

Abstract
High salt diet induces renal medullary COX2 expression. Selective blockade of renal medullary COX2 activity in rats causes salt sensitive hypertension, suggesting a role for renal medullary COX2 in maintaining systemic sodium balance. The present study characterized the cellular location of COX2 induction in the kidney of mice following high salt diet and examined the role of NFκB in mediating this COX2 induction in response to increased dietary salt. High salt diet (8% NaCl) for 3 days markedly increased renal medullary COX2 expression in C57Bl/6J mice. Co-immunofluorescence using a COX2 antibody and antibodies against AQP2, ClC-K, AQP1 and CD31 showed that high salt diet-induced COX2 was selectively expressed in renal medullary interstitial cells. By using NFκB reporter transgenic mice, we observed a 7 fold increase of luciferase activity in the renal medulla of the NFκB-luciferase reporter mice following high salt diet, and a robust induction of EGFP expression mainly in renal medullary interstitial cells of the NFκB-EGFP reporter mice following high salt diet. Treating high salt diet fed C57Bl/6J mice with selective IκB kinase inhibitor IMD-0354 (8mg/kg bw) substantially suppressed COX2 induction in renal medulla, and also significantly reduced urinary PGE2. These data therefore suggest that renal medullary interstitial cell NFκB plays an important role in mediating renal medullary COX2 expression and promoting renal PGE2 synthesis in response to increased dietary sodium.
Keywords: kidney, salt, prostaglandin, mice
Introduction
The renal medulla is essential for renal regulation of body electrolyte/water balance and maintenance of blood pressure[8,13,9,1]. Bromoethylamine (BEA) that causes renal papillary-medullary lesion induces salt sensitive hypertension in rats[17], supporting a critical role of renal medulla in mediating sodium balance. The renal medulla is also known as a major site of cyclooxygenase (COX) derived prostanoid biosynthesis[15,12,2,14]. Inhibition of endogenous prostaglandin biosynthesis by COX inhibitors (including COX2 selective inhibitors) results in systemic hypertension or compromise the control of blood pressure in patients with preexisting salt-sensitive hypertension [11,23,40,41], implying a critical role of COX derived prostanoids in the maintenance of body sodium balance and blood pressure in humans. High salt diet is shown to induce abundant COX2 expression in the renal medulla of rodents together with significantly increased renal PGE2 synthesis[42,44,43]. In contrast, COX1 is constitutively expressed in renal medullary collecting duct cells as well as interstitial cells, and not altered following high salt diet [43]. Importantly, intramedullary infusion of COX2 selective inhibitor or blockage of COX2 expression in renal medulla leads to hypertension in high salt diet fed rats[44,43], consistent with a potential role for renal medullary COX2 in mediating sodium balance.
The molecular mechanisms by which renal medullary COX2 expression is increased following high salt diet are incompletely defined. COX2 is known as an important mediator in cellular response to diverse stressors[38,20]. The 5′ flanking region of the COX2 gene possesses consensus sequences for several transcriptional factors, including CRE, NFκB and NF-IL6[21]. Regulation of COX2 gene expression by these transcription factors is cell type and stressor specific [20,16,6]. Activation of NFκB has been shown to be required for COX2 induction in renal medullary interstitial cells following hypertonic stress in culture and in water deprived animals [16], suggesting a critical role for NFκB signaling in mediating renal medullary interstitial cell COX2 expression following hypertonic challenge.
The present study carefully examined the cellular location of COX2 expression in high salt die fed mice and revealed an essential role of NFκB in mediating renal medullary interstitial cell COX2 induction following high salt diet.
Methods
Experimental Animals
Male C57Bl/6J mice were purchased from Jackson Laboratory (Bar Harbour, ME). The mice were maintained on standard rodent chow and allowed free access to water prior to experiments. To examine the effect of high salt diet on renal medullary COX expression, mice were fed with either high salt diet (8% NaCl, Research Diet) or kept on normal salt diet (0.4% NaCl) for 1 to 7 days. At the end of experiments, mice were sacrificed under anesthesia and the kidneys were harvested for immunoblot, in situ hybridization and immunohistochemistry.
The effect of high salt diet on renal medullary NFκB activity was examined in transgenic mice carrying a luciferase reporter driven by an NFκB response promoter, HIV long-terminal-repeat (LTR) (HLL mice) [16]. HLL mice were fed with either normal salt diet or high salt diet for 3 days, after which renal medullary luciferase activity was determined using a commercial luciferase assay kit, according to the manufacture’s protocol (Promega Corp, Madison, WI). Luciferase activity was quantified with a luminometer (Monolight 3010, PharMingen, San Diego, CA) and adjusted for the total amount of proteins [16].
The cellular location of NFκB activation was examined using transgenic mice that carry an enhanced green fluorescent protein (EGFP) fusion protein under the control of an NFκB response promoter LTR [7]. EGFP was detected by immunofluorescent staining using an anti-EGFP antibody (Invitrogen, Carlsbad, CA) as previously described [7].
To test if NFκB is responsible for mediating high salt diet induced COX2 expression in the renal medulla, mice on normal salt diet were pretreated with an NFκB inhibitor, IMD-0354 (Sigma, St. Louis, MO) or vehicle for 2 days, followed by high salt diet for 3 days. IMD-0354, dissolved in 0.5% carboxymethylcellulose (CMC; Sigma), was administered by gavage once daily at the dosage of 8mg/kg bw, which is reported to effectively block NFκB activation [10,22,31,35,36].
A tenascin-C promoter driven Cre-ER-IRES-EGFP mouse line (TNC-CreER, unpublished) was used to examine site of COX2 induction following a high salt diet. The site of COX2 expression was assessed by co-labeling COX2 and TNC reporter EGFP.
A metabolic cage experiment was performed to examine the effect of NFkB inhibition on sodium excretion. The mice were provided with the same amount of gel food (8g containing 3.2g chow food with 0.4% NaCl) every day. After 7 days of accommodation, mice were treated with IMD-0354 or vehicle for 2 days. Then the mice were switched to high salt diet (8% NaCl) for 3 days. Daily water intake, urine volume and urinary sodium excretion was determined.
All animal experiments were approved by the Institutional Animal Care and Use Committee of Vanderbilt University.
Immunoblot
Renal medullary COX2 and COX1 expression was examined in mice fed with normal or high salt diet for 1, 2, 3 and 7 days. After mice were sacrificed, the renal medulla was isolated, and proteins were extracted. Protein concentration was determined using the bicinchoninic acid protein assay (Sigma, St. Louis, Missouri, USA). 30 micrograms of protein extract was loaded in each lane of a 10% SDS-PAGE mini-gel and run at 120 V. Proteins were transferred to a PVDF membrane at 100 V for 1 hour on ice. The membrane was washed 3 times with TBST (50 mM Tris, pH 7.5, 150 mM NaCl, 0.05% Tween-20), incubated in blocking buffer (150 mM NaCl, 50 mM Tris, 0.05% Tween-20, and 5% Carnation nonfat dry milk, pH 7.5) for 1 hour at room temperature, and then incubated with primary antibody in blocking buffer overnight at 4°C. The primary antibodies used for immunoblot studies were: anti-COX2 antibody (1:1000), anti-COX1 antibody (1:1000) from Cayman Chemical Corp. (Ann Arbor, MI); anti-β-actin antibody (Jackson ImmunoResearch Laboratories mouse monoclonal, 1:5000); anti-α-tubulin antibody (Sigma mouse monoclonal, 1:2000). After washing for 3 times, the membrane was incubated with horseradish peroxidase–conjugated secondary antibody (1:2,000-1:20,000, Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 hour at room temperature, followed by 3 washes. Antibody labeling was visualized by the addition of chemiluminescence reagent (Renaissance, PerkinElmer Life Sciences) and the membrane was exposed to Kodak XAR-5 film.
Immunofluorescent Staining
Kidney tissues were fixed in 4% paraformaldehyde and incubated in 30% sucrose overnight. Cryostat sections (5 μm) were blocked with 10% normal donkey serum for 20 min. The blocking buffer from the M.O.M. kit (Vector Laboratories, Burlingame, CA) was used with mouse monoclonal primary. Sections were then incubated with primary antibody for 60 minutes at room temperature. After washing in PBS, the sections were incubated in Cy2 or Cy3 conjugated anti-IgG secondary antibody (Jackson Immunoresearch Laboratories) for 30 minutes. Sections were viewed and imaged with a Zeiss Axioskop and spot-cam digital camera (Diagnostic Instruments) or co-focal microscopy (Zeiss LSM510). The primary antibodies used for immunofluorescent studies were: anti-COX2 antibody (1:1000) from Cayman Chemical Corp. (Ann Arbor, MI); anti-CD31 antibody (1:100, clone MEC13.3) from Pharmingen (Sanprego, CA); anti-aquaporin-2 (AQP2) antibody (1:1000) from Alpha Diagnostic International (San Antonio, TX); anti-aquaporin-1 (AQP1) antibody (1:100), anti-ClC-K antibody (1:100) from Santa Cruz Bio (Santa Cruz, CA); anti-Tamm Horsfall protein (THP) antibody (1:1000) from MP Biomedical goat polyclonal.
In Situ Hybridization
In situ hybridization was performed as described previously [16,27]. A 597-bp COX2 fragment and a 450-bp COX1 fragment were generated from the 3′ untranslated region of mouse COX2 and COX1 cDNAs respectively, using PCR [28]. The COX2 and COX1 fragments were used to synthesize 35S-UTP-labeled sense and antisense riboprobes.
Mouse kidneys were fixed in 4% paraformaldehyde and then embedded in paraffin. Sections (7 μm) were cut and hybridized at 50–55°C for approximately 18 hours. After hybridization, sections were washed at 50°C in 50% formamide, 2′ SSC, and 100mM b-mercaptoethanol for 60 minutes, treated with RNase A (10 mg/ml) at 37°C for 30 minutes, followed by wash es in 19 mM Tris, 5 mM EDTA, 500 mM NaCl (37°C), 2′ SSC (50°C), and 0.1′ S SC (50°C). Slides were dehydrated with ethanol containing 300 mM ammonium acetate. Photomicrographs were taken from slides dipped in K5 emulsion (Ilford Ltd., Knutsford, Cheshire, United Kingdom) diluted 1:1 with 2% glycerol/water and exposed for 7 days at 4°C. After development in Kod ak XAR-5 film, slides were counterstained with hematoxylin. Photomicrographs were taken with a Zeiss Axioskop microscope.
Quantitative RT-PCR
Total RNA was extracted from renal medullary tissues using TRIZOL reagent (Invitrogen). Reverse transcription was performed using a high capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative real time PCR was performed using Taqman gene expression assay system (Applied biosystems). The probes used were: Mm00478374_m1 (mouse COX2), Mm00477214_m1 (mouse COX1). Probes for eukaryotic 18S rRNA (4319413E) were used as endogenous control. Gene expression values were calculated based on the comparative threshold cycle (Ct) method detailed in Applied Biosystems User Bulletin Number 2. COX2 and COX1 expression values were normalized to the expression values of 18S rRNA. Data are displayed as fold induction relative to control (vehicle treated mice on normal salt diet).
Prostaglandin E2 measurement
Twenty four hour urine samples of mice on normal salt diet or high salt diet for days were centrifuged for 5 min at 10,000 rpm and diluted 1:1 with enzyme immunoassay buffer. Urinary PGE2 was determined using Cayman Prostaglandin E2 ELISA Kit-Monoclonal (Cat# 514010). Data are presented as fold induction relative to control (vehicle treated mice on normal salt diet).
Statistical Analysis
Data are shown as mean±SEM. Statistical analysis was performed using Microsoft Excel 2007. An unpaired two-tailed student t test was used to determine the significant differences. P<0.05 was considered to be significant.
Results
High salt diet induced COX2 expression is exclusively localized to renal medullary interstitial cells
High salt diet (8% NaCl) dramatically induced COX2 expression in the renal medulla of mice (Figure 1a, P<0.05). COX2 expression was increased as early as day 2 following high salt diet, and remained elevated throughout the study (from day 2 to day 7 following high salt diet) (Figure 1). In contrast, COX1 immunoreactive protein level was constitutively high, and not altered following high salt diet (Figure 1b).
Fig. 1. Effect of high sodium diet on renal medullary COX1 and COX2 expression.

(a): Immunoblots of COX2. C57BL6/j mice were fed with a high salt (8% NaCl) or a normal salt (0.4% NaCl) for 3 days; (b) Immunoblots of COX1 and COX2 expression for 0 to 7 days following a high sodium diet as indicated in the figure (n=5 in each time points). (c): In situ hybridization of COX1 (A, B, C) and COX2 (D. E, F). C57BL6/j mice with fed with normal (A, D) Dr high salt diet (B, C, E and F) for 5 days. COX1 and COX2 mRNA expression in the renal medulla was determined by in situ hybridization. NS, normal salt diet; HS, high salt diet.
To examine the cellular location of COX2 expression in the renal medulla of mice following high salt diet, in situ hybridization was performed. COX2 mRNA expression was dramatically increased in the renal medulla of mice on high salt diet (Figure 1c, E) when compared to mice on normal salt diet (Figure 1c, D). High power picture further showed COX2 mRNA expression was primarily located in the renal medullary interstitium between renal tubules (Figure 1c, F). In contrast to COX2, high levels of COX1 mRNA expression were detected in the renal medulla of mice on both normal salt diet (Figure 1c, A) and high salt diet (Figure 1c, B), and it was mainly located in the collecting ducts (Figure 1c, C, Figure 2D,G,K).
Figure 2. COXs expression in the kidney following a high salt diet.

C57BL/6J mice were fed with normal salt diet (A, E, H) or high salt diet (B, F, I, D, G and K) for three days. COX2 expression was examined by immunofluorescence (red). COX1 mRNA expression was examined by in situ hybridization (D, G, K).
Immunofluorescent study shows high salt diet-induced COX2 expression is restricted in the inner medulla (Figure 2). Co-immunofluorescent staining was performed using antibodies against COX2 and renal medullary segment markers: AQP2 for collecting duct, ClC-K channel for thin ascending limb of Henle’s loop, AQP1 for thin descending limb of Henle’s loop, CD31 for vasa recta, and Tamm-Horsfall protein for thick ascending limb in outer medulla. COX2 expression (red) was observed in subpopulation of renal medullary cells that are arranged in rows (Figure 3). COX2 immunofluorescence did not co-localize with any of the renal segmental markers used (green), consistent with COX2 expression exclusively located in renal medullary interstitial cells. COX2 expression was co-localized with tenascin-C reporter EGFP in the TNC reporter transgenic mice, further supporting COX2 expression in the stromal cells (Figure 4). In addition, COX2 immunofluorescence was not detected in the region where Tamm-Horsfall protein was detected, suggesting that COX2 is induced in the inner medullary interstitial cells but not in the outer medulla.
Figure 3. COX2 expression in renal medulla.

C57BL/6J mice were fed with high salt diet for three days. COX2 expression was examined by immunofluorescence (red). The structures in the renal medulla were co-labeled with their distinct markers by immunofluorescence (green): AQP2 for collecting duct (A and B); AQP1 for thin descending limb (E and F); CIC-K for thin ascending Iirnb(C and D) and CD31 for vasa recta (G and H). VR, vasa recta; * endothelial cell body.
Figure 4. Co-expression of COX2 with Tenastin-C in renal medulla.
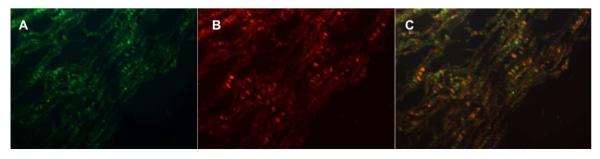
Tenasci n-C reporter mice were fed with high salt diet for three days. COX2 expression was examined by immunofluorescence (red). Tenascin-C promoter driven reporter EGFP was visualized in green.
NFκB is activated in the renal medullary interstitial cells following high salt diet
Transgenic mice carrying an NFκB response promoter driven luciferase reporter were fed with normal salt diet or high salt diet for 3 days. High salt diet significantly increased luciferase reporter activity in the renal medullary tissues by 7 fold when compared to normal salt diet (Figure 4a, 3626±1045 vs 513±248 unit/mg protein, P<0.05), suggesting that NFκB was activated in renal medulla following high salt diet.
To determine the cellular location of NFκB activation, cryostat sections of the kidneys from transgenic mice carrying an NFκB response promoter driven EGFP reporter either on normal salt diet or high salt diet were examined by immunofluorescent staining using an anti-EGFP antibody. EGFP immunofluorescence was only detected in mice fed with high salt diet, but not in mice on normal salt diet (Figure 4b). In addition, the EGFP expression was mainly located in the renal medullary interstitial cells that are arranged in rows (Figure 4b, right panel). Interstitial cell NFκB activation is supported by immunohistochemistry of activated p65 (Figure 5D).
Figure 5. Effect of high salt diet on NFκB activity.

NFκB promoter driven luciferase (A) or EGFP (B) reporter transgenic mice were treated with high salt diet or normal salt diet for three days. Luciferase activity in renal medulla was determined to assess endogenous NFκB activity CA, n= 10). The location of NFκB activation was determined by imrnunofuorescence for NFκB reporter EGFP (B). NFκB activity was also examined by activated p65 immunohistochemistry (D). White arrows and red arrows indicate respectively the EGFP expressing cells and acti vated p65 positive cells arranged in a laddering pattern. *p<0.05 vs normal salt.
NFκB activation mediates the increase of renal medullary COX2 expression and renal PGE2 synthesis following high salt diet
To test whether NFκB mediates COX2 induction in the renal medullary interstitial cells following high salt diet, a selective IκB kinase inhibitor IMD-0354 was used to block NFκB activation in mice. Immunoblot showed treatment with the NFκB inhibitor IMD-0354 dramatically suppressed high salt diet induced renal medullary COX2 expression (Figure 5a, P<0.0001). qRT-PCR further showed markedly attenuated COX2 mRNA induction in renal medullary tissues of IMD-0354 treated mice on high salt diet (Figure 5b, P<0.01), suggesting a critical role for NFκB activation in mediating COX2 induction. In contrast, neither high salt diet nor IMD-0354 altered COX1 expression (Figure 7). Furthermore, urinary PGE2 dramatically increased following high salt diet (Figure 5c, P<0.001), suggesting increased renal PGE2 biosynthesis. The increase of urinary PGE2 following high salt diet was partially but significantly attenuated in mice treated with the NFκB inhibitor (Figure 5c, P<0.05), consistent with blocked renal medullary COX2 induction.
Figure 7. Effect of NFκB inhibitor on renal medullary COX1 expression.

C57Bl/6J mice were pretreated with NFκB inhibitor IMD-03S4 or vehicle for 2 days, then additionally fed with high salt diet or kept on normal salt diet for another 3 days. (a) COX1 expression in the renal medullary tissues was determined by immunoblot. (b) COX1 mRNA expression in the renal medullary tissues was determined by qRT-PCR. Black bar: vehicle treated mice, Grey bar: IMD-0354 treated mice.
To examine the role NFκB in sodium excretion after high salt diet, we performed metabolic cage studies to measure sodium balance. As the mice were provided with the same amount of gel food (8g containing 3.2g chow food with 0.4% NaCl) every day, we assume these mice consume the same amount of sodium each day. Thus daily urinary sodium excretion was compared. As shown in Figure 8, following high salt diet, mice treated with NFκB inhibitor IMD-0354 show a tendency to excrete less sodium when compared to vehicle. However, statistical analysis using two-tailed unpaired student t test failed to demonstrate a significant difference in sodium excretion on either day 1, day 2 or day 3 following high salt diet.
Figure 8. The effect of NFκB inhibitor on body weight and sodium balance.

10 C57Bl 6J male mice were housed in metabolic cages. Each mouse was fed with 8g gel food containing 3.6 g normal chow food (0.4%NaCl) every day. Daily water intake (b), urine volume (c) and body weight (d) was recorded. 24hour urinary sodium excretion Twenty-four hour urine was collected for measuring urinary sodium output. Starting from day7, mice were administered with NFκB inhibitor IMD-0354(8mg/kgbw) or vehicle daily by gavage. Two days later, all the mice were switched to high salt diet (8%NaCl) for another 3 days. Black:vehicle treated mice, Red:IMD-0354 treated mice.
Discussion
The present study has shown that renal medullary interstitial cells are the major sites of COX2 induction in mice following a high salt diet. The mechanism of this COX2 induction appears to require activation of NFκB in renal medullary interstitial cells. The present finding therefore implicates a role for NFκB-COX2 pathway in renal response to increased dietary sodium.
Our studies demonstrated in mice that COX2 expression significantly increased in the renal medulla from day 2 to day 7 following high salt diet. Previous studies show elevated COX2 expression in the renal medulla on day 14 following high salt diet [44,43]. Thus these observations together suggest a continuous COX2 induction in the renal medulla in response to salt loading. High salt diet induced COX2 expression in rats is found to be predominantly located in renal medullary interstitial cells [43]. The present study carefully examined the cellular location of COX2 induction in high salt diet fed mice and demonstrated that renal medullary interstitial cells are the major sites of COX2 induction in mice. Induced COX2 expression was not detected in the region where Tamm-Horsfall protein was detected, consistent with COX2 induction in the inner medullary interstitial cells. Whether COX2 gene expression in human renal medullary interstitial cells also responds to systemic sodium loading remains to be investigated [26,25,37].
Synthesis of prostanoids requires co-localization of COX with prostanoid synthases within the same cell[14,3]. Previous studies show PGE2 synthase mPGES1 expression in mouse renal medullary interstitial cells, and high salt diet significantly increased renal medullary mPGES1 expression[5], suggesting that mPGES1 also responds to sodium loading. Therefore renal medullary interstitial cell COX2 is very likely to couple with mPGES1 to promote the production of PGE2 following dietary sodium loading. The mechanism by which renal medullary COX derived prostanoids modulate sodium excretion and maintains blood pressure, however, is not fully understood. Inhibition of COX2 has been reported to reduce renal medullary blood flow[34], and the reduction of renal medullary blood flow is associated with sodium retention and hypertension though incompletely defined mechanisms [1]. Previous studies have also demonstrated a critical role of renal medullary PGE2-EP2 receptor signaling in maintaining normotension in the setting of high salt intake[5]. Since EP2 receptor is reported to locate at vasa recta [37], PGE2 derived from renal medullary interstitial cell COX2 may modulate renal medullary blood flow through EP2 receptor on adjacent vasa recta and promote renal sodium excretion following high salt diet.
COX2 expression is regulated at multiple levels, including transcriptional and post-transcriptional levels [20,32,24]. CRE, NFκB, and NF-IL6 are known important transcriptional regulators of COX2 expression, and they display variable efficacy in a cell or stimulus specific manner[39,30,4]. Among these transcription factors, activation of NFκB is reported to be required for COX2 induction in renal medullary interstitial cells following hypertonic stress in culture and also in water deprived animals [16]. This NFκB-COX2 pathway is further demonstrated to confer cytoprotection in renal medullary interstitial cells against hypertonic stress in culture and in water deprived animals. In the present studies, high salt diet dramatically increased renal medullary NFκB activity, and blockage of NFκB activation by a selective IκB kinase inhibitor IMD-0354 substantially suppressed high salt diet induced renal medullary COX2 expression, suggesting that the NFκB-COX2 pathway in renal medullary interstitial cells also responds to systemic sodium loading. Interestingly, known as a stress resistant molecule and a metabolic master switcher, a NAD+ dependent histone/protein deacetylase Sirt1 is also shown to be preferentially expressed in the inner medullary interstitial cells where it exerts cytoprotection against oxidative stress through mediating COX2 induction[18]. However, the role of Sirt1 in mediating renal medullary interstitial cell COX2 induction following sodium loading remains to be investigated.
The present study show that following NFκB inhibitor IMD-0354 treatment, high salt diet induced COX2 expression was almost completely blocked, but renal PGE2 synthesis is only partially reduced, implicating involvement of COX2 independent PGE2 synthesis following a high salt diet. As aforementioned, COX1 is constitutively expressed in renal medullary collecting duct cells as well as interstitial cells at high levels. mPGES1 is also expressed in the collecting duct and induced by high salt diet (5). Ye et al. have shown that inhibition of either COX2 or COX1 in renal medulla results in increased blood pressure in high salt diet fed rats, and that high salt diet fed COX1 knockout mice exhibit a significant increase of blood pressure which is associated with suppressed urinary PGE2 excretion [43].
Although our data show a tendency of reduced sodium excretion in IMD-0354 treated mice, the difference did not reach statistical significance. Several possibilities may account for this: Incomplete block of PGE2 synthesis as discussed above may attenuate the anti-diuretic effect of COX2 blockade; The very scattered nature of the data, which is characteristic in sodium balance study, particularly in small animals, may also be a possible reason.
The molecular basis of NFκB activation following salt loading, however, remains unclear. Cell culture studies have shown that NFκB is activated in the renal medullary interstitial cells by NaCl and mannitol but not by the membrane permeable osmole urea [16], suggesting stimulation of NFκB activation by increased tonicity. Interestingly, high salt diet is reported to increase renal medullary NaCl concentration [29,33,19]. Thus the mechanism by which NFκB signaling responds to dietary sodium loading is probably in part through sensing the increase of tonicity in renal medullary interstitium.
In conclusion, the present studies have demonstrated that high salt diet induces COX2 expression exclusively in renal medullary interstitial cells in mice. Nuclear factor NFκB plays a critical role in mediating this COX2 induction. Induced COX2 together with constitutive COX1 further increases PGE2 biosynthesis in the renal medulla, thus promoting renal sodium excretion and blood pressure is stabilized.
Perspectives and Significance.
The present study has provided further insights into the roles of COX isoforms in response to increased dietary sodium. Further studies may investigate the molecular mechanism by which NFκB is activated in the renal medullary interstitial cells following sodium loading. By identifying a role for NFκB in renal response to increased dietary sodium intake through mediating renal medullary COX2 induction, the present study has provided information in understanding the molecular basis of altered body electrolyte/water balance in patients administered compounds that inhibit NFκB (such as Bortezomib).
Figure 6. NFκB inhibitor suppresses renal medullary COX2 induction following high salt diet.

C57Bl/6J mice were pretreated with NFκB inhibitor IMD-0354 or vehicle for 2 days. then additionally fed with high salt diet or kept on normal salt diet for 3 days. (a) COX2 expression in the renal medullary tissues was determined by immunoblot. (n=3, *P<0.0001 versus vehicle treated mice on high salt) (b) COX2 mRNA expression in the renal medull ary ti ssues was determined by qRT-PCR. (n=5, *p <0.01 versus vehicle treated mice on normal salt, **P<0.01 versus vehicle treated mice on high salt) (c) Twenty four hour urinary PGE2 was measured using Cayman PGE2 ELISA kit. (n=8, *P<0.001 versus vehicle treated mice on normal salt, **P<0.001 versus TMD-0354 treated mice on normal salt, †P<0.05 versus vehicle treated mice on high salt)
Acknowledgments
Immunofluorescence experiments were performed in part through the use of the VUMC Cell Imaging Shared Resource. Source(s) of Funding: These studies were supported by National Institute of Diabetes and Digestive and Kidney Disease grant DK071876 to CM Hao.
Footnotes
Disclosure none
References
- 1.Bergstrom G, Evans RG. Mechanisms underlying the antihypertensive functions of the renal medulla. Acta Physiol Scand. 2004;181(4):475–486. doi: 10.1111/j.1365-201X.2004.01321.x. [DOI] [PubMed] [Google Scholar]
- 2.Breyer MD, Hao C, Qi Z. Cyclooxygenase-2 selective inhibitors and the kidney. Curr Opin Crit Care. 2001;7(6):393–400. doi: 10.1097/00075198-200112000-00005. [DOI] [PubMed] [Google Scholar]
- 3.Breyer RM, Kennedy CR, Zhang Y, Breyer MD. Structure-function analyses of eicosanoid receptors. Physiologic and therapeutic implications. Ann N Y Acad Sci. 2000;905:221–231. doi: 10.1111/j.1749-6632.2000.tb06552.x. [DOI] [PubMed] [Google Scholar]
- 4.Chen G, Wood EG, Wang SH, Warner TD. Expression of cyclooxygenase-2 in rat vascular smooth muscle cells is unrelated to nuclear factor-kappaB activation. Life Sci. 1999;64(14):1231–1242. doi: 10.1016/s0024-3205(99)00055-7. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, Zhao M, He W, Milne GL, Howard JR, Morrow J, Hebert RL, Breyer RM, Chen J, Hao CM. Increased dietary NaCl induces renal medullary PGE2 production and natriuresis via the EP2 receptor. Am J Physiol Renal Physiol. 2008;295(3):F818–825. doi: 10.1152/ajprenal.90253.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, Zhao M, Rao R, Inoue H, Hao CM. C/EBP{beta} and its binding element are required for NF{kappa}B-induced COX2 expression following hypertonic stress. J Biol Chem. 2005;280(16):16354–16359. doi: 10.1074/jbc.M411134200. [DOI] [PubMed] [Google Scholar]
- 7.Connelly L, Robinson-Benion C, Chont M, Saint-Jean L, Li H, Polosukhin VV, Blackwell TS, Yull FE. A transgenic model reveals important roles for the NF-kappa B alternative pathway (p100/p52) in mammary development and links to tumorigenesis. J Biol Chem. 2007;282(13):10028–10035. doi: 10.1074/jbc.M611300200. [DOI] [PubMed] [Google Scholar]
- 8.Curtis JJ, Luke RG, Dustan HP, Kashgarian M, Whelchel JD, Jones P, Diethelm AG. Remission of essential hypertension after renal transplantation. N Engl J Med. 1983;309(17):1009–1015. doi: 10.1056/NEJM198310273091702. [DOI] [PubMed] [Google Scholar]
- 9.Dahl LK, Heine M. Primary role of renal homografts in setting chronic blood pressure levels in rats. Circ Res. 1975;36(6):692–696. doi: 10.1161/01.res.36.6.692. [DOI] [PubMed] [Google Scholar]
- 10.Fan M, Ahmed KM, Coleman MC, Spitz DR, Li JJ. Nuclear factor-kappaB and manganese superoxide dismutase mediate adaptive radioresistance in low-dose irradiated mouse skin epithelial cells. Cancer Res. 2007;67(7):3220–3228. doi: 10.1158/0008-5472.CAN-06-2728. [DOI] [PubMed] [Google Scholar]
- 11.Fierro-Carrion GA, Ram CV. Nonsteroidal anti-inflammatory drugs (NSAIDs) and blood pressure. Am J Cardiol. 1997;80(6):775–776. doi: 10.1016/s0002-9149(97)00514-6. [DOI] [PubMed] [Google Scholar]
- 12.Guan Y, Chang M, Cho W, Zhang Y, Redha R, Davis L, Chang S, DuBois RN, Hao CM, Breyer M. Cloning, expression, and regulation of rabbit cyclooxygenase-2 in renal medullary interstitial cells. Am J Physiol. 1997;273(1 Pt 2):F18–26. doi: 10.1152/ajprenal.1997.273.1.F18. [DOI] [PubMed] [Google Scholar]
- 13.Guyton AC. Blood pressure control--special role of the kidneys and body fluids. Science. 1991;252(5014):1813–1816. doi: 10.1126/science.2063193. [DOI] [PubMed] [Google Scholar]
- 14.Hao CM, Breyer MD. Physiologic and pathophysiologic roles of lipid mediators in the kidney. Kidney Int. 2007;71(11):1105–1115. doi: 10.1038/sj.ki.5002192. [DOI] [PubMed] [Google Scholar]
- 15.Hao CM, Komhoff M, Guan Y, Redha R, Breyer MD. Selective targeting of cyclooxygenase-2 reveals its role in renal medullary interstitial cell survival. Am J Physiol. 1999;277(3 Pt 2):F352–359. doi: 10.1152/ajprenal.1999.277.3.F352. [DOI] [PubMed] [Google Scholar]
- 16.Hao CM, Yull F, Blackwell T, Komhoff M, Davis LS, Breyer MD. Dehydration activates an NF-kappaB-driven, COX2-dependent survival mechanism in renal medullary interstitial cells. J Clin Invest. 2000;106(8):973–982. doi: 10.1172/JCI9956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haugan K, Shalmi M, Petersen JS, Marcussen N, Spannow J, Christensen S. Effects of renal papillary-medullary lesion on the antihypertensive effect of furosemide and development of salt-sensitive hypertension in Dahl-S rats. J Pharmacol Exp Ther. 1997;280(3):1415–1422. [PubMed] [Google Scholar]
- 18.He W, Wang Y, Zhang MZ, You L, Davis LS, Fan H, Yang HC, Fogo AB, Zent R, Harris RC, Breyer MD, Hao CM. Sirt1 activation protects the mouse renal medulla from oxidative injury. J Clin Invest. 120(4):1056–1068. doi: 10.1172/JCI41563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herrera M, Garvin JL. A high-salt diet stimulates thick ascending limb eNOS expression by raising medullary osmolality and increasing release of endothelin-1. Am J Physiol Renal Physiol. 2005;288(1):F58–64. doi: 10.1152/ajprenal.00209.2004. [DOI] [PubMed] [Google Scholar]
- 20.Herschman HR. Prostaglandin synthase 2. Biochim Biophys Acta. 1996;1299(1):125–140. doi: 10.1016/0005-2760(95)00194-8. [DOI] [PubMed] [Google Scholar]
- 21.Hla T, Bishop-Bailey D, Liu CH, Schaefers HJ, Trifan OC. Cyclooxygenase-1 and -2 isoenzymes. Int J Biochem Cell Biol. 1999;31(5):551–557. doi: 10.1016/s1357-2725(98)00152-6. [DOI] [PubMed] [Google Scholar]
- 22.Inayama M, Nishioka Y, Azuma M, Muto S, Aono Y, Makino H, Tani K, Uehara H, Izumi K, Itai A, Sone S. A novel IkappaB kinase-beta inhibitor ameliorates bleomycin-induced pulmonary fibrosis in mice. Am J Respir Crit Care Med. 2006;173(9):1016–1022. doi: 10.1164/rccm.200506-947OC. [DOI] [PubMed] [Google Scholar]
- 23.Jackson EK. Relation between renin release and blood pressure response to nonsteroidal anti-inflammatory drugs in hypertension. Hypertension. 1989;14(5):469–471. doi: 10.1161/01.hyp.14.5.469. [DOI] [PubMed] [Google Scholar]
- 24.Kang KB, Wang TT, Woon CT, Cheah ES, Moore XL, Zhu C, Wong MC. Enhancement of glioblastoma radioresponse by a selective COX-2 inhibitor celecoxib: inhibition of tumor angiogenesis with extensive tumor necrosis. Int J Radiat Oncol Biol Phys. 2007;67(3):888–896. doi: 10.1016/j.ijrobp.2006.09.055. [DOI] [PubMed] [Google Scholar]
- 25.Khan KN, Alden CL, Gleissner SE, Gessford MK, Maziasz TJ. Effect of papillotoxic agents on expression of cyclooxygenase isoforms in the rat kidney. Toxicol Pathol. 1998;26(1):137–142. doi: 10.1177/019262339802600116. [DOI] [PubMed] [Google Scholar]
- 26.Komhoff M, Grone HJ, Klein T, Seyberth HW, Nusing RM. Localization of cyclooxygenase-1 and -2 in adult and fetal human kidney: implication for renal function. Am J Physiol. 1997;272(4 Pt 2):F460–468. doi: 10.1152/ajprenal.1997.272.4.F460. [DOI] [PubMed] [Google Scholar]
- 27.Komhoff M, Jeck ND, Seyberth HW, Grone HJ, Nusing RM, Breyer MD. Cyclooxygenase-2 expression is associated with the renal macula densa of patients with Bartter-like syndrome. Kidney Int. 2000;58(6):2420–2424. doi: 10.1046/j.1523-1755.2000.00425.x. [DOI] [PubMed] [Google Scholar]
- 28.Komhoff M, Wang JL, Cheng HF, Langenbach R, McKanna JA, Harris RC, Breyer MD. Cyclooxygenase-2-selective inhibitors impair glomerulogenesis and renal cortical development. Kidney Int. 2000;57(2):414–422. doi: 10.1046/j.1523-1755.2000.00861.x. [DOI] [PubMed] [Google Scholar]
- 29.Nakanishi T, Uyama O, Nakahama H, Takamitsu Y, Sugita M. Determinants of relative amounts of medullary organic osmolytes: effects of NaCl and urea differ. Am J Physiol. 1993;264(3 Pt 2):F472–479. doi: 10.1152/ajprenal.1993.264.3.F472. [DOI] [PubMed] [Google Scholar]
- 30.Newton R, Kuitert LM, Bergmann M, Adcock IM, Barnes PJ. Evidence for involvement of NF-kappaB in the transcriptional control of COX-2 gene expression by IL-1beta. Biochem Biophys Res Commun. 1997;237(1):28–32. doi: 10.1006/bbrc.1997.7064. [DOI] [PubMed] [Google Scholar]
- 31.Onai Y, Suzuki J, Maejima Y, Haraguchi G, Muto S, Itai A, Isobe M. Inhibition of NF-{kappa}B improves left ventricular remodeling and cardiac dysfunction after myocardial infarction. Am J Physiol Heart Circ Physiol. 2007;292(1):H530–538. doi: 10.1152/ajpheart.00549.2006. [DOI] [PubMed] [Google Scholar]
- 32.Park JW, Qi WN, Cai Y, Urbaniak JR, Chen LE. Proteasome inhibitor attenuates skeletal muscle reperfusion injury by blocking the pathway of nuclear factor-kappaB activation. Plast Reconstr Surg. 2007;120(7):1808–1818. doi: 10.1097/01.prs.0000287245.17319.57. [DOI] [PubMed] [Google Scholar]
- 33.Peterson DP, Murphy KM, Ursino R, Streeter K, Yancey PH. Effects of dietary protein and salt on rat renal osmolytes: covariation in urea and GPC contents. Am J Physiol. 1992;263(4 Pt 2):F594–600. doi: 10.1152/ajprenal.1992.263.4.F594. [DOI] [PubMed] [Google Scholar]
- 34.Qi Z, Hao CM, Langenbach RI, Breyer RM, Redha R, Morrow JD, Breyer MD. Opposite effects of cyclooxygenase-1 and -2 activity on the pressor response to angiotensin II. J Clin Invest. 2002;110(1):61–69. doi: 10.1172/JCI14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanaka A, Konno M, Muto S, Kambe N, Morii E, Nakahata T, Itai A, Matsuda H. A novel NF-kappaB inhibitor, IMD-0354, suppresses neoplastic proliferation of human mast cells with constitutively activated c-kit receptors. Blood. 2005;105(6):2324–2331. doi: 10.1182/blood-2004-08-3247. [DOI] [PubMed] [Google Scholar]
- 36.Tanaka A, Muto S, Konno M, Itai A, Matsuda H. A new IkappaB kinase beta inhibitor prevents human breast cancer progression through negative regulation of cell cycle transition. Cancer Res. 2006;66(1):419–426. doi: 10.1158/0008-5472.CAN-05-0741. [DOI] [PubMed] [Google Scholar]
- 37.Therland KL, Stubbe J, Thiesson HC, Ottosen PD, Walter S, Sorensen GL, Skott O, Jensen BL. Cycloxygenase-2 is expressed in vasculature of normal and ischemic adult human kidney and is colocalized with vascular prostaglandin E2 EP4 receptors. J Am Soc Nephrol. 2004;15(5):1189–1198. doi: 10.1097/01.asn.0000124673.79934.24. [DOI] [PubMed] [Google Scholar]
- 38.Wadleigh DJ, Reddy ST, Kopp E, Ghosh S, Herschman HR. Transcriptional activation of the cyclooxygenase-2 gene in endotoxin-treated RAW 264.7 macrophages. J Biol Chem. 2000;275(9):6259–6266. doi: 10.1074/jbc.275.9.6259. [DOI] [PubMed] [Google Scholar]
- 39.Warnock LJ, Hunninghake GW. Multiple second messenger pathways regulate IL-1 beta-induced expression of PGHS-2 mRNA in normal human skin fibroblasts. J Cell Physiol. 1995;163(1):172–178. doi: 10.1002/jcp.1041630120. [DOI] [PubMed] [Google Scholar]
- 40.Whelton A, White WB, Bello AE, Puma JA, Fort JG. Effects of celecoxib and rofecoxib on blood pressure and edema in patients > or =65 years of age with systemic hypertension and osteoarthritis. Am J Cardiol. 2002;90(9):959–963. doi: 10.1016/s0002-9149(02)02661-9. [DOI] [PubMed] [Google Scholar]
- 41.White WB, Kent J, Taylor A, Verburg KM, Lefkowith JB, Whelton A. Effects of celecoxib on ambulatory blood pressure in hypertensive patients on ACE inhibitors. Hypertension. 2002;39(4):929–934. doi: 10.1161/01.hyp.0000014323.99765.16. [DOI] [PubMed] [Google Scholar]
- 42.Yang T, Singh I, Pham H, Sun D, Smart A, Schnermann JB, Briggs JP. Regulation of cyclooxygenase expression in the kidney by dietary salt intake. Am J Physiol. 1998;274(3 Pt 2):F481–489. doi: 10.1152/ajprenal.1998.274.3.F481. [DOI] [PubMed] [Google Scholar]
- 43.Ye W, Zhang H, Hillas E, Kohan DE, Miller RL, Nelson RD, Honeggar M, Yang T. Expression and function of COX isoforms in renal medulla: evidence for regulation of salt sensitivity and blood pressure. Am J Physiol Renal Physiol. 2006;290(2):F542–549. doi: 10.1152/ajprenal.00232.2005. [DOI] [PubMed] [Google Scholar]
- 44.Zewde T, Mattson DL. Inhibition of cyclooxygenase-2 in the rat renal medulla leads to sodium-sensitive hypertension. Hypertension. 2004;44(4):424–428. doi: 10.1161/01.HYP.0000140924.91479.03. [DOI] [PubMed] [Google Scholar]