CNTN2 | NCAM1 | NCAN | 0.402 | |
| CNTN2 | NFASC | NRCAM | 0.117 | |
| KI2L4 | LIRB2 | HLA-G (2) | 0.063 | X |
| KI2L1 | KI3S1 | HLA-C (3) | 0.072 | X |
| KI3S1 | LIRB2 | HLA-C, HLA-A (8) | 0.072 | X |
| LIRB1 | LIRB2 | HLA-A, HLA-C, HLA-F HLA-G | 0.019 | X |
| MERTK | TYRO3 | growth arrest-specific 6 | 0.192 | |
| MERTK | UFO | growth arrest-specific 6 | 0.192 | |
| TYRO3 | UFO | growth arrest-specific 6 | 0.124 | |
| CSPG2 | PGCA | fibulin-1, filbulin-2 | 0.141 | |
| CSPG2 | PGCB | filbulin-2 | 0.082 | |
| DCC | NEO1 | Netrin-1 | 0.074 | |
| FCG2A | FCG2B | C-reactive protein | 0.011 | X |
| FCG2A | FCGR1 | C-reactive protein | 0.034 | X |
| FGFR1 | FGFR2 | fibroblast growth factor 4, keratinocyte growth factor | 0.135 | |
| FGFR1 | FGFR3 | fibroblast growth factor 4, keratinocyte growth factor | 0.162 | |
| FGFR1 | FGFR4 | fibroblast growth factor 4 | 0.162 | |
| FGFR2 | FGFR3 | fibroblast growth factor 4, keratinocyte growth factor | 0.162 | |
| FGFR2 | FGFR4 | fibroblast growth factor 4 | 0.162 | |
| FGFR3 | FGFR4 | fibroblast growth factor 4 | 0.141 | |
| HLAF | HLAG | LIRB1 LIRB2 | 0.009 | |
| ICAM1 | ICAM2 | MAC-1 (Integrin αMβ2) | 0.017 | X |
| ICAM1 | ICAM3 | LFA-1 (Integrin αLβ2) | 0.017 | X |
| ICAM1 | ICAM5 | LFA-1 (Integrin αLβ2) | 0.017 | X |
| ICAM3 | ICAM5 | LFA-1 (Integrin αLβ2) | 0.010 | X |
| L1CAM | NCAM1 | NCAN | 0.402 | |
| NRG1 | NRG2 | Receptor tyrosine-protein kinase erbB-3 Receptor tyrosine-protein kinase erbB-4 |
0.070 | |
| NTRK1 | NTRK2 | Neurotrophin-3 | 0.138 | |
| NTRK1 | NTRK3 | Neurotrophin-3 | 0.129 | |
| NTRK2 | NTRK3 | Neurotrophin-3 | 0.138 | |
| PD1L1 | PD1L2 | PDCD1 | 0.060 | X |
| PGCA | PGCB | Fibulin-2 | 0.141 | |
| PGFRA | PGFRB | Platelet-derived growth factor subunit B | 0.086 | X |
| PVRL3 | PVRL4 | PVRL1 | 0.107 | X |
| PVRL3 | TACT | PVR | 2.925 | X |
| PVR | PVRL2 | CD226 PVRL3 | 0.044 | X |
| ROBO1 | ROBO2 | Slit homolog 2 protein | 0.070 | |
| SEM3A | SEM3B | Neuropilin-1 | 0.028 | |
| SEM3A | SEM3C | Neuropilin-1 | 0.021 | |
| SEM3A | SEM3F | Neuropilin-1 | 0.021 | |
| SEM3B | SEM3C | Neuropilin-1, Neuropilin-2 | 0.028 | |
| SEM3B | SEM3F | Neuropilin-1, Neuropilin-2 | 0.028 | |
| SEM3C | SEM3F | Neuropilin-1, Neuropilin-2 | 0.016 | |
| SEM3E | SEM4A | Neuropilin-2 | 0.048 | |
| SEM4A | SEM4D | Plexin-D | 0.046 | |
| SHPS1 | SIRPG | CD47 | 0.019 | X |
| UNC5A | UNC5B | Netrin-1 | 0.036 | |
| UNC5B | UNC5C | Netrin-1 (2) | 0.036 | |
| VGFR2 | VGFR3 | Vascular endothelial growth factor C Vascular endothelial growth factor D |
0.073 | |
| WFKN1 | WFKN2 | Growth/differentiation factor 11 | 0.019 |
We also extracted a ‘negative’ training set of 36,066 non-redundant IgSF pairs that are not known to bind any common ligand. This negative training set is an approximation of the true negative set, because it is not possible to definitively establish that two IgSFs do not share any common ligand. This is because (i) there is an enormous number of possible common ligands to check; (ii) such binding experiments might not have been performed; (iii) negative binding results are not recorded in protein interaction databases; (iv) the existence of false negatives - even when two proteins were reported not to interact, subsequent experiments could prove otherwise. As an example of this latter issue, myelin-associated glycoprotein was reported to be unable to bind fibronectin32; however, a subsequent paper reported otherwise33. For these reasons, our negative training set includes IgSF pairs that, in the future, could be shown to share common ligands when more experimental data become available.
We generated a PICTree clustering for the 477 IgSF proteins from our dissimilarity matrix computed (see Methods). We define a measure, h-value, which is the node-to-node distance between two IGSF proteins within the graph tree, to characterize functional similarity. The distributions of h-values within PICTree for the 55 IgSF pairs in our positive and the 36,066 pairs in our negative training set are well separated (Fig. 1). Out of the 55 positive common-ligand IgSF pairs, 50 have h values less than 0.2, while the remaining five outliers (Table 1, in bold) have h values between 0.402 to 2.925. In contrast, the negative dataset has h ranging from 0 to 21.02, with 95% of them between 0.5–5.0. Overall, h values for the full set of 477 IgSF proteins studied ranged from 0 to 28.6. To determine an optimal cutoff for delineating functional families, we plotted the sensitivity and specificity of our predictions as a function of various h cutoffs (Fig. 2). We aimed to identify an optimal cutoff that achieves greater than 90% sensitivity, while maximizing the specificity. The optimal trade-off is achieved at h = 0.192, corresponding to a sensitivity of 90.9% and a specificity of 99.2% with an upper bound on the false discovery rate at 0.8%. Fig. 3 shows the performance of the PICTree method on positive training set at the selected cutoff.
Figure 1.

Distribution of PICTree node-to-node distances for the training sets. Green solid bars: node-to-node distance distribution of the positive dataset of 55 common-ligand IgSF pairs; red shaded bars: distribution of a representative subset from the negative training set of IgSF pairs with no known common ligands (see Methods).
Figure 2.

Specificity (black circles) and sensitivity (red crosses) of the PICTree method at various node-to-node distance cutoff values.
Figure 3.

Performance of PICTree method on 55 non-redundant common-ligand IgSF pairs that share experimentally verified common ligand from the STRING database. An edge between two IgSF proteins signifies that they share at least one common verified trans-binding ligand. Green edges denote linkages that can be predicted using PICTree method (true positives); red edges denote outliers above the chosen height cutoff of hc = 0.192 (false negatives).
The methodology was applied to assign all 477 human extracellular IgSF proteins into functional groups. IgSF pairs with pairwise PICTree node-to-node distance h less than the cutoff hc = 0.192 are assigned to the same functional family. We predict 390 IgSFs to share common ligand(s) with at least one other IgSF protein (Fig. 4 and Supplemental Table 2), forming 83 functionally-related clusters; while 87 IgSFs remained unclustered singletons. Our positive training set of 55 common-ligand IgSF pairs is distributed across 26 different clusters. A detailed list of the clusters is available in Supplementary Table 2.
Figure 4.

Clustering of 477 human IgSF proteins into functionally related IgSF clusters using our PICTree-based method. Of the 477 proteins (denoted as nodes), 390 can be assigned to functionally related clusters, while 87 remained singletons.
Figure 5(a) shows the distribution of the cluster size from PICTree. Fifty-three out of the 83 multi-member clusters are between 2 to 4 members. The three largest clusters are dominated by members from gene clusters: Cluster 3 (30 out of 34 members from the Leukocyte Receptor Complex34); Cluster 18 (20 out of 20 members from the Pregnancy-Specific Glycoprotein gene cluster35;), and Cluster 10 (7 out of 17 members from the Butyrophilin gene cluster36). Figure 5(b) shows the variance of extracellular segment sequence length amongst cluster members as a function of cluster size. The lack of correlation between length variance and cluster size suggests that IgSFs with different domain architectures are capable of binding the same ligand using a common N-terminal domain. For instance, T-cell immunoglobulin and ITIM domain protein (TIGIT)37, which has one single Ig-V domain, shares the same ligand (PVR) with the nectins, which extracellular segment comprise of a single N-terminal Ig-V domain and two proximal Ig-C domains. Hence, a large length variance is not necessary indicative of functional mis-assignment.
Figure 5.


Analysis of PICTree cluster sizes. (a) Distribution of PICTree cluster sizes, (b) Variance in sequence length amongst cluster members against cluster size.
Comparison with other clustering algorithms
We compared PICTree to three other subfamily clustering methods, SCI-PHY16, FunFams18, and BAR+19. For SCI-PHY, we used both versions 1.0 and 3.0 (‘SCIPHY1’ and SCIPHY3) and three different algorithms (MUSCLE38, CLUSTALW39, and MAFFT40) to generate the required input multiple sequence alignments (MSAs). Performances were benchmarked against the positive set of 55 common-ligand IgSF pairs and the negative set of 36,066 pairs with no known common ligand. Since FunFams assignments are domain-based, IgSF sequences were scanned against the CATH Ig superfamily to download FunFams assignment for the Ig domains only. To provide a fair comparison, performance was measured on a smaller, ‘Ig-only’ subset involving only IgSF pairs with only Ig domains in their extracellular segments (see Methods).
The performances of all clustering schemes are shown in Table 2. Interestingly the performance of SCI-PHY 1.0 is significantly better than that of SCI-PHY 3.0. Both versions showed a strong dependence on the choice of generating the input MSA, however SCI-PHY 3.0 provided extreme results by clustering the IgSFs into either one single family (Table 2a: SCIPHY3_with MAFFT or MUSCLE inputs) or into mostly singletons (SCIPHY3_with CLUSTALW input). The older version, SCI-PHY 1.0 gave a more consistent performance, with comparable specificity to PICTree. It was unclear what caused the discrepancy in performance between the two SCI-PHY versions – although one documented difference between the two versions is the switch from SAM-HMM41 in version 1.0 to HMMER42 in version 3.0. BAR+ has slightly superior specificity over PICTree, but has poor sensitivity (50.91% for ‘all’ and 15% for ‘ig-only’ test sets). A key reason could be that clusters in the BAR+ database are based on the entire length of proteins, whereas our current PICTree clustering is based on the extracellular segment of proteins, because we are interested in extracellular binding function. Overall, PICTree has the best sensitivity amongst all methods tested for both ‘all’ and ‘Ig-only’ benchmarks (excluding the single-cluster results of SCIPHY3_MAFFT and SCIPHY3_MUSCLE).
Table 2.
Comparison of PICTree against other cluster methods.
| (a) Number of clusters generated. | |||
|---|---|---|---|
| Method | # of multimember clusters | # of singletons | total |
| PICTREE | 83 | 87 | 170 |
| ORENGO (FunFams) | 33 | 36 | 69 |
| SCIPHY1_CLUSTALW | 85 | 135 | 220 |
| SCIPHY1_MAFFT | 117 | 74 | 191 |
| SCIPHY1_MUSCLE | 97 | 174 | 271 |
| SCIPHY3_CLUSTALW | 44 | 399 | 443 |
| SCIPHY3_MAFFT | 1 | 0 | 1 |
| SCIPHY3_MUSCLE | 1 | 0 | 1 |
| BAR+ | 69 | 245 | 314 |
| (b) Performance against positive and negative data set | ||||||||
|---|---|---|---|---|---|---|---|---|
| method | nTP | nFN | nPos | nFP | nTN | nNeg | Sensitivity | Specificity |
| Benchmark set : alla | ||||||||
| PICTREE | 50 | 5 | 55 | 287 | 35779 | 36066 | 90.91 | 99.20 |
| SCIPHY1_CLUSTALW | 39 | 16 | 55 | 121 | 35945 | 36066 | 70.91 | 99.66 |
| SCIPHY1_MAFFT | 44 | 11 | 55 | 133 | 35933 | 36066 | 80.00 | 99.63 |
| SCIPHY1_MUSCLE | 22 | 33 | 55 | 53 | 36013 | 36066 | 40.00 | 99.85 |
| SCIPHY3_CLUSTALW | 5 | 50 | 55 | 14 | 36052 | 36066 | 9.09 | 99.96 |
| SCIPHY3_MAFFT | 55 | 0 | 55 | 36066 | 0 | 36066 | 100.00 | 0.00 |
| SCIPHY3_MUSCLE | 55 | 0 | 55 | 36066 | 0 | 36066 | 100.00 | 0.00 |
| BAR+ | 28 | 27 | 55 | 39 | 36027 | 36066 | 50.91 | 99.89 |
| Benchmark set : Ig-onlyb | ||||||||
| PICTREE | 17 | 3 | 20 | 120 | 8863 | 8983 | 85.00 | 98.66 |
| ORENGO (FunFams) | 15 | 5 | 20 | 634 | 8349 | 8983 | 75.00 | 92.94 |
| SCIPHY1_CLUSTALW | 11 | 9 | 20 | 43 | 8940 | 8983 | 55.00 | 99.52 |
| SCIPHY1_MAFFT | 13 | 7 | 20 | 36 | 8947 | 8983 | 65.00 | 99.60 |
| SCIPHY1_MUSCLE | 7 | 13 | 20 | 25 | 8958 | 8983 | 35.00 | 99.72 |
| SCIPHY3_CLUSTALW | 0 | 20 | 20 | 0 | 8983 | 8983 | 0.00 | 100.00 |
| SCIPHY3_MAFFT | 20 | 0 | 20 | 8983 | 0 | 8983 | 100.00 | 0.00 |
| SCIPHY3_MUSCLE | 20 | 0 | 20 | 8983 | 0 | 8983 | 100.00 | 0.00 |
| BAR+ | 3 | 17 | 20 | 12 | 8971 | 8983 | 15.00 | 99.87 |
all: Benchmark comprises of all 55 pairs in positive set and 36066 pairs in negative set
Ig-only: Benchmark involves only 264 IgSFs that have exclusively Ig domains in their extracellular segments, corresponding to a subset of 20 pairs in positive set and 8983 pairs in negative set.
We next compared our PICTree clustering results to that from the Brotherhood algorithm23(Fig. 6). Of the 14 functional families assigned by the Brotherhood algorithm, PICTree obtained the same assignments for two families (TIM, semaphorins); and further divided nine families (B7/ butyrophilin, SLAM, nectin/nectin-like, PGFR, SIGLEC, CD28, MHC-II, CTX, CEACAM/PSG) into subclusters or as singletons. This subdivision is not surprising because PICTree uses a more stringent cutoff hc based on IgSF pairs binding identical ligands. For instance, the Brotherhood algorithm assigned CD80 and CD86 to the same family as the butyrophilins, whereas PICTree considers CD80 and CD86 as a separate family, in line with the experimental observation that CD80 and CD86 share the same binding partners, CD28 and CTLA4, while butyrophilins do not bind CD28 or CTLA4.
Figure 6.

PICTree clustering of 14 functional families predicted by Brotherhood algorithm23. A subset of the PICTree clustering involving the 14 Brotherhood algorithm-predicted families is shown. Family designations from Brotherhood algorithm are denoted by color: yellow: Leukocyte immunoglobulin-like receptor (LIR), pink: Killer cell immunoglobulin-like receptor (KIR), dark blue: CEACAM/PSG, green: B7/butyrophilin, orange: semaphorin, olive: sialic acid-binding immunoglobulin-type lectins (SIGLEC), light blue: MHC-I, turquoise: MHC-II, brown: cortical thymocyte marker in Xenopus(CTX), light grey: nectin/nectin-like, dark grey: T-cell immunoglobulin and mucin domain-containing (TIM), red: Signaling lymphocytic activation molecule (SLAM), purple: Platelet-derived growth factor receptor (PGFR), black: CD28.
In the remaining cases, PICTree either combined or added new members to the Brotherhood families. For instance, KIR and LIR were combined in the PICTree clustering along with newly added members. The joining of KIR and LIR families by PICTree made sense because KIR and LIR families both contain MHC-I binding proteins. The new members LAIR1, LAIR2, FCAR, OSCAR, GPVI, and NCTR1 are all co-located with KIR and LIR on the leukocyte receptor complex (LRC) on chromosome 19q13.4 in human, sharing ancient homology with KIR and LIR that likely arisen from gene duplication, although their known ligands do not include MHC-I34. In another example, the MHC-I family was also assigned new members HMR1, ZAGL1, ZA2G, HFE, MICA, MICB, FCGRN. Of these, HMR1 is shown to have antigen-presenting activity43. Finally, PICTree assigned to the B7/ butyrophilin family a new member, selection and upkeep of intraepithelial T-cells protein 1 homolog (SKIT1). Mouse SKIT1 is expressed in thymus and skin epithelia and is essential for positive selection of Vγ5+Vδ1+ T cells44, although the exact mechanism of its interaction with T cell is not known.
Node-to-node distances within PICTree are a better discriminator of functional similarity than pairwise sequence identity
A key issue is the accuracy of PICTree in quantifying functional relationships between proteins compared to the performance of traditional approaches based on pairwise sequence identity. The pairwise sequence identities of the 55 verified common-ligand pairs range between 20.5–93.5%. In contrast, their PICTree h values have a tight range between 0–2.925, out of a possible observed range of 0-28.6 (Fig. 7). If we omit the five pairs that were not properly assigned by PICTree (red circles in Fig. 7 and bold in Table 1), the remaining 50 verified common-ligand IgSF pairs (black circles) have an even tighter range of h (0–0.192), even though their sequence identities are widely distributed between 26–94%. This observation suggests that PICTree node-to-node distance is a more discriminating predictor of common-binding functionality.
Figure 7.

Comparison of PICTree and pairwise sequence identity methods to predict common-ligand IgSF pairs. Sequence identity is plotted against PICTree node-to-node distances for 55 non-redundant IgSF protein pairs sharing common binding partners (black and red circles) and 74,710 non-redundant pairwise combinations of other IgSF proteins (brown crosses). Two optimal cutoff criteria are shown: one for sequence identity (blue dashed line) and one for PICTree node-to-node distance (green dashed-dotted line).
We illustrate this practical advantage of PICTree’s by estimating the number of testable IgSF:X interactions (ntest) generated by PICTree and pairwise sequence identity clustering results respectively. We first note that the exact number of known trans-binding IgSF:X interactions for all 477 IgSFs is currently not known – our manually curated list of 175 IgSF:X interactions is intended for training (see Methods) and represents just a small, high-quality subset of all reported interactions that could potentially be screened. Second, a poor clustering method that over-merges functional families could result in multiple known ligands within a cluster. To get an upper estimate of ntest, we consider here an extreme case where for each non-redundant IgSF pair (i,j) in a cluster, there is a known unique ligand for i that we need to test against j. The number of testable ligand-receptor interactions is then estimated by where Ncluster is the number of mulit-member clusters generated (i.e. excluding singletons), and N(i,j)redundant,c the number of non-redundant IgSF pairs in cluster c.
When no clustering is performed (i.e. all 477 IgSFs are in a single super-cluster), there are 74,710 non-redundant IgSF pairs (Fig. 7, brown crosses) and hence ntest = 74,710. If pairwise sequence identity is used as the metric for functional relatedness, an identity of ≥ 26% is required to correctly capture the 50 verified common-ligand IgSF pairs to attain 90.2% sensitivity. Clustering by sequence identity at this criterion will result in 6,338 non-redundant, clustered IgSF pairs (Fig. 7, brown crosses above blue dashed line) for experimental verification. In contrast, a PICTree node-to-node distance cutoff of hc = 0.192 captures the same 50 verified common-ligand pairs but predicts a more manageable ntest =1,072 (Fig. 7, brown crosses left of green dashed-dotted line). This number is comparable to a recently reported large scale screen involving ~1000 ligand-receptor interactions that resulted in the identification of poliovirus receptor (PVR), and PVR-like proteins 2 and 3 as binding partners for TIGIT37.
PICTree method can predict novel functional assignments: the case of VSIG8 protein
The clustering of IgSF members into functional families allows us to generate hypotheses regarding previously uncharacterized ligand-receptor interactions. If at least one member of a family has a known ligand or distinct binding feature, one can speculate that the other family members share this feature. VSIG8 (V-set containing and immunoglobulin domain containing protein 8) provides one such example. VSIG8 is a 414-residue transmembrane protein found in stratified epithelia of hair follicle, nail and oral cavity45; 46, with no trivial relationship to other IGSFs. The PICTree method assigned VSIG8 to Cluster No. 4 (see Supplementary Table 2) in which all other constituents are known members of the CTX gene family (Fig. 8). As with VSIG8, all other members of this PICTree cluster are single-pass membrane proteins with two extracellular Ig domains. Coxsackievirus and adenovirus receptor (CXAR) and endothelial cell-selective adhesion molecule (ESAM) are found in cellular tight junctions: CXAR is an essential component of tight junctions in simple epithelial cells, but is not found in stratified epithelia; whereas ESAM is selectively expressed in cultured human and murine vascular endothelial cells47. In addition, it has been shown that along with ESAM47 and CXAR48, two additional Cluster 4 members, adipocyte adhesion molecule (ACAM)49 and immunoglobulin superfamily member 11 (IGS11)50, also mediate cell-cell adhesion through homophilic dimerization. We therefore propose that VSIG8 is the counterpart to CXAR, serving the analogous function of maintaining tight junctions in stratified epithelia through homophilic trans-dimerization.
Figure 8.
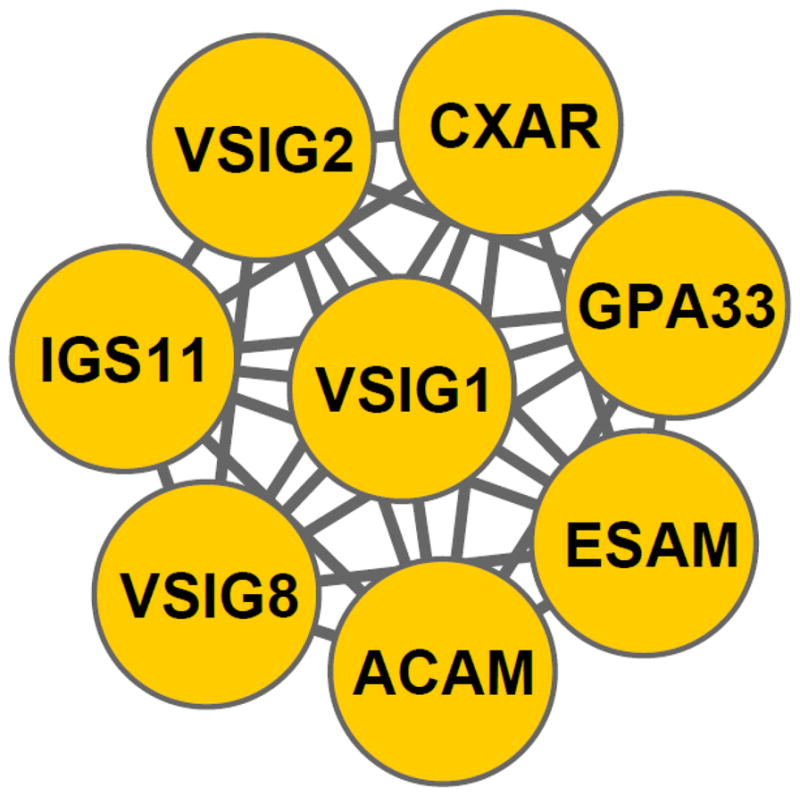
Novel assignment of VSIG8 to the cortical thymocyte marker in Xenopus (CTX) gene family
Analysis of five mis-annotated IgSF pairs
It is important to examine the five common-ligand IgSF pairs in the positive training set that PICTree clustering failed to predict correctly (Table 1, bold). The first two pairs, contactin-2 (CNTN2)/neural cell adhesion molecule 1 (NCAM1) [common ligand: neurocan (NCAN)], and L1CAM / NCAM1 [common ligand: neurocan], involved neural cell adhesion proteins critical for neuronal development. NCAM1 and L1CAM can bind both the chondroitin sulphate chains and the core protein of NCAN51, whereas CNTN2 only binds to the NCAN core protein52. For NCAM1 and L1CAM, their chondroitin sulphate binding sites are both located on Ig domains (second Ig domain for NCAM1 53 and first Ig for L1CAM54). Both binding sites involve strands C and G, although the NCAM1’s binding site is more extensive. Hence evidence points to L1CAM and NCAM1 binding chrodroitin sulphate chains on NCAN via similar mode. On the other hand, the core-NCAN-binding sites on CNTN2, and NCAM1 are unknown, so there is not enough evidence to ascertain if CNTN2 and NCAM1 bind core NCAN protein via the same mode.
The remaining three common-ligand IgSF pairs that escaped PICTree prediction are: CD226-PVRL3 [common ligand: PVR and PVRL2], CD226-T-cell-activated increased late expression protein (TACT) [common ligand: PVR], and PVRL3-TACT [common ligand: PVR], all belong to the nectin/nectin-related family23. While PVRL3, TACT and CD226 all bind to PVR via their N-terminal variable Ig domain 55; 56; 57, there is no further biochemical or structural information to pinpoint the exact PVR-binding patches.
Altogether, in four out of the five cases, further binding site information is required to definitively establish if the IgSF pairs bind via common binding mechanisms, before we can conclude whether these cases as true or false negatives. Given the large sequence dissimilarities computed from PICTree, it would be of interest to further elucidate the binding mechanisms in these four cases to confirm that they indeed bind via similar modes.
Large clusters most likely have several shared ligands
Our clustering yielded several large clusters with upwards of 10 members (Fig. 4). While it has been experimentally established that some of these clusters do indeed bind the same common ligand (e.g. all killer-cell immunoglobulin-like receptors (KIRs) and leukocyte immunoglobulin-like receptors (LIRs) bind to major histocompatibility complex (MHC)-I, and the sialic acid binding Ig-like lectins (SIGLECs) all bind sialic-acids), it is more likely that members of large clusters have several ligands that are shared among different members of the clusters and the cluster is formed by transitivity criteria. For instance, in the contactin-NCAM family, our common-ligand dataset (Table 1) showed that contactin 2 (CNTN2) shared a common ligand, neuronal cell adhesion molecule (NRCAM), with neurofascin (NFASC), whereas CNTN2 shared a different common ligand, NCAN, with L1CAM. Because of this situation, the identity of an IgSF’s ligand cannot be unambiguously deduced from the clustering results, however the clustering nonetheless drastically reduces the search field for cognate ligands from a unfeasibly large number of combinations to a handful of candidates that are experimentally tractable.
Success of N-terminal alignment criterion
While there is a preponderance of data supporting the claim that trans-binding receptor-ligand interaction frequently involves the N-terminal domain, there are some notable exceptions, such as NCAM1 that binds NCAN using its second Ig domain53. In addition, for secreted proteins, there is no reason to assume that the N-terminal should be more involved in binding than the rest of the protein. In a few cases this assumption will cause some functionally related connections to be missed, as they will be assigned to different clusters when they should really be in the same. A future improvement to the PICTree method would be to incorporate protein-specific binding site information into the scoring criterion whenever available.
Conclusion
Functional clustering of proteins is an essential tool to form testable hypotheses. In the case of extracellular IgSF proteins it can provide information about binding preferences and consequently about their possible role in regulating innate and adaptive immune responses within the immunological synapse. Sequence identity-based clustering is inherently difficult in case of exploring a single superfamily such as IgSF proteins that evolved relatively recently. Given that some known common-ligand IgSF pairs have sequence identities as low as 26%, the PICTree method presented here provides a highly sensitive approach to scan for such hard-to-detect binding pairs. The method was applied to all 477 IgSF proteins of interest in the human IgSF, of which 390 IgSFs were assigned to a functional family that binds the same ligand. The potential use of the method is illustrated by the suggested relationship between VSIG8 and the CTX family of proteins, which could provide actionable predictions to elucidate VSIG8 function. The method can be readily adapted to handle other classes of proteins, and can be easily updated to include additional empirical information about protein binding modes.
Materials and Methods
Dataset of human extracellular IgSF proteins
The set of extracellular human IgSF proteins was previously identified23. Briefly, human IgSF proteins were identified from the Uniprot database58, retained if they are membrane-integral or secreted according to predictions by the Phobius program59, and their Interpro60 identifiers correspond to Ig domains. Antibodies and T-cell receptors were excluded, and the highly polymorphic MHC I/II proteins are represented by only one protein per gene. This resulted in an IgSF dataset with 477 proteins (Supplementary Table 1).
Since this clustering focuses on the extracellular binding function of IgSF proteins, only sequences of extracellular protein segments were considered. For integral membrane proteins, we used the boundaries denoted by the Uniprot annotation line “Regions:Topological Domain: Extracellular” to extract sequences of the extracellular fragment. For secreted proteins, we removed the N-terminal signal peptide segment specified under the Uniprot annotation line “Molecular Processing: Signal peptide”.
Generating PICTree clustering using novel dissimilarity measure and protein interaction data
Our hidden Markov model-based, hierarchical tree clustering method (‘PICTree’) is detailed in Fig. 9. In step 1, a hidden Markov model is generated for each input IgSF sequence using programs from the HHsearch 1.5.1 software package26. Using the buildali.pl script a PSI-BLAST search was performed for each input sequence against the non-redundant protein sequence database filtered at 70% sequence identity. The default PSI-BLAST parameters in HHsearch were calibrated with the intent of detecting “distant homologous relationships”26. For our specific purpose of functional clustering, we needed a more stringent criteria and explored different parameter sets for the number of iterations (niter = 1, 8), minimum sequence identity (idmin= 0%, 20%, 25%, 30%). Using 9 curated sequence families as benchmark (see Supplementary table 3), we selected the final parameters niter = 1, idmin = 30%, and maximum E-value = 10−4. Each resulting multiple sequence alignment, along with secondary structure predictions using PSIPRED61, was then converted into a hidden Markov model using hhmake26. In step 2, all-to-all pairwise alignments of these HMMs were performed using hhalign26, allowing up to 10 alternative alignments. For each pairwise alignment, the following metrics of alignment quality provided by HHalign were monitored: HMM alignment score (Sali), secondary structure agreement score (Sss), alignment length (Lali), query alignment range (qstart, qend), and template alignment range (tstart, tend). Details of the metrics are described in Ref 26. Briefly, Sali comprised of the log-sum-of-odds score for aligning two HMMs, and a correlation score that measures the correlation in column scores between the two HMMs; Sss sums up the log-odds scores of PSIPRED prediction agreement between the two protein sequences in each column. Sali and Sss depend on the alignment length and have no fixed value range. For our 477x477 HMM-HMM alignments Sali and Sss ranged from −19 to 18378 and −2.7 to 682 respectively. In step 3, these metrics were used to generate a dissimilarity (or distance) score for each HMM-HMM alignment. Our IgSF ligand-binding dissimilarity scoring function has three components:
| (1) |
Figure 9.

Flowchart of functional clustering using PICTree.
The first component, dnormScore = Lali/(Sali + Sss), accounts for the alignment quality per aligned residue. Our corresponding dnormscore range is −0.22 – 4.42. The second component, dNali, incorporates the biological propensity for N-terminal domains to be involved in binding. If the alignment starts at the N-terminus then dNali has a value of 0, otherwise it is 1. The start of the first domain is determined using the Conserved Domain Database62. Alignment is considered to start at the N-terminal if both qstart and tstart are within 40 residues of the start of the first domain both in the query and template sequences. The third component, dminlen, is 0 if the alignment length is longer than a minimum alignment length of 60 residues, and 1 otherwise. A 477-by-477 dissimilarity (‘distance’) matrix was generated, using the smallest distance d among all alternative alignments for each HMM-HMM pair. Elements in each row i and column j then underwent the transformation dij′ = dij − 0.5 (dii+djj). This ensures that all diagonal elements (i.e., self-self alignment distances) are zero, by offsetting each matrix element dij by the average raw self-alignment score of proteins i and j. In step 4, using this distance matrix, we performed a hierarchical clustering of 477 IgSFs using average linkage clustering as implemented in R’s hclust package63. This ensures that if two proteins A and B are considered similar, then for a third protein C to join the group it must be similar to both A and B. Finally in step 5, from the resulting tree we obtained the node-to-node height h for all combinations of IgSF pairs using the distance() function in BioPERL treeio module64.
To guide the selection of the optimal cutoff in PICTree hierarchical clusters so that the resulting sub-trees contain IgSF proteins with common ligand binding preferences, we compiled positive and negative training sets of IgSF pairs that bind (or do not bind) common ligands from the STRING 9.0 database (downloaded Aug 25 2011) (Fig. 10). The 477 IgSF proteins were first mapped to human STRING identifiers using BLAST11, requiring a minimum of 95% sequence identity and 70% coverage of the query sequence. When multiple IgSF queries are mapped to the same STRING identifier, the STRING identifier was assigned to the IgSF protein with the highest sequence identity. Conversely, multiple STRING identifiers can be assigned to the same IgSF protein. For IgSFs returning no hits with these criteria, we searched the STRING database using their associated Uniprot accession codes, and assigned matching identifiers to these queries as long as they had not been previously assigned. This process resulted in the assignment of 463 IgSF proteins to 604 STRING identifiers. We then extracted IgSF:X interactions in STRING where the first interactor is one of these STRING IgSF identifiers, and the second interactor (X) can be any protein. Out of the 448,692,034 interactions recorded in STRING, 3,281,414 were human interactions, and from these 92,326 IgSF:X interactions were extracted. These interactions were then filtered based on their STRING confidence scores to build the positive and negative training sets, as described below.
Figure 10.

Extracting common-ligand IgSF pairs from STRING.
(i) Positive training set: Common-ligand IgSF pairs
The positive training set was comprised of IgSF pairs that shared at least one common extracellular, trans-binding partner (“common-ligand IgSF pairs”). We first extracted 2,440 high quality IgSF:X interactions with an experimental confidence score (STRING) above 0.5, and an overall confidence score above 0.7, and filtered these interactions to remove intracellular interactors using Phobius and signalP59; 65. From the remaining 674 IgSF:X interactions, we identified 510 putative common-ligand IgSF pairs that bind at least one common ligand. For each of these IgSF pairs, we manually verified the underlying IgSF:X interactions from the original publications cited in STRING. For a common-ligand pair IgSF1-IgSF2 to be valid, at least one of the common ligands must have manually verified IgSF1:X and IgSF2:X interactions. Conversely, for an IgSF pair to be completely negated, all putative common ligands must be manually confirmed as invalid. We removed 242 invalid IgSF:X interactions: 129 cis-associations (IgSF interacting with ligands from the same cell surface); 50 interactions not supported by the reference cited by STRING (due to incorrect name assignment by STRING or our identifier mapping), 37 MHC-I:beta-2-microglobulin(B2MG) association (because MHC-I:B2MG functions as an obligate complex), 7 intracellular interactions, 12 interactions with antibodies or uncharacterized proteins, and 6 interactions with proteolytic enzymes. We verified 175 trans-binding IgSF:X interactions, supporting 113 common-ligand IgSF pairs.
From this set, we removed 28 IgSF pairs where evidence exists in literature that they bind their common ligand(s) via different modes. For instance, both sialic acid-binding Ig-like lectin 6 (SIGL6) and leptin receptor (LEPR) are verified to bind leptin. The second cytokine receptor (CK) domain in LEPR is essential for its leptin-binding66, while SIGL6 does not contain any CK domain, thereby excluding the possibility that it binds leptin in a similar fashion as LEPR. If there is insufficient information to prove or disprove common binding modes, the common-ligand IgSF pair is retained in our dataset. We also removed three cases where the binding is facilitated on the IgSF side through a non-protein moiety. For instance, both carcinoembryonic antigen-related cell adhesion molecule (CEACAM1) and intercellular cell adhesion molecule (ICAM1) bind to the dendritic cell specific c-lectin CD209 via a Lewis-X oligosacchharide. Since the occurrence of such non-protein moieties is not predictable by sequence alone, we omitted these cases from our training set. Lastly, in cases where the common ligands are integrins, we required that the common-ligand IgSF pair bind both the same alpha and beta integrin subunits (e.g. αLβ2), since integrins are obligate heterodimers and the binding interface typically involves both subunits67. We removed 14 cases where only one integrin subunit is listed as a common interactor. The remaining 68 verified common-ligand IgSF pairs were filtered at 70% redundancy, and the resulting 55 IgSF pairs (Table 1) were retained as our positive training set.
(ii) Negative training set: IgSF pairs with no known common ligand
To establish a lower bound for our h cutoff, we compiled a negative training set, that is, IgSF pairs that do not bind any common ligand, with the caveat that it is not possible to definitively establish that two IgSFs do not share any common ligand (see Results and Discussion). To approximate a negative dataset, we compiled a list of IgSF pairs that have no known common ligands in STRING. We first extracted 5164 human IgSF:X interactions from STRING that have confidence scores greater than zero in either experimental, database, experiment-homology or database-homology categories. This represents all known human IgSF:X interactions recorded in STRING, involving 324 IgSF proteins. From these 5164 interactions, we identified 6643 common-ligand pairs, which represent all putative common-ligand pairs that can be inferred from STRING. We separately generated 52,003 pairwise combinations of the 324 IgSF proteins, and cross-filtered out the 6643 pairs that could putatively bind common ligands. After removing redundancy at 70% sequence identity, we are left with 36,066 non-redundant IgSF pairs that are not known to bind any common ligand (based on current STRING data), which served as our proxy for a negative training set.
Clustering by other algorithms
(A) SCI-PHY
Multiple sequence alignment (MSA) of sequences of the extracellular segments of 477 IgSF proteins were performed using CLUSTALW239, MAFFT40, and MUSCLE38 with default settings. For each MSA method, clustering was performed using both SCI-PHY 1.0 and 3.0, using default settings.
(B) FunFams
Each of the 477 IgSF sequences was scanned against the CATH Ig superfamily (CATH code: 2.60.40.10) using the “Sequence Scan” option from the CATH-Gene3D database (version 3.5.0, http://gene3d.biochem.ucl.ac.uk/Gene3DScanSvc/FunfamScan/Simple) to obtain its FunFam assignments. IgSFs with the same FunFams assignments are considered to be in a functional cluster.
(C) BAR+
We queried the BAR+ database (http://bar.biocomp.unibo.it/bar2.0/index.html) using Uniprot accession numbers of the 477 IgSF proteins. At the time of manuscript preparation, two of the IgSF proteins (TRML3_HUMAN and VSIG7_HUMAN) have been deleted from the latest Uniprot database, so BAR+ returned 475 cluster assignments (‘cluster-ID’). IgSFs with the same cluster-ID assignments are considered to be in a functional cluster.
Computing performance measures
We computed the following metrics:
where TP is the number of true positive predicted from the positive training dataset, and TN is the number of true negatives predicted from the negative training dataset, and The false discovery rate (FDR) is given by FDR=1−Specificity. For the full training set (‘all’) NPositive = 55 and NNegative = 36,066.
The representative node-to-node distance distribution of negative training set shown in Fig. 1 is generated by randomly drawing 55 IgSF pairs from the negative training set of 36,066 repeatedly for 36,066 / 55 ≈ 650 times, and computing the mean and standard deviation of the resulting 650 histograms.
To evaluate the performance of FunFams, we extracted a smaller ‘Ig-only’ set from the full training set. First, we identified 264 (out of 477) IgSF proteins that have only Ig domain(s) in their extracellular segments based on domain assignments by CDD62 and Interpro60. Next, we extracted from the full positive and negative sets only those pairs where both proteins belong to this subset. For the ‘Ig-only’ benchmarking set, NPositive = 20 (listed in Table 1) and NNegative = 8,983.
Removing redundancy in IgSF-IgSF pairs within the test datasets
To remove redundant IgSF pairs in our datasets, we first clustered all 477 IgSF proteins at 70% sequence identity using CD-HIT12, resulting in 413 clusters. We then mapped both IgSFs in each pair to their respective cluster representatives, and group together pairs that share the same two representatives. Finally, we chose from each group a representative pair that has the largest PICTree cutoff.
Calculation of sequence identity
As an alternative predictor of functional similarity, we computed all-to-all pairwise sequence identities for all 477 IgSF proteins based on their extracellular sequences. Each IgSF pair is aligned using CLUSTALW39 and the number of identical amino acids is divided by the shorter of the two sequences to give the sequence identity.
Supplementary Material
Highlights.
Most secreted IgSF proteins that mediate immune response have no known ligand
Extracellular IgSF proteins are clustered by similar ligand binding preferences
Function is predicted using protein interaction calibrated sequence profile data
82% of the 477 known IgSFs can be assigned to a functional family
The novel assignment of VSIG8 to CTX family generates hypothesis on VSIG8 function
Acknowledgments
This work was supported by grants GM094665, GM094662, GM096041.
Abbreviations
- Ig
immunoglobulin
- IgSF
immunoglobulin superfamily
- CTLA-4
Cytotoxic T-lymphocyte protein 4
- IGDC4
DCC subclass member 4
- L1CAM
neural cell adhesion molecule L1
- CTX
cortical thymocyte marker in Xenopus family
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Williams AF, Barclay AN. The Immunoglobulin Superfamily - Domains for Cell-Surface Recognition. Annual Review of Immunology. 1988;6:381–405. doi: 10.1146/annurev.iy.06.040188.002121. [DOI] [PubMed] [Google Scholar]
- 3.Barclay AN. Membrane proteins with immunoglobulin-like domains - a master superfamily of interaction molecules. Seminars in Immunology. 2003;15:215–223. doi: 10.1016/s1044-5323(03)00047-2. [DOI] [PubMed] [Google Scholar]
- 4.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annual Review of Immunology. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 5.Moreland L, Bate G, Kirkpatrick P. Abatacept. Nature Reviews Drug Discovery. 2006;5:185–186. doi: 10.1038/nrd1989. [DOI] [PubMed] [Google Scholar]
- 6.Larsen CP, Pearson TC, Adams AB, Tso P, Shirasugi N, Strobert E, Anderson D, Cowan S, Price K, Naemura J, Emswiler J, Greene J, Turk LA, Bajorath J, Townsend R, Hagerty D, Linsley PS, Peach RJ. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant. 2005;5:443–53. doi: 10.1111/j.1600-6143.2005.00749.x. [DOI] [PubMed] [Google Scholar]
- 7.Weber J. Review: anti-CTLA-4 antibody ipilimumab: case studies of clinical response and immune-related adverse events. Oncologist. 2007;12:864–72. doi: 10.1634/theoncologist.12-7-864. [DOI] [PubMed] [Google Scholar]
- 8.Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo YX, Lu LF, Gondek D, Wang Y, Fava RA, Fiser A, Almo S, Noelle RJ. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. Journal of Experimental Medicine. 2011;208:577–592. doi: 10.1084/jem.20100619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bork P, Sander C, Valencia A. Convergent Evolution of Similar Enzymatic Function on Different Protein Folds - the Hexokinase, Ribokinase, and Galactokinase Families of Sugar Kinases. Protein Science. 1993;2:31–40. doi: 10.1002/pro.5560020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu G, Fiser A, ter Kuile B, Sali A, Muller M. Convergent evolution of Trichomonas vaginalis lactate dehydrogenase from malate dehydrogenase. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:6285–6290. doi: 10.1073/pnas.96.11.6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li WZ, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 13.Rost B. Protein structures sustain evolutionary drift. Folding & Design. 1997;2:S19–S24. doi: 10.1016/s1359-0278(97)00059-x. [DOI] [PubMed] [Google Scholar]
- 14.Toguchida J, Nakamata T, Murakami H, Nakayama T, Nakamura T. DDBJ/EMBL/GenBank databases. 2000. Up-regulation of a ras effector and down-regulation of a cell adhesion molecule are associated with transformation of osteoblasts. [Google Scholar]
- 15.Hlavin ML, Lemmon V. Molecular-Structure and Functional Testing of Human L1cam - an Interspecies Comparison. Genomics. 1991;11:416–423. doi: 10.1016/0888-7543(91)90150-d. [DOI] [PubMed] [Google Scholar]
- 16.Brown DP, Krishnamurthy N, Sjölander K. Automated protein subfamily identification and classification. PLoS computational biology. 2007;3:e160. doi: 10.1371/journal.pcbi.0030160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee DA, Rentzsch R, Orengo C. GeMMA: functional subfamily classification within superfamilies of predicted protein structural domains. Nucleic Acids Research. 2010;38:720–737. doi: 10.1093/nar/gkp1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sillitoe I, Cuff AL, Dessailly BH, Dawson NL, Furnham N, Lee D, Lees JG, Lewis TE, Studer RA, Rentzsch R, Yeats C, Thornton JM, Orengo CA. New functional families (FunFams) in CATH to improve the mapping of conserved functional sites to 3D structures. Nucleic Acids Research. 2013;41:D490–D498. doi: 10.1093/nar/gks1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Human mutation. 2009;30:1237–1244. doi: 10.1002/humu.21047. [DOI] [PubMed] [Google Scholar]
- 20.Kaplan N, Sasson O, Inbar U, Friedlich M, Fromer M, Fleischer H, Portugaly E, Linial N, Linial M. ProtoNet 4.0: a hierarchical classification of one million protein sequences. Nucleic acids research. 2005;33:D216–D218. doi: 10.1093/nar/gki007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Research. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pegg SCH, Brown SD, Ojha S, Seffernick J, Meng EC, Morris JH, Chang PJ, Huang CC, Ferrin TE, Babbitt PC. Leveraging enzyme structure-function relationships for functional inference and experimental design: the structure-function linkage database. Biochemistry. 2006;45:2545–2555. doi: 10.1021/bi052101l. [DOI] [PubMed] [Google Scholar]
- 23.Rubinstein R, Ramagopal UA, Nathenson SG, Almo SC, Fiser A. Functional classification of immune regulatory proteins. Structure. 2013;21:766–76. doi: 10.1016/j.str.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radivojac P, Clark WT, Oron TR, Schnoes AM, Wittkop T, Sokolov A, Graim K, Funk C, Verspoor K, Ben-Hur A. A large-scale evaluation of computational protein function prediction. Nature methods. 2013 doi: 10.1038/nmeth.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engelhardt BE, Jordan MI, Srouji JR, Brenner SE. Genome-scale phylogenetic function annotation of large and diverse protein families. Genome research. 2011;21:1969–1980. doi: 10.1101/gr.104687.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 27.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, von Mering C. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Research. 2011;39:D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Staunton DE, Dustin ML, Erickson HP, Springer TA. The Arrangement of the Immunoglobulin-Like Domains of Icam-1 and the Binding-Sites for Lfa-1 and Rhinovirus. Cell. 1990;61:243–254. doi: 10.1016/0092-8674(90)90805-o. [DOI] [PubMed] [Google Scholar]
- 29.Klickstein LB, York MR, deFougerolles AR, Springer TA. Localization of the binding site on intercellular adhesion molecule-3 (ICAM-3) for lymphocyte function-associated antigen 1 (LFA-1) Journal of Biological Chemistry. 1996;271:23920–23927. doi: 10.1074/jbc.271.39.23920. [DOI] [PubMed] [Google Scholar]
- 30.Brummendorf T, Rathjen FG. Cell-Adhesion Molecules.1. Immunoglobulin Superfamily. Protein Profile. 1995;2:963. [PubMed] [Google Scholar]
- 31.Michener CD, Sokal RR. A Quantitative Approach to a Problem in Classification. Evolution. 1957;11:32. [Google Scholar]
- 32.Fahrig T, Landa C, Pesheva P, Kuhn K, Schachner M. Characterization of Binding-Properties of the Myelin-Associated Glycoprotein to Extracellular-Matrix Constituents. Embo Journal. 1987;6:2875–2883. doi: 10.1002/j.1460-2075.1987.tb02590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strenge K, Brossmer R, Ihrig P, Schauer R, Kelm S. Fibronectin is a binding partner for the myelin-associated glycoprotein (siglec-4a) Febs Letters. 2001;499:262–267. doi: 10.1016/s0014-5793(01)02566-2. [DOI] [PubMed] [Google Scholar]
- 34.Barrow AD, Trowsdale J. The extended human leukocyte receptor complex: diverse ways of modulating immune responses. Immunol Rev. 2008;224:98–123. doi: 10.1111/j.1600-065X.2008.00653.x. [DOI] [PubMed] [Google Scholar]
- 35.Teglund S, Olsen A, Khan WN, Frångsmyr L, Hammarström S. The pregnancy-specific glycoprotein (PSG) gene cluster on human chromosome 19: fine structure of the 11 PSG genes and identification of 6 new genes forming a third subgroup within the carcinoembryonic antigen (CEA) family. Genomics. 1994;23:669–684. doi: 10.1006/geno.1994.1556. [DOI] [PubMed] [Google Scholar]
- 36.Rhodes D, Stammers M, Malcherek G, Beck S, Trowsdale J. The Cluster of BTN Genes in the Extended Major Histocompatibility Complex. Genomics. 2001;71:351–362. doi: 10.1006/geno.2000.6406. [DOI] [PubMed] [Google Scholar]
- 37.Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, Tom I, Ivelja S, Refino CJ, Clark H, Eaton D, Grogan JL. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nature Immunology. 2009;10:48–57. doi: 10.1038/ni.1674. [DOI] [PubMed] [Google Scholar]
- 38.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larkin M, Blackshields G, Brown N, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 40.Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Briefings in bioinformatics. 2008;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- 41.Krogh A, Brown M, Mian IS, Sjölander K, Haussler D. Hidden Markov models in computational biology: Applications to protein modeling. Journal of molecular biology. 1994;235:1501–1531. doi: 10.1006/jmbi.1994.1104. [DOI] [PubMed] [Google Scholar]
- 42.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 43.Miley MJ, Truscott SM, Yu YY, Gilfillan S, Fremont DH, Hansen TH, Lybarger L. Biochemical features of the MHC-related protein 1 consistent with an immunological function. J Immunol. 2003;170:6090–8. doi: 10.4049/jimmunol.170.12.6090. [DOI] [PubMed] [Google Scholar]
- 44.Boyden LM, Lewis JM, Barbee SD, Bas A, Girardi M, Hayday AC, Tigelaar RE, Lifton RP. Skint1, the prototype of a newly identified immunoglobulin superfamily gene cluster, positively selects epidermal gammadelta T cells. Nat Genet. 2008;40:656–62. doi: 10.1038/ng.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee YJ, Rice RH, Lee YM. Proteome analysis of human hair shaft - From protein identification to posttranslational modification. Molecular & Cellular Proteomics. 2006;5:789–800. doi: 10.1074/mcp.M500278-MCP200. [DOI] [PubMed] [Google Scholar]
- 46.Rice RH, Phillips MA, Sundberg JP. Localization of the novel hair shaft protein VSIG8 in the hair follicle, nail unit, and oral cavity. Journal of Investigative Dermatology. 2011;131:S57–S57. doi: 10.1038/jid.2011.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hirata K, Ishida T, Penta K, Rezaee M, Yang E, Wohlgemuth J, Quertermous T. Cloning of an immunoglobulin family adhesion molecule selectively expressed by endothelial cells. Journal of Biological Chemistry. 2001;276:16223–16231. doi: 10.1074/jbc.M100630200. [DOI] [PubMed] [Google Scholar]
- 48.Cohen CJ, Shieh JTC, Pickles RJ, Okegawa T, Hsieh JT, Bergelson JM. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:15191–15196. doi: 10.1073/pnas.261452898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eguchi J, Wada J, Hida K, Zhang H, Matsuoka T, Baba M, Hashimoto I, Shikata K, Ogawa N, Makino H. Identification of adipocyte adhesion molecule (ACAM), a novel CTX gene family, implicated in adipocyte maturation and development of obesity. Biochemical Journal. 2005;387:343–353. doi: 10.1042/BJ20041709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harada H, SS, Hayashi Y, Okada S. BT-IgSF, a novel immunoglobulin superfamily protein, functions as a cell adhesion molecule. J Cell Physiol. 2005;204:8. doi: 10.1002/jcp.20361. [DOI] [PubMed] [Google Scholar]
- 51.Friedlander DR, Milev P, Karthikeyan L, Margolis RK, Margolis RU, Grumet M. Neuronal Chondroitin Sulfate Proteoglycan Neurocan Binds to the Neural Cell-Adhesion Molecules Ng-Cam/L1/Nile and N-Cam, and Inhibits Neuronal Adhesion and Neurite Outgrowth. Journal of Cell Biology. 1994;125:669–680. doi: 10.1083/jcb.125.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Milev P, Maurel P, Haring M, Margolis RK, Margolis RU. TAG-1/axonin-1 is a high-affinity ligand of neurocan, phosphacan/protein-tyrosine phosphatase-zeta/beta, and N-CAM. Journal of Biological Chemistry. 1996;271:15716–15723. doi: 10.1074/jbc.271.26.15716. [DOI] [PubMed] [Google Scholar]
- 53.Kulahin N, Rudenko O, Kiselyov V, Poulsen FM, Berezin V, Bock E. Modulation of the homophilic interaction between the first and second Ig modules of neural cell adhesion molecule by heparin. Journal of Neurochemistry. 2005;95:46–55. doi: 10.1111/j.1471-4159.2005.03338.x. [DOI] [PubMed] [Google Scholar]
- 54.Oleszewski M, Gutwein P, von der Lieth W, Rauch U, Altevogt P. Characterization of the L1-neurocan-binding site - Implications for L1-L1 homophilic binding. Journal of Biological Chemistry. 2000;275:34478–34485. doi: 10.1074/jbc.M004147200. [DOI] [PubMed] [Google Scholar]
- 55.Meyer D, Seth S, Albrecht J, Maier MK, du Pasquier L, Ravens I, Dreyer L, Burger R, Gramatzki M, Schwinzer R, Kremmer E, Foerster R, Bernhardt G. CD96 Interaction with CD155 via Its First Ig-like Domain Is Modulated by Alternative Splicing or Mutations in Distal Ig-like Domains. Journal of Biological Chemistry. 2009;284:2235–2244. doi: 10.1074/jbc.M807698200. [DOI] [PubMed] [Google Scholar]
- 56.Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, Cantoni C, Grassi J, Marcenaro S, Reymond N, Vitale M, Moretta L, Lopez M, Moretta A. Identification of PVR (CD155) and nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. Journal of Experimental Medicine. 2003;198:557–567. doi: 10.1084/jem.20030788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mueller S, Wimmer E. Recruitment of nectin-3 to cell-cell junctions through trans-heterophilic interaction with CD155, a vitronectin and poliovirus receptor that localizes to alpha(v)beta(3) integrin-containing membrane microdomains. Journal of Biological Chemistry. 2003;278:31251–31260. doi: 10.1074/jbc.M304166200. [DOI] [PubMed] [Google Scholar]
- 58.Apweiler R, Martin MJ, O’Donovan C, Magrane M, Alam-Faruque Y, Antunes R, Barrell D, Bely B, Bingley M, Binns D, Bower L, Browne P, Chan WM, Dimmer E, Eberhardt R, Fedotov A, Foulger R, Garavelli J, Huntley R, Jacobsen J, Kleen M, Laiho K, Leinonen R, Legge D, Lin Q, Liu WD, Luo J, Orchard S, Patient S, Poggioli D, Pruess M, Corbett M, di Martino G, Donnelly M, van Rensburg P, Bairoch A, Bougueleret L, Xenarios I, Altairac S, Auchincloss A, Argoud-Puy G, Axelsen K, Baratin D, Blatter MC, Boeckmann B, Bolleman J, Bollondi L, Boutet E, Quintaje SB, Breuza L, Bridge A, deCastro E, Ciapina L, Coral D, Coudert E, Cusin I, Delbard G, Doche M, Dornevil D, Roggli PD, Duvaud S, Estreicher A, Famiglietti L, Feuermann M, Gehant S, Farriol-Mathis N, Ferro S, Gasteiger E, Gateau A, Gerritsen V, Gos A, Gruaz-Gumowski N, Hinz U, Hulo C, Hulo N, James J, Jimenez S, Jungo F, Kappler T, Keller G, Lachaize C, Lane-Guermonprez L, Langendijk-Genevaux P, Lara V, Lemercier P, Lieberherr D, Lima TD, Mangold V, Martin X, Masson P, Moinat M, Morgat A, Mottaz A, Paesano S, Pedruzzi I, Pilbout S, Pillet V, Poux S, Pozzato M, Redaschi N, et al. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Research. 2010;38:D142–D148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kall L, Krogh A, Sonnhammer ELL. A combined transmembrane topology and signal peptide prediction method. Journal of Molecular Biology. 2004;338:1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 60.Apweiler R, Attwood TK, Bairoch A, Bateman A, Birney E, Biswas M, Bucher P, Cerutti L, Corpet F, Croning MDR. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic acids research. 2001;29:37–40. doi: 10.1093/nar/29.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. Journal of Molecular Biology. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 62.Marchler-Bauer A, Lu SN, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke ZX, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang DC, Zhang NG, Zheng CJ, Bryant SH. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Research. 2011;39:D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Team, R. D. C. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2008. [Google Scholar]
- 64.Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, Fuellen G, Gilbert JGR, Korf I, Lapp H, Lehvaslaiho H, Matsalla C, Mungall CJ, Osborne BI, Pocock MR, Schattner P, Senger M, Stein LD, Stupka E, Wilkinson MD, Birney E. The bioperl toolkit: Perl modules for the life sciences. Genome Research. 2002;12:1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nature Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 66.Fong TM, Huang RRC, Tota MR, Mao C, Smith T, Varnerin J, Karpitskiy VV, Krause JE, Van der Ploeg LHT. Localization of leptin binding domain in the leptin receptor. Molecular Pharmacology. 1998;53:234–240. doi: 10.1124/mol.53.2.234. [DOI] [PubMed] [Google Scholar]
- 67.Arnaout MA, Goodman SL, Xiong JP. Coming to grips with integrin binding to ligands. Current Opinion in Cell Biology. 2002;14:641–651. doi: 10.1016/s0955-0674(02)00371-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.