

Abstract
We have recently gained a remarkable understanding of the mutational landscape of head and neck squamous cell carcinoma (HNSCC). However, the nature of the dysregulated signaling networks contributing to HNSCC progression is still poorly defined. Here, we have focused on the role of the family of mitogen activated kinases (MAPKs), extracellular regulated kinase (ERK), c‐Jun terminal kinase (JNK) and p38 MAPK in HNSCC. Immunohistochemical analysis of a large collection of human HNSCC tissues revealed that the levels of the phosphorylated active form of ERK1/2 and JNK were elevated in less than 33% and 16% of the cases, respectively. Strikingly, however, high levels of active phospho‐p38 were observed in most (79%) of hundreds of tissues analyzed. We explored the biological role of p38 in HNSCC cell lines using three independent approaches: treatment with a specific p38 inhibitor, SB203580; a retro‐inhibition strategy consisting in the use of SB203580 combined with the expression of an inhibitor‐insensitive mutant form of p38α; and short‐hairpin RNAs (shRNAs) targeting p38α. We found that specific blockade of p38 signaling significantly inhibited the proliferation of HNSCC cells both in vitro and in vivo. Indeed, we observed that p38 inhibition in HNSCC cancer cells reduces cancer growth in tumor xenografts and a remarkable decrease in intratumoral blood and lymphatic vessels. We conclude that p38α functions as a positive regulator of HNSCC in the context of the tumor microenvironment, controlling cancer cell growth as well as tumor‐induced angiogenesis and lymphangiogenesis.
Keywords: p38 MAPK, Head and neck cancer, Angiogenesis, Lymphangiogenesis, p38 Inhibitor, MAPK, JNK
1. Introduction
Head and neck squamous cell carcinomas (HNSCCs) are one of the six most prevalent cancers in the developing world, and represent around 6% of all new cancers in the United States (Siegel et al., 2012b). Most of the HNSCC cases are diagnosed at late stages, which often leads to poor prognosis and patient survival. In spite of the progress made in the understanding of the processes involved in HNSCC initiation and progression and the use of new treatment options, the survival of patients HNSCC has only improved marginally in the last 40 years (Siegel et al., 2012a). The standard treatment options for HNSCC are surgical excision and radiation alone, or combined with chemotherapy. These have severe consequences and affect the quality of life of the HNSCC patients (Forastiere et al., 2001; Specenier and Vermorken, 2009). Thus, the current emphasis is in the development of novel molecular targeted treatment strategies, aimed at improving the survival and quality of life of HNSCC patients.
The family of mitogen‐activated protein kinases (MAPKs) represents a highly conserved group of protein kinases that play an essential role in signal transduction by modulating gene transcription in the nucleus in response to changes in the cellular environment (Pearson et al., 2001; Schaeffer and Weber, 1999; Turjanski et al., 2007). In humans, there are at least 11 members of the MAPK superfamily, which can be divided into multiple groups. The most studied MAPKs are the extracellular signal‐regulated protein kinases (ERK1 and ERK2), c‐Jun N‐terminal kinases (JNK1, JNK2, JNK3), and p38 MAPKs (p38α, p38β, p38γ, p38δ) (Pearson et al., 2001). Each group of MAPKs can be stimulated by a separate signal transduction pathway in response to different extracellular stimuli. In turn, MAPKs exhibit distinct substrate specificities and biological functions. ERK1 and ERK2 (collectively referred to as ERK) are the MAPKs most frequently associated with cancer cell proliferation, as these kinases can be activated by normal and oncogenically active mutants of tyrosine kinase growth factor receptors, and the Ras and Raf oncogenes, among others (Schaeffer and Weber, 1999). Early studies using antibodies recognizing the activated forms of ERK, however, revealed that in HNSCC this particular MAPK is activated in recurrent disease but not in most primary HNSCC lesions (Albanell et al., 2001). These observations raised the possibility that rather than ERK, other MAPK family members may contribute to HNSCC progression.
In this study, we took advantage of antibodies recognizing the phosphorylated active form of MAPKs and the availability of tissue microarrays including hundreds of HNSCC lesions collected as part of an international initiative (Molinolo et al., 2007) to examine the status of activation of MAPKs in HNSCC, and its relationship to other well established events in HNSCC. Of interest, we found that most HNSCC cases exhibit activation of the p38 MAPK, in contrast to very few cases showing accumulation of the active form of ERK and JNK. Activation of p38 was more prominent in less differentiated HNSCC cases, which are often associated with poor prognosis (Thomas et al., 2013). To test whether p38 signaling could control cell proliferation in HNSCC, we blocked p38 signaling in a panel of HNSCC cells by three complementary strategies: using short hairpin RNAs (shRNA), small molecule inhibitors, and a retro‐inhibition approach. We found that the blockade of p38 signaling significantly inhibited the proliferation of cancer cells both in vitro and in vivo, the latter concomitant with a remarkable decrease in tumor induced angiogenesis and lymphangiogenesis. These studies may provide the foundation for the future evaluation of p38 inhibitors in clinical trials targeting p38 signaling in HNSCC, alone or in combination with currently available treatment options.
2. Materials and methods
2.1. Cell lines and reagents
Human HNSCC cell lines were maintained as previous described (Amornphimoltham et al., 2008, 2008). Briefly, the cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and incubated at 37 °C in 5% CO2. Normal oral keratinocytes (NOK) were obtained from healthy volunteer under the National Institute of Dental and Craniofacial Research (NIDCR) Clinical Protocol No. 06‐D‐0144. The cells were isolated and cultured as previously described (Leelahavanichkul and Gutkind, 2012). Briefly, the tissue was incubated in trypsin overnight and dispersed from the connective tissue. The oral keratinocytes were recovered and cultured in keratinocytes serum‐free medium (K‐SFM, Invitrogen, Carlsbad, CA) at 37 °C in 5% CO2. Interleukin‐1β (IL‐1β) was obtained from R&D system (Minneapolis, MN), and the inhibitor SB203580 from Sigma–Aldrich (St. Louis, MO) and LC laboratory (Woburn, MA).
2.2. Antibodies
Antibodies against phospho p44/42 MAPK (ERK1/2) XP™, p44/42 MAPK (ERK1/2), phospho‐p38 MAPK XP™, p38 MAPK, phospho‐JNK, were obtained from Cell Signaling Technology (Danvers, MA). Anti‐JNK and Ki‐67 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and R&D system (Minneapolis, MN), respectively. For the staining of blood and lymphatic vessels in human tumor xenografts, we used anti‐CD31 (BD Pharmingen San Diego, CA) and LYVE1 (Abcam, Cambridge, MA), respectively. In human tissues, we used PECAM1 (CD31 Santa Cruz Biotechnology) and podoplanin (Abcam), together with tyramide signal amplification system (Dako).
2.3. Immunohistochemistry staining
The tissue sections were deparaffinized and hydrated through graded alcohols, and then rinsed with PBS. Antigen retrieval was done using a citrate buffer (pH 6.0) in a microwave for 2 min at 100% power and 15 min at 10% power. Sections were incubated in 3% hydrogen peroxide in PBS for 10 min and incubated in blocking solution (2% bovine serum albumin in PBS–1% Tween 20) for 1 h at room temperature. Sections were incubated with the primary antibody at 4 °C overnight. After washing, the sections were sequentially incubated in the biotinylated secondary antibody (Vector Laboratories, Burlingame, CA) for 30 min followed by the ABC complex (Vector Stain Elite®, ABC kit, Vector Laboratories, Burlingame, CA) for 30 min at room temperature. The sections were developed in 3,3‐diaminobenzidine (Sigma–Aldrich, St. Louis, MO) under microscopic observation and counterstained with Mayer's hematoxylin. The images were scanned using a ScanScope (Aperio Technologies Inc., Vista, CA). The sections were examined and classified based on the percentage of positive cells (0, <10%; 1, 10–25%; 2, 25–50%; 3, 50–75%; and 4, 75–100% of positive cells) and staining intensity (0, negative; 1, weak; 2, moderate and 3, strong) from two persons as previous described (Molinolo et al., 2007). Results were scored by multiplying the percentage of positive cells by the intensity.
2.4. Hierarchical cluster analysis and data visualization
The staining scores that resulted from immunohistochemistry were converted into scaled values centered on zero (e.g. a score of 0 is converted to −2, 1 to 0, and 4 to 3) in Microsoft Excel. The hierarchical cluster analysis was done using Cluster software (Eisen Software) with average linkage based on Pearson's correlation coefficient as the selection variable on the unsupervised approach. The results were visualized using the Java TreeView software. The clustered data were arranged with markers on the horizontal axis and tissue samples on the vertical axis as recently described (Molinolo et al., 2007). Two biomarkers with a close relationship were located next to one another.
2.5. Western blotting
Cell lines were homogenized in RIPA buffer and sonicated for 20 s. Protein yields were quantified using protein assay kit (Bio‐Rad, Hercules, CA). Similar amounts of protein (50 μg) were loaded, separated by SDS‐PAGE and electro transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked for 1 h in blocking buffer (5% nonfat dry milk in 0.1% Tween 20–TBS), then replaced by the primary antibody and incubated overnight at 4 °C. Primary antibody was detected using horseradish peroxidase‐linked goat anti‐mouse or goat anti‐rabbit IgG antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and visualized with SuperSignal West Pico chemiluminescent substrate (Pierce Biotechnology, Rockford, IL).
2.6. Cell proliferation assay
For Click‐iT® EdU (Invitrogen, Carlsbad, CA) assays, the cells were seeded on coverslips 24 h before treatment. After 20 h of treatment, they were incubated in modified nucleoside solution, EdU (5‐ethynyl‐2′‐deoxyuridine), which is incorporated during DNA synthesis and labeled with Alexa Fluor® 594, according to manufacturer's instruction. The coverslips were mounted on slides and images were randomly captured from six different areas using Axio Plane II (Zeiss, Thornwood, NY) in six different areas. Morphometric analysis was performed using Image J (NIH) and calculated by the average pixel value.
2.7. [3H]‐thymidine incorporation assay
The cells were seeded in 24‐well plates in triplicates and treated with SB203580 (10 μM) at 37 °C in a 5% CO2 atmosphere. After 20 h, the cells were incubated with [3H]‐thymidine (1 μCi/mL) (PerkinElmer, Waltham, MA) for 4 h at 37 °C with 5% CO2, then cells were washed with PBS and 10% TCA, and incubated in 0.3 N NaOH for 1 h. The radioactivity incorporated into the cellular DNA was evaluated in a scintillation counter. The results represented the percentage of [3H]‐thymidine incorporation in average±standard error (SEM) as compared with control cells.
2.8. RNAi technology
To knockdown the endogenous p38α, shRNA targeted MAPK14 (p38α) was used. Lentiviral pGIPZ shRNA targeting MAPK14 and control (OpenBiosystem, Huntsville, AL) were transfected into packaging 293FT cells, using TurboFect™ (Fermentus Inc., Glen Burnie, MD). After 48 h, supernatants were collected, passed through 0.45 μm filters, and then used to infect CAL27 cells at 37 °C with 5% CO2 in the presence of 8 μg/mL of polybrene (hexadimethrine bromide; Sigma). After 24h, the cells were washed twice with PBS, returned to normal medium and selected by cell sorting, taking advantage of GFP that is expressed from the pGIPZ vector.
2.9. Retro‐inhibition approach using retroviral gene expression
To confirm the specific role of p38 in HNSCC, CAL27 cells were stably expressed an inhibitor‐insensitive mutant forms of p38α, p38 T106M, p38α, and a mutant p38α tagged with HA, as described previously (Marinissen et al., 1999; Sanchez‐Prieto et al., 2000). The latter includes a mutation in threonine 106 for a methionine that renders p38α insensitive to the p38 inhibitor. All plasmids were cloned into pLPCX as a retroviral vector, and then were transfected to packaging cells, GP2‐293 cells, using TurboFect™ (Fermentus Inc., Glen Burnie, MD). After 48 h, supernatants were collected and passed through filters, and then used to infect CAL27 cells at 37 °C with 5% CO2 in the presence of polybrene (hexadimethrine bromide; Sigma). After 24 h, the cells were washed twice with PBS, returned to normal medium, and selected by puromycin.
2.10. Treatment of tumor xenografts in athymic Nu/Nu mice
All animal studies were carried out according to NIH‐approved protocols, in compliance with the Guide for the Care and Use of Laboratory Animals. Six‐week‐old female atymic Nu/Nu mice were obtained from NCI Frederick, housed in appropriate sterile filter‐capped cages, and fed and watered ad libitum. All handling, tumor induction, and treatment procedures were conducted in a laminar flow biosafety hood. Two million CAL27 and retroviral‐infected CAL27 cells were used to induce HNSCC tumor xenografts, and tumor growth analysis was performed as previous described (Amornphimoltham et al., 2008, 2008). Drug treatment was initiated when tumor volume reached 60 mm3. For each experiment, 10 tumor‐bearing animals were randomly divided into two groups and intra peritoneal injected with SB203580 (5 mg/kg/d) or vehicle control daily for 16 consecutive days, and monitored three times a week thereafter, for tumor growth. For RNAi, 2 million lentiviral‐infected CAL27 cells were used to induce tumor xenografts and tumor growth was monitored three times a week. At the indicated time points, animals were sacrificed, and tissues were collected. Tissues were either fixed in buffered zinc formalin (Z‐fix, Anatech) and embedded in paraffin, or frozen and embedded in optimal cutting temperature (OCT) compound. Unpaired t test was used to analyze the differences of tumor burden between experimental groups. Data analysis was done with GraphPad Prism version 5.03 for Windows (GraphPad Software); p values of <0.05 were considered statistically significant.
2.11. Immunofluorescence double staining
The tissues were embedded in OCT media and cut at 20 μm. The cryosections were hydrated in distilled water, and washed with PBS. The sections were incubated in the blocking solution (5% normal goat serum in 0.1% Tween‐20 in PBS) for 1 h at room temperature and incubated with primary antibody in blocking solutions at 4 °C overnight. After washing with PBS, the slides were incubated with the Alexa Flour® 488 and 594 conjugated secondary antibody (Invitrogen, Carlsbad, CA) for 45 min, then mounted in Vecta Shield mounting medium with 4′,6‐diamidino‐2‐phenylindole (DAPI; Vector Laboratories). The fluorescent imaging was performed using an inverted confocal microscope (model IX81, Olympus America, Center Valley, PA) and an UPlanSApo ×10 objective lens, numerical aperture (NA) 0.40 (Olympus America).
2.12. Microvessel analysis
Micro blood vessels and lymph vessels were identified by CD31 and Lyve1 immunofluorescence double staining, respectively, as recently described (Patel et al., 2011). Images were randomly captured by Axio Plane II (Zeiss, Thornwood, NY) in six different areas in each section. Cell counting was performed using Image J (NIH) and calculated by the average value. For the analysis of microvessel CD31 and podoplanin in HNSCC tissue arrays, the whole slides were captured by ScanScope (Aperio Technologies Inc., Vista, CA). The digital images were analyzed using microvessel analysis algorithm for Scanscope. Staining results are calculated from the number of vessels per total analysis area.
2.13. Circulating human cytokines levels in mice bearing human HNSCC xenografts
The retro‐orbital blood collection from medial canthus was done as described in NIH animal protocol and guideline. Briefly, a microhematocrit tube was inserted through the conjunctiva and into the orbital sinus by quickly rotating the tube. After the required amount of blood was obtained, the tube was withdrawn and blood was collected into serum separation tubes (BD Biosciences, Franklin Lakes, NJ) and bleeding ceased by the eye pressure. After clotting, blood samples were spun at 400 g at room temperature for 15 min. Serum samples were collected in new tubes and kept at −80 °C. Human cytokine analysis was performed at the cytokine core facility, University of Maryland, Baltimore, using specific assays for human cytokines (Malhotra et al., 2012).
2.14. Statistical analysis
Data analysis was performed using GraphPad Prism 5.03 (GraphPad Software). All values were expressed as the means ± standard error (SEM). Differences were considered significant at p value <0.05. Student's t‐test was used to analyze the differences between control and treatment groups.
3. Results
3.1. Activation of p38 MAPK in human HNSCCs
Initially, we examined the status of phosphorylation of ERK1/2, JNK and p38, using antibodies recognizing their dual phosphorylated, active form, in a representative set of human HNSCC and normal oral mucosa controls. Examples of HNSCC tumor biopsies that reacted positively for these phosphorylated kinases are shown (Figure 1 A). No phosphorylation of any of these MAPKs was detected in normal tissues. To gain an insight into the prevalence of MAPK activation in HNSCC, we explored the presence of active MAPKs in a large collection (more than 400 cores) of human HNSCC tissues (Figure 1 B and C). Phosphorylated ERK1/2 (p‐ERK1/2) and JNK (p‐JNK) were observed in only 33% and 16% of the cases, respectively. In stark contrast, we found high level of p‐p38 MAPK in 79% of the HNSCC cases, along with p‐AKTT308 (70%) and p‐S6 (83%), reflecting the activation of the AKT and mTOR pathway, respectively, and COX2 (60%) and EGFR (73%), which were highly overexpressed, as previously reported in this international HNSCC tissue array (Molinolo et al., 2007).
Figure 1.

Activation of MAPK cascades in human head and neck squamous cell carcinoma (HNSCC). A. Examples of representative HNSCC tissue biopsies in which immunohistochemistry revealed the expression of the active, phosphorylated forms of ERK1/2, JNK and p38 in oral squamous cell carcinoma (OSCC) lesions, but not in normal tissues. B. The expression of p‐ERK, p‐p38 and p‐JNK is evident in the nuclei of representative tissue cores from the International HNSCC Tissue Microarray (n = 293) (200×). C. Percentage of positive cases for each protein from the HNSCC tissue microarray. The bar graph represents the percentage of positive cases for each protein in the HNSCC tissue microarray. The immunohistochemistry results were quantified and classified as described (0, <10%; 1, 10–25%; 2, 25–50%; 3, 50–75%; and 4, 75–100% of positive cells). D. The relationship between the different pathways is depicted in the heat map. The closeness of the columns directly indicates correlation. There is a strong correlation between p‐ERK and p‐JNK. Phopho‐p38 showed high correlation with proteins of the AKT–mTOR pathway, EGFR and COX2 whereas p53 and pEGFR segregate independently. The percentage of cells stained in each tissue core was classified from, 0 to 4 as above, and represented in the indicated color scale (green to red). Missing stained cores for each sample were represented in gray.
A heat map approach was used to represent the percentage of stained cells according to the scoring systems used for Figure 1 C. Tissues scored as 0 were represented as green; 1, as black; 2, as dark red; and 3 as red. No available data was represented as gray (Figure 1 D). The hierarchical cluster analysis showed high correlation between phospho‐ERK and phospho‐JNK, but not phospho‐p38. Interestingly, phospho‐p38 showed strong correlation with the phosphorylation of proteins of the AKT–mTOR pathway, EGFR and COX2 overexpression. No significant correlation between p53 and EGFR or pEGFR was observed, as previously reported (Molinolo et al., 2007). Overall, activation of p38 MAPK is a frequent event in HNSCC, and most HNSCC cases display increased expression of EGFR and COX2, and overactive AKT, mTOR, and p38 MAPK pathways.
3.2. Activation of p38 MAPK is associated with disease progression and increased angiogenesis and lymphangiogenesis in human HNSCCs
We next explored whether p38 MAPK activation is associated with HNSCC disease progression. For this analysis, we used commercial HNSCC tissue arrays constructed from normal oral squamous epithelium and HNSCC tissues including well, moderately, and poorly differentiated lesions (Biomax US, Rockville), considering that the status of differentiation often correlates inversely with disease prognosis (Thomas et al., 2013). Of interest, the phosphorylation of p38 increased already in well differentiated tissues, however there was a remarkable heterogeneity among the different lesions, and hence this difference was not statistically significant (Figure 2). In contrast, p‐p38 expression was clearly increased in moderately differentiated and even further increased in poorly differentiated HNSCC. Increased angiogenesis, and more specifically lymphangiogenesis, can be good predictors of poor prognosis (Hasina and Lingen, 2001). Indeed, angiogenesis and lymphangiogenesis, as judged by CD31 and podoplanin staining, respectively, increased in well differentiated and moderately differentiated HNSCC lesions with respect to normal oral epithelium (Figure 2). In the case of poorly differentiated lesions, angiogenesis was much more diffuse, which was not significant with respect to normal controls due to its heterogeneous distribution in the tissue array cores analyzed. Intratumoral lymphangiogenesis was clearly increased with respect to normal controls. Considering the widespread distribution of p‐p38, CD31, and podoplanin staining in a large collection of well differentiated HNSCC tissues (n = 56), we used the opportunity to explore the potential correlation between p38 MAPK activation and tumor angiogenesis. We found that the correlation between p‐p38 and CD31 staining did not reach significance in our sample set (p = 0.1404) but p‐p38 staining correlated significantly with the accumulation of podoplanin+ lymphatic cells (p = 0.0236), hence suggesting the possible contribution of p38 MAPK activation to tumor lymphangiogenesis. Overall, p38 activation is increased particularly in the less differentiated HNSCC lesions and correlates with enhanced lymphangiogenesis, both of which are often associated with cancer severity and poor prognosis (Hasina and Lingen, 2001; Thomas et al., 2013).
Figure 2.

Activation of p38 MAPK is inversely associated with the status of differentiation in HNSCC lesions. The levels of active, phosphorylated form of p38 MAPK (p‐p38) were analyzed in normal oral epithelium (Normal) and HNSCC tissue biopsies of distinct differentiation status (WD: well differentiated; MD: moderately differentiated; PD: poorly differentiated) in HNSCC tissue microarrays (n = 102) (upper panel). A similar analysis was performed for the endothelial marker CD31 (middle panel) and lymphatic endothelial marker, podoplanin (bottom panel). The graph represents the scores of the individual tissues expressed as arbitrary units, and the corresponding average ± the standard error of the mean (SEM) (*p < 0.05; **p < 0.01; ***p < 0.001). Representative tissue sections stained for the corresponding markers are included (40× magnification).
3.3. Inhibition of p38 decreases the proliferation of HNSCC cells in vitro
The basal status of activation of MAPK pathways was examined in a variety of HNSCC cell lines. Although almost all HNSCC cell lines show intrinsic elevated levels of pERK1/2 and pJNK, only a few cell lines showed activated p‐p38 (Figure S1). Thus, elevated activity of p38 appears not to represent an intrinsic event in HNSCC, but instead may result from the presence of the inflammatory tumor microenvironment in vivo. Nonetheless, a limited activity of p38 may contribute to HNSCC cell proliferation in vitro. To address this possibility, we used the highly specific p38 MAPK inhibitor SB203580 (Davies et al., 2000). Using CAL27 as a representative cell line, we observed that SB203580 blocked p‐p38 accumulation, whereas the increase in p‐p38 in response to IL‐1β served as a positive control (Supplmentary Figure 1, right panel). Furthermore, SB203580 diminished the accumulation of the phosphorylated form of MK2, a direct downstream target of p38, which appears to be already maximal in these cells as it cannot be further increased by IL‐1β. Thus, SB203580 was effective to block p38 at concentrations (10 μM) similar to those reported to be highly specific for this MAPK (Davies et al., 2000). The p38 inhibitor decreased the proliferation of a variety of HNSCC cell lines, as judged the direct incorporation of [3H]‐thymidine into cellular DNA. The values represent the percentage of [3H]‐thymidine incorporation in triplicates with respect to control, vehicle treated cells (Figure 3). The p38 inhibitor reduced DNA‐synthesis in all HNSCC cell tested, even in those exhibiting reduced p38 activity (HN12). This suggested that a limited activity of p38 may be required for HNSCC proliferation, while raising the need to validate these observations by experimental approaches complementing the use of p38 small molecule inhibitors.
Figure 3.
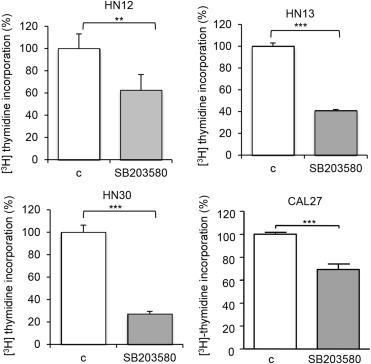
Cell proliferation in HNSCC cell lines. A–E. All HNSCC cell lines showed a significant decrease in cell proliferation when treated with the p38 inhibitor SB203580 (10 μM) as judged by [3H]‐thymidine incorporation assays (treatment time 48 h). The values represent mean value of triplicates experiments (**p < 0.01; ***p < 0.001).
3.4. Role of p38 MAPK in HNSCC tumorigenicity using RNAi technology
To determine whether p38 MAPK has a specific effect in tumorigenesis, we generated p38α (MAPK14) knockdown CAL27 cells and determined the p38 MAPK activity by immunoblotting for p‐MK2, which had been shown to be one of the direct targets of p38 MAPK (Figure 4 A). The cells were transplanted into nude mice and tumor burden was monitored. It was clear that p38 knockdown resulted in significant lower tumor burden (Figure 4 B). We further examined the number of proliferating cells in the xenograft tissues using the Ki‐67 marker (Figure 4 C). The immunohistochemical staining revealed that p38‐knockdown tumors had significantly less proliferative capacity. p38 is often activated in tumors by inflammatory cytokines, and in turn p38 regulates the expression of pro‐angiogenic mediators (Cuenda and Rousseau, 2007; Wagner and Nebreda, 2009; Yoshizuka et al., 2012). Thus, in light of these observations and our findings in human HNSCC lesions, we used CD31 and LYVE1 as markers to investigate whether p38 inhibition affects tumor angiogenesis and lymphangiogenesis, respectively. Indeed, the staining showed that p38‐knockdown tumors had reduced angiogenesis and lymphangiogenesis (Figure 4 D).
Figure 4.
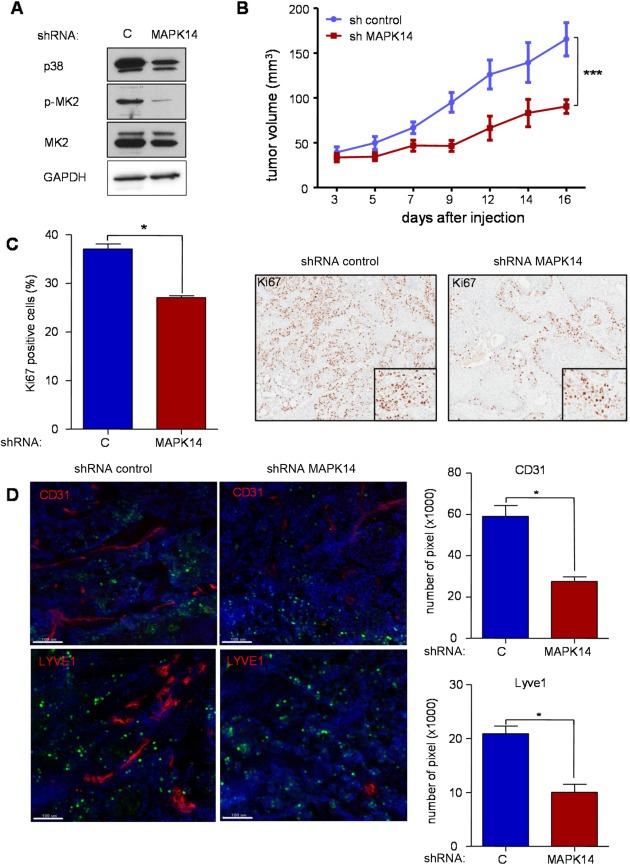
A . Knockdown of p38 MAPK decreased cell proliferation, tumorigenesis, and angiogenesis in HNSCC cells. A. Western blot analysis depicts MAPK14 (p38α) protein knock‐down in CAL27 cells using siRNA (shRNA), concomitant with reduced phosphorylated MK2, p‐MK2, a direct target of p38, without affecting overall MK2 expression levels. GAPDH was used as a loading control. B. CAL27 cells expressing shRNA control or shRNA targeting MAPK14 were transplanted into nude mice and tumor growth monitored every other day. After 2 weeks, CAL27‐sh MAPK14 tumors are significantly smaller than the shRNA control (n = 10; p < 0.001). C. Cell proliferation, as evaluated by Ki67 immunohistochemistry, reflects the in vivo growth curves. The histological pictures (right; 10×) depict nuclear Ki‐67 staining in representative areas of both shRNA control and shRNA MAPK14 expressing cells. D. The development of both blood and lymphatic vessels is inhibited in CAL27‐sh MAPK14 tumors, as judged by immunofluorescence stainings of CD31 and LYVE1, respectively (scale bar 100 μm). The bar graph shows the quantification values of the immunostainings (*p < 0.05; ***p < 0.001).
3.5. A retro‐inhibition approach to study p38 MAPK function
As pharmacological inhibitors are expected to affect both HNSCC cells and their stroma, as well as exhibit possible non‐targeted effects, we challenge the contribution of p38 in each compartment, and the drug specificity, by engineering a mutated p38 MAPK tagged with HA, p38 T106M, which is insensitive to the SB203580 and hence can rescue p38 function in the cells even in the presence of endogenous p38 that remains sensitive to the p38 blocker (Sanchez‐Prieto et al., 2000). We stably expressed p38 wild type (WT) and its T106M mutant in CAL27 cells (Figure 5 A). Immunoblotting was used to confirm the expression of HA and overexpression of p38 (Figure 5 B). EdU staining showed that the proliferative cells were significantly affected by SB203580 in parental CAL27 and CAL27 expressing p38WT. Overexpression of p38WT may partially reduce the effect of the drug due to its high expression, thus limiting the availability of the p38 inhibitor to block all p38 molecules. However, the expression p38TM nearly abolished the effect of the p38 inhibitor in cell proliferation in CAL27 cells (Figure 5 C and D).
Figure 5.
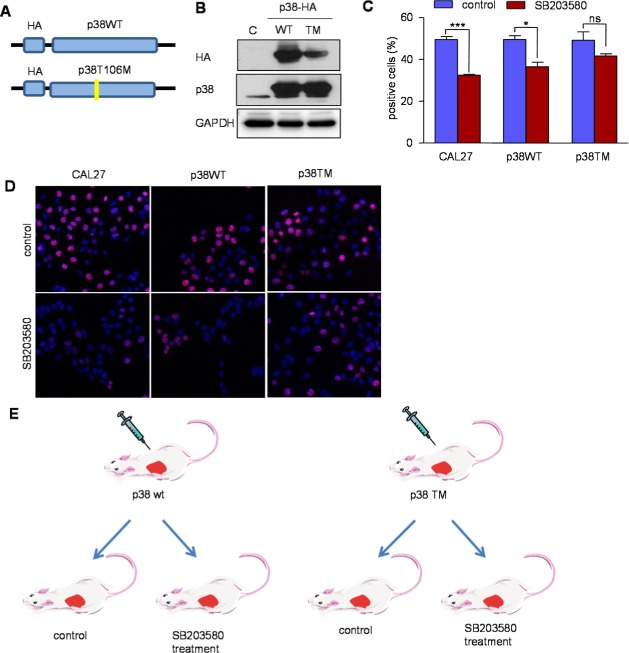
A retroinhibition approach to examine the specificity of p38 inhibition in HNSCC growth and tumorigenesis. A. Retroviral p38WT and p38TM constructs tagged with HA were engineered and used to generate stable expressing CAL27 cells. B, expression of p38WT and p38TM was confirmed by Western blotting using HA and p38 antibodies. C. The cells, CAL27, CAL27 p38WT and CAL27 p38TM were incubated with EdU (ClickIT®) and Alexa Flour 594 (red) after treating with the p38 inhibitor. A limited effect of SB203580 is shown on p38TM cells (*p < 0.05; ***p < 0.001; ns: not significant). D. Proliferating cells are shown purple in these representative images; nuclei are labeled with DAPI (blue) (40×). E. Scheme of the experimental system: CAL27 p38WT and p38TM were transplanted into nude mice, and tumor burden was monitored every other day. After the tumors reached approximately 60 mm3, the mice were treated with SB203580 (5 mg/kg) or vehicle (control). F. After 2 weeks of SB203580 treatment, CAL27 p38WT tumors were significantly smaller; CAL27 p38TM tumors were not affected by the p38 inhibitor (n = 10) (G). H. Ki67 staining revealed the antiproliferative effect of SB203580 treatment in p38WT, but not in p38TM tumors. Representative images showed Ki67 expression in the nucleus (right; 10×). I. Immunofluorescent double staining showed limited effects in blood vessels (CD31, red) and lymphatic vessels (LYVE1, green) in p38TM tumors (scale bar 100 μm). The bar graph shows the quantification values of the immunostainings (*p < 0.05; ns: not significant).
Figure 5.

(continued).
3.6. The impact on tumorigenesis in vivo of using a p38 retro‐inhibition approach
The CAL27 cells expressing p38WT and p38TM were grafted into nude mice and tumor formation was evaluated. Daily SB203580 treatment (5 mg/kg/day) started after tumor reached 60 mm3 (Figure 5 E–G). No differences in the tumorigenicity of cells were observed. However, after 2 weeks of SB203580 treatment, p38WT tumors showed a significantly smaller tumor burden when compared with p38TM tumors that were treated in parallel (Figure 5 F and G). Upon SB203580 administration, p38WT tumors also revealed fewer proliferating cells detected by Ki‐67 staining. In contrast, SB203580‐treated p38TM tumors were resistant to the antitumor effect of the p38 inhibitor (Figure 5 H). This provided an opportunity to examine the role of cancer cells and stromal p38 in angiogenesis and lymphangiogenesis by a complementary approach. We performed double immunostaining which detected endothelial cells and lymph vessels. Treatment with the p38 MAPK inhibitor resulted in significantly reduced blood vessels in p38WT tumors (Figure 5 I, red). In contrast, SB203580 had minimal effect in blood vessels in p38TM tumors. Moreover, SB203580 treatment reduced lymph vessel growth (Figure 5 I, green), while expression of p38TM prevented this effect. Overall, while the limited effect of the p38 MAPK inhibitor in p38TM cells can be explained due to the direct impact of the inhibitor in the normal endothelial cells recruited to the tumor, our data strongly suggest that p38 function in the tumor cells may dictate the angiogenic and lymphangiogenic response in HNSCC tumors.
3.7. p38 MAPK controls cytokine and chemokine production in vivo
The remarkable impact of p38 inhibition in tumor‐induced angiogenesis promoted us to explore whether this MAPK contributes to the production of inflammatory and pro‐angiogenic mediators that contribute to the formation of a permissive tumor microenvironment. Indeed, multiple cytokines are highly elevated in the serum of HNSCC patients, some of which are currently being investigated as biomarkers for monitoring disease progression or recurrence (Chen et al., 1999; Pries and Wollenberg, 2006; Rusling et al., 2010). For these experiments, mice bearing HNSCC xenografts were treated for 2 days with SB203580 or its vehicle control, and sera collected for human cytokine analysis. At this time point no major differences in tumor volume are observed yet, hence preventing that the reduction of the tumor mass may act as a confounding factor. As expected, in control experiments we did not detect human cytokines in normal mice (not shown), but multiple cytokines and chemokines, including IL6, IL8, VEGF, IL1β, and GN‐CSF were readily detected in the serum of mice harboring CAL27 xenografts (Figure 6 A–E). All of these circulating human cytokines were remarkably reduced after the treatment of mice with the p38 inhibitor, suggesting that p38 plays an important role in HNSCC proliferation as well as in the release of tumor‐associated cytokines that may promote a permissive tumor microenvironment (Figure 6 F).
Figure 6.

p38 MAPK controls cytokine and chemokine production in HNSCC cells in vivo. CAL27 cells were transplanted into nude mice, and after the tumors reached approximately 200 mm3, the mice were treated with SB203580 (5 mg/kg) or vehicle (control). The level of circulating human IL6 (A), IL8 (B), VEGFA (C), IL‐1β (D), and GM‐CSF (E), were rapidly decreased in the serum of tumor bearing mice as early as 2 days after p38 inhibition (n = 4, *p < 0.05; **p < 0.01). F, Proposed roles of p38 controlling the phosphorylation and activity of transcription factors (TFs) regulating cell proliferation as well as the expression and release of multiple tumor‐derived cytokines. The latter may in turn promote angiogenesis and lymphangiogenesis, thereby favoring the establishment of a permissive tumor microenvironment. This may be disrupted by the inhibition of p38 using small molecule inhibitors.
4. Discussion
As part of comprehensive effort aimed at investigating the nature of the dysregulated signaling networks in a large sample collection of human HNSCC lesions (Molinolo et al., 2007), we have now investigated the activation in the MAPK cascades in this frequent human malignancy. Surprisingly, whereas ERK is marginally activated in HNSCC (Albanell et al., 2001), and we observed that JNK is also not often activated in this cancer type, strikingly most of the HNSCC lesions show highly activated p38. Whereas the increased activation of p38 has been reported in breast and bladder cancers (Davidson et al., 2006; Kumar et al., 2010), our results may represent the first demonstration of a highly active level of p38 in HNSCC. These findings may also explain prior reports showing elevated p38α protein levels in the serum of HNSCC patients, which decline after cancer control by radiotherapy (Gill et al., 2012).
The p38 MAPK is rapidly activated by various stimuli including inflammatory cytokines, growth factors and physical and chemical stresses resulting in cellular proliferation, differentiation, migration, and/or apoptosis (Cuenda and Rousseau, 2007; Wagner and Nebreda, 2009). The activation of p38 is primarily regulated by MAP kinase kinase (MKK) 3 and MKK6 (Wagner and Nebreda, 2009). Substrates of p38α include protein kinases such as MAPK‐activated protein kinases 2 (MK2), and multiple transcription factors, such as ATF2 (Cuenda and Rousseau, 2007; Wagner and Nebreda, 2009). Of interest, p38α can negatively regulate cell proliferation under physiological conditions in a variety of cell types in vivo, as it can promote cell differentiation (Hui et al., 2007b; Wagner and Nebreda, 2009). Genetic deletion of different members of this pathway revealed that p38α could even function as a tumor suppressor (Cuenda and Rousseau, 2007, 2007, 2007, 2009). On the other hand, several studies have shown that p38 plays an important role in hematological malignancies (Gaundar and Bendall, 2010), and in the growth and progression of a number of solid tumors such as breast (Chen et al., 2009), prostate (Rodriguez‐Berriguete et al., 2012), and lung cancers (Greenberg et al., 2002). These seemingly contradictory biological responses of inhibiting or stimulating cell growth in a tumor dependent fashion can likely be explained by the fact that many cancer cells exhibit a limited ability to engage genetic programs involved in cell differentiation or senescence due to genetic and epigenetic alterations. In line with this possibility, HNSCC harbor mutations in Notch genes that drive squamous differentiation, as well as loss of heterozygosity and epigenetic silencing of the remaining allele of the tumor suppressive and senescence initiator gene p16 ink4A (Agrawal et al., 2011; Stransky et al., 2011). Thus, we can speculate that HNSCC and other cancer types in which p38 is activated, this MAPK may contribute to tumor growth due to the loss of mechanisms triggering cell differentiation in response to p38 activation during the carcinogenic process.
Indeed, in HNSCC p38 can promote cancer cell growth both in vitro and in vivo, as its inhibition by a highly selective pharmacological agent (Davies et al., 2000) causes decreased tumor cell proliferation. Moreover, the direct role of p38 in this process was confirmed by a retro‐inhibition approach, as expression of a p38α form that is insensitive to SB203580 rendered this p38 inhibitor ineffective in vitro as well as in vivo. This was also further supported by genetic strategies enabling the decreased expression of p38α, specifically using lentiviral delivered shRNAs in HNSCC cells. Overall, we can conclude that while in normal cells p38 may drive cell differentiation or death, HNSCC cells may have reprogrammed their intracellular circuitries to resist the antiproliferative effects of p38, and instead grow after the activation of p38 and its downstream targets, or the release of p38 regulated inflammatory cytokines.
In addition to the role of p38 HNSCC in cell growth, this MAPK may also promote cancer cell invasion and migration (Kumar et al., 2010), and can contribute to the production of tumor‐related cytokines such as interleukin‐6 (IL‐6) (Vanderkerken et al., 2007). Here, we focused our attention on tumor‐induced angiogenesis and lymphangiogenesis, which have been linked to increase metastasis leading to decreased survival of patients with HNSCC (see in Patel et al., 2011 and references therein). In this regard, p38 activation was associated with decreased HNSCC differentiation and increased angiogenesis and lymphangiogenesis, both predictive markers of poor prognosis (Hasina and Lingen, 2001; Thomas et al., 2013), in a large collection of HNSCC tissues. The p38 pathway has been shown to control the release of several cytokines and chemokines including IL‐6, IL8, PDGF and VEGF which are potent proangiogenic factors (Gaundar et al., 2009; Wagner and Nebreda, 2009). In fact, our studies demonstrated that inhibition of p38 reduced both angiogenesis and lymphangiogenesis in mice in a tumor‐cell autonomous manner, as judged by the fact that p38 knockdown in cancer cells is sufficient to decrease both processes. Furthermore, the retro‐inhibition approach that renders cancer cells but not the host lymphatic and vascular endothelial cells insensitive to the p38 inhibitor prevented the ability of SB203580 to reduce intratumoral angiogenesis when used systemically. Consistent with these findings, p38 inhibition resulted in a remarkable reduction in the production of tumor‐derived cytokines and chemokines, which are likely involved in the tumor‐induced angiogenesis and lymphangiogenesis. As depicted in Figure 6 F, the emerging results support the possibility that the antitumor activity by p38 inhibition might reflect a direct effect on cancer cell proliferation, as well as the normalization of the tumor microenvironment by preventing the excess production of pro‐angiogenic factors, and hence reducing vascular and lymphatic endothelial cell recruitment and growth.
Taken together, the present observations suggest that p38α functions as a positive regulator of HNSCC cell proliferation and tumor angiogenesis and lymphangiogenesis. Whereas the precise biologically relevant downstream targets for p38 in HNSCC warrant further investigation, our collective findings provide a strong rationale for the early evaluation of the potential clinical benefits of p38 inhibitors in HNSCC patients that exhibit elevated activities of p38. Indeed, preventing the aberrant activation of the p38 MAPK pathway may lead to reduced tumor‐induced inflammation and angiogenesis, and ultimately to halt HNSCC growth and metastasis. Hence, these findings support that p38 may represent a potential novel molecular target for adjuvant therapy in HNSCC.
Funding source
The funding agency did not play a role in the study design, collection, analysis and interpretation of data, in the writing of the report; and in the decision to submit the article for publication.
Supporting information
The following is the supplementary data related to this article:
Supplemental Figure 1 Right: Activation of MAPK pathways in HNSCC cell lines. The Western blot shows the basal expression of MAPK family members and their status of activation (phosphorylation) in a variety of HNSCC cell lines. Normal oral keratinocytes (NOK) stimulated with EGF and IL‐1β were used as a control. Only HN30 HNSCC cells display elevated levels of p‐ERK and p‐JNK, while three of the HNSCC cell lines show remarkably increased basal p‐p38 levels, which could be seen in additional cells as well upon prolonged exposure of the film (not shown). p38 may be activated by tumor‐associated cytokines, as shown here in normal cells in response to IL‐1β as a control. Left: Similarly, IL‐1β stimulates p38 activation in CAL27 HNSCC cells, which is prevented by p38 inhibition with SB203580 (SB) (10 μM). SB also decreases the phosphorylation of MK2, a direct target of p38, which is already highly phosphorylated in these cancer cells.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Dental and Craniofacial Research, project Z01DE00558.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.10.003.
Leelahavanichkul Kantima, Amornphimoltham Panomwat, Molinolo Alfredo A., Basile John R., Koontongkaew Sittichai and Gutkind J. Silvio, (2014), A role for p38 MAPK in head and neck cancer cell growth and tumor‐induced angiogenesis and lymphangiogenesis, Molecular Oncology, 8, doi: 10.1016/j.molonc.2013.10.003.
References
- Agrawal, N. , Frederick, M.J. , Pickering, C.R. , Bettegowda, C. , Chang, K. , Li, R.J. , Fakhry, C. , Xie, T.X. , Zhang, J. , Wang, J. , Zhang, N. , El-Naggar, A.K. , Jasser, S.A. , Weinstein, J.N. , Trevino, L. , Drummond, J.A. , Muzny, D.M. , Wu, Y. , Wood, L.D. , Hruban, R.H. , Westra, W.H. , Koch, W.M. , Califano, J.A. , Gibbs, R.A. , Sidransky, D. , Vogelstein, B. , Velculescu, V.E. , Papadopoulos, N. , Wheeler, D.A. , Kinzler, K.W. , Myers, J.N. , 2011. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 333, 1154–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanell, J. , Codony-Servat, J. , Rojo, F. , Del Campo, J.M. , Sauleda, S. , Anido, J. , Raspall, G. , Giralt, J. , Rosello, J. , Nicholson, R.I. , Mendelsohn, J. , Baselga, J. , 2001. Activated extracellular signal-regulated kinases: association with epidermal growth factor receptor/transforming growth factor alpha expression in head and neck squamous carcinoma and inhibition by anti-epidermal growth factor receptor treatments. Cancer Res.. 61, 6500–6510. [PubMed] [Google Scholar]
- Amornphimoltham, P. , Leelahavanichkul, K. , Molinolo, A. , Patel, V. , Gutkind, J.S. , 2008. Inhibition of mammalian target of rapamycin by rapamycin causes the regression of carcinogen-induced skin tumor lesions. Clin. Cancer Res.. 14, 8094–8101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amornphimoltham, P. , Patel, V. , Leelahavanichkul, K. , Abraham, R.T. , Gutkind, J.S. , 2008. A retroinhibition approach reveals a tumor cell-autonomous response to rapamycin in head and neck cancer. Cancer Res.. 68, 1144–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L. , Mayer, J.A. , Krisko, T.I. , Speers, C.W. , Wang, T. , Hilsenbeck, S.G. , Brown, P.H. , 2009. Inhibition of the p38 kinase suppresses the proliferation of human ER-negative breast cancer cells. Cancer Res.. 69, 8853–8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. , Malhotra, P.S. , Thomas, G.R. , Ondrey, F.G. , Duffey, D.C. , Smith, C.W. , Enamorado, I. , Yeh, N.T. , Kroog, G.S. , Rudy, S. , McCullagh, L. , Mousa, S. , Quezado, M. , Herscher, L.L. , Van Waes, C. , 1999. Expression of proinflammatory and proangiogenic cytokines in patients with head and neck cancer. Clin. Cancer Res.. 5, 1369–1379. [PubMed] [Google Scholar]
- Cuenda, A. , Rousseau, S. , 2007. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta. 1773, 1358–1375. [DOI] [PubMed] [Google Scholar]
- Davidson, B. , Konstantinovsky, S. , Kleinberg, L. , Nguyen, M.T. , Bassarova, A. , Kvalheim, G. , Nesland, J.M. , Reich, R. , 2006. The mitogen-activated protein kinases (MAPK) p38 and JNK are markers of tumor progression in breast carcinoma. Gynecol. Oncol.. 102, 453–461. [DOI] [PubMed] [Google Scholar]
- Davies, S.P. , Reddy, H. , Caivano, M. , Cohen, P. , 2000. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J.. 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forastiere, A. , Koch, W. , Trotti, A. , Sidransky, D. , 2001. Head and neck cancer. N. Engl. J. Med.. 345, 1890–1900. [DOI] [PubMed] [Google Scholar]
- Gaundar, S.S. , Bendall, L.J. , 2010. The potential and limitations of p38MAPK as a drug target for the treatment of hematological malignancies. Curr. Drug Targets. 11, 823–833. [DOI] [PubMed] [Google Scholar]
- Gaundar, S.S. , Bradstock, K.F. , Bendall, L.J. , 2009. p38MAPK inhibitors attenuate cytokine production by bone marrow stromal cells and reduce stroma-mediated proliferation of acute lymphoblastic leukemia cells. Cell Cycle. 8, 2975–2983. [PubMed] [Google Scholar]
- Gill, K. , Mohanti, B.K. , Ashraf, M.S. , Singh, A.K. , Dey, S. , 2012. Quantification of p38alphaMAP kinase: a prognostic marker in HNSCC with respect to radiation therapy. Clin. Chim. Acta. 413, 219–225. [DOI] [PubMed] [Google Scholar]
- Greenberg, A.K. , Basu, S. , Hu, J. , Yie, T.A. , Tchou-Wong, K.M. , Rom, W.N. , Lee, T.C. , 2002. Selective p38 activation in human non-small cell lung cancer. Am. J. Respir. Cell Mol. Biol.. 26, 558–564. [DOI] [PubMed] [Google Scholar]
- Hasina, R. , Lingen, M.W. , 2001. Angiogenesis in oral cancer. J. Dent. Educ.. 65, 1282–1290. [PubMed] [Google Scholar]
- Hui, L. , Bakiri, L. , Mairhorfer, A. , Schweifer, N. , Haslinger, C. , Kenner, L. , Komnenovic, V. , Scheuch, H. , Beug, H. , Wagner, E.F. , 2007. p38alpha suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat. Genet.. 39, 741–749. [DOI] [PubMed] [Google Scholar]
- Hui, L. , Bakiri, L. , Stepniak, E. , Wagner, E.F. , 2007. p38alpha: a suppressor of cell proliferation and tumorigenesis. Cell Cycle. 6, 2429–2433. [DOI] [PubMed] [Google Scholar]
- Kumar, B. , Koul, S. , Petersen, J. , Khandrika, L. , Hwa, J.S. , Meacham, R.B. , Wilson, S. , Koul, H.K. , 2010. p38 Mitogen-activated protein kinase-driven MAPKAPK2 regulates invasion of bladder cancer by modulation of MMP-2 and MMP-9 activity. Cancer Res.. 70, 832–841. [DOI] [PubMed] [Google Scholar]
- Leelahavanichkul, K. , Gutkind, J.S. , 2012. Oral and pharyngeal epithelial cell culture. In Randell S.H., Methods in Molecular Biology. second ed. Humana Press; [DOI] [PubMed] [Google Scholar]
- Malhotra, R. , Patel, V. , Chikkaveeraiah, B.V. , Munge, B.S. , Cheong, S.C. , Zain, R.B. , Abraham, M.T. , Dey, D.K. , Gutkind, J.S. , Rusling, J.F. , 2012. Ultrasensitive detection of cancer biomarkers in the clinic by use of a nanostructured microfluidic array. Anal. Chem.. 84, 6249–6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinissen, M.J. , Chiariello, M. , Pallante, M. , Gutkind, J.S. , 1999. A network of mitogen-activated protein kinases links G protein-coupled receptors to the c-jun promoter: a role for c-Jun NH2-terminal kinase, p38s, and extracellular signal-regulated kinase 5. Mol. Cell Biol.. 19, 4289–4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinolo, A.A. , Hewitt, S.M. , Amornphimoltham, P. , Keelawat, S. , Rangdaeng, S. , Meneses Garcia, A. , Raimondi, A.R. , Jufe, R. , Itoiz, M. , Gao, Y. , Saranath, D. , Kaleebi, G.S. , Yoo, G.H. , Leak, L. , Myers, E.M. , Shintani, S. , Wong, D. , Massey, H.D. , Yeudall, W.A. , Lonardo, F. , Ensley, J. , Gutkind, J.S. , 2007. Dissecting the Akt/mammalian target of rapamycin signaling network: emerging results from the head and neck cancer tissue array initiative. Clin. Cancer Res.. 13, 4964–4973. [DOI] [PubMed] [Google Scholar]
- Patel, V. , Marsh, C.A. , Dorsam, R.T. , Mikelis, C.M. , Masedunskas, A. , Amornphimoltham, P. , Nathan, C.A. , Singh, B. , Weigert, R. , Molinolo, A.A. , Gutkind, J.S. , 2011. Decreased lymphangiogenesis and lymph node metastasis by mTOR inhibition in head and neck cancer. Cancer Res.. 71, 7103–7112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, G. , Robinson, F. , Beers Gibson, T. , Xu, B.E. , Karandikar, M. , Berman, K. , Cobb, M.H. , 2001. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev.. 22, 153–183. [DOI] [PubMed] [Google Scholar]
- Pries, R. , Wollenberg, B. , 2006. Cytokines in head and neck cancer. Cytokine Growth Factor Rev.. 17, 141–146. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Berriguete, G. , Fraile, B. , Martinez-Onsurbe, P. , Olmedilla, G. , Paniagua, R. , Royuela, M. , 2012. MAP kinases and prostate cancer. J. Signal Transduct.. 2012, 169170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusling, J.F. , Kumar, C.V. , Gutkind, J.S. , Patel, V. , 2010. Measurement of biomarker proteins for point-of-care early detection and monitoring of cancer. Analyst. 135, 2496–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Prieto, R. , Rojas, J.M. , Taya, Y. , Gutkind, J.S. , 2000. A role for the p38 mitogen-activated protein kinase pathway in the transcriptional activation of p53 on genotoxic stress by chemotherapeutic agents. Cancer Res.. 60, 2464–2472. [PubMed] [Google Scholar]
- Schaeffer, H.J. , Weber, M.J. , 1999. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol. Cell Biol.. 19, 2435–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel, R. , Desantis, C. , Virgo, K. , Stein, K. , Mariotto, A. , Smith, T. , Cooper, D. , Gansler, T. , Lerro, C. , Fedewa, S. , Lin, C. , Leach, C. , Cannady, R.S. , Cho, H. , Scoppa, S. , Hachey, M. , Kirch, R. , Jemal, A. , Ward, E. , 2012. Cancer treatment and survivorship statistics, 2012. CA Cancer J. Clin.. 62, 220–241. [DOI] [PubMed] [Google Scholar]
- Siegel, R. , Naishadham, D. , Jemal, A. , 2012. Cancer statistics, 2012. CA Cancer J. Clin.. 62, 10–29. [DOI] [PubMed] [Google Scholar]
- Specenier, P.M. , Vermorken, J.B. , 2009. Current concepts for the management of head and neck cancer: chemotherapy. Oral Oncol.. 45, 409–415. [DOI] [PubMed] [Google Scholar]
- Stransky, N. , Egloff, A.M. , Tward, A.D. , Kostic, A.D. , Cibulskis, K. , Sivachenko, A. , Kryukov, G.V. , Lawrence, M.S. , Sougnez, C. , McKenna, A. , Shefler, E. , Ramos, A.H. , Stojanov, P. , Carter, S.L. , Voet, D. , Cortes, M.L. , Auclair, D. , Berger, M.F. , Saksena, G. , Guiducci, C. , Onofrio, R.C. , Parkin, M. , Romkes, M. , Weissfeld, J.L. , Seethala, R.R. , Wang, L. , Rangel-Escareno, C. , Fernandez-Lopez, J.C. , Hidalgo-Miranda, A. , Melendez-Zajgla, J. , Winckler, W. , Ardlie, K. , Gabriel, S.B. , Meyerson, M. , Lander, E.S. , Getz, G. , Golub, T.R. , Garraway, L.A. , Grandis, J.R. , 2011. The mutational landscape of head and neck squamous cell carcinoma. Science. 333, 1157–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, B. , Stedman, M. , Davies, L. , 2013. Grade as a prognostic factor in oral squamous cell carcinoma: a population based analysis of the data. Laryngoscope. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- Turjanski, A.G. , Vaque, J.P. , Gutkind, J.S. , 2007. MAP kinases and the control of nuclear events. Oncogene. 26, 3240–3253. [DOI] [PubMed] [Google Scholar]
- Vanderkerken, K. , Medicherla, S. , Coulton, L. , De Raeve, H. , Willems, A. , Lawson, M. , Van Camp, B. , Protter, A.A. , Higgins, L.S. , Menu, E. , Croucher, P.I. , 2007. Inhibition of p38alpha mitogen-activated protein kinase prevents the development of osteolytic bone disease, reduces tumor burden, and increases survival in murine models of multiple myeloma. Cancer Res.. 67, 4572–4577. [DOI] [PubMed] [Google Scholar]
- Wagner, E.F. , Nebreda, A.R. , 2009. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer. 9, 537–549. [DOI] [PubMed] [Google Scholar]
- Yoshizuka, N. , Chen, R.M. , Xu, Z. , Liao, R. , Hong, L. , Hu, W.Y. , Yu, G. , Han, J. , Chen, L. , Sun, P. , 2012. A novel function of p38-regulated/activated kinase in endothelial cell migration and tumor angiogenesis. Mol. Cell Biol.. 32, 606–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplemental Figure 1 Right: Activation of MAPK pathways in HNSCC cell lines. The Western blot shows the basal expression of MAPK family members and their status of activation (phosphorylation) in a variety of HNSCC cell lines. Normal oral keratinocytes (NOK) stimulated with EGF and IL‐1β were used as a control. Only HN30 HNSCC cells display elevated levels of p‐ERK and p‐JNK, while three of the HNSCC cell lines show remarkably increased basal p‐p38 levels, which could be seen in additional cells as well upon prolonged exposure of the film (not shown). p38 may be activated by tumor‐associated cytokines, as shown here in normal cells in response to IL‐1β as a control. Left: Similarly, IL‐1β stimulates p38 activation in CAL27 HNSCC cells, which is prevented by p38 inhibition with SB203580 (SB) (10 μM). SB also decreases the phosphorylation of MK2, a direct target of p38, which is already highly phosphorylated in these cancer cells.