

Abstract
Background
Chronic Myeloid Leukemia (CML) may progress to blast phase (BP) at the rate of 1–1.5% per year. With the use of single-agent tyrosine kinase inhibitors (TKI), median overall survival (OS) ranges between 7 and 11 months.
Methods
We analyzed the outcome of 42 patients with lymphoid BP-CML treated with hyperfractionated cyclophosphamide, vincristine, adriamycin, dexamethasone (HCVAD) plus imatinib or dasatinib.
Results
Complete hematological response (CHR) was achieved in 90% of patients, complete cytogenetic remission (CCyR) in 58%, and complete molecular remission (CMR) in 25%. Flow cytometry Minimal Residual Disease (FC MRD) negativity was achieved by 42% of evaluable patients after induction. Eighteen patients received allogeneic stem cell transplant (SCT) while in first CHR. Median remission duration was 14 months and was longer among SCT recipients (p=0.01) on multivariate analysis. Median OS was 17 (7–27) months and was longer among SCT recipients (p<0.001) and patients treated with dasatinib (p=0.07) on multivariate analysis. Although a high rate of hematologic toxicity (100%) and infectious complications (59%) were observed, the related rate of treatment discontinuation was low (7 and 9%, respectively).
Conclusions
HCVAD combined with TKI is an effective regimen for the management of CML-BP, particularly when followed by allogeneic SCT.
Keywords: Chronic Myeloid Leukemia, Blast Phase, Lymphoid Variant, HyperCVAD, Tyrosine Kinase Inhibitors
Introduction
Blast phase of chronic myeloid leukemia (BP-CML) is defined by the presence of more than 30% blasts in the peripheral blood or the bone marrow or by the presence of extramedullary disease1. More recently, the World Health Organization has proposed a blast count of 20% to define BP-CML2. CML progresses to advanced phase at the rate of 1–1.5% per year3. Very rarely CML-BP may present without a recognized antecedent chronic phase (CP). Myeloid blast phase occurs in 50–60% of cases of BP-CML whereas lymphoid blast phase is seen in about 20–30% of cases; 15–20% of cases have an undifferentiated phenotype. Up to 80% of patients affected by BP-CML show additional cytogenetic abnormalities (ACA)4. Tyrosine kinase inhibitors (TKI) have demonstrated clinical activity in BP-CML although, when used by themselves, responses are usually short lived5. Imatinib, used at the dose of 600 mg, induced complete hematological remission (CHR) in 31% of patients and complete cytogenetic remission (CCyR) in 10%. The median overall survival (OS) was 7 months, with no differences in outcome by immunophenotypic subtype6. Dasatinib, used at a dose of 70 mg, induced CHR in 35% of patients and CCyR in 26% (up to 46% in the lymphoid variant). However, median OS was only 8 months (longer in the myeloid subtype)7. In view of the short remission duration, allogeneic stem cell transplant (SCT) has been used in 1st remission to improve outcome8.
The transient nature of BP-CML response to TKIs is likely the result of the complex molecular milieu of BP-CML. Moreover, even in CP, the earliest hematopoietic progenitors are not eliminated by TKIs, a fact that likely contributes to the recurrence of the disease9. To overcome this, TKIs have been combined with chemotherapy in the advanced stages, with mixed results10–15. In the present study we analyzed the outcome of patients with lymphoid BP-CML treated with a standard ALL chemotherapy regimen combined with imatinib or dasatinib.
Methods
Patients and eligibility criteria
All patients 15 years of age or older with lymphoid BP-CML who received therapy with hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with methotrexate and cytarabine (HCVAD) plus imatinib or dasatinib at MD Anderson Cancer Center (MDACC), whether as initial therapy or as salvage treatment, were included in this analysis. BP-CML was defined by the presence of ≥30% blasts in the peripheral blood or the bone marrow or by the presence of extramedullary disease. Patients affected by acute lymphoblastic leukemia (ALL) Philadelphia-positive (Ph+) were excluded from this analysis. Pregnant and nursing women were excluded, and those of childbearing potential were required to practice effective methods of contraception. All patients signed an informed consent prior to the start of therapy. Patients were included in studies approved by the Institutional Review Board of MDACC and conducted in accordance with the Declaration of Helsinki.
Treatment and monitoring
HCVAD was administered as previously described. The odd courses (cycles 1,3,5 and 7) consisted of Cyclophosphamide 300 mg/m2 IV over 3 hours every 12 hours for 6 doses on days 1, 2 and 3; Doxorubicin 50 mg/m2 IV over 24 hours on day 4; Vincristine 2mg IV on days 1 and 11; Dexamethasone 40mg IV or PO daily on days 1–4 and 11–14; Methotrexate (MTX) 12 mg intrathecally on day 2 and cytarabine (araC) 100 mg intrathecally on day 7. Even courses (cycles 2,4,6 and 8) consisted of: MTX 200 mg/m2 IV over 2 hours followed by 800 mg/m2 over 22 hours on day 1; Solumedrol 40 mg IV over 2 hours every 12 hours for 6 doses on days 1–3; Cytarabine 3 gm/m2 IV over 2 hours every 12 hours for 4 doses on days 2–3; Filgrastim 10 μg/kg/day was given until neutrophil recovery (1×109/L). Maintenance consisted of vincristine 2mg IV on day 1 every 28 days and prednisone 200 mg PO on days 1–5 every 28 days and continued for 24 months.
Imatinib was administered at a starting dose of 400 mg in 3 patients, 600 mg in 23 patients, and 800 mg in 1 patient. The starting dose of dasatinib was 50 mg (2 patients), 70 mg (2 patients), 100 mg (9 patients) or 140 mg (2 patients). Both imatinib and dasatinib were administered from day 1 to day 14 during the first cycle and continuously during the following cycles, unless toxicity. During maintenance, imatinib was administered at the dose of 800 mg orally daily and dasatinib at the dose of 100 mg orally daily, as tolerated. Toxicities were reported according to CTCAE v3.0.
Before the start of therapy, all patients had a complete history and physical examination, a complete blood count, a comprehensive biochemistry panel, and a bone marrow (BM) aspiration with flow cytometry (FC), cytogenetics and real-time polymerase chain reaction (RT-PCR) analysis for BCR-ABL transcript. BM for FC and/or cytogenetics and RT-PCR analyses were collected at least every 3 months for the first 12 months of therapy and at least every 6 months thereafter while on therapy. Patients were followed for progression and survival at least every 3 months.
Complete hematologic response (CHR) was defined as <5% blasts in the bone marrow with peripheral blood neutrophils ≥1×109/L and platelets ≥100×109/L. Complete cytogenetic remission (CCyR) was defined as absence of Ph+ metaphases by conventional cytogenetic analysis in 20 metaphases. Major molecular response was defined as BCR-ABL transcripts ≤0.1% in the international scale (IS), and complete molecular response (CMR) as the absence of detectable BCR-ABL transcript with a minimum sensitivity of at least 5-logs.
FC analyses, cytogenetics and quantitative RT-PCR
MRD assessment by FC was performed on whole bone marrow specimens using a standard stain-lyse-wash procedure. 1 × 106 cells were stained per analysis tube, and data were acquired on at least 2 × 105 cells when specimen quality permitted. Specimens were excluded as inadequate if fewer than 5 × 104 cells were available for analysis. In the first part of the study, a subset of patients were studied and data on four-color staining combinations were acquired on FACSCalibur cytometers using CellQuest software (BD Biosciences, San Diego, CA) and analyzed using FlowJo (TreeStar, Ashland, OR). Starting in 3/2009, patients were studied more systematically and data on six-color stains were acquired on FACSCanto cytometers using FACSDiva software (BD Biosciences) and analyzed using FCS Express (De Novo Software, Los Angeles, CA). Four-color combinations contained CD34-FITC or CD34-PerCP-Cy5.5 as well as CD19-APC in all tubes, with additional antigens conjugated to FITC and PE including CD10, CD13, CD15, CD20, CD22, CD25, CD33, CD38, CD45, CD58, CD66c, and CD81 (all antibodies from BD except CD10 from Beckman Coulter, Fullerton CA and CD66c from Immunotech, Marseilles, France). Six-color combinations included CD34-PerCP-Cy5.5, CD10-PE-Cy7, and CD19-V450 or later CD19-BV421 in each tube, with the additional antigens listed above conjugated to FITC, PE and APC. MRD was identified in comparison with the known patterns of antigen expression by normal maturing CD19+ B cells, as described by Weir et al16. A distinct cluster of at least 20 cells showing altered antigen expression was regarded as an aberrant population, yielding a sensitivity for both four-color and six-color assays of 1 in 104 or 0.01% (for adequate specimens where at least 2 × 105 cells could be collected). The sensitivity was at least 4 in 104 cells (0.04%) for all specimens. We required aberrant expression of at least 2 antigens for confirmation of positive MRD.
Cytogenetic analysis was performed by standard G-banding techniques, and at least 20 metaphases were analyzed. Marrow specimens were examined on direct 24-hour cultures.
BCR-ABL transcripts were detected by quantitative RT-PCR analysis on BM aspirate. Following lysis of red blood cells, RNA was isolated from 10–20 million white blood cells in 100 uL elution volume using Qiagen’s QIAsymphony extractor (Germantown, MD) and their QIAsymphony RNA kit. A total of 2.85 ug of total RNA at 100 ng/ul concentration was then reverse transcribed in a 60 uL final volume using Superscript II reverse transcriptase enzyme (Life Technologies, Carlsbad, CA). Specimen was considered suboptimal if ABL copies were below < 10,000 and BCR-ABL fusion transcripts were not detected.
Statistical considerations
Differences between variables were compared by the chi2 test and Mann-Whitney U test for categorical and non-categorical variables, respectively. Remission duration (RD) was calculated from the time of CHR achievement until CHR loss. OS was calculated from the time of start of therapy until death from any cause or last follow-up. Survival curves were estimated by the Kaplan-Meier method and compared by the log-rank test. Cox regression with a backward and forward stepwise method was used for multivariate analysis.
Results
Study group
A total of 42 patients were treated with HCVAD plus imatinib (n=27) or dasatinib (n=15) between 2001 and 2011. The baseline characteristics of the 42 patients are presented in Table 1. Four patients (10%) had BP as initial manifestation of CML. Thirty-eight (90%) of the 42 patients had a previous CP of CML and had received a median of 1 (range, 1–5) different prior treatments. Thirty (71%) patients had received prior therapy for CP with TKI (including imatinib, nilotinib and dasatinib). Other prior therapies included interferon alone in 8 patients, or in combination with cytarabine in 8 patients. Five patients had received prior therapy for BP, including single agent imatinib (n=3), single agent dasatinib (n=1), and allogeneic SCT (n=1). Eight of 16 patients assessed for ABL kinase domain mutations had a detectable mutation at the time of BP, namely Y253H, T315I, Q252H, F317L, E255K, M244V, M351T and Y253H/F359V/E459K (one each). Nineteen of 33 (58%) evaluable patients had additional chromosomal abnormalities (ACA) at the start of therapy besides the Philadelphia chromosome, most frequently abnormalities of chromosome 1 (32%), 7 (42%) and 8 (26%). Two patients had an e1a2 BCR-ABL rearrangement, 17 had a b2a2, 18 had a b3a2, four had a co-expression of b2a2 and b3a2, and 1 a b3a3. The median time from diagnosis of CML to BP was 25 months (11–39 months).
Table 1.
Patient characteristics (N=42)
| No. (%) or Median [range] | Imatinib (n=27) | Dasatinib (n=15) | |
|---|---|---|---|
| No. males | 29 (69%) | 17 (63%) | 12 (80%) |
|
| |||
| Median age, y | 48 [22–74] | 49 [22–74] | 47 [27–72] |
|
| |||
| No. de novo BP (%) | 4 (10%) | 4 (15%) | 0 (0%) |
|
| |||
| Months from CML to BP (n=38) | 25 [11–39] | 27 [4–25] | 9 [2–263] |
|
| |||
| No. prior CML therapies (n=38) | 1 [1–8] | 2 [1–8] | 1 [1–4] |
|
| |||
| No. patients with prior BP therapy (%) | 5 (12%) | 3 (11%) | 2 (13%) |
|
| |||
| No. patients with prior TKI | 31 (74%) | 18 (67%) | 13 (87%) |
|
| |||
| Median white blood cells, ×109/L | 23 [1–165] | 22 [1–145] | 26 [2–374] |
|
| |||
| Median hemoglobin, g/dL | 10.6 [6.3–16.4] | 10.6 [6.3–16.4] | 10.9 [7.1–15.7] |
|
| |||
| Median platelets, ×109/L | 46 [6–526] | 54 [6–526] | 28 [5–310] |
|
| |||
| Median peripheral blood blasts, % | 40 [0–91] | 26 [0–91] | 62 [0–87] |
|
| |||
| Median peripheral blood basophils, % | 0 [0–2] | 0 [0–2] | 0 [0–2] |
|
| |||
| Median creatinine, mg/dL | 1 [0.6–1.5] | 1 [0.6–1.5] | 0.9 [0.8–1.3] |
|
| |||
| Median bilirubin, mg/dL | 0.5 [0.2–3.4] | 0.5 [0.2–1.7] | 0.5 [0.2–3.4] |
|
| |||
| Median ALT, UI/L | 32 [12–446] | 44 [12–446] | 23 [12–115] |
|
| |||
| Median bone marrow blasts, % | 79 [30–99] | 75 [32–95] | 85 [30–99] |
|
| |||
| Median bone marrow basophils, % | 0 [0–4] | 0 [0–4] | 0 [0–0] |
|
| |||
| No. with ABL kinase mutations at BP | 8/17 (47%) | 2/7 (29%) | 6/10 (60%) |
|
| |||
| Additional chromosomal aberrations (ACA) | 19/33 (58%) | 10/21 (48%) | 9/12 (75%) |
| chromosome 1 | 6/19 (32%) | 2/10 (20%) | 4/9 (44%) |
| chromosome 7 | 8/19 (42%) | 4/10 (40%) | 4/9 (44%) |
| chromosome 8 | 5/19 (26%) | 4/10 (40%) | 1/9 (12%) |
Response to treatment
Responses to treatment are reported in Table 2 and Table 3. CHR was achieved in 38 (90%) patients. Twenty-nine (69%) patients achieved CHR after 1 cycle and 9 (21%) patients after 2 or more cycles. Of the 4 patients who did not achieve CHR, 3 (7%) were primary refractory after 1 or 2 cycles, and one patient interrupted treatment because of an acute vascular event (stroke) after 1 cycle, not having achieved a CHR.
Table 2.
Responses to Therapy by used TKI.
| N (%) | |||
|---|---|---|---|
| Overall (n=42) | Imatinib (n=27) | Dasatinib (n=15) | |
| Complete Hematological Remission (CHR) | 38 (90) | 23 (85) | 15 (100) |
| Complete Cytogenetic Remission (CCyR) | 24 (59) | 11 (41) | 13 (87) |
| Complete Molecular Remission (CMR) | 8/32 (25) | 7/18 (39) | 1/14 (7)* |
| Major Molecular Remission (MMR) | 16/32 (50) | 8/18 (44) | 8/14 (57) |
| Minimal Residual Disease (MRD) negative by FC | 7/12 (58) | 2/5 (40) | 5/7 (71) |
among patients achieving MMR, SCT was performed before CMR achievement in 4/8 (50%) of patients on dasatinib and 1/8 (12%) of patients on imatinib.
Table 3.
Responses and outcome by subtype and previous therapy.
| N (%), median [range] | ||||||||
|---|---|---|---|---|---|---|---|---|
| De novo (n=4) | Not De novo (n=38) | Prior TKI (n=31) | No prior TKI (n=11) | Prior BP therapy (n=5) | No prior BP therapy (n=38) | Same TKI as previous (n=20) | No same TKI as previous (n=22) | |
| CHR | 3 (75) | 35 (92) | 28 (90) | 10 (91) | 5 (100) | 34 (89) | 17 | 21 |
| CCyR | 2 (50) | 22 (58) | 17 (55) | 7 (64) | 2 (40) | 22 (58) | 7 | 17 |
| CMR | 0/3 (0) | 8/29 (28) | 4/23 (17) | 4/9 (44) | 0/3 (0) | 8/29 (28) | 3/12 | 5/20 |
| MMR | 0/3 (0) | 16/29 (56) | 10/23 (43) | 6/9 (67) | 0/3 (0) | 16/29 (55) | 4/12 | 12/20 |
| FC MRD (−) | NA | 7/12 (58) | 6/10 (60) | 1/2 (50) | NA | 7/12 (58) | 7/10 | 14/16 |
| RD (months) | 14 [5–68] | NR | 67 [38–62] | NR | 14 [3–25] | NR | 14 [5–23] | NR |
| OS (months) | 10 [1–30] | 17 [5–29] | 17 [6–28] | 17 [1–61] | 14 [5–23] | 17 [1–61] | 12 [7–17] | 59 [3–134] |
NA: not assessable; NR: not reached; RD: remission duration; OS: overall survival
CCyR was achieved in 24 (57%) patients after a median of 2 cycles (range, 1 to 5). At the time CCyR was achieved, the median BCR-ABL transcript levels were 0.19% (range, 0 to 14.8%) and decreased to 0.001% (range, 0 to 91%) after a median of 2 months (1–17 months) (32 patients assessed). Sixteen of 32 (50%) patients achieved a MMR, after a median of 4 cycles (range, 1 to 8) of therapy. Eight of 32 (25%) patients achieved a CMR, 4 of them after induction. The median number of cycles to achievement of CMR was 2 (range, 1 to 5). Flow cytometry data were available for 12 patients (all previously untreated for BP-CML) and 7 (58%) became negative at some point for MRD. Five (42%) of 12 patients had undetectable FC MRD after the first induction cycle.
Eighteen (47%) patients had an allogeneic SCT while in CHR, 10 (37%) in the imatinib-treated group and 8 (53%) in the dasatinib-treated group.
Among patients who had not received prior therapy for BP-CML (n=38), a CHR was achieved in 34 (89%), a CCyR in 22 (58%) and CMR in 8/29 (28%).
Imatinib, nilotinib and dasatinib had all been used as therapy for CP in various patients, whereas only imatinib and dasatinib were used in previous treatment of BP. Fifteen (36%) patients who received imatinib and 2 (5%) patients who received dasatinib during the study period had already received the same TKI previously. Patients who received on study a previously used TKI had a significantly lower probability of achieving CCyR (35% vs 77%, p=0.01). However, there was no significant association with the probability of achieving CHR (p=0.33), MRD by flow cytometry (p=0.34), MMR (p=0.27) or CMR (p=1).
Remission Duration and Overall Survival
After a median follow-up of 15 (1–134) months, 15 patients have relapsed, including 5 after SCT. Three out of 5 FC MRD-positive patients relapsed, whereas none of the FC MRD-negative patients did. Given the limited number of samples evaluable for MRD by FC, this factor was not included in survival analysis. The median remission duration for the 38 patients that achieved CHR was 14 months (range, 1 to 62) (Figure 1A). On univariate analysis, RD was significantly longer for patients that received a SCT (not reached vs 7 months, p=0.01)(Figure 1B), for patients that achieved a CCyR (not reached vs 8 months, p< 0.015). Although not statistically significant, there was a trend for a longer RD for patients on dasatinib (not reached vs 14 months, p=0.15) and for patients receiving on study a TKI not previously used (not reached vs 14 months, p=0.09). On multivariate analysis of meaningful characteristics, the association with RD was confirmed only for SCT (OR 4.3, p=0.04).
Figure 1.
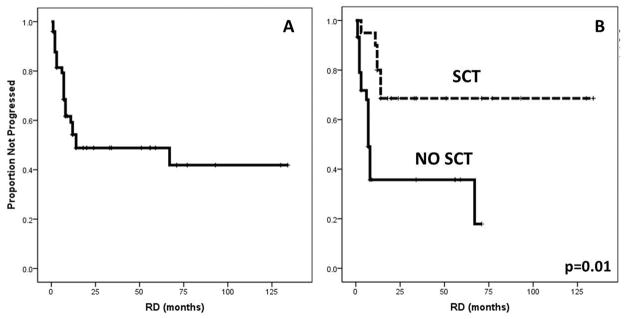
Remission Duration (RD). A. Median RD in 38 patients achieving CHR was 14 (1–62 months). B. Longer RD was associated with Stem Cell Transplant (SCT)(not reached vs 7 months, p=0.001).
Twenty-six (62%) patients have died. Eighteen (69%) died due to refractory disease or relapse and 2 (8%) died after SCT while in CHR from non-relapse causes. The remaining 6 (23%) patients died while in CHR, from infectious complications (4 while on treatment, one died 2 months after the last cycle and one died 5 years later, after developing therapy-related myelodysplastic syndrome). Median OS was 17 months (range, 7 to 27 months) (Figure 2A). OS was significantly better in SCT recipients (median 93 vs 9 months, p<0.001)(Figure 2B), in patients achieving CCyR (77 vs 9 months, p<0.001), in patients with baseline bone marrow basophils <1% (23 vs 7 months, p=0.01), in patients treated with dasatinib (compared to imatinib) in association with HCVAD (not reached vs 12 months, p=0.007)(Figure 2C), and in patients receiving a TKI not previously used (59 vs 12 months, p=0.009). On multivariate analysis receiving SCT (OR 10.1, p=0.02) and treatment with dasatinib (OR 5.8, p=0.02) were independently associated with prolonged survival. Of interest, ACA, prior use of TKI and previous therapy for BP were not significant on univariate analysis for RD neither OS.
Figure 2.
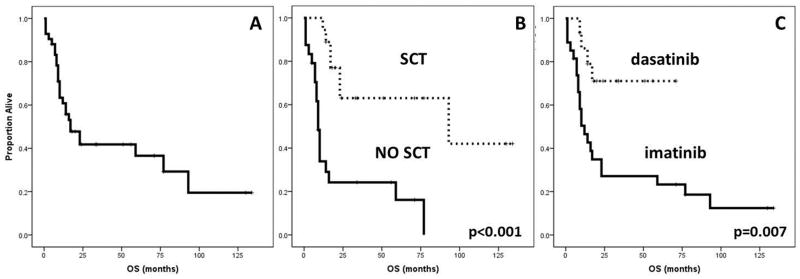
Overall Survival (OS). A. Median OS in 42 patients was 17 (7–27) months. B. Longer OS was associated with Stem Cell Transplant (SCT)(93 vs 9 months, p<0.001). C. Longer OS was associated with the use of dasatinib compared to imatinib (not reached vs 12 months, p=0.007).
Treatment characteristics and toxicity
The median number of administered cycles was 4 (1–8), with only 3 patients completing the planned 8 cycles of therapy. Toxicity was similar to the historical experience with HCVAD alone17–19. All patients experienced grade (G) 3-4 neutropenia and thrombocytopenia during treatment, and 88% of them had G3-4 anemia. However, hematological toxicity prompted treatment discontinuation in only 3 (7%) patients. G3-4 infections were observed in 59% of patients and prompted treatment discontinuation in 4 (9%) of patients. G3-4 hepatotoxicity (elevation in alanine aminotransferase and/or total bilirubin) was reported in 29% of patients, and G3-4 nephrotoxicity (transient) in 2% of patients. Other reasons for not completing 8 cycles included: SCT (18), relapse (9), refractoriness (3, after a median of 2 cycles), stroke (1), and severe diarrhea (1). Early death (within 60 days from the start of therapy) occurred in 4 (9%) patients.
Discussion
In this report we show the efficacy of HCVAD combined with a TKI for the treatment of lymphoid BP-CML. BP-CML with a lymphoid phenotype is less common than the myeloid subtype and has unique biological and clinical features. Occasionally, lymphoid BP-CML can occur de novo (i.e., not preceded by a recognized CP)20. This phenomenon was identified in 10% of the patients described in this series. Differential diagnosis between de novo lymphoid BP-CML and ALL Ph+ is challenging. The four patients in our study with no preceding CP CML had morphologic features of CML in the bone marrow samples, including dwarf megakaryocites, left shifted myeloid predominance, eosinophilia and/or basophilia, which supported the former diagnosis. For patients with preceding CP, time from CML to BP was short (25 months) in line with what has been previously reported2.
ACA are commonly reported in BP-CML. Trisomy 8, additional Ph-chromosome, isochromosome (17q) and trisomy 19 are called major route ACA: they are considered relevant for pathogenesis and have a negative prognostic impact21. Chromosome 3 aberrations, loss of the Y-chromosome and other rarer aberrations are less frequent and indicate genetic instability22–24. In our series, ACA occurred in 58% of all patients, more frequently involving chromosome 7 (42%), 1 (32%) and 8 (26%), and mostly represented by monosomies. Further investigation is warranted in order to clarify the biological significance of these abnormalities and to identify potential molecular therapeutic targets.
BP represented an inevitable fate for CML in the pre-imatinib era, with an annual rate of transformation of 20%. Lymphoid BP-CML was treated in the past with various vincristine-prednisone-based regimens, with disappointing short-lasting remission durations25,26. TKIs have dramatically changed the natural history of CML, with fewer patients transforming to blast phase, at a rate of 5–6% per year in the first 2–3 years, and less than 1% per year thereafter. Furthermore, it appears that the rate of transformation is decreased with the use of second generation TKI as initial therapy of the disease27,28. It has been suggested that Ph+ disease with lymphoid phenotype might have a greater dependence on activation of Src family of kinase29. Interestingly, up to 50% of patients have been reported to achieve a CCyR with single-agent dasatinib (a Src inhibitor). However, with the use of single-agent imatinib or dasatinib responses are of short duration and the median OS of patients affected by BP-CML has improved only modestly from 3–4 months to approximately 7 months30. A major insight into the biology and molecular mechanisms regulating this entity is then necessary, in order to identify more effective therapies. BCR-ABL could be responsible for progressive genomic instability or epigenetic changes occurring in CML stem cells leading to BP31. This is indirectly suggested not only by the higher incidence of BP prior to the TKI era, but also by the correlation between presence of BCR-ABL mutations and clonal evolution32,33. Once progressed to BP, the disease becomes more molecularly complex and only partially dependent from BCR-ABL, thus the need of combination treatment. The combination of chemotherapy + TKI has been used extensively in Ph+ ALL. We have previously published on the combination of HCVAD + imatinib18 or dasatinib19 in this setting. Twenty (both treated and untreated) Ph+ ALL patients were treated with the HCVAD plus imatinib: CR rate was 100% with a median duration of 20 months; 10 patients were transplanted in first CR and only 1 relapsed thereafter. Thirty-five untreated Ph+ ALL patients were treated with HCVAD plus dasatinib and CR rate was 93%; with a median follow-up of 14 months, median PFS and OS were not reached.
There is limited experience in the literature using TKI-based combinations in lymphoid CML-BP. In our previous series14 2 patients with lymphoid BP-CML were treated with HCVAD plus imatinib and one with HCVAD plus dasatinib. One patient treated with HCVAD plus imatinib obtained a CHR lasting 12 months, but the others 2 were refractory14. A single case report was later published showing a PFS of 19 months for a patient with lymphoid BP-CML treated with analogous chemotherapy scheme followed by allogeneic SCT15. These data highlighted the potential efficacy of such combination treatment. In the present report, the first large series of chemotherapy combined with TKI for BP-CML, HCVAD plus imatinib or dasatinib induced a CHR in 90% of patients and CCyR in 59%. These results are clearly superior to what is reported with imatinib or dasatinib as single agents (CHR rate <35%)6,7. Despite the use of TKI added to chemotherapy, there is no evidence of increased toxicity compared to what is expected with chemotherapy alone, as reflected by a low rate of early mortality and a low rate of discontinuations related to cytopenia or infectious complications.
One of the benefits of an effective therapy in this setting is the ability to take patients to SCT. In transplanted patients, median remission duration has not been reached and OS at 7 years is 61%. Of interest, in a recent report from the German CML Study Group, the 3-year survival of 28 patients pretreated with imatinib-based regimens and transplanted for advanced phase CML (25 with BP-CML) was 59%8. In our series, 59% of patients achieved CCyR, with a median time to CCyR of 2 (1–5) cycle. An important consideration is the proper timing for SCT on a patient that has achieved CHR but not yet CCyR. While continued chemotherapy to aim for CCyR may prove beneficial for the long term outcome in some patients, others may not be able to reach a SCT because of relapse or emerging co-morbidities that preclude a transplant. Further studies are needed to better settle this clinical dilemma. However, in the absence of additional data and as highlighted by the results of our multivariate analysis, it might be appropriate to proceed to SCT whenever a patient has reached at least a CHR if other conditions are optimal for transplant.
As predicted by its use as a single agent7, the use of HCVAD plus dasatinib produced an improved overall survival, with a trend for improved remission duration, although the later did not achieve statistical significance probably because of the small sample size. Prospective, randomized trials with a larger number of patients are needed in order to confirm any potential superiority compared to imatinib. Given its association with a higher CCyR rate and OS (although not confirmed on MVA), further investigation is also needed in order to establish whether using a previously employed TKI may affect the efficacy of this regimen.
In conclusion, HCVAD plus imatinib or dasatinib is an effective and safe regimen for patients with lymphoid BP-CML. Better results are achieved combining allogeneic SCT. More extensive prospective studies are warranted to confirm these observations.
Acknowledgments
This research is supported in part by the MD Anderson Cancer Center Support Grant CA016672 and the NIH Grant P01 CA049639. F.R. received research funding and honoraria from BMS. J.C. received research support from BMS, Novartis, Ariad, Pfizer, and Chemgenex; and is consultant for Ariad, Pfizer and Teva. E.J. received honoraria from BMS, Novartis, Pfizer, and Ariad. The remaining authors declare no competing financial interests.
References
- 1.Karanas A, Silver RT. Characteristics of the terminal phase of chronic granulocytic leukemia. Blood. 1968;32(3):445–459. [PubMed] [Google Scholar]
- 2.Silver RT. The blast phase of chronic myeloid leukaemia. Best Pract Res Clin Haematol. 2009;22(3):387–394. doi: 10.1016/j.beha.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 3.Druker BJ, Guilhot F, O’Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 4.Cortes J, Di Cosimo S, Climent MA, et al. Nonpegylated liposomal doxorubicin (TLC-D99), paclitaxel, and trastuzumab in HER-2-overexpressing breast cancer: a multicenter phase I/II study. Clin Cancer Res. 2009;15(1):307–314. doi: 10.1158/1078-0432.CCR-08-1113. [DOI] [PubMed] [Google Scholar]
- 5.Hehlmann R. How I treat CML blast crisis. Blood. 2012;120(4):737–747. doi: 10.1182/blood-2012-03-380147. [DOI] [PubMed] [Google Scholar]
- 6.Palandri F, Castagnetti F, Testoni N, et al. Chronic myeloid leukemia in blast crisis treated with imatinib 600 mg: outcome of the patients alive after a 6-year follow-up. Haematologica. 2008;93(12):1792–1796. doi: 10.3324/haematol.13068. [DOI] [PubMed] [Google Scholar]
- 7.Cortes J, Jabbour E, Kantarjian H, et al. Dynamics of BCR-ABL kinase domain mutations in chronic myeloid leukemia after sequential treatment with multiple tyrosine kinase inhibitors. Blood. 2007;110(12):4005–4011. doi: 10.1182/blood-2007-03-080838. [DOI] [PubMed] [Google Scholar]
- 8.Saussele S, Lauseker M, Gratwohl A, et al. Allogeneic hematopoietic stem cell transplantation (allo SCT) for chronic myeloid leukemia in the imatinib era: evaluation of its impact within a subgroup of the randomized German CML Study IV. Blood. 2010;115(10):1880–1885. doi: 10.1182/blood-2009-08-237115. [DOI] [PubMed] [Google Scholar]
- 9.Graham SM, Jorgensen HG, Allan E, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99(1):319–325. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- 10.Fruehauf S, Topaly J, Buss EC, et al. Imatinib combined with mitoxantrone/etoposide and cytarabine is an effective induction therapy for patients with chronic myeloid leukemia in myeloid blast crisis. Cancer. 2007;109(8):1543–1549. doi: 10.1002/cncr.22535. [DOI] [PubMed] [Google Scholar]
- 11.Oki Y, Kantarjian HM, Gharibyan V, et al. Phase II study of low-dose decitabine in combination with imatinib mesylate in patients with accelerated or myeloid blastic phase of chronic myelogenous leukemia. Cancer. 2007;109(5):899–906. doi: 10.1002/cncr.22470. [DOI] [PubMed] [Google Scholar]
- 12.Quintas-Cardama A, Kantarjian H, Garcia-Manero G, et al. A pilot study of imatinib, low-dose cytarabine and idarubicin for patients with chronic myeloid leukemia in myeloid blast phase. Leuk Lymphoma. 2007;48(2):283–289. doi: 10.1080/10428190601075973. [DOI] [PubMed] [Google Scholar]
- 13.Fang B, Li N, Song Y, Han Q, Zhao RC. Standard-dose imatinib plus low-dose homoharringtonine and granulocyte colony-stimulating factor is an effective induction therapy for patients with chronic myeloid leukemia in myeloid blast crisis who have failed prior single-agent therapy with imatinib. Ann Hematol. 2010;89(11):1099–1105. doi: 10.1007/s00277-010-0991-4. [DOI] [PubMed] [Google Scholar]
- 14.Verma D, Kantarjian HM, Jones D, et al. Chronic myeloid leukemia (CML) with P190 BCR-ABL: analysis of characteristics, outcomes, and prognostic significance. Blood. 2009;114(11):2232–2235. doi: 10.1182/blood-2009-02-204693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawano N, Okuda S, Yoshida S, et al. Successful treatment of lymphoid blastic crisis in chronic myelogenous leukemia with the additional bcr/abl transcript using imatinib-combined chemotherapy and high-dose chemotherapy with allogeneic bone marrow stem cell transplantation. Int J Hematol. 2011;94(6):561–566. doi: 10.1007/s12185-011-0956-y. [DOI] [PubMed] [Google Scholar]
- 16.Weir EG, Cowan K, LeBeau P, Borowitz MJ. A limited antibody panel can distinguish B-precursor acute lymphoblastic leukemia from normal B precursors with four color flow cytometry: implications for residual disease detection. Leukemia. 1999;13(4):558–567. doi: 10.1038/sj.leu.2401364. [DOI] [PubMed] [Google Scholar]
- 17.Kantarjian HM, O’Brien S, Smith TL, et al. Results of treatment with hyper-CVAD, a dose-intensive regimen, in adult acute lymphocytic leukemia. J Clin Oncol. 2000;18(3):547–561. doi: 10.1200/JCO.2000.18.3.547. [DOI] [PubMed] [Google Scholar]
- 18.Thomas DA, Faderl S, Cortes J, et al. Treatment of Philadelphia chromosome-positive acute lymphocytic leukemia with hyper-CVAD and imatinib mesylate. Blood. 2004;103(12):4396–4407. doi: 10.1182/blood-2003-08-2958. [DOI] [PubMed] [Google Scholar]
- 19.Ravandi F, O’Brien S, Thomas D, et al. First report of phase 2 study of dasatinib with hyper-CVAD for the frontline treatment of patients with Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia. Blood. 2010;116(12):2070–2077. doi: 10.1182/blood-2009-12-261586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kantarjian H, O’Brien S, Cortes J, et al. Sudden onset of the blastic phase of chronic myelogenous leukemia: patterns and implications. Cancer. 2003;98(1):81–85. doi: 10.1002/cncr.11477. [DOI] [PubMed] [Google Scholar]
- 21.Fabarius A, Leitner A, Hochhaus A, et al. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: long-term observation of 1151 patients from the randomized CML Study IV. Blood. 2011;118(26):6760–6768. doi: 10.1182/blood-2011-08-373902. [DOI] [PubMed] [Google Scholar]
- 22.Mitelman F, Nilsson PG, Levan G, Brandt L. Non-random chromosome changes in acute myeloid leukemia. Chromosome banding examination of 30 cases at diagnosis. Int J Cancer. 1976;18(1):31–38. doi: 10.1002/ijc.2910180106. [DOI] [PubMed] [Google Scholar]
- 23.Alimena G, De Cuia MR, Diverio D, Gastaldi R, Nanni M. The karyotype of blastic crisis. Cancer Genet Cytogenet. 1987;26(1):39–50. doi: 10.1016/0165-4608(87)90131-2. [DOI] [PubMed] [Google Scholar]
- 24.Johansson B, Fioretos T, Mitelman F. Cytogenetic and molecular genetic evolution of chronic myeloid leukemia. Acta Haematol. 2002;107(2):76–94. doi: 10.1159/000046636. [DOI] [PubMed] [Google Scholar]
- 25.Koller CA, Miller DM. Preliminary observations on the therapy of the myeloid blast phase of chronic granulocytic leukemia with plicamycin and hydroxyurea. N Engl J Med. 1986;315(23):1433–1438. doi: 10.1056/NEJM198612043152301. [DOI] [PubMed] [Google Scholar]
- 26.Thomas ED, Clift RA, Fefer A, et al. Marrow transplantation for the treatment of chronic myelogenous leukemia. Ann Intern Med. 1986;104(2):155–163. doi: 10.7326/0003-4819-104-2-155. [DOI] [PubMed] [Google Scholar]
- 27.Kantarjian HM, Hochhaus A, Saglio G, et al. Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet Oncol. 2011;12(9):841–851. doi: 10.1016/S1470-2045(11)70201-7. [DOI] [PubMed] [Google Scholar]
- 28.Kantarjian HM, Shah NP, Cortes JE, et al. Dasatinib or imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: 2-year follow-up from a randomized phase 3 trial (DASISION) Blood. 2012;119(5):1123–1129. doi: 10.1182/blood-2011-08-376087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu Y, Liu Y, Pelletier S, et al. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet. 2004;36(5):453–461. doi: 10.1038/ng1343. [DOI] [PubMed] [Google Scholar]
- 30.Jabbour E, Kantarjian H, O’Brien S, et al. Sudden blastic transformation in patients with chronic myeloid leukemia treated with imatinib mesylate. Blood. 2006;107(2):480–482. doi: 10.1182/blood-2005-05-1816. [DOI] [PubMed] [Google Scholar]
- 31.Ilaria RL., Jr Pathobiology of lymphoid and myeloid blast crisis and management issues. Hematology Am Soc Hematol Educ Program. 2005:188–194. doi: 10.1182/asheducation-2005.1.188. [DOI] [PubMed] [Google Scholar]
- 32.O’Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348(11):994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 33.Willis SG, Lange T, Demehri S, et al. High-sensitivity detection of BCR-ABL kinase domain mutations in imatinib-naive patients: correlation with clonal cytogenetic evolution but not response to therapy. Blood. 2005;106(6):2128–2137. doi: 10.1182/blood-2005-03-1036. [DOI] [PubMed] [Google Scholar]