

Abstract
ATP-dependent proteases exist in all cells and are crucial regulators of the proteome. These machines consist of a hexameric, ring-shaped motor responsible for engaging, unfolding, and translocating protein substrates into an associated peptidase for degradation. Here, we discuss recent work that has established how the six motor subunits coordinate their ATP-hydrolysis and translocation activities. The closed topology of the ring and the rigidity of subunit/subunit interfaces cause conformational changes within a single subunit to drive motions in other subunits of the hexamer. This structural effect generates allostery between the ATP-binding sites, leading to a preferred order of binding and hydrolysis events among the motor subunits as well as a unique biphasic mechanism of translocation.
ATP-dependent proteases maintain the proteome
All cells rely on ATP-powered proteolytic machines to degrade targeted protein substrates for quality control and regulation [1]. These molecular machines share a common architecture, consisting of a ring-shaped protein unfoldase and a barrel-shaped compartmental peptidase. The unfoldase ring is composed of six AAA+ subunits (ATPases associated with various cellular activities) [2, 3] that serve as a motor, converting the energy from ATP hydrolysis into the mechanical work required to unfold and translocate protein substrates through its central pore. The unfoldase docks to one or both axial faces of the barrel-shaped peptidase, which contains proteolytic active sites sequestered in an internal chamber. Access to these active sites is restricted by two narrow axial pores with a diameter too small to allow entry of even the smallest folded proteins. The central pore of the docked unfoldase aligns with the pore of the peptidase, enabling the ATPase ring to deliver substrates into the peptidase chamber for degradation.
The protein degradation pathway follows a similar trajectory for all ATP-dependent proteases (Fig. 1). Substrate specificity is usually determined through placement of degradation tags, or degrons. Degrons can be intrinsic peptide sequences that are hidden within a correctly folded protein and become exposed only upon protein damage or misfolding, for instance during bacterial heat shock [4]. They can also be appended to a protein during certain checkpoints, for example, in the ssrA-tagging system for stalled protein synthesis. In this pathway, the ribosome is rescued by a modified transfer-RNA that includes a message to co-translationally append the 11-residue ssrA degron to the nascent chain [5]. However, not all degrons are small peptide sequences. For instance, the eukaryotic 26S proteasome recognizes protein substrates via polyubiquitin chains covalently attached to surface-exposed lysines [6]. Recognition of degrons can occur in many different ways: the degron can bind directly to the pore of the unfoldase [7], to an auxiliary site somewhere else on the protease [8], or to a cofactor that in turn delivers the substrate to the protease [9]. Because recognition logic is not the focus of this review, we refer the reader to some excellent discussions of this topic [10, 11].
Figure 1. Protein degradation by ATP-dependent proteases.

A protein substrate is specifically recognized by the unfoldase and engaged by residues located in the central pore. Downstream ATP-dependent steps lead to the unfolding of the substrate and the subsequent translocation of the unfolded polypeptide through the central pore of the unfoldase into the degradation chamber of the peptidase for proteolysis.
Once bound to the protease, a protein substrate must become engaged with the translocation machinery in the central pore of the unfoldase to allow mechanical unraveling of folded structures and threading of the polypeptide into the peptidase. Cycles of ATP hydrolysis lead to conformational changes within the ATPase subunits of the ring, generating a vectorial force to propel the substrate through the pore and unravel folded domains that are too large to pass [12, 13]. Recent studies have provided exciting new insight into the dynamics of actively-translocating AAA+ rings, and we will discuss the current state of knowledge surrounding the mechanism of mechanical force generation and polypeptide translocation. Many of these detailed discussions will include studies of the homohexameric ClpX from Escherichia coli. ClpX has been established as a model unfoldase system that currently encompasses the most comprehensive and advanced body of experimental data regarding AAA+ protease mechanisms.
Subunit conformational changes drive substrate translocation via pore loop contacts
It has become evident that loops protruding from every ATPase subunit into the central pore (pore-1 loops) are at least in part responsible for transmitting the ATP-dependent conformational changes of the unfoldase ring to the protein substrate [14–18]. For all AAA+ protein translocases, the pore-1 loop contains a highly conserved aromatic residue, and cross-linking experiments with the homohexameric ClpX unfoldase from Escherichia coli have established that its loop tyrosine directly interacts with polypeptide substrates [16]. Mutation of this tyrosine to alanine or substitution of the neighboring valine residue to alanine or phenylalanine has been shown to change the rate of ATP hydrolysis and severely affect the speed as well as efficiency of protein unfolding and translocation [16, 19]. Interestingly, the nucleotide state of the subunit carrying the pore-loop mutation significantly affected the observed phenotype. When analyzing ClpX mutant hexamers with combinations of active and inactive subunits, loop mutations exhibited more deleterious effects when placed in ATP-hydrolyzing versus hydrolysis-deficient subunits [16], consistent with a model where the subunit’s hydrolysis cycle drives loop motion and thus substrate translocation.
The interaction between the pore-1 loops and the polypeptide substrates must be surprisingly promiscuous, given the many different cellular proteins with highly variable amino-acid compositions that need to be grabbed and translocated. In principle, the loop could sense regularities of the peptide bond or even nonspecifically engage the side-chains of individual residues, much like the teeth on the chain ring of a bicycle grip the chain. Remarkably, the apparent substrate indifference of AAA+ proteases during translocation extends well beyond the requirements necessary to carry out their natural protein translocation tasks. For instance, recent studies have shown that polypeptide translocation can not only occur from either terminus, but also irrespective of D or L chirality and with little regard for side-chain chemistry or peptide-bond spacing [20, 21]. However, the side-chain composition and thus complexity or “slipperiness” of a particular substrate portion has been found to affect the probability of unraveling subsequent folded domains with high thermodynamic stability [22]. Certain proteins, such as the transcription factors NFκB, Spt23, Mga2, Gli2, Gli3 and others, contain internal degradation-stop signals with low-complexity sequences that reduce the grip of the unfoldase and result in partial degradation and the release of protein fragments with new functions [23–25]. It remains unclear what particular polypeptide feature the pore-1 loops grab on to, and future studies will have to establish the molecular details of this interaction.
Regardless of how the pore-1 loop interacts with polypeptide, it seems clear that its movement is coupled to the ATP-hydrolysis cycle of the respective subunit. Given the hexameric architecture of the ring, understanding the coordination among all six hydrolysis cycles would thus provide critical insights into the mechanism of translocation. Three general classes of inter-subunit coordination for unfoldases have been previously proposed: sequential, concerted, and stochastic firing [26]. In the sequential model, the hydrolysis cycle proceeds successively in a spatial and temporal order around the ring. In the concerted model, all subunits complete individual steps of the hydrolysis cycle at the same time. Finally, in the stochastic model, each subunit progresses through its hydrolysis cycle independently of the others. Using single-chain variants of ClpX in which all subunits were covalently linked, it has been shown that diverse arrangements of active and inactive subunits still support protein unfolding and polypeptide translocation, though at reduced rates [27]. These results immediately rule out strictly sequential or concerted models, because they violate the tight synchrony required for such mechanisms. Although these results generally favored a more stochastic model, the ATP-hydrolysis and substrate-translocation rates did not simply correlate with the number of active subunits, but showed a dependence on the arrangement of nucleotide states in the ring, indicating a certain degree of inter-subunit coordination [27]. Recent work suggests that much of the coordination among unfoldase subunits is due to the closed-ring topology of the hexamer. In the following sections, we paint a structural picture of the ring and discuss its implications on the translocation cycle using a convergence of recent structural, biochemical, and biophysical data.
Structural constraints of unfoldase rings
The architecture of unfoldase rings limits their possible modes of operation. In order to understand the origins of these constraints, we first focus on the structure of the individual ATPase subunits. Each subunit contains at least one AAA+ module, which is composed of a large AAA+ domain and an α-helical small AAA+ domain, connected by a covalent linkage known as the N-linker [28]. The nucleotide-binding pocket is formed by conserved motifs located at the interface between the large and small AAA+ domains (Walker A, Walker B, sensor I, sensor II, and Box VII; see Figure 2a) [29]. Interestingly, nucleotide hydrolysis requires an arginine residue that is donated by the neighboring subunit’s large AAA+ domain. The presence of this “arginine finger” indicates that the hydrolysis capability of the nucleotide-binding site requires oligomerization. Although we omit a detailed discussion of the AAA+ ATP-binding pocket here, we refer the curious reader to more thorough reviews of its structure and function [2, 30].
Figure 2. Architecture of unfoldase rings.
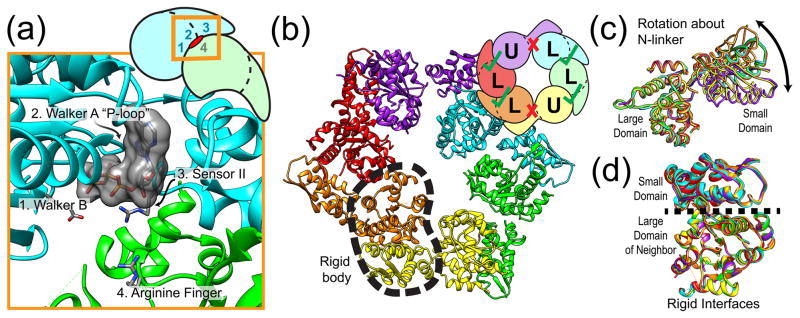
(A) Organization of a single ClpX ATP binding pocket showing the position of characteristic functional motifs (PDB ID 4I81). The side-chain positioning of the sensor-2 arginine was not resolved in the original crystal structure and has been modeled here for clarity. The inset shows the location of the binding pocket at the interface between two subunits, highlighting that both subunits contribute active-site residues. (B) Hexameric structure of ClpX shows two major conformational classes. Subunits labeled U (for “unloadable”) are in an open conformation leading to a destroyed ATP binding site (red X). Subunits labeled L (for “loadable”) are in a closed conformation and form an intact ATP binding pocket (green check). Different subunits have distinct colors. One “rigid body” between a small AAA+ domain and the large AAA+ domain of the clockwise-next neighbor is indicated by a dashed outline. (C) Structural alignment of the large domains of every subunit reveals a nucleotide-dependent hinge-like motion of the same subunit’s small domain about its N-linker. (D) Structural alignment of the large domains of every subunit shows the existence of a rigid binding interface with the previous subunit’s small domain. Subunit colors for (C) and (D) are as shown in (B).
ATP hydrolysis requires oligomerization, and the structural organization of the hexameric ring provides insight into the conformational degrees of freedom that are important for function. Recent crystal structures of the bacterial unfoldase ClpX from E. coli have revealed a ring architecture with distinct structural asymmetry, despite being composed of six identical ATPase subunits [31, 32]. Two major classes of subunits were observed within the hexamer (Fig. 2B). For one class, termed “loadable” (L), the relative orientation of the large and small domains is compatible with an intact nucleotide-binding pocket. In the other class, termed “unloadable” (U), an ~80° rotation about the N-linker between the domains destroys the nucleotide binding pocket (Fig. 2C). An asymmetric arrangement of L/U/L/L/U/L was observed for the majority of crystallized ClpX variants. The stoichiometry of four L subunits within the hexamer is consistent with binding studies showing that a maximum of four nucleotides can occupy the ring at saturation [33]. Intriguingly, the interface between the small domain of a given subunit and the large domain of its clockwise-next neighbor, as viewed from the top of the ring, is invariant regardless of whether the subunits are “loadable” or “unloadable”, leading to the formation of six “rigid bodies” (Fig. 2D). The static subunit interfaces persist during protein unfolding and translocation [34], indicating that substrate translocation may be driven by nucleotide-dependent movements of the rigid bodies about their connecting N-linker “hinges”.
Whether or not the structural asymmetry observed for ClpX extends as a general feature of AAA+ unfoldases remains to be established. Crystal structures of other unfoldases such as HslU and Lon show a more symmetric ring structure in which all subunits adopt a similar conformation [35, 36]. These symmetric structures may imply a different mechanism of function; however, they seem at odds with existing biochemical data reporting that the hexameric rings of the HslU, PAN, and ClpX unfoldases can only bind four nucleotides at saturation [33, 37, 38]. It is possible that these structures of HslU and Lon represent non-functional conformations or apo states in the absence of nucleotide and/or substrate. Symmetry breaking for these unfoldases could arise after ATP loads on the ring or substrate enters the pore, as evidence for both of these modes of induced conformational changes is emerging. Crystal structures of FtsH in the apo- and nucleotide-bound states show that the unfoldase ring breaks from a six-fold symmetry to a pseudo two-fold symmetric structure comparable to the ClpX hexamer [39]. In a similar fashion, recent cryo-EM structures of the eukaryotic 26S proteasome in the absence and presence of protein substrate show two distinct conformations [40–42]. Comparison of these structures reveals that substrate engagement triggers the formation of rigid bodies reminiscent of those observed in the ClpX hexamer, suggesting that substrate translocation by the proteasome also relies on constrained motions of rigid bodies about their N-linker hinges.
Topology of the ring provides allosteric means of inter-subunit coordination
Due to the rigid bodies formed between neighboring ATPase subunits and their limited degrees of freedom imposed by the connecting linkers, motions of a single subunit likely also cause global conformational flexing of the entire ring (Fig. 3A). Direct observation of this behavior has been possible using a technique called tmFRET, which relies on the distance-dependent quenching of a fluorescent dye in nanometer-scale proximity to a transition-metal ion [43]. By carefully choosing the placement of the probe within a single ClpX subunit, the tmFRET signal could report on either the binding of nucleotide or the conformational state of the labeled subunit within an active hexamer [32]. Surprisingly, the nucleotide occupancy of a particular subunit did not strictly correlate with its conformational state, implying that the conformation of empty subunits can be influenced by nucleotides bound somewhere else in the ring (Fig. 3B). This result suggests a certain degree of coordination among subunits in their ATP-hydrolysis and substrate-translocation cycles. A recent in silico study arrived at a similar conclusion purely by enforcing the structural constraints described above [44]. By simulating faux ClpX hexamers composed of same-type subunits (L or U) with obligatory rigid interfaces, it was discovered that the resulting structures adopt a helical “open lockwasher” conformation. Only when mixtures of L and U-type subunits were allowed to coexist, it was possible to recover the closed ring architecture known to be present during ClpX operation [34]. Although these same rules were not observed to apply for the symmetric structures of HslU, the AAA+ motor domain of cytoplasmic dynein appears to also require mixtures of subunits in different conformations to form a closed ring [44], and increasing experimental evidence for conformational switching dynamics in the dynein motor is emerging from structural [45] as well as mechanistic studies [46]. Indications of a conformational switching requirement to form closed, functional rings has also been observed in the asymmetric crystal structure of the related RecA-type T7 gene 4 helicase [47]. These observations illustrate that strong structural constraints do not only apply to AAA+ protein unfoldases like ClpX but also extend to other ring-shaped motors with significantly different functions.
Figure 3. Topological constraints necessitate conformational switching.
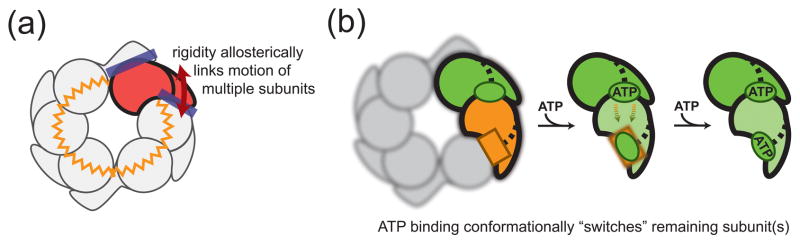
(A) The rigid interfaces between neighboring subunits as well as the closed-ring topology cause conformational changes of a single subunit to also change the conformation of other subunits. The conformational coupling of subunits throughout the hexamer is indicated by a zigzag circle. (B) ATP binding to a subunit results in a hinge-like motion of the small domain about its N-linker (see Fig. 2C). Other subunits within the ring are forced to switch their conformation (orange-colored subunit changes green) and, equivalently, change their nucleotide binding affinity (square binding site changes its shape). The spatial order of conformational switching and the detailed conformation of switched subunits remain unclear. This illustration depicts the simplest case involving adjacent subunits, with the remainder of the ring blurred from view.
A preferred order of hydrolysis events around the ring
Conformational switching of empty subunits raises the possibility that a subunit’s nucleotide affinity or hydrolysis activity may be modulated by the nucleotide occupancy and the state of the hydrolysis cycle elsewhere in the ring. Different classes of subunits with correspondingly different nucleotide affinities have been observed in the hexamers of various ATP-dependent unfoldases, including ClpX [33], PAN [48], and HslU [37]. Thus, it is plausible that a preferred order of nucleotide loading, hydrolysis, and product release steps exists among neighboring subunits as a result of their structurally constrained geometry. Consistently, the ATP-hydrolysis rate of a particular ClpX subunit has been shown to significantly depend on the nucleotide state of its neighbors [27]. An active subunit that was flanked by a permanently empty-state subunit on the counter-clockwise side and a trapped ATP-bound, pre-hydrolysis-state subunit on the clockwise side had a 3-fold higher hydrolysis rate than when the order of mutant subunits was reversed. Although diverse arrangements of active and inactive subunits in the hexamer were observed to support ATP hydrolysis and even protein unfolding and translocation, these results support a model in which subunits of wild-type ClpX progress through their ATPase cycles in a clockwise order during normal operation, with the ability to skip subunits and take alternative paths in a probabilistic manner when necessary or beneficial [27].
Cryo-EM reconstructions of the 26S proteasome also point to a preferred order of nucleotide states around its heterohexameric ATPase ring during active substrate translocation [42] or in the presence of the slowly hydrolysable ATPγS [49]. Rigid bodies between neighboring subunits were found to be tilted to different extents within the plane of the unfoldase ring, leading to a continuous spiral-staircase arrangement of the six pore-1 loops. The fixed position of each distinct subunit within this spiral may originate from the heterohexameric nature of the ring or the asymmetry imposed by surrounding ubiquitin-interacting modules and the proteasomal peptidase. Substrate translocation could thus in theory proceed through only local pore-1 loop motions with subunits otherwise statically positioned in the spiral arrangement. Alternatively, the spiral staircase observed in the EM reconstructions may represent a long-lived state of an ATPase ring, whose hydrolysis cycles rely on a biphasic mechanism with rapid subunit motions. In support of this idea, the highly coordinated translocation mechanism of the structurally related ϕ29 DNA packaging motor [50, 51] and emerging single-molecule data on the translocation mechanism of ClpX (discussed in the following section) [12, 13, 52] point towards a long-lived “dwell” state that is adopted by the ATPase ring before or after coordinated hydrolysis events lead to a rapid wave of subunit conformational changes around the ring and a “burst” of substrate translocation. The burst phase may thus involve large-scale motions of individual subunits that rapidly progress through the registers of the spiral staircase. As the ring resets, the initial spiral conformation would return and the ring would be primed for the next burst cycle. The ϕ29 DNA packaging motor as well as the ClpX unfoldase have been observed to spend more than 90% of their time in the dwell state and less than 10% with conformational changes in the burst. A potentially similar behavior of the proteasome would explain why its ATPase subunits appear to be arranged in a static spiral staircase when thousands of particles are averaged for a cryo-EM reconstruction. Notably, the pseudo two-fold symmetric structure of ClpX shows a different organization of pore-1 loops, in which each half ring contains a separate three-step staircase [31]. However, the ClpX structure was solved in the absence of substrate, and it is conceivable that this motor adopts a continuous spiral staircase, similar to the proteasome unfoldase, as soon as substrate is engaged and being translocated. Of course, it cannot be ruled out that ClpX and the proteasome use different mechanisms for translocation, and future studies will be necessary to definitively distinguish between these possibilities. Besides the cryo-EM reconstruction of the 26S proteasome, the structures of only three other substrate-bound ring translocases have been solved to date: the AAA+ E1 helicase [53] as well as the structurally related RecA-type Rho and DnaB helicases [54, 55]. Interestingly, similar to the proteasome, each of these structures shows a continuous spiral arrangement of subunits around the ring (Fig. 4A–D), further suggesting that unfoldases like ClpX may also adopt such a staircase arrangement upon substrate engagement in the central pore.
Figure 4. Spiral staircase arrangements of subunits in structures of substrate-bound ring translocases.

(A) Unfoldase (base) subunits of the 26S proteasome show a pronounced spiral staircase arrangement of pore-1 loops in the substrate-bound cryo-EM structure (EMD-5669). Colored spheres highlight the position of the pore-1 loop phenylalanine for each subunit. Inset shows the docking of the PAN crystal structure (3H4M) into the proteasome cryo-EM electron density. (B) Crystal structure of the RecA-type Rho helicase (3ICE) exhibits a similar spiral staircase arrangement of RNA binding loops. Colored spheres show the location of a threonine residue in the so-called “Q-loop”, which is important for making contacts with the phosphate backbone. The side-chain positioning of the Q-loop threonine in the left-most subunit was poorly resolved in the crystal structure and has been modeled here. All other key RNA-binding residues form a similar spiral staircase arrangement, but are omitted for clarity. (C) E1 helicase shows a spiral staircase arrangement of DNA-binding loops in the substrate-bound crystal structure (2GXA). Colored spheres show the location of a conserved histidine within the principle DNA binding loop important for making crucial phosphate-backbone hydrogen bonds. Although all other pore/substrate contacts form a similar spiral staircase, they have been omitted for clarity. (D) The DNA-binding loops of the DnaB helicase form a spiral staircase arrangement in the substrate-bound crystal structure (4ESV). Colored spheres show the location of an arginine residue located within the DNA binding loop that is responsible for making side-chain hydrogen bonds with the phosphate backbone of the DNA substrate. Other residues within the DNA-binding loop form a similar spiral staircase arrangement, but have been omitted for clarity.
Substrate translocation is biphasic and highly coordinated among multiple subunits
The emerging structural picture of the unfoldase ring provides a valuable new perspective to interpret available single-molecule data on ClpX. Remarkably, the use of optical tweezers has enabled the direct monitoring of ClpX-mediated substrate processing at a sensitivity that resolves separate phases of the translocation cycle [12, 13]. The dynamics of unfolding and translocation could thereby be observed as changes in the motor position along the protein substrate. Unfolding of three-dimensional structures results in a rapid increase in substrate extension that subsequently gradually decreases as the unfolded polypeptide chain gets translocated into the protease (Fig. 5A). Detailed analyses revealed that this translocation exhibits a step-like pattern, consisting of a dwell phase, during which the ClpX position on the substrate chain remains fixed, and a burst phase, in which ClpX rapidly translocates a certain length of polypeptide through its central pore (Fig. 5B). Interestingly, burst-size variability was observed, but it remained unclear whether this was due to the coordinated firing of a variable number of ClpX subunits or to the heterogeneity of the flexible polypeptide substrate.
Figure 5. Single-molecule studies reveal flexibility in ATP usage, but high coordination during firing.

(A) Optical tweezers allow the direct observation of ClpX-mediated protein unfolding and polypeptide translocation into its associated peptidase ClpP. The cartoon schematic on the left illustrates the events occurring during an experiment. The plot on the right shows an example of a typical single-molecule trace, with the extension between the beads plotted as a function of time. Different features within the trace are color-coded according to the corresponding event, as depicted in the cartoon schematic. Protein unfolding is observed as a rapid increase in extension, and subsequent polypeptide translocation is observed as a gradual decrease in extension with time, the zoom-in of which exhibits a step-wise pattern. (B) Top: Polypeptide translocation occurs via discrete steps composed of a long-lived dwell phase and a rapid burst phase. Bottom left: The burst size exhibits a dependence on the ATP concentration, with 2, 3, or 4 subunits firing at saturation and only 2 or 3 subunits firing at ATP concentrations near the Km. Bottom right: The mean dwell duration between bursts exhibits no dependence on ATP concentration. Data for the bottom left and right graphs were taken from reference [52]. (C) Proposed model of translocation based on single-molecule results. The ClpX ring can bind at most four ATP molecules (the two subunits that cannot bind ATP are shown in grey). Two of the remaining subunits have a high affinity for ATP (blue outline) and the other two subunits have a low affinity (green outline). Before the burst phase initiates, at least the two high affinity subunits have to bind ATP during the dwell phase. The duration of this dwell phase is determined by at least two slow steps regardless of the occupancy of the low-affinity sites. At high ATP concentrations the low-affinity subunits can bind additional ATP molecules, whereas at low concentrations the translocation burst occurs before all of the low-affinity subunits can fill.
Recent single-molecule experiments have now established that the translocation burst is driven by the phosphate-release step of the ATPase cycle, that ClpX subunits are highly coordinated during this burst phase, and that the variability in burst sizes indeed results from the variable number of hydrolyzing subunits [52]. At saturating ATP concentrations, the distribution of burst sizes corresponded to the coordinated firing of 2, 3, or 4 subunits, whereas at limiting ATP the burst-size distribution shifted to lower values equivalent to the participation of only 2 or 3 subunits (Fig. 5C). Firing bursts involving 4 subunits are again consistent with the maximal ATP-binding capacity of the ring [33], while the predominance of 2-subunit bursts and the almost complete absence of single-subunit bursts at low ATP concentrations implies that at least two subunits must bind ATP to initiate translocation. These two subunits must have an affinity for ATP much higher than the affinity of the titratable subunits, a finding that is in agreement with both the known structure of ClpX [31, 32] as well as the different classes of binding sites found within the ClpX ring [33]. The frequent occurrence of smaller-size bursts and the fact that the burst size can be titrated with the ATP concentration suggests that normal motor function does not always use the full capacity of the ring, even when ATP is present at saturating amounts. Based on the conformational switching models of inter-subunit coordination described in the previous section, it is feasible that the sub-saturated ring could still drive global conformational changes sufficient to produce an effective translocation burst. Because substrate contacts require bound nucleotide [33, 56], only filled subunits might contribute to the burst size. Future mutational studies involving combinations of hydrolysis-inactivated subunits with subunits housing compromised pore-1 loops will be required to test this model.
The ring has a hard-wired ‘rhythm’
Despite the ATP-titratability of the burst size, the distribution of completion times for the dwell-burst cycle remained constant over the entire accessible ATP-concentration range (Fig. 5B) [52]. Statistical analysis of the dwell-time distribution revealed that at least two rate-limiting transitions must happen before the burst of translocation occurs (Fig. 5C). Because this requirement is independent of the ATP concentration, these dwell-determining transitions are not coupled to ATP binding the low-affinity subunits. Although the identity of these transitions remains unknown, the structural framework described above provides testable candidates for future studies. For example, within the established conformational switching framework, existing data might suggest that the arrangement of subunit conformations in the nucleotide-loaded ring must be reset via U-to-L and L-to-U isomerizations during each individual translocation cycle [32]. It is also possible that the dwell-determining transitions follow the binding of ATP and involve setting up at least one of the subunits for catalysis, perhaps by correctly positioning the arginine finger from the neighboring subunit [57]. The rigid interfaces within the hexamer could then facilitate the coordinated firing of the remaining nucleotide-bound subunits.
Concluding remarks
Remarkable progress has been made in understanding the translocation mechanism of ATP-dependent proteases in recent years. The emerging models have required the convergence of interdisciplinary fields spanning state-of-the-art approaches in structural biology, bulk biochemistry, and single-molecule force spectroscopy. Fundamental questions still persist, such as revealing the identity of the transitions that control the dwell time during substrate translocation and mapping out the detailed spatiotemporal ordering of ATP binding, hydrolysis, and product release events within ATPase hexamers. Addressing these questions using ClpX as the most advanced model system will likely provide important new insight into operating principles applicable to other ATP-dependent proteases and ring-shaped motors serving different functions in the cell. However, it will critical to determine whether the evolving ClpX mechanism can indeed be generalized. Recent hints from related motors of the AAA+ and RecA families, such as the 26S proteasome and DNA helicases, suggest considerable similarities in basic function, and we are excited about the prospect of a mechanistic convergence of the ATP-dependent motor field.
Highlights to manuscript entitled: “Marching to the beat of the ring: polypeptide translocation by AAA+ proteases”.
ATP-dependent proteases contain a hexameric unfoldase for protein translocation.
Rigidity within unfoldase ring leads to preferred order of ATP hydrolysis events.
Subunits in the hexamer are coordinated in their conformational changes.
Acknowledgments
We thank members of the Martin and Bustamante laboratories for helpful discussions. K.N. acknowledges support by the National Science Foundation Graduate Research Fellowship. A.M. thanks the National Science Foundation (grant NSF-MCB-1150288) and the National Institutes of Health (grant R01-GM094497) for funding.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sauer RT, Baker TA. AAA+ Proteases: ATP-Fueled Machines of Protein Destruction. Annu Rev Biochem. 2010:1–26. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 2.Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Bio. 2005;6:519–29. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 3.Neuwald AF, et al. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- 4.Truscott KN, et al. Unfolded protein responses in bacteria and mitochondria: a central role for the ClpXP machine. IUBMB Life. 2011;63:955–63. doi: 10.1002/iub.526. [DOI] [PubMed] [Google Scholar]
- 5.Giudice E, Gillet R. The task force that rescues stalled ribosomes in bacteria. Trends Biochem Sci. 2013;38:403–11. doi: 10.1016/j.tibs.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 6.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin A, et al. Diverse pore loops of the AAA+ ClpX machine mediate unassisted and adaptor-dependent recognition of ssrA-tagged substrates. Mol Cell. 2008;29:441–50. doi: 10.1016/j.molcel.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matyskiela ME, Martin A. Design Principles of a Universal Protein Degradation Machine. J Mol Biol. 2012:1–15. doi: 10.1016/j.jmb.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirstein J, et al. Adapting the machine: adaptor proteins for Hsp100/Clp and AAA+ proteases. Nat Rev Micro. 2009;7:589–599. doi: 10.1038/nrmicro2185. [DOI] [PubMed] [Google Scholar]
- 10.Inobe T, Matouschek A. Protein targeting to ATP-dependent proteases. Curr Opin Struct Biol. 2008;18:43–51. doi: 10.1016/j.sbi.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baker TA, Sauer RT. ATP-dependent proteases of bacteria: recognition logic and operating principles. Trends Biochem Sci. 2006;31:647–53. doi: 10.1016/j.tibs.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maillard RA, et al. ClpX(P) Generates Mechanical Force to Unfold and Translocate Its Protein Substrates. Cell. 2011;145:459–469. doi: 10.1016/j.cell.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aubin-Tam M, et al. Single-Molecule Protein Unfolding and Translocation by an ATP-Fueled Proteolytic Machine. Cell. 2011;145:257–67. doi: 10.1016/j.cell.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, et al. Crystal structures of the HslVU peptidase-ATPase complex reveal an ATP-dependent proteolysis mechanism. Structure (London, England : 1993) 2001;9:177–84. doi: 10.1016/s0969-2126(01)00570-6. [DOI] [PubMed] [Google Scholar]
- 15.Park E, et al. Role of the GYVG pore motif of HslU ATPase in protein unfolding and translocation for degradation by HslV peptidase. J Biol Chem. 2005;280:22892–8. doi: 10.1074/jbc.M500035200. [DOI] [PubMed] [Google Scholar]
- 16.Martin A, et al. Pore loops of the AAA+ ClpX machine grip substrates to drive translocation and unfolding. Nature Structural & Molecular Biology. 2008;15:1147–51. doi: 10.1038/nsmb.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koga N, et al. Paddling mechanism for the substrate translocation by AAA+ motor revealed by multiscale molecular simulations. P Natl A Sci USA. 2009;106:18237–18242. doi: 10.1073/pnas.0904756106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang F, et al. Mechanism of substrate unfolding and translocation by the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol Cell. 2009;34:485–96. doi: 10.1016/j.molcel.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siddiqui SM, et al. Role of the processing pore of the ClpX AAA + ATPase in the recognition and engagement of specific protein substrates. Genes Dev. 2004;18:369–374. doi: 10.1101/gad.1170304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barkow SR, et al. Polypeptide translocation by the AAA+ ClpXP protease machine. Chem Biol. 2009;16:605–12. doi: 10.1016/j.chembiol.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee C, et al. ATP-Dependent Proteases Degrade Their Substrates by Processively Unraveling Them from the Degradation Signal. Mol Cell. 2001;7:627–637. doi: 10.1016/s1097-2765(01)00209-x. [DOI] [PubMed] [Google Scholar]
- 22.Too PH, et al. Slippery substrates impair function of a bacterial protease ATPase by unbalancing translocation versus exit. J Biol Chem. 2013;288:13243–57. doi: 10.1074/jbc.M113.452524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rape M, Jentsch S. Taking a bite: proteasomal protein processing. Nat Cell Biol. 2002;4:E113–E116. doi: 10.1038/ncb0502-e113. [DOI] [PubMed] [Google Scholar]
- 24.Palombella VJ, et al. The ubiquitin proteasome pathway is required for processing the NF-κB1 precursor protein and the activation of NF-κB. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 25.Schrader EK, et al. A three-part signal governs differential processing of Gli1 and Gli3 proteins by the proteasome. J Biol Chem. 2011;286:39051–39058. doi: 10.1074/jbc.M111.274993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyubimov AY, et al. The nuts and bolts of ring-translocase structure and mechanism. Curr Opin Struct Biol. 2011;21:240–8. doi: 10.1016/j.sbi.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin A, et al. Rebuilt AAA + motors reveal operating principles for ATP-fuelled machines. Nature. 2005;437:1115–20. doi: 10.1038/nature04031. [DOI] [PubMed] [Google Scholar]
- 28.Smith GR, et al. A link between sequence conservation and domain motion within the AAA+ family. J Struct Biol. 2004;146:189–204. doi: 10.1016/j.jsb.2003.11.022. [DOI] [PubMed] [Google Scholar]
- 29.Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct. 2006;35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 30.Wendler P, et al. Structure and function of the AAA+ nucleotide binding pocket. Biochim Biophys Acta. 2012;1823:2–14. doi: 10.1016/j.bbamcr.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 31.Glynn SE, et al. Crystal structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell. 2010;139:744–756. doi: 10.1016/j.cell.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stinson BM, et al. Nucleotide Binding and Conformational Switching in the Hexameric Ring of a AAA+ Machine. Cell. 2013;153:628–39. doi: 10.1016/j.cell.2013.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hersch GL, et al. Asymmetric interactions of ATP with the AAA+ ClpX6 unfoldase: allosteric control of a protein machine. Cell. 2005;121:1017–27. doi: 10.1016/j.cell.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 34.Glynn SE, et al. Dynamic and static components power unfolding in topologically closed rings of a AAA+ proteolytic machine. Nat Struct Mol Biol. 2012:1–8. doi: 10.1038/nsmb.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bochtler M, et al. The structures of HslU and the ATP-dependent protease HslU-HslV. Nature. 2000;403:800–805. doi: 10.1038/35001629. [DOI] [PubMed] [Google Scholar]
- 36.Cha S, et al. Crystal structure of Lon protease: molecular architecture of gated entry to a sequestered degradation chamber. EMBO J. 2010;29:3520–30. doi: 10.1038/emboj.2010.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yakamavich JA, et al. Asymmetric nucleotide transactions of the HslUV protease. J Mol Biol. 2008;380:946–57. doi: 10.1016/j.jmb.2008.05.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horwitz AA, et al. ATP-induced structural transitions in PAN, the proteasome-regulatory ATPase complex in Archaea. J Biol Chem. 2007;282:22921–9. doi: 10.1074/jbc.M702846200. [DOI] [PubMed] [Google Scholar]
- 39.Bieniossek C, et al. The crystal structure of apo-FtsH reveals domain movements necessary for substrate unfolding and translocation. Proc Natl Acad Sci U S A. 2009;106:21579–84. doi: 10.1073/pnas.0910708106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lander GC, et al. Complete subunit architecture of the proteasome regulatory particle. Nature. 2012;482:186–91. doi: 10.1038/nature10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beck F, et al. Near-atomic resolution structural model of the yeast 26S proteasome. Proc Natl Acad Sci U S A. 2012;109:14870–5. doi: 10.1073/pnas.1213333109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matyskiela ME, et al. Conformational switching of the 26S proteasome enables substrate degradation. Nat Struct Mol Biol. 2013 doi: 10.1038/nsmb.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taraska JW, et al. Mapping the structure and conformational movements of proteins with transition metal ion FRET. Nat Meth. 2009;6:532–537. doi: 10.1038/nmeth.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hwang W, Lang MJ. Nucleotide-dependent control of internal strains in ring-shaped AAA+ motors. Cell Mol Bioeng. 2013;6:65–73. doi: 10.1007/s12195-012-0264-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmidt H, et al. Insights into dynein motor domain function from a 3.3-Å crystal structure. Nat Struct Mol Biol. 2012;19:492–7. S1. doi: 10.1038/nsmb.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang J, et al. Lis1 Acts as a “Clutch” between the ATPase and Microtubule-Binding Domains of the Dynein Motor. Cell. 2012;150:975–86. doi: 10.1016/j.cell.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singleton MR, et al. Crystal structure of T7 gene 4 ring helicase indicates a mechanism for sequential hydrolysis of nucleotides. Cell. 2000;101:589–600. doi: 10.1016/s0092-8674(00)80871-5. [DOI] [PubMed] [Google Scholar]
- 48.Smith DM, et al. ATP Binds to Proteasomal ATPases in Pairs with Distinct Functional Effects, Implying an Ordered Reaction Cycle. Cell. 2011;144:526–538. doi: 10.1016/j.cell.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.ŒledŸ P, et al. Structure of the 26S proteasome with ATP-γS bound provides insights into the mechanism of nucleotide-dependent substrate translocation. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1305782110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moffitt JR, et al. Intersubunit coordination in a homomeric ring ATPase. Nature. 2009;457:446–50. doi: 10.1038/nature07637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chistol G, et al. High Degree of Coordination and Division of Labor among Subunits in a Homomeric Ring ATPase. Cell. 2012;151:1017–1028. doi: 10.1016/j.cell.2012.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sen M, et al. The ClpXP protease unfolds substrates with a constant pulling frequency but using different gears. Cell. 2013 doi: 10.1016/j.cell.2013.09.022. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Enemark EJ, Joshua-Tor L. Mechanism of DNA translocation in a replicative hexameric helicase. Nature. 2006;442:270–5. doi: 10.1038/nature04943. [DOI] [PubMed] [Google Scholar]
- 54.Thomsen ND, Berger JM. Running in reverse: the structural basis for translocation polarity in hexameric helicases. Cell. 2009;139:523–34. doi: 10.1016/j.cell.2009.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Itsathitphaisarn O, et al. The hexameric helicase DnaB adopts a nonplanar conformation during translocation. Cell. 2012;151:267–77. doi: 10.1016/j.cell.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bolon DN, et al. Nucleotide-dependent substrate handoff from the SspB adaptor to the AAA+ ClpXP protease. Mol Cell. 2004;16:343–50. doi: 10.1016/j.molcel.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 57.Kainov DE, et al. Structural basis of mechanochemical coupling in a hexameric molecular motor. J Biol Chem. 2008;283:3607–17. doi: 10.1074/jbc.M706366200. [DOI] [PubMed] [Google Scholar]