

Abstract
Background
Previous studies indicate that as many as six genes within the PARK10 region (RNF11, UQCRH, HIVEP3, EIF2B3, USP24, ELAVL4) might modify susceptibility or age at onset in Parkinson’s disease (PD).
Methods
We sought to identify new PD susceptibility genes and to validate previously nominated candidate genes within the PARK10 region using a two-stage design. We used data from a large, publicly-available genome-wide association study (GWAS) in the discovery stage (n=2000 cases and 1986 controls) and data from three independent studies for the replication stage (total n=2113 cases and 2095 controls). Marker density was increased by imputation using HapMap3 and 1000 Genomes reference panels, and over 40,000 single nucleotide polymorphisms (SNPs) were used in the final analysis. The association between each SNP and PD was modeled using logistic regression with an additive allele dosage effect and adjusted for sex, age, and axes of geographical variation.
Results
Although the discovery stage yielded promising findings for SNPs in several novel genes, including DAB1, none of the results were validated in the replication stage. Furthermore, in meta-analyses across all datasets no genes within PARK10 reached significance after accounting for multiple testing.
Conclusion
Our results suggest that common variation in the PARK10 region is not associated with PD risk. However, additional studies are needed to assess the role of PARK10 in modifying age at onset and to determine whether rare variants in this region might affect PD susceptibility.
Keywords: PARK10, Parkinson’s Disease, Replication, GWAS
1. Introduction
Parkinson’s disease (PD) is the second-most common neurodegenerative disorder with a lifetime risk estimated at 1.6% [1]. Linkage and association studies have implicated over 20 susceptibility loci for PD, though in many instances the specific disease gene has not been clearly identified [2, 3]. In 2002, two independent studies reported evidence of linkage to PD on chromosome 1p32; one to PD risk [4] and the other to age at onset [5]. This region, which was designated PARK10, spans 19.2 megabases (Mb) (40.2–59.4 Mb on NCBI build 36) and contains approximately 200 genes. Several subsequent studies [6–8] nominated potential PD genes within the PARK10 region including the: (1) ring-finger protein 11 gene (RNF11), (2) ubiquinol-cytochrome c reductase hinge protein gene (UQCRH), (3) human immunodeficiency virus enhancer-binding protein 3 gene (HIVEP3), (4) gamma-subunit of the translation initiation factor EIF2B gene (EIF2B3), (5) ubiquitin-specific protease 24 gene (USP24), and (6) embryonic lethal, abnormal vision, Drosophila-like 4 gene (ELAVL4). However, attempts to replicate these candidate genes have yielded mixed results [9–11] and no markers within the PARK10 region have reached genome-wide significance in any of the recently published PD genome-wide association studies [3, 12–19]. Thus, there is considerable uncertainty as to whether the PARK10 region actually harbors PD susceptibility genes.
We re-examined the PARK10 region in the publicly available NeuroGenetics Research Consortium (NGRC) Genome-Wide Association Study (GWAS) dataset enriched with additional markers imputed from the HapMap 3 and 1000 Genomes reference panels. Furthermore, we utilized two other publicly available GWAS datasets [15, 18] and unpublished data from a third case-control study [20] for replication. Our primary goals were to identify new PD-associated genes that might have been previously missed due to low genotyping coverage, and to verify associations with the aforementioned candidate genes.
2. Methods
2.1 Studies
The discovery phase for novel risk variants used data from the NGRC Genome-Wide Association Study of Parkinson Disease: Genes and Environment (phs000196.v1.p1) with 2000 subjects with PD and 1986 controls that were recruited from movement disorder clinics in Oregon, Washington, Georgia, and New York [14]. All subjects were genotyped on the IlluminaHumanOmni1-Quad_v1-0_B array and data were downloaded from dbGaP. Details of the quality control processes employed in the analysis are provided (Appendix A. Supplementary methods).
The replication phase used two publicly available GWASs, the Center for Inherited Disease Research (CIDR) GWAS in Familial PD (phs000126.v1.p1) and the National Institute of Neurological Disorders and Stroke (NINDS) Genome-Wide Genotyping in PD Study (phs000089.v3.p2), and a population-based study of PD at the University of Washington and Group Health Cooperative (GHC-UW) [20]. Information on both the CIDR [15] and NINDS [18] GWAS datasets has been published elsewhere. Because controls for both GWASs came from the NINDS Human Genetics Repository (http://ccr.coriell.org/Sections/Collections/NINDS), we performed identity-by-descent (IBD)-estimation using 287,341 independent single nucleotide polymorphisms (SNPs) to elucidate any overlap in controls that showed evidence of being duplicates or were a result of cryptic relatedness (PI≥0.5). We found evidence that nine controls overlapped between the NINDS and CIDR GWAS datasets, and these individuals were subsequently omitted from the NINDS dataset. The GHC-UW sample consisted of 758 non-Hispanic, white subjects (321 cases, 437 controls) who were genotyped for 1,138 tagging SNPs spanning the PARK10 region and 440 ancestry informative markers (AIMs) using custom Affymetrix GeneChip Universal 3K Tag Arrays. The AIMs were unlinked SNPs selected to distinguish intercontinental population structure [21, 22] as well as European substructure [23]. Before imputation, SNPs were omitted if Hardy-Weinberg equilibrium was violated (P < 0.001 using an exact test) in the combined sample or if the genotyping rate was less than 90%. The project was approved by the Veterans Affairs Puget Sound Health Care System and University of Washington institutional review boards, and all subjects provided written informed consent.
Patients in the NGRC, CIDR, and NINDS studies met UK PD Society Brain Bank clinical diagnostic criteria for PD [14, 15, 18]. Patients in the GHC-UW study met similar clinical diagnostic criteria for PD as previously described [20]. The age at onset of PD was similar across studies with the following mean +/− SD onset age: 58.3+/−11.9 years (NGRC), 61.9+/−10.9 years (CIDR), 58.4+/−13.2 years (NINDS). In the GHC-UW study, age at onset data were not collected, but the mean +/− SD age at diagnosis was 66.2 +/− 10.4 years.
2.2 Genotype enrichment and imputation
A subset of 499 subjects from the NGRC study population was previously genotyped using the same custom Affymetrix 3K Array as was used in the GHC-UW study. Prior to imputation, these genotype data were merged into the NGRC dataset using the default consensus genotype call approach in PLINK [24]. Additionally, standard data-cleaning methods (e.g. strand flipping) were performed. We then used IMPUTE2 [25] to enrich the PARK10 region with imputed markers. To ensure that rare variants were adequately covered, we used two phased reference panels from both HapMap3 and the 1000 Genomes pilot data, with release dates of Feb 2009 and Jun 2010, respectively, and we imputed genotypes for every 5-Mb interval in the 40–60 Mb region of chromosome 1 for each study separately. We omitted SNPs with an information metric less than 0.30 and noted those SNPs with information metrics between 0.30 and 0.50. After genotype imputation, the number of SNPs (NSNP-STUDY) analyzed in the PARK10 region for the NGRC, CIDR, NINDS, and GHC-UW studies were: NSNP-NGRC = 43,799, NSNP-CIDR = 43,243, NSNP-NINDS = 43,362 and NSNP-GHC-UW = 40,351, respectively, with 43,744 SNPs common to two or more studies.
2.3 Statistical analysis
Failure to recognize population structure can lead to confounding results. Thus, we accounted for differences in ancestry between cases and controls in our analyses using a principal components analysis (PCA) approach as implemented in the EigenStrat program [26]. For the three GWASs, we obtained a subset of uncorrelated markers by pruning the genome-wide SNPs to approximate linkage equilibrium as described in the Supplement. These markers were then used to calculate the axes of geographical variance or principal components (PCs) that describe the genetic variation. The first 28, 10, and 9 PCs were found to be significant (Tracy-Widom P-value < 0.05) for the NGRC, NINDS, and CIDR studies, respectively. However, for the GHC-UW dataset, we used the 440 AIMs to perform PCA, and although Tracy-Widom statistics may not be applicable to AIMs, we conservatively chose the top six PCs (Tracy-Widom P-value < 0.05).
We used PLINK [24] to analyze the genotyped and imputed SNPs based on the genotype probabilities with an additive dosage model in a logistic regression analysis adjusted for sex, age, and the significant PCs from EigenStrat PCA analysis. The asymptotic P-value was obtained from the Z-test assessing the significance of the association with PD. SimpleM [27] was used to infer the effective number of independent tests (Meff) after accounting for LD between SNPs with a minor allele frequency (MAF) of at least 0.005; Meff was 15,572 in the NGRC study. Thus, after accounting for multiple testing using a Bonferroni adjustment on the effective number of tests, we set the significance threshold for the overall study to P < 3.2 × 10−6 (i.e., 0.05/15,572). However, in selecting SNPs from the discovery phase for replication we used a less stringent threshold of P < 5 × 10−4 to reduce the probability of false negative results. Additionally, we performed a meta-analysis for each SNP across all studies using the classical approach of pooling effect size estimates and standard errors using a random effects model in PLINK. Cochrane’s Q statistic was used to test for the presence of heterogeneity, and I2 was calculated to examine the degree of heterogeneity present, and values of 25%, 50%, and 75% generally represent low, moderate, and high levels of heterogeneity, respectively.
Power was calculated using Quanto (http://hydra.usc.edu/gxe) and assuming a 1.5% prevalence of PD, a significance of 5×10−4 and a rare variant under a log-additive genetic model with a minor allele frequency (MAF) of 0.05 and 0.25.
3. Results
In the NGRC dataset, SNPs in five genes (DAB1, SLFNL1, OMA1, SSBP3, and AGBL4) met the predefined, exploratory significance threshold of 5 × 10−4 (Figure 1). The two SNPs with the lowest P-values resided near DAB1 (Table 1) (rs61781879, P=1.4 × 10−6; rs61781882, P=1.8 × 10−6). The next most significant marker occurred within SLFNL1 (rs11576546, P = 9.4 × 10−5). In the NGRC dataset, we did not replicate the association findings of any of the previously nominated PARK10 candidate genes (Table 2), though the P-values for two SNPs in HIVEP3 approached the exploratory significance threshold (rs4600038, P = 7.8 × 10−4; rs6680824, P = 9.3 × 10−4). In the discovery phase for variants with a MAF of 0.05 and 0.25, there was at least 80% power to detect risk alleles with odds ratios (ORs) of 1.50 and 1.25, respectively, and protective alleles with ORs of 0.60 and 0.80, respectively.
Figure 1. Manhattan plot of the PARK10 region in the discovery phase.
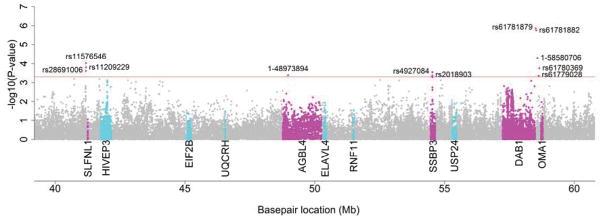
Each dot represents −log10(P-value) (y-axis) for a given SNP at a given location in 106 base pairs (Mb) in the PARK10 region on chromosome 1 (x-axis). SNPs above the exploratory significance threshold (P < 5 × 10−4) are individually labeled. Grey dots represent SNPs from genes lacking any significant markers. SNPs from novel genes having one or more markers above the threshold are indicated by magenta dots. Cyan dots indicate SNPs from previously nominated candidate genes.
Table 1.
Association results for novel PARK10 genes identified in the discovery phase
| Discovery | Replication | Meta-Analysis | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||
| NGRC (2000 cases/1986 controls) | GHC-UW (316 cases/436 controls) | CIDR (857 cases/867 controls) | NINDS (940 cases/792 controls) | All Studies (4113 cases/4081 controls) | ||||||||||||
|
| ||||||||||||||||
| Gene | SNP | Positiona | MAb | MAFb | OR(95%CI) | P | OR(95%CI) | P | OR(95%CI) | P | OR(95%CI) | P | ORR | PR | PQ | I2 |
| DAB1 | rs61781879 | 58514900 | C | .04 | .35(.23,.53) | 1.4×10−6 | .51(.17,1.49) | .22 | 1.62(.78,3.37) | .20 | .79(.45,1.39) | .41 | .68 | .28 | .003 | 79 |
| rs61781882 | 58535050 | T | .04 | .35(.23,.54) | 1.8×10−6 | .50(.17,1.47) | .21 | 1.6(.77,3.31) | .21 | .79(.45,1.39) | .41 | .68 | .28 | .003 | 78 | |
| 1-58580706 | 58580706 | G | .02 | .44(.29,.65) | 5.2×10−5 | .66(.27,1.61) | .36 | 1.05(.54,2.01) | .89 | .83(.47,1.46) | .52 | .68 | .08 | .093 | 53 | |
|
| ||||||||||||||||
| SLFNL1 | rs11576546 | 41189495 | G | .21 | .78(.68,.88) | 9.4×10−5 | .89(.68,1.16) | .38 | 1.09(.91,1.32) | .35 | .83(.70,1.00) | .05 | .88 | .12 | .031 | 66 |
| rs11209229 | 41187945 | G | .20 | .78(.69,.89) | 1.6×10−4 | .87(.66,1.14) | .32 | 1.08(.89,1.3) | .44 | .84(.71,1.01) | .06 | .88 | .09 | .056 | 60 | |
| rs28691006 | 41183208 | C | .21 | .80(.71,.90) | 2.5×10−4 | .89(.69,1.15) | .39 | 1.09(.91,1.31) | .33 | .84(.71,1.00) | .05 | .89 | .13 | .041 | 64 | |
|
| ||||||||||||||||
| OMA1 | rs61780369 | 58653409 | T | .02 | .39(.23,.63) | 1.8×10−4 | .60(.21,1.68) | .33 | 1.99(.73,5.4) | .18 | .62(.29,1.33) | .22 | .67 | .24 | .039 | 64 |
| rs61779028 | 58631461 | A | .02 | .50(.34,.73) | 4.4×10−4 | .82(.33,2.03) | .67 | 1.42(.69,2.92) | .34 | .96(.56,1.66) | .89 | .82 | .43 | .046 | 63 | |
|
| ||||||||||||||||
| SSBP3 | rs4927084 | 54532654 | G | .30 | 1.21(1.09,1.34) | 2.9×10−4 | .77(.61,.96) | .02 | 1.05(.9,1.22) | .56 | .96(.82,1.11) | .57 | 1.00 | 1.00 | .001 | 81 |
| rs2018903 | 54531693 | C | .30 | 1.21(1.09,1.34) | 4.4×10−4 | .77(.61,.96) | .02 | 1.05(.9,1.23) | .55 | .95(.82,1.11) | .55 | 1.00 | .99 | .001 | 81 | |
|
| ||||||||||||||||
| AGBL4 | rs72904830c | 48973894 | A | .02 | .41(.25,.68) | 4.1×10−4 | - | - | 1.48(.64,3.44) | .36 | .75(.41,1.36) | .35 | .72 | .35 | .029 | 72 |
CI, confidence interval; OR, odds ratio; ORR, pooled odds ratio for random effects model; P, P-value corresponding to OR; PR, P-value corresponding to ORR in random effects model; PQ, P-value for Cochran’s Q statistic for heterogeneity; I2, percentage of the total variability in a set of effect sizes due to heterogeneity; MA, minor allele; MAF, minor allele frequency; SNP, single-nucleotide polymorphism
Results within grey cells are for SNPs with an information metric between 0.30 and 0.50. The information metric is a measure of the SNP imputation certainty ranging from 0 to 1 with increasing certainty.
Based on NCBI build 36.
In the NGRC study.
The results for rs72904830 in the GHC-UW study were not shown because the information metric was less than 0.20, and the resulting meta-analysis only used results from the other three studies.
Table 2.
Association results for previously nominated PARK10 candidate genes
| Discovery | Replication | Meta-Analysis | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||
| NGRC (2000 cases/1986 controls) | GHC-UW (316 cases/436 controls) | CIDR (857 cases/867 controls) | NINDS (940 cases/792 controls) | All Studies (4113 cases/4081 controls) | ||||||||||||
|
| ||||||||||||||||
| Gene | SNP | Positiona | MAb | MAFb | OR(95%CI) | P | OR(95%CI) | P | OR(95%CI) | P | OR(95%CI) | P | ORR | PR | PQ | I2 |
| HIVEP3 | rs4600038c | 42007887 | A | 0.42 | .85(.77,.93) | 7.8×10−4 | 1.02(.82,1.27) | .84 | 1.00(.86,1.18) | .96 | .96(.82,1.11) | .56 | .93 | .16 | .16 | 41 |
| rs6680824c | 42008591 | T | 0.45 | 1.17(1.07,1.29) | 9.3×10−4 | .99(.81,1.22) | .93 | 1.03(.88,1.19) | .73 | 1.02(.89,1.18) | .75 | 1.08 | .09 | .22 | 32 | |
| rs661225d | 42082752 | G | 0.05 | .98(.82,1.17) | .80 | .88(.71,1.09) | .23 | 1.04(.90,1.21) | .58 | 1.04(.91,1.20) | .55 | 1.00 | .94 | .54 | 0 | |
| rs648178d | 41774750 | G | 0.10 | .99(.88,1.11) | .81 | .93(.74,1.17) | .55 | .97(.81,1.15) | .73 | 1.03(.88,1.22) | .68 | .99 | .75 | .90 | 0 | |
|
| ||||||||||||||||
| ELAVL4 | rs2480684c | 50392177 | G | 0.29 | .84(.73,.96) | .01 | .97(.79,1.20) | .79 | .92(.74,1.13) | .42 | 1.04(.85,1.27) | .73 | .92 | .08 | .33 | 12 |
| rs7532927c | 50397630 | G | 0.28 | 1.15(1.03,1.28) | .02 | 1.09(.83,1.42) | .54 | 1.11(.94,1.32) | .22 | .98(.84,1.16) | .85 | 1.10 | .02 | .51 | 0 | |
| rs967582d | 50354759 | C | 0.35 | 1.07(.97,1.18) | .19 | .84(.67,1.04) | .11 | 1.02(.88,1.18) | .81 | .89(.77,1.03) | .13 | .97 | .58 | .08 | 55 | |
|
| ||||||||||||||||
| USP24 | 1-55362603c | 55362603 | C | 0.05 | .77(.62,.96) | .02 | .80(.46,1.39) | .42 | .91(.63,1.34) | .64 | 1.10(.78,1.54) | .59 | .86 | .08 | .38 | 3 |
| rs17111652c | 55363053 | T | 0.05 | .77(.62,.96) | .02 | .76(.45,1.28) | .30 | .94(.67,1.32) | .72 | 1.13(.82,1.56) | .45 | .89 | .21 | .25 | 28 | |
| rs287235d | 55451087 | G | 0.24 | 1.03(.93,1.15) | .56 | 1.11(.87,1.42) | .41 | .99(.84,1.17) | .94 | .90(.76,1.06) | .19 | 1.00 | .99 | .43 | 0 | |
| rs1165226d | 55380593 | C | 0.38 | .99(.90,1.09) | .82 | 1.08(.88,1.34) | .46 | 1.00(.86,1.16) | 1 | .90(.78,1.04) | .17 | .98 | .57 | .53 | 0 | |
|
| ||||||||||||||||
| RNF11 | 1-51505913c | 51505913 | T | 0.01 | 1.85(1.06,3.22) | .03 | .26(.05,1.40) | .12 | 2.73(1.04,7.15) | .04 | 1.37(.6,3.11) | .46 | 1.50 | .21 | .11 | 50 |
| rs72692296c | 51500183 | T | 0.01 | .55(.31,1.00) | .05 | .79(.24,2.59) | .70 | .67(.33,1.37) | .27 | 1.11(.47,2.59) | .81 | .70 | .06 | .62 | 0 | |
|
| ||||||||||||||||
| UQCRH | rs55762739c | 46554315 | T | 0.04 | 1.38(1.02,1.86) | .04 | 1.19(.5,2.82) | .70 | 1.43(.87,2.34) | .16 | .82(.49,1.36) | .44 | 1.23 | .09 | .34 | 11 |
| 1-46545090c | 46545090 | G | 0.01 | .65(.39,1.08) | .09 | 2.45(.93,6.5) | .07 | 1.63(.80,3.35) | .18 | 1.14(.58,2.23) | .71 | 1.20 | .52 | .05 | 62 | |
|
| ||||||||||||||||
| EIF2B3 | 1-45102457c | 45102457 | T | 0.02 | 1.54(1.02,2.33) | .04 | 1.35(.36,5.08) | .66 | .76(.35,1.65) | .49 | 1.01(.56,1.83) | .96 | 1.23 | .19 | .38 | 2 |
| rs72214904c | 45139896 | T | 0.01 | 1.96(1.01,3.79) | .05 | .17(.04,.83) | .03 | 1.33(.59,3.00) | .49 | 1.63(.63,4.20) | .31 | 1.17 | .69 | .05 | 62 | |
| rs546354d | 45204251 | G | 0.18 | .98(.87,1.11) | .77 | 1.09(.83,1.43) | .53 | 1.11(.92,1.34) | .28 | .94(.79,1.13) | .54 | 1.01 | .84 | .58 | 0 | |
CI, confidence interval; OR, odds ratio; ORR, pooled odds ratio for random effects model; P, P-value corresponding to OR; PR, P-value corresponding to ORR in random effects model; PQ, P-value for Cochran’s Q statistic for heterogeneity; I2, percentage of the total variability in a set of effect sizes due to heterogeneity; MA, minor allele; MAF, minor allele frequency; SNP, single-nucleotide polymorphism
Results within grey cells are for SNPs with an information metric between 0.30 and 0.50. The information metric is a measure of the SNP imputation certainty ranging from 0 to 1 with increasing certainty.
Based on NCBI build 36.
In the NGRC study.
Among the top two SNPs for the corresponding gene in the discovery phase.
Results from the replication phase for new variants are shown in Table 1. None of the SNPs discovered in the NGRC dataset were replicated in the CIDR, NINDS, or GHC-UW studies. Although two SNPs (rs4927084, rs2018903) in SSBP3 were weakly associated with PD in the GHC-UW study, the direction of the effect was opposite to that seen in the NGRC study. Several of the imputed SNPs chosen for replication had marginal information metrics (0.3 –0.5), including DAB1 variants rs61781879 and rs61781882 (Table 1). In the meta-analysis, none of the SNPs examined were associated with PD (P > 0.05). However, we observed evidence of heterogeneity across studies as indicated by I2 > 50% for all SNPs and a significant Cochran’s Q statistic (PQ < 0.05) for all but two SNPs (1-58580706 in DAB1 and rs11209229 in SLFNL1).
We also examined previously nominated PARK10 candidate genes in more detail. From each of these genes, we selected the two SNPs with the lowest P-values in the NGRC and candidate SNPs in previously published studies [7–11] to test for association with PD in the CIDR, NINDS, and GHC-UW datasets (Table 2). However, no significant associations were observed in any of the three studies or in the meta-analysis across studies. In the meta-analysis, there were varying levels of heterogeneity, with I2 values ranging from 0–62%, but Cochran’s Q statistic was not significant for any of the SNPs (PQ ≥ 0.05).
4. Discussion
We sought to identify PD susceptibility genes within the PARK10 region using data on over 40,000 markers in a two-stage design. Despite promising results in the discovery phase for SNPs in several novel genes, especially DAB1, none of these findings were validated in the replication stage. Furthermore, in the meta-analyses across all datasets, no genes within PARK10 reached significance after accounting for multiple testing. One consideration in interpreting the results observed for DAB1 is that the top-ranked SNPs were all imputed and in most instances the information metric was between 0.30 and 0.50. Thus, findings for this gene might simply represent false positives due to inaccuracies in imputation.
Oliveira and colleagues performed association mapping of the PARK10 region with 284 SNPs in 267 multiplex PD families [8]. Using both an orthogonal model (OM) and the Monks-Kaplan method (MKM) they observed an association with age at onset for two genes, EIF2B3 (rs546354, POM = 0.01 and PMKM = 0.0004) and USP24 (rs287235, POM = 0.001 and PMKM = 0.004). The authors also reported that SNPs in HIVEP3 were associated with PD risk (rs648178, P = 0.008; rs661225, P = 0.004). USP24 was later reported to associate with PD risk in a small case-control study [11]. In a sample containing both multiplex and singleton PD families (n=643), Noureddine et al reported an association between ELAVL4 (located within the PARK10 region) and age at onset (rs967582, P = 0.006) but not PD risk [7]. Subsequent attempts to replicate this finding in case-control studies have yielded mixed results. Markers within ELAVL4 were found to associate with PD risk in the GenePD Study [9] and in an Irish PD cohort [10], but not in case-control series from Norway [10] or the United States [10]. Two genes, RNF11 and UQCRH, have gained consideration as PARK10 candidate genes based on indirect evidence. In a comprehensive analysis of gene expression patterns in substantia nigra, both genes were highly differentially expressed in PD patients versus controls (RNF11, P = 8.9 × 10−7; UQCRH, P = 3.0 × 10−6). Furthermore, RNF11 is expressed at high levels in neurons and is contained within Lewy bodies [28]. However, SNPs in these two genes were not associated with PD risk or age at onset in the study by Oliveira and colleagues [8] and to our knowledge these genes have not been included in other published candidate gene analyses. Subsequent to the aforementioned studies, a number of PD GWASs have been published. Most focused on PD risk [12–19] but one examined age at onset [29]. However, no genes within the PARK10 region reached genome-wide significance or were among the highest ranking genes listed in any study.
Overall, we did not observe evidence of association with PD risk for any of the previously nominated PARK10 candidate genes (Table 2). There are several possible explanations for our failure to replicate these findings. First, because our study populations included only unrelated cases, we chose to test for association with risk but not age at onset. Using this approach we might have missed genes that primarily modify age at onset but not susceptibility. Second, our study might have lacked adequate power to detect risk alleles of small effect or of low frequency. This limitation is especially relevant for rare variants that were imputed, as imputation accuracy tends to be lower for low frequency markers. Third, the subjects used to generate all of the datasets except CIDR represented a mixture of familial and sporadic PD which might have confounded results if the effects of PARK10 variants are largely confined to familial PD. In contrast, the study which provided the most robust evidence in favor of specific candidate genes within the PARK10 region utilized only multiplex PD families [8]. However, there is evidence to suggest that familial and sporadic PD share common genetic pathways in many instances [30]. Finally, it is possible that previous studies reporting that variants in the PARK10 region affect PD risk or age at onset simply represent spurious findings.
Our results suggest that common variation in the PARK10 region is not associated with PD risk. However, additional studies are needed to assess the role of PARK10 in modifying age at onset and to determine whether rare variants in the PARK10 region affect PD susceptibility. Family-based analyses utilizing whole exome or whole genome sequencing and possibly focusing on pedigrees with evidence of linkage to the region might have the highest likelihood of success.
Supplementary Material
Acknowledgments
We thank Brian Fish for computing support and Nirupama Shridhar and Taryn O. Hall for technical support. This work was supported by funding from the Department of Veterans Affairs (1I01BX000531), National Institutes of Health (P50 NS062684, P42 ES004696, and R01 NS065070), and the University of Washington Royalty Research Fund (3677).
Appendix A. Supplementary methods
Supplementary methods related to this project can be found online.
Footnotes
Financial disclosure
Ms. Wan receives salary support from the NIH.
Dr. Edwards is funded by grants from the NIH.
Dr. Hutter received funding from University of Washington Royalty Research Fund and grants from the NIH.
Dr. Mata is funded by grants from the Department of Veterans Affairs, NIH, and Parkinson’s Disease Foundation.
Dr. Samii has received honoraria from Teva and Boehringer-Ingelheim.
Dr. Roberts has no financial disclosures.
Dr. Agarwal has received honoraria as a speaker for Teva, GSK, UCB, Impax, and consultancy for Merz and has been provided research support from Allergan, Prana Biotechnology Limited, MERZ, NeuroSearch, Solstice Neurosciences, Ipsen, Adamas, CHDI, HP Therapeutics Foundation, Inc., Addex Therapeutics, and the NIH.
Dr. Checkoway is funded by grants from the NIH and the Multiple Sclerosis Society. He also serves as a paid consultant for the Alcoa Co., Pittsburgh, PA, the Electric Power Research Institute, Palo Alto, CA, ENVIRON, Amherst, MA, and the University of Minnesota Dept. of Environmental Health, Minneapolis, MN
Dr. Farin is funded by grants from the NIH.
Ms. Yearout receives salary support from the Department of Veterans Affairs and NIH.
Dr. Zabetian is funded by grants from the Department of Veterans Affairs, NIH, the American Parkinson Disease Association, Department of Veterans Affairs, NIH, and Parkinson’s Disease Foundation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Elbaz A, Bower JH, Maraganore DM, McDonnell SK, Peterson BJ, Ahlskog JE, et al. Risk tables for Parkinsonism and Parkinson’s disease. J Clin Epidemiol. 2002;55:25–31. doi: 10.1016/s0895-4356(01)00425-5. [DOI] [PubMed] [Google Scholar]
- 2.Bekris LM, Mata IF, Zabetian CP. The genetics of Parkinson disease. J Geriatr Psychiatry Neurol. 2010;23:228–42. doi: 10.1177/0891988710383572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, et al. International Parkinson Disease Genomics C. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377:641–9. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hicks AA, Petursson H, Jonsson T, Stefansson H, Johannsdottir HS, Sainz J, et al. A susceptibility gene for late-onset idiopathic Parkinson’s disease. Ann Neurol. 2002;52:549–55. doi: 10.1002/ana.10324. [DOI] [PubMed] [Google Scholar]
- 5.Li YJ, Scott WK, Hedges DJ, Zhang F, Gaskell PC, Nance MA, et al. Age at onset in two common neurodegenerative diseases is genetically controlled. Am J Hum Genet. 2002;70:985–93. doi: 10.1086/339815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noureddine MA, Li YJ, van der Walt JM, Walters R, Jewett RM, Xu H, et al. Genomic convergence to identify candidate genes for Parkinson disease: SAGE analysis of the substantia nigra. Mov Disord. 2005;20:1299–309. doi: 10.1002/mds.20573. [DOI] [PubMed] [Google Scholar]
- 7.Noureddine MA, Qin XJ, Oliveira SA, Skelly TJ, van der Walt J, Hauser MA, et al. Association between the neuron-specific RNA-binding protein ELAVL4 and Parkinson disease. Hum Genet. 2005;117:27–33. doi: 10.1007/s00439-005-1259-2. [DOI] [PubMed] [Google Scholar]
- 8.Oliveira SA, Li YJ, Noureddine MA, Zuchner S, Qin X, Pericak-Vance MA, et al. Identification of risk and age-at-onset genes on chromosome 1p in Parkinson disease. Am J Hum Genet. 2005;77:252–64. doi: 10.1086/432588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeStefano AL, Latourelle J, Lew MF, Suchowersky O, Klein C, Golbe LI, et al. Replication of association between ELAVL4 and Parkinson disease: the GenePD Study. Hum Genet. 2008;124:95–9. doi: 10.1007/s00439-008-0526-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haugarvoll K, Toft M, Ross OA, Stone JT, Heckman MG, White LR, et al. ELAVL4, PARK10, and the Celts. Mov Disord. 2007;22:585–7. doi: 10.1002/mds.21336. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Schrodi S, Rowland C, Tacey K, Catanese J, Grupe A. Genetic evidence for ubiquitin-specific proteases USP24 and USP40 as candidate genes for late-onset Parkinson disease. Hum Mutat. 2006;27:1017–23. doi: 10.1002/humu.20382. [DOI] [PubMed] [Google Scholar]
- 12.Do CB, Tung JY, Dorfman E, Kiefer AK, Drabant EM, Francke U, et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 2011;7:e1002141. doi: 10.1371/journal.pgen.1002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edwards TL, Scott WK, Almonte C, Burt A, Powell EH, Beecham GW, et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet. 2010;74:97–109. doi: 10.1111/j.1469-1809.2009.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet. 2010;42:781–5. doi: 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pankratz N, Wilk JB, Latourelle JC, DeStefano AL, Halter C, Pugh EW, et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet. 2009;124:593–605. doi: 10.1007/s00439-008-0582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saad M, Lesage S, Saint-Pierre A, Corvol JC, Zelenika D, Lambert JC, et al. Genome-wide association study confirms BST1 and suggests a locus on 12q24 as the risk loci for Parkinson’s disease in the European population. Hum Mol Genet. 2011;20:615–27. doi: 10.1093/hmg/ddq497. [DOI] [PubMed] [Google Scholar]
- 17.Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–7. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 18.Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41:1308–12. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simon-Sanchez J, van Hilten JJ, van de Warrenburg B, Post B, Berendse HW, Arepalli S, et al. Genome-wide association study confirms extant PD risk loci among the Dutch. Eur J Hum Genet. 2011;19:655–61. doi: 10.1038/ejhg.2010.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Checkoway H, Powers K, Smith-Weller T, Franklin GM, Longstreth WT, Jr, Swanson PD. Parkinson’s disease risks associated with cigarette smoking, alcohol consumption, and caffeine intake. Am J Epidemiol. 2002;155:732–8. doi: 10.1093/aje/155.8.732. [DOI] [PubMed] [Google Scholar]
- 21.Enoch MA, Shen PH, Xu K, Hodgkinson C, Goldman D. Using ancestry-informative markers to define populations and detect population stratification. J Psychopharmacol. 2006;20:19–26. doi: 10.1177/1359786806066041. [DOI] [PubMed] [Google Scholar]
- 22.Lao O, van Duijn K, Kersbergen P, de Knijff P, Kayser M. Proportioning whole-genome single-nucleotide-polymorphism diversity for the identification of geographic population structure and genetic ancestry. Am J Hum Genet. 2006;78:680–90. doi: 10.1086/501531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seldin MF, Shigeta R, Villoslada P, Selmi C, Tuomilehto J, et al. European population substructure: clustering of northern and southern populations. PLoS Genet. 2006;2:e143. doi: 10.1371/journal.pgen.0020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 27.Gao X, Becker LC, Becker DM, Starmer JD, Province MA. Avoiding the high Bonferroni penalty in genome-wide association studies. Genet Epidemiol. 2010;34:100–5. doi: 10.1002/gepi.20430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson LR, Betarbet R, Gearing M, Gulcher J, Hicks AA, Stefansson K, et al. PARK10 candidate RNF11 is expressed by vulnerable neurons and localizes to Lewy bodies in Parkinson disease brain. J Neuropathol Exp Neurol. 2007;66:955–64. doi: 10.1097/nen.0b013e3181567f17. [DOI] [PubMed] [Google Scholar]
- 29.Latourelle JC, Pankratz N, Dumitriu A, Wilk JB, Goldwurm S, Pezzoli G, et al. Genomewide association study for onset age in Parkinson disease. BMC Med Genet. 2009;10:98. doi: 10.1186/1471-2350-10-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corti O, Lesage S, Brice A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol Rev. 2011;91:1161–218. doi: 10.1152/physrev.00022.2010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.