

Abstract
Purpose of the study
Many blinding diseases of the inner retina are associated with degeneration and loss of retinal ganglion cells (RGCs). Recent evidence implicates several new signaling mechanisms as causal agents associated with RGC injury and remodeling of the optic nerve head. Ion channels such as Transient receptor potential vanilloid isoform 4 (TRPV4), pannexin-1 (Panx1) and P2X7 receptor are localized to RGCs and act as potential sensors and effectors of mechanical strain, ischemia and inflammatory responses. Under normal conditions, TRPV4 may function as an osmosensor and a polymodal molecular integrator of diverse mechanical and chemical stimuli, whereas P2X7R and Panx1 respond to stretch- and/or swelling-induced adenosine triphosphate release from neurons and glia. Ca2+ influx, induced by stimulation of mechanosensitive ion channels in glaucoma, is proposed to influence dendritic and axonal remodeling that may lead to RGC death while (at least initially) sparing other classes of retinal neuron. The secondary phase of the retinal glaucoma response is associated with microglial activation and an inflammatory response involving Toll-like receptors (TLRs), cluster of differentiation 3 (CD3) immune recognition molecules associated with the T-cell antigen receptor, complement molecules and cell type-specific release of neuroactive cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β). The retinal response to mechanical stress thus involves a diversity of signaling pathways that sense and transduce mechanical strain and orchestrate both protective and destructive secondary responses.
Conclusions
Mechanistic understanding of the interaction between pressure-dependent and independent pathways is only beginning to emerge. This review focuses on the molecular basis of mechanical strain transduction as a primary mechanism that can damage RGCs. The damage occurs through Ca2+-dependent cellular remodeling and is associated with parallel activation of secondary ischemic and inflammatory signaling pathways. Molecules that mediate these mechanosensory and immune responses represent plausible targets for protecting ganglion cells in glaucoma, optic neuritis and retinal ischemia.
Keywords: ATP, calcium, cytokines, glaucoma, glia, inflammation, mechanosensation, retinal ganglion cells
INTRODUCTION
The spatiotemporal properties of retinal ganglion cell (RGC) action potentials within the axons of the optic nerve represent the entire visual output projected from the eye to the brain. Loss of RGCs therefore culminates in vision loss in debilitating blinding diseases such as ischemia, diabetic retinopathy and glaucoma. 1 While the biological mechanisms that compromise RGC viability in retinal disease are currently under intense experimental scrutiny, potentially useful insights into the disease etiology might be obtained from the observation that RGC degeneration may, at least initially, occur without injury to other classes of retinal neurons.1,2 Many studies have investigated the anatomical and molecular mechanisms that could account for the selective vulnerability of RGCs in glaucoma and diabetes. It has been suggested that RGCs are uniquely vulnerable to disruptions in energy supply3 mitochondrial function4 and axonal transport5 due to the need to support metabolically expensive long-distance axons. This need is tended through continuous supply of glucose, oxygen and signaling molecules across the blood–retina barrier (BRB), which in turn requires intact function of pericytes, astroglia, microglia and Müller cell endfeet that interface between the vascular endothelium and RGC somata/axons. Astroglia contribute to the metabolic homeostasis of RGCs through glucose/lactate transport mediated by monocarboxylate transporters (MCTs), glucose transporters (SLC2A), glutamate transporters, Gamma-aminobutyric acid (GABA) transporters and the proposed glutamate-glutamine shuttle driven by the bidirectional System N (SN1) transporter.6–8 While blood vessels and glia maintain the ocular immune privilege by shielding the retina from systemic inflammation, glaucomatous RGC dysfunction might also involve the breakdown of the glial-vascular-immune interface, resulting in increased vascular permeability, hypoxia/ischemia, release of free radicals, cytokines, eicosanoids and growth factors and access of auto-immune molecules.5,9 Importantly, RGCs are uniquely susceptible to trauma and biomechanical strain, leading to their selective loss in several debilitating blinding diseases.9,10
Excessive mechanical stress compromises the viability of many sensory systems, including hearing, somatosensation and vision.11,12 Glaucoma, the blinding disease most commonly associated with pathological mechanical stress in the eye, is a designation that covers many distinct eye diseases unified by anterior chamber dysfunction, optic neuropathy and glial activation. Its etiology is linked to many known risk factors that include mechanical, genetic (monogenic or polygenic), epigenetic and environmental factors, and possibly combinations thereof.13 A major risk factor for developing primary open angle glaucoma (which accounts for the majority of glaucoma patients) is ocular hypertension9,14 caused by increased production or decreased outflow of aqueous humor within the anterior chamber.15 Positive correlations between intraocular pressure (IOP) levels and RGC loss, and between duration of elevated IOP and RGC axon loss, have been reported for glaucomatous mice, rats, primates and humans.9,14,16,17 Currently, pharmacological targeting of increased IOP represents by far the most common clinical treatment of glaucoma. Because the disease is too often identified by the time when axonal atrophy and somatic degeneration reach the irreversible terminal stage, there is an increasing interest in early diagnosis and neuroprotective strategies that will complement IOP reduction within the anterior eye.18 Both require us to understand the mechanotransduction mechanism at the target (RGCs). As discussed below, application of pressure/stretch triggers influx of calcium ions into RGCs through several classes of putative mechanosensitive ion channels. Given that excessive calcium entry overloads cells with calcium and drives neuronal death in many neurodegenerative diseases in the retina and the brain,19–21 mechanosensitive channels represent obvious neuroprotection targets in glaucoma.
Although pharmacological targeting of IOP-elevations represents by far the most common clinical treatment of glaucoma, recent studies suggest that the disease also involves pressure-independent mechanisms mediated by vascular, glial and immune cells. Reactive astroglia and microglia appear to regulate RGC survival through parallel and intersecting pathways that encompass elevated levels of the vasoconstrictor endothelin-1, inflammatory chemokines and cytokines (e.g. TNF-α, IL-1β and IL-18), ATP, eicosanoids and/or damage-associated molecular patterns (DAMPs) released by injured and dying RGCs, which in turn activate multiple RGC targets, including TNF-α, TGF-β, IL receptors, the inflammasome and T-cell antigen receptor (TCR)/major histocompatibility complex (MHC) immune complexes.22–27 Secondary insults, triggered by molecules released from injured RGCs, together with cytokines released from reactive glia and immune molecules arriving from leaky blood vessels may further disrupt the blood retina barrier and facilitate additional infiltration of circulating immune cells,28,29 thereby fueling RGC damage inflicted by the primary mechanical stress.22,30,31 Obviously, diagnosis and treatment of glaucoma will need to simultaneously address the primary (pressure-related) and secondary (inflammatory and ischemic) symptoms as well as consider the possibility that glaucomatous injury is exacerbated through feedback interactions between primary and secondary pathways.
The aim of this mini-review is to describe recent developments in glaucoma research, focusing on genetic, physiological and pharmacological studies, many from the authors’ laboratories. We introduce molecular mechanisms that underlie intrinsic RGC mechanosensation, showcase the intimate relationship between immune and inflammatory pathways in glaucoma and conclude by identifying a novel retinal immune recognition mechanism that might contribute to glaucomatous remodeling in the inner retina.
INTRAOCULAR PRESSURE AND GLAUCOMA
All cells and organisms live within mechanically active environments in which they must sense and adapt to physical forces such as hydrostatic pressure, osmotic swelling/shrinkage, shear flow and developmentally driven tissue stretch.32,33 Cells in the eye are additionally exposed to intraocular pressure, the magnitude of which reflects the elasticity of ocular tissues and the balance between production and drainage of aqueous humor within the anterior chamber. Biomechanical strain, exerted by the IOP, was suggested to play a central role in the normal development of the vertebrate retina through scleral expansion and continuous stretching of the eye. Consistent with this view, IOP dissipation blocked ocular expansion even as the neural retina continued to grow.34,35 Furthermore, Coulombre showed that IOP-deprived chick retinas are forced to increase their thickness, suggesting that mechanical stress is required for proper establishment of retinal circuits. Thus, as observed in other tissues,33,36 morphogenesis, migration, adhesion, osmoregulation and contractility of developing ocular cells are likely to be influenced by mechanical forces that include IOP.
Given that vision loss in animal glaucoma models and humans reflects the magnitude and time course of IOP elevations,9,14,16,17,37 it would appear that the identification of mechanosensitive mechanisms within RGCs, retinal vasculature and glia should represent a priority target in glaucoma research. Potential candidate mechanisms might include pressure- and/or stretch-sensitive ion channels, enzymes, cytoskeletal proteins, extracellular matrix proteins or combinations thereof. Force-induced stretching of focal adhesion junctions could, for example, reveal intracellular binding sites for cytoskeletal proteins and influence activation of mechanosensitive ion channels.38,39 Another possibility is that the pressure gradient across axons within the optic nerve mechanically and/or biochemically impairs axonal transport between the cell body and midbrain synapses, depriving RGCs of critical “neurotrophic factors”.40–44 The perfusion pressure difference between arm-measured blood pressure and IOP is a strong risk factor for incidence and progression of open angle glaucoma.18 Variants of the “vascular hypothesis”45,46 suggest that the primary defect in glaucoma is due to vasoconstriction and insufficient blood supply, caused by compromised arterial flow through the capillaries connected to the peripapillary choroid and the circle of Zinn–Haller. There is, however, little clear evidence that chronic IOP increases observed in most ocular hypertensive patients directly affect ocular blood flow. The biomechanics of this process, especially with respect to the late remodeling of the optic nerve head, has been reviewed elsewhere.24,47
The observations that some of the earliest actions of increased IOP target the dendritic field size, the number of synapses as well as light-evoked responses of RGCs48–53 suggested that the primary retinal pressure sensors may be RGCs themselves. Consistent with this view, pressure-induced reductions in axon thickness and deformation of the optic nerve head in primate glaucoma models appear later than abnormalities in the dendritic arbors.54 How do RGCs sense mechanical stress? Ocular hypertension could affect the cells through compressive forces (force/cross sectional area) and/or tensile strain (local stretch of the tissue). A major role for compression is doubtful given that the neural retina is entirely enclosed within the eye. However, even in healthy eyes IOP-driven increases in ocular volume could exacerbate tensile forces that impinge on pre-stressed extracellular matrix, cytoskeleton and plasma membrane structures. The pressure–volume relationship described by Friedenwald’s ocular rigidity coefficient (“resistance exerted against distending forces”) is between 0.0126 mm Hg/μL and 0.0224 mm Hg/ μL.55,56 According to Pallikaris et al.,57, 20 mm Hg increase in IOP ought to increase the volume of the human eye by ~30 microliters whereas tonometric measurements from living eyes give a larger volume increment of ~45 microliters,58 which leads to the prediction that a 20 mm Hg increase in IOP would expand the ocular volume by ~1%. Showing that retinal cells respond to 1% stretch would confirm that they are directly sensitive to the tensile forces driven by IOP changes that are commonly observed in glaucoma. Ocular rigidity is lower in glaucoma compared to healthy subjects.59 Thus, an increase in IOP will provoke larger tensile stretch forces across membrane/matrix proteins in glaucomatous RGCs compared to healthy cells and should be more efficacious in crossing the thresholds of intrinsic mechanosensitive mechanisms. Mechanical forces and submicrometer displacements generated by IOP elevations are comparable to the measured free energies of known mechanosensitive channels.60,61
MECHANOSENSATION, TRPV4 SIGNALING AND RGC NEURODEGENERATION
The long-standing question in glaucoma research has been whether RGCs are themselves capable of transducing mechanical stimuli generated by physiological changes in IOP amplitude. In vitro, in vivo and preclinical evidence published in recent years shows that RGCs are themselves highly sensitive to mechanical forces.9,62–67 RGC viability has been shown to be affected by physical compression, tensile stretch, prolonged swelling and IOP elevations, which, in intact preparations, were able to induce changes in the molecular composition and synaptic organization within hours to weeks.63,68–70
The recently identified Transient Receptor Potential (TRP) and Piezo channels represent obvious candidates for retinal IOP transducers. While little is known about the Piezo family, the seven subfamilies of the TRP superfamily – so named after their Drosophila homolog, which plays a key role in phototransduction – are crucial for the perception of sensory information in vertebrates and invertebrates.11 Most TRP isoforms are nonselective cation channels that are permeable to Ca2+, therefore their activation serves as suitable trigger for many different types of intracellular signaling events. Members of four TRP subfamilies, specifically of the vanilloid (TRPV), ankyrin (TRPA), polycystin (TRPP), and canonical (TRPC) families are relevant to mechanosensation. These channels are only weakly sensitive to depolarization but open in response to a wide variety of mechanical, osmotic, chemical and thermal stimuli.22 RGCs express mechanosensitive TRPC1, 3-, 6-, 7- and TRPV1- and 4-channel isoforms.66,71–73 TRPV4 is a particularly attractive candidate as a glaucoma mechanosensor because, while strongly expressed in RGCs, it is excluded from other types of retinal neuron.66 Selective TRPV4 agonists, such as 4α-PDD and GSK1016790A, induce calcium influx into RGCs and increase the rate of spontaneous RGC firing, whereas excessive TRPV4 stimulation induces RGC apoptosis but spares other retinal neurons.66,74 Mechanosensitive TRPV4-mediated responses could account for the increased excitability and reduced RGC survival induced by experimental elevation of IOP or membrane stretch.66 The precise mechanism through which membrane tension activates RGC TRPV4 channels is unclear. The mutually not incompatible mechanisms include direct activation by lipid stretch,75 phospholipase A2 or through mechano-chemical feedback involving β1 integrins and/or focal adhesion kinases.76,77
It remains to be determined whether excessive calcium influx through TRPV4 channels contributes to calcium dysregulation that has been linked to the pathogenesis of glaucoma in animal studies and clinical trials.23,78,79 Interestingly, the risk for developing the disease in humans is increased by taking high daily doses of calcium supplements80 or by not taking calcium channel blockers.78 At the very least, calcium ions are going to play a central role in cytoskeletal reorganization that underpins dendritic/axonal remodeling in glaucoma. According to the model shown in Figure 1, local Ca2+ influx driven by excessive TRPV4 activation contributes to increased baseline [Ca2+]i levels and RGC hyperexcitability. This leads to activation of Ca2+-dependent genes belonging to NFAT (nuclear factor of activated T-cells), c-fos, DREAM (DRE antagonistic modulator protein) and/or CREB (cAMP response element-binding protein) families and triggers Ca2+-dependent catabolic enzymes such as calcineurin and calpains, as well as the cytoskeletal remodeling pathways involving actin and/or microtubular assemblies.8,23,81–86 Calcium levels also modulate the opening probability of purinergic channels and pannexin hemichannels and could affect their responsiveness to mechanical stimuli. Consistent with this, TRPV4-mediated Ca2+ entry was shown to regulate ATP release, which, via P2X7 receptors, could exacerbate mechanically-induced cell injury and facilitate release cytoactive molecules such as endothelin and/or TNF-α.87,88
FIGURE 1.
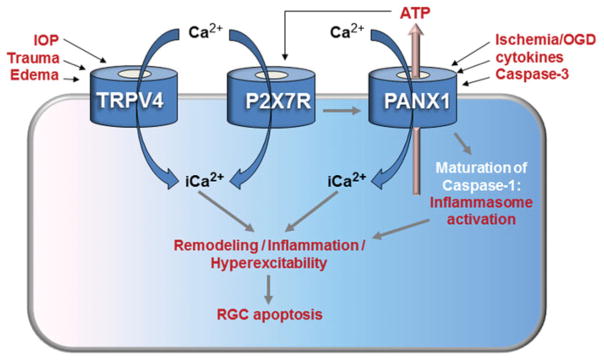
Proposed model for RGC mechanotransduction. Pressure-induced membrane stretch activates plasma membrane TRPV4 channels leading to calcium entry, activation of Panx1 and ATP release. This leads to secondary activation of P2X channels and P2Y receptors on neurons and glial cells. Calcium dysregulation may then trigger dendritic and axonal remodeling, inflammation, glial reactivity, RGC hyperexcitability, and eventually, apoptosis.
The emergence of new models of mechanical gating33,89 suggests that force transduction cannot be disentangled from intracellular biochemistry. Hence, while the direct mechanosensory function of TRPV4 figures most prominently, the polymodal features of TRPV4 activation, such as sensitivity to swelling and inflammatory agents75,76 place the channel squarely within the crossroads of mechanosensing, inflammatory signaling and anatomical remodeling. Inflammatory mediators would exacerbate RGC injury, induced by mechanically generated TRP-mediated Ca2+ overload (Figures 1 and 2). According to this view, TRPV4 signaling represents an epicenter that links primary, pressure-induced RGC damage to secondary pathophysiological mechanisms mediated by glialvascular inputs (delineated below). Pannexin channels link the mechanosensitive release of ATP to P2X7 receptor-mediated death of RGCs.
FIGURE 2.
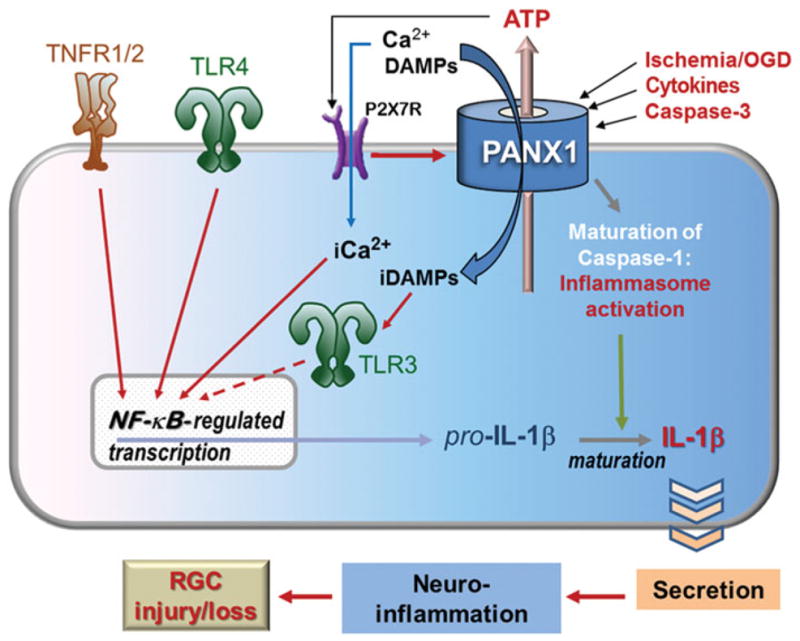
Schematic diagram of signaling mediated by surface receptors and Panx1 in the injured retina. Although signaling by TNFRs, TLR4 (signals through MyD88 and/or TRIF) and Ca2+ can feed directly into NF-κB activation, TLR3 signaling via TRIF first results in the transcription of type 1 interferon genes. These can also promote NF-κB-regulated transcription (dashed arrow). Ultimately, the maturation of pro-IL-1β into IL-1β via inflammasome pathways perpeturates inflammation and worsens the glaucomatous damage.
MECHANOSENSITIVE RELEASE OF ATP VIA PANNEXINS AND AUTOSTIMULATION OF P2X7R RECEPTORS ON RGCs
In the eye, the mechanical strains resulting from increased IOP are also associated with ATP release. This ATP can mediate physiological and pathological responses through binding to purinergic P2 receptors, the ligand-gated ion channels activated by ATP.90–92 Several isoforms of ionotropic P2X receptors, including P2X3-5 and P2X7 were reported to be expressed in RGCs.93 Increased concentrations of extracellular ATP are present in the aqueous humor of human patients with acute94 or chronic95 glaucoma. Extracellular ATP is also elevated in the retina following acute elevations in IOP from rat and bovine retina,68,96 and preliminary data suggest a prolonged increase in retinal ATP occurs in primate and rat models of chronic glaucoma.97,98 Because extracellular ATP is rapidly degraded in the central nervous system (CNS) by ecto-ATPases, such sustained increase is the evidence of a prolonged release from stressed neural cells. While the ATP released in response to mechanical strain can act at multiple receptors, the P2X7 receptor is of particular interest given its ability to initiate both inflammatory responses and neuronal death.99,100 Since RGCs express P2X7Rs, stimulation with its agonist, 2′(3′)-O-(4-Benzoylbenzoyl) adenosine-5′-triphosphate (BzATP), elevates intracellular Ca2+ and kills RGCS in vitro.101 BzATP also kills RGCs in vivo; this death is inhibited by P2X7R blockers MRS 2540 and Brilliant blue G.102 This suggests that the mechanosensitive release of ATP accompanying elevation of IOP may influence ganglion cell health in acute and chronic glaucoma.103,104
Given the pathological effects of excess extracellular ATP on ganglion cells, the cellular source of this released ATP and the signaling pathways leading to this release are of interest. Although Müller cells release ATP into the region surrounding RGCs upon mechanical stimulation, its rapid dephosphorylation into adenosine may limit the concentrations reaching RGC membranes.105,106 In a healthy retina with little mechanically-evoked ATP release, ATP dephosphorylation regulates P2X7 receptor activity, because P2X7R requires relatively high concentrations of ATP for activation.107 In disease, sufficient ATP to activate the P2X7 receptors may come from the efflux of ATP through channels in close proximity to the P2X7 receptor on the membrane, with local concentrations high enough to autostimulate the receptors. The pannexin-1 (Panx1) channel, which can be recruited and directly activated by P2X7R, has been recently implicated in this role.67,108–110
Pannexins are membrane channels with a high single channel conductance of 500 pS that are permeable to molecules over 1 kDa.111 Unlike connexin gap junction proteins, pannexin channels are not coupled to partners in adjacent cells but instead act as pores connecting the cell interior to extracellular space; opening of the pore is tightly regulated to maintain cellular integrity.109 Pannexins are widely distributed and have important implications for inflammation, as discussed below. However, pannexins have two characteristics essential to the current context; they are highly permeable to ATP and open upon application of mechanical strain to the membrane.67 Whether pannexins are themselves the primary mechanosensor or activated by other upstream mechanosensitive sensors through Rho kinase,112 their ability to release ATP in proximity to P2X7 receptors upon stretch of the membrane implicates them in connecting elevated IOP with activation of the p2X7 receptors.
This pannexin/P2X7R system was recently found to translate mechanical strain into receptor activation in ganglion cells.113 RGCs strained by stretching on a silicone substrate, or swollen with hypotonic solution, released ATP. This release was inhibited by pannexin blockers carbenoxolone, probenecid and inhibitory peptide, 10 Panx, implicating the pannexin channel in the efflux. Importantly, this mechanosensitive release came from isolated immunopanned cells, identifying RGCs themselves as a cellular source of releasable ATP. Whole cell ion currents activated by swelling were reduced by pannexin channel blockers by removal of extracellular ATP with apyrase or by P2X7R blockers A438079, AZ10606120 and zinc. Together, these observations strongly support a model whereby the mechanosensitive release of ATP through pannexin channels on RGCs autostimulates P2X7 receptors on the cells.
The consequences of this mechanosensitive auto-stimulation are likely to be more complex than originally thought. Although stimulation of the P2X7R is widely associated with cell death, the expression of pannexins114 and P2X7 receptors115,116 in healthy adult RGCs suggests that death is not a necessarily consequence of receptor stimulation. However, the massive ATP release that accompanies excessive mechanical strain may push the system into a pathological state. The location of the pannexin/P2X7 receptors may also influence the function of this mechanosensitive ATP release and autostimulation. According to immunohistochemical analysis, Panx1 and P2X7 proteins are expressed on both the soma and neurites of isolated ganglion cells.113 As much of the mechanical strain in glaucoma occurs at or near the optic nerve head,117 expression of this mechanosensitive signaling pair along neurites suggests the system may translate strain in the optic nerve head to local damage in ganglion cell axons.
THE ROLE OF PANNEXIN1-ACTIVATED PATHWAYS IN RGC INJURY
The Panx1 protein forms large non-selective membrane channels and is implicated in paracrine signaling and regulation of the inflammasome. Compared to connexin hemichannels, Panx1 channels are less sensitive to extracellular Ca2+, and open when intra-cellular Ca2+ increases, suggesting them to serve as an additional pathway for Ca2+ influx across membrane in pathological conditions.118–120 Importantly, Panx1 membrane channels have superior permeability to ATP, which prompted referring to them as “the ATP channels” suitable for paracrine signaling in astrocytes, neurons and other cell types.88 As described above, pannexins are implicated in massive ATP release by glial and blood endothelial cells is typically observed in various pathologies.89–92 Moreover, Panx1 channels were shown to regulate the inflammasome.121,122 The Panx1-mediated pathway activates faster in pathogenic conditions, i.e. after stress, injury and cytokine exposure, when cells were shown to decrease the number of gap junctions in the plasma membrane in favor of hemichannels made of either connexins or pannexins.123–125 Mechanistically, the abnormal opening of Panx1 channels is facilitated by a combination of pathogenic stimuli typically released after stress or injuries, including mechanical stress, extracellular K+, ATP, glutamate, cytokines and Zn2+ and proteolysis by active caspase-3.109,126–129
Recent studies utilizing gene knockdown and knockout mouse models suggested that Panx1 plays a key role in ischemic death of multiple types of neurons.130,131 Several reports showed that over-stimulation of the Panx1 channels facilitate neuronal loss in models of stroke, glaucoma, retinal ischemia, spreading depression and enteric colitis.99,130–135 Because RGCs express high levels of Panx1 and are extremely susceptible to ischemic injury, we tested the hypothesis that activation of Panx1 directly facilitates rapid and selective loss of RGC neurons in ischemia. Our data generated using the Panx1 knockout mice, which are significantly protected from ischemic injury, showed that two distinct neurotoxic processes are mediated by these channels in ischemic conditions.130
As revealed by dye transfer and calcium imaging assays, one mechanism involves permeation of RGC plasma membranes. This causes an imbalance of small molecules, and, in particular, an influx of Ca2+ and the efflux of ATP.130,132,136 Ca2+ overload, which activates Ca2+-dependent proteases and facilitates apoptosis, can occur directly via Ca2+-insensitive Panx1 channels (Figures 1 and 2). In addition, an opening of Panx1 channels can occur via the P2X7R-dependent mechanism in response to several external and internal stimuli of physiological or pathological nature, which prompted researchers to name P2X7R-Panx1 a “death complex”.99,120,137–139 It is plausible that prolonged opening of Panx1 can be triggered by a combination of pathological factors such as increases in extracellular concentration of known agonists including ATP, K+, Zn2+, glutamate and pro-inflammatory cytokines. 67,109,124,128,140 Such a combination is common in retinal or brain ischemia (stroke) and other CNS injuries.99,130,131
The second process that is interrupted in the Panx1 knockout is the activation of caspase-1 and inflammasome-mediated production of IL-1β and IL-18.84–86 The inflammasome is a macromolecular complex, first characterized in macrophages108,141 and, more recently, in glia and neurons.122 The well-known pathway for transcriptional activation of IL-1β and IL-18 genes involves stimulation of TNF and Toll-like receptors (TLRs),28,142–146 which causes the activation of NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells)-dependent gene transcription (Figures 2 and 3). Thus, production of these ILs depends on two independent pathways: (1) transcriptional activation via the NF-κB pathway and (2) proteolytic processing of IL precursors by the inflammasome.
FIGURE 3.

Retinal signaling and cellular remodeling mediated by TLR, TNF and immune molecules involves neuronal-glial circuits. The activation of TLR4, TNFRs and TCRs can promote NF-κB-regulated transcription, generating pro-IL-1β, which is then processed into IL-1β and secreted. This drives glial reactivity and furthers inflammation (e.g. the release of more TNF-α), thus perpetuating the inflammatory cycle. As RGCs are damaged and killed, they may release DAMPS (e.g. certain HSPs) that activate innate immune receptors such as TLR4 and thereby worsen glaucoma. The activation of TCR/CD3 may be involved in glaucomatous dendritic remodeling, which may disturb normal circuit functions and degrade the fidelity of visual processing.
How does Panx1 contribute to inflammasome activation? One feasible mechanism is a direct interaction with the inflammasome complex that facilitates proteolytic processing of caspase-1 and results in Il-1β release, as reported by Pelegrin and Surprenant.141 The second mechanism of Panx1-mediated inflammasome activation involves transcriptional activation of IL-1β, since the large pore of the Panx1 channel can provide a gateway for the entry of pro-inflammatory molecules into the cell, leading to stimulation of intracellular membrane receptors such as TLR3.86,108,141 TLR3, as well as surface receptors TLR4 and TNFR, cause transcriptional activation of NF-κB (Figure 2) and have been recently implicated in several neurodegenerations, including glaucoma.147–149 In a similar fashion, the activation Panx1 and, subsequently, the inflammasome can be triggered via stimulation of P2X7 receptors by extracellular ATP.150,151 It was demonstrated that Panx1 is essential for P2X7R-induced proteolytic cleavage of caspase-1 and subsequent IL-1β maturation/release, which can be blocked by pharmacological blockade of the Panx1 channels with small interfering RNA, mimetic peptide or carbenoxolone.108,121,151,152 Likewise, genetic ablation of Panx1 resulted in a robust neuroprotection in mouse models of enteric colitis and traumatic brain injury.39,43 Importantly, a study of neuron-specific Panx1 knockout mice demonstrated that Panx1-mediated neurotoxicity is facilitated by the endogenous, neuronal inflammasome.84,122 Neuronal types expressing high levels of Panx1, such as RGCs, are vulnerable to Panx1-mediated death in response to certain pathological and pro-inflammatory stimuli. Opening of Panx1 channels was shown to be independent of TLR activation.86 Combined with our own results,130 this finding allowed us to propose a model where Panx1 acts in parallel, not downstream of TLRs. This model implies synergy between the MyD88-NF-κB pathway and Panx1-mediated processes for IL-1β processing and secretion. Indeed, cytokine maturation appears to be a crucial step in the neurotoxic pro-inflammatory program that is activated in injured CNS via the MyD88-NF-κB pathway.122,130,153 Consistent with our hypothesis, the extent of neuroprotection in Panx1 knockout mice is similar to that observed in the knockouts of caspase-1, P2X7 receptor, TNF receptors 1/2, TLR3/4 and conditional knockouts of NF-κB.130,142,154–158 In a similar fashion, pharmacological blockade of P2X7R, NALP1/ 3 or ASC subunits of inflammasome showed robust neuroprotection in various CNS injuries,84,85,151,159 a strong evidence for neurotoxic effects of inflammasome activation.
TNFR and TLR Signaling Promote Inflammation through NF-κB, Driving RGC Degeneration
Increased glial production of TNF-α in the glaucomatous human retina and optic nerve has been implicated in RGC death and inflammatory processes through the TNFR signaling.28,160–162 High-throughput characterization of the retinal proteome has recently indicated a prominent up-regulation of TNFR-mediated apoptosis pathway and inflammation signaling in human glaucoma.161 Retinal proteins exhibiting increased expression in glaucoma have included TNF-α, TNFR1 and various downstream adaptor/ interacting proteins, such as TNFR1-associated death domain protein (TRADD) and the members of the TNFR-associated factor (TRAF) family, and protein kinases involved in TNF-α/TNFR1 signaling. Proteomics data support that a complex crosstalk relationship between multiple signaling pathways determines diverse bioactivities of TNF-α.28 Besides the proteolytic caspase cascade, co-activation of calpain-mediated pathways, mitochondrial dysfunction and endoplasmic reticulum stress may reinforce each other during RGC apoptosis in glaucoma. Regarding TNFR-mediated inflammation signaling, proteomics analysis of the glaucomatous human retina has produced data supporting NF-κB activation, JAK/STAT signaling and inflammasome assembly.161 Proteomics analysis of RGC and astrocyte samples has also showed cell-specific regulation of TNF-α signaling in experimental glaucoma, such as caspase activation leading to apoptosis in RGCs, but NF-κB activation promoting cell survival and inflammation in astrocytes.162 In addition, the type of receptor preferentially used is important in determining the outcomes of TNF-α signaling. This multifunctional cytokine can bind two different receptors of the TNFR superfamily, TNFR1 (p55) and TNFR2 (p75). TNFR1 appears to be the primary receptor for both neurodegenerative and inflammatory consequences of TNF-α signaling in glaucoma.161 This is because a death domain present on the intracellular region of TNFR1, but not present in TNFR2, leads to apoptotic cell death, while signaling through TNFR2 leads primarily to cell proliferation. TNFR1 is also the primary signaling receptor responsible for the majority of TNF-α-mediated inflammatory responses, particularly those mediated by the soluble TNF-α required for inflammation.28,163
The glaucomatous human retina also exhibits up-regulation of TLR signaling149 (Figures 2 and 3). Innate immune activity in the CNS can be triggered by numerous pathways after recognition of invading pathogens or tissue stress/injury by pattern recognition receptors,164 which include TLRs and nucleotide-binding oligomerization domain-like receptors (NLRs). Although TLRs are membrane-spanning receptors, NLRs are cytoplasmic sensors that form a platform for the assembly of the inflammasome, a multiprotein complex that processes pro-ILs into their mature forms via proteolytic cleavage by caspase-1,165 as is evident in glaucoma.161,162 TLRs recognize a wide variety of pathogen-associated molecular patterns and also the DAMPs expressed during tissue stress or injury.166 Recent proteomics studies of human glaucoma and animal models have revealed that glial cells, including both microglia and astroglia, are the main cell types that express a repertoire of TLRs, as well as several inflammasome-related molecules.149,162 In addition, there is in vitro evidence indicating that glaucomatous stress-related intrinsic ligands, such as heat shock proteins (HSPs) and oxidation products, can activate glial TLRs and stimulate T cells.149 After recognizing specific molecular patterns, TLRs recruit adaptor proteins, such as MyD88, and activate NF-κB (Figure 2), a major transcription factor for the expression of pro-inflammatory cytokines.167 Proteomic data from human glaucoma and animal models, along with the findings of in vitro treatment experiments, support the notion that the glial TLR signaling initiated by glaucomatous stress-related ligands includes MyD88-dependent pathways.149
NF-κB activation after binding TNFRs and TLRs triggers the transcriptional activation of pro-ILs that are processed into their active forms by the inflammasome.165 Glaucomatous retinal proteome exhibits increased glial expression of specific kinases involved in the NF-κB activation pathway, such as receptor-interacting serine-threonine kinase (RIPK), NF-κB-inducing kinase (NIK), and inhibitory kappa B (IκB) kinases (IκKs), including a master regulator, IκKgamma (NF-κB essential modulator), and phosphorylation of NF-κB subunits, NF-κB1-p105/p50 and p65.161,162 Although NF-κB regulates neuronal survival programs, including in the retina and optic nerve,168 this transcription factor is a master regulator of the inflammatory responses leading to secondary neurodegenerative processes.167,169 The NF-κB pathway may similarly play a major role in regulation of glia-driven pro-inflammatory processes during glaucomatous neurodegeneration.162 As implicated in other neurodegenerative diseases,170 TNF-α/TNFR signaling, TLR signaling and the inflammasome together exhibit the potent inflammatory capacity with beneficial and detrimental outcomes in glaucoma. NF-κB, as being a common player of inflammation through TNFR or TLR signaling, appears to be a promising treatment target to provide immunomodulation in glaucoma171 and deserves further studies.
Function and Possible Mechanisms of Activation of Immune Molecules in the Retina
Recent studies demonstrated that genes typically associated with the immune system, such as those in the MHC and complements, are expressed by neurons in various regions of the CNS, including retina, and may play important roles in synapse formation during normal development and pathogenesis in CNS diseases.25,172–177 Consistent with this notion, genetic deletion or mutation of a number of MHC class I genes (such as a MHCI co-subunit β2-microglobulin or a key component of MHCI receptor complex CD3ζ), complements or complement receptors result in the failure of development of the eye-specific segregation of RGC axonal projections to the dLGN.25,174,178,179 On the other hand, over-expression of MHCI molecules caused effects on the retinogeniculate projections opposite to that of MHCI or complement mutations.180 Furthermore, the expression of complements is up-regulated in glaucomatous retinas,181 and over-expression of MHCI molecules significantly enhanced the recovery of locomotor abilities after spinal cord injury.181
The precise molecular mechanisms of how MHCI molecules and complement cascade expressed by CNS neurons are activated, and how they regulate the normal development and pathogenesis of CNS diseases are unclear. It was suggested that the expression and activity of MHCI molecules and complement cascade are regulated by neuronal activity. Consistently, retinal activity was found to regulate the level of mRNA of MHCI molecules in the dorsal lateral geniculate nucleus (dLGN).173 In retina, the effects of CD3ζ seem to be cell-type and neurotransmitter-specific. Xu et al.,25 reported that CD3ζ is specifically expressed by RGCs and mice with genetic mutation of CD3ζ exhibited a selective reduction of glutamate receptor-mediated synaptic transmission of RGCs. It was also postulated that activation of immune molecules in neurons could produce similar intracellular signals as those generated in immune cells but with different ultimate effects, such as altering synaptic development, strength, neuronal morphology or circuit properties downstream of synaptic activity25,182,183 (Figure 3). In the immune system, activation of CD3ζ triggers several downstream cascades, including a Ras-MAPK pathway and actin-based cytoskeleton reorganization, which regulates immune cell polarization, migration and dendritic growth.184–186 It has been shown that most components of these cascades are expressed in the CNS and implicated in activity-dependent synaptic plasticity.182,187 In addition, direct activation of CD3ζ on hippocampal neurons affects cell morphology by promoting dendritic pruning through a tyrosine-based phosphorylation signaling motif common to the immune system.172 Furthermore, neuronal activity in the retina is also suggested to regulate the complement-dependent activation of the resident immune cells in retina, microglia, which in turn regulates the developmental remodeling of RGC axonal projection during normal development through a process similar to “phagocytosis”.178 Recent studies also implied that retinal microglia might play an important role in RGC death in glaucomatous retinal degeneration.188–190 These observations strongly support the possibility that the immune molecules and cells could regulate the neuronal structure and function through mechanisms similar to those in the immune system.
CONCLUDING REMARKS
Analysis of signaling pathways associated with RGC injury points at intracellular involvement of ubiquitous messenger molecules such as Ca2+ and ATP, which could participate in intrinsic mechanosensation and drive feedback pathways associated with secondary glial and vascular mechanisms. Because [Ca2+]i maintenance is critically important for the regulation of excitability, cytoskeletal integrity, metabolism and synaptic function, disruption of Ca2+ homeostasis would ultimately lead to anatomical and physiological remodeling observed in glaucoma. Recent evidence suggests that such Ca2+ overloads in RGCs could be mediated by TRPV4, P2X7R and/or pannexin channels. The effect of mechanical stress on resident mechanosensitive channels has to be placed within the larger context encompassing secondary inflammatory and immune responses driven by feedback loops between injured RGCs, astrocytes, microglia and the vascular endothelium. Inflammatory cytokines and complement molecules released from glial and endothelial cells could drive the plasma membrane P2X7R-Panx1 complex as well as complex arrays of immune/ inflammatory signaling molecules that might include TNF-α, IL-1β, Toll-like and T-cell receptors. The ensuing reconfiguration of intracellular signals is proposed to involve the NF-κB pathway and activation of the inflammasome complex.
Current glaucoma treatments are limited to minimization of mechanical impact mediated by elevated IOP and lack tools that would protect RGCs by targeting the mechanosensing mechanisms and/or secondary inflammatory/immune pathways within the retina. Thus, development of novel neuroprotective treatments depends on our ability to characterize the force transduction mechanisms that mediate retinal IOP sensing (TRPV4, Panx1 and P2X7R discussed here) together with the role of secondary interactions between RGCs and the surrounding vascular endothelial cells, pericytes, astrocytes, Müller cells and microglia.
Acknowledgments
Supported by NIH grants T32DC008553 (DAR), EY14232, EY021517 (VIS), EY013434, EY015537 (CHM), EY13870, EY022076 (DK), EY018666 and GM060019 (VZS), EY013813 and EY017131 (GT), Center Grants P30 EY014801 (VIS) and P30 EY14800 (DK), Department of Defense (VIS, DK), Russian Federal Special Program Grant 2012-1.5-12-000-1002-018 (V.I.S.), University of Utah Translational Seed grant (DK), State of Utah TCIP (DK), Foundation Fighting Blindness (DK) and unrestricted grants from RPB to Bascom Palmer Eye Institute (Miami) and Moran Eye Institute (Salt Lake City). The authors would like to thank Dr. Harilaos Ginis (University of Crete, Greece) for helpful comments.
Footnotes
DECLARATION OF INTEREST
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- 1.Harwerth RS, Crawford ML, Frishman LJ, Viswanathan S, Smith EL, 3rd, Carter-Dawson L. Visual field defects and neural losses from experimental glaucoma. Prog Retin Eye Res. 2002;21:91–125. doi: 10.1016/s1350-9462(01)00022-2. [DOI] [PubMed] [Google Scholar]
- 2.Jakobs TC, Libby RT, Ben Y, John SW, Masland RH. Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J Cell Biol. 2005;171:313–325. doi: 10.1083/jcb.200506099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baltan S, Inman DM, Danilov CA, Morrison RS, Calkins DJ, Horner PJ. Metabolic vulnerability disposes retinal ganglion cell axons to dysfunction in a model of glaucomatous degeneration. J Neurosci. 2010;30:5644–5652. doi: 10.1523/JNEUROSCI.5956-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tezel G. Oxidative stress in glaucomatous neurodegeneration: mechanisms and consequences. Prog Retin Eye Res. 2006;25:490–513. doi: 10.1016/j.preteyeres.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quigley HA, Addicks EM. Chronic experimental glaucoma in primates. I. Production of elevated intraocular pressure by anterior chamber injection of autologous ghost red blood cells. Invest Ophthalmol Vis Sci. 1980;19:126–136. [PubMed] [Google Scholar]
- 6.Umapathy NS, Li W, Mysona BA, Smith SB, Ganapathy V. Expression and function of glutamine transporters SN1 (SNAT3) and SN2 (SNAT5) in retinal Muller cells. Invest Ophthalmol Vis Sci. 2005;46:3980–3987. doi: 10.1167/iovs.05-0488. [DOI] [PubMed] [Google Scholar]
- 7.Sullivan RK, Woldemussie E, Macnab L, Ruiz G, Pow DV. Evoked expression of the glutamate transporter GLT-1c in retinal ganglion cells in human glaucoma and in a rat model. Invest Ophthalmol Vis Sci. 2006;47:3853–3859. doi: 10.1167/iovs.06-0231. [DOI] [PubMed] [Google Scholar]
- 8.Reichenbach A, Bringmann A. Müller cells in the healthy and diseased retina. New York: Springer; 2010. p. xiv.p. 417. [DOI] [PubMed] [Google Scholar]
- 9.Bonomi L, Marchini G, Marraffa M, Morbio R. The relationship between intraocular pressure and glaucoma in a defined population. Data from the Egna-Neumarkt Glaucoma Study. Ophthalmologica. 2001;215:34–38. doi: 10.1159/000050823. [DOI] [PubMed] [Google Scholar]
- 10.Blanch RJ, Ahmed Z, Berry M, Scott RA, Logan A. Animal models of retinal injury. Invest Ophthalmol Vis Sci. 2012;53:2913–2920. doi: 10.1167/iovs.11-8564. [DOI] [PubMed] [Google Scholar]
- 11.Nilius B, Owsianik G. Transient receptor potential channelopathies. Pflugers Arch. 2010;460:437–450. doi: 10.1007/s00424-010-0788-2. [DOI] [PubMed] [Google Scholar]
- 12.Christensen AP, Corey DP. TRP channels in mechanosensation: direct or indirect activation? Nat Rev Neurosci. 2007;8:510–521. doi: 10.1038/nrn2149. [DOI] [PubMed] [Google Scholar]
- 13.Wiggs JL. The cell and molecular biology of complex forms of glaucoma: updates on genetic, environmental, and epigenetic risk factors. Invest Ophthalmol Vis Sci. 2012;53:2467–2469. doi: 10.1167/iovs.12-9483e. [DOI] [PubMed] [Google Scholar]
- 14.Sommer A, Tielsch JM, Katz J, Quigley HA, Gottsch JD, Javitt J, et al. Relationship between intraocular pressure and primary open angle glaucoma among white and black Americans. The Baltimore Eye Survey. Arch Ophthalmol. 1991;109:1090–1095. doi: 10.1001/archopht.1991.01080080050026. [DOI] [PubMed] [Google Scholar]
- 15.Stamer WD, Acott TS. Current understanding of conventional outflow dysfunction in glaucoma. Curr Opin Ophthalmol. 2012;23:135–143. doi: 10.1097/ICU.0b013e32834ff23e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.John SW, Smith RS, Savinova OV, Hawes NL, Chang B, Turnbull D, et al. Essential iris atrophy, pigment dispersion, and glaucoma in DBA/2J mice. Invest Ophthalmol Vis Sci. 1998;39:951–962. [PubMed] [Google Scholar]
- 17.Chauhan BC, Pan J, Archibald ML, LeVatte TL, Kelly ME, Tremblay F. Effect of intraocular pressure on optic disc topography, electroretinography, and axonal loss in a chronic pressure-induced rat model of optic nerve damage. Invest Ophthalmol Vis Sci. 2002;43:2969–2976. [PubMed] [Google Scholar]
- 18.Quigley HA. Clinical trials for glaucoma neuroprotection are not impossible. Curr Opin Ophthalmol. 2012;23:144–154. doi: 10.1097/ICU.0b013e32834ff490. [DOI] [PubMed] [Google Scholar]
- 19.Sharma AK, Rohrer B. Sustained elevation of intracellular cGMP causes oxidative stress triggering calpain-mediated apoptosis in photoreceptor degeneration. Curr Eye Res. 2007;32:259–269. doi: 10.1080/02713680601161238. [DOI] [PubMed] [Google Scholar]
- 20.Vallazza-Deschamps G, Fuchs C, Cia D, Tessier LH, Sahel JA, Dreyfus H, et al. Diltiazem-induced neuroprotection in glutamate excitotoxicity and ischemic insult of retinal neurons. Doc Ophthalmol. 2005;110:25–35. doi: 10.1007/s10633-005-7341-1. [DOI] [PubMed] [Google Scholar]
- 21.Bezprozvanny I, Hayden MR. Deranged neuronal calcium signaling and Huntington disease. Biochem Biophys Res Commun. 2004;322:1310–1317. doi: 10.1016/j.bbrc.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 22.Wax MB, Tezel G. Immunoregulation of retinal ganglion cell fate in glaucoma. Exp Eye Res. 2009;88:825–830. doi: 10.1016/j.exer.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 23.Crish SD, Calkins DJ. Neurodegeneration in glaucoma: progression and calcium-dependent intracellular mechanisms. Neuroscience. 2011;176:1–11. doi: 10.1016/j.neuroscience.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burgoyne CF. A biomechanical paradigm for axonal insult within the optic nerve head in aging and glaucoma. Exp Eye Res. 2011;93:120–132. doi: 10.1016/j.exer.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu HP, Chen H, Ding Q, Xie ZH, Chen L, Diao L, et al. The immune protein CD3zeta is required for normal development of neural circuits in the retina. Neuron. 2010;65:503–515. doi: 10.1016/j.neuron.2010.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steele MR, Inman DM, Calkins DJ, Horner PJ, Vetter ML. Microarray analysis of retinal gene expression in the DBA/2J model of glaucoma. Invest Ophthalmol Vis Sci. 2006;47:977–985. doi: 10.1167/iovs.05-0865. [DOI] [PubMed] [Google Scholar]
- 27.Stasi K, Nagel D, Yang X, Wang RF, Ren L, Podos SM, et al. Complement component 1Q (C1Q) upregulation in retina of murine, primate, and human glaucomatous eyes. Invest Ophthalmol Vis Sci. 2006;47:1024–1029. doi: 10.1167/iovs.05-0830. [DOI] [PubMed] [Google Scholar]
- 28.Tezel G. TNF-alpha signaling in glaucomatous neurode-generation. Prog Brain Res. 2008;173:409–421. doi: 10.1016/S0079-6123(08)01128-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang W, Xing W, Ryskamp DA, Punzo C, Krizaj D. Localization and phenotype-specific expression of ryanodine calcium release channels in C57BL6 and DBA/2J mouse strains. Exp Eye Res. 2011;93:700–709. doi: 10.1016/j.exer.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lebrun-Julien F, Duplan L, Pernet V, Osswald I, Sapieha P, Bourgeois P, et al. Excitotoxic death of retinal neurons in vivo occurs via a non-cell-autonomous mechanism. J Neurosci. 2009;29:5536–5545. doi: 10.1523/JNEUROSCI.0831-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tezel G. The immune response in glaucoma: a perspective on the roles of oxidative stress. Exp Eye Res. 2011;93:178–186. doi: 10.1016/j.exer.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delmas P. Polycystins: from mechanosensation to gene regulation. Cell. 2004;118:145–148. doi: 10.1016/j.cell.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 33.Eyckmans J, Boudou T, Yu X, Chen CS. A hitchhiker’s guide to mechanobiology. Dev Cell. 2011;21:35–47. doi: 10.1016/j.devcel.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coulombre AJ, Coulombre JL. The role of intraocular pressure in the development of the chick eye. IV. Corneal curvature. AMA Arch Ophthalmol. 1958;59:502–506. doi: 10.1001/archopht.1958.00940050058005. [DOI] [PubMed] [Google Scholar]
- 35.Coulombre AJ, Coulombre JL. Lens development. I. Role of the lens in eye growth. J Exp Zool. 1964;156:39–47. doi: 10.1002/jez.1401560104. [DOI] [PubMed] [Google Scholar]
- 36.Vogel V, Sheetz M. Local force and geometry sensing regulate cell functions. Nat Rev Mol Cell Biol. 2006;7:265–275. doi: 10.1038/nrm1890. [DOI] [PubMed] [Google Scholar]
- 37.Morrison JC, Moore CG, Deppmeier LM, Gold BG, Meshul CK, Johnson EC. A rat model of chronic pressure-induced optic nerve damage. Exp Eye Res. 1997;64:85–96. doi: 10.1006/exer.1996.0184. [DOI] [PubMed] [Google Scholar]
- 38.del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, Sheetz MP. Stretching single talin rod molecules activates vinculin binding. Science. 2009;323:638–641. doi: 10.1126/science.1162912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stewart AP, Smith GD, Sandford RN, Edwardson JM. Atomic force microscopy reveals the alternating subunit arrangement of the TRPP2-TRPV4 heterotetramer. Biophys J. 2010;99:790–797. doi: 10.1016/j.bpj.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quigley HA, Green WR. The histology of human glaucoma cupping and optic nerve damage: clinicopathologic correlation in 21 eyes. Ophthalmology. 1979;86:1803–1830. doi: 10.1016/s0161-6420(79)35338-6. [DOI] [PubMed] [Google Scholar]
- 41.Danias J, Lee KC, Zamora MF, Chen B, Shen F, Filippopoulos T, et al. Quantitative analysis of retinal ganglion cell (RGC) loss in aging DBA/2NNia glaucomatous mice: comparison with RGC loss in aging C57/BL6 mice. Invest Ophthalmol Vis Sci. 2003;44:5151–5162. doi: 10.1167/iovs.02-1101. [DOI] [PubMed] [Google Scholar]
- 42.Schlamp CL, Li Y, Dietz JA, Janssen KT, Nickells RW. Progressive ganglion cell loss and optic nerve degeneration in DBA/2J mice is variable and asymmetric. BMC Neurosci. 2006;7:66. doi: 10.1186/1471-2202-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adalbert R, Coleman MP. Axon pathology in age-related neurodegenerative disorders. Neuropathol Appl Neurobiol. 2013;39:90–108. doi: 10.1111/j.1365-2990.2012.01308.x. [DOI] [PubMed] [Google Scholar]
- 44.Crish SD, Sappington RM, Inman DM, Horner PJ, Calkins DJ. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc Natl Acad Sci USA. 2010;107:5196–5201. doi: 10.1073/pnas.0913141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Begg IS, Drance SM. Progress of the glaucomatous process related to recurrent ischaemic changes at the optic disc. Exp Eye Res. 1971;11:141. doi: 10.1016/s0014-4835(71)80081-7. [DOI] [PubMed] [Google Scholar]
- 46.Fechtner RD, Weinreb RN. Mechanisms of optic nerve damage in primary open angle glaucoma. Surv Ophthalmol. 1994;39:23–42. doi: 10.1016/s0039-6257(05)80042-6. [DOI] [PubMed] [Google Scholar]
- 47.Fortune B, Burgoyne CF, Cull GA, Reynaud J, Wang L. Structural and functional abnormalities of retinal ganglion cells measured in vivo at the onset of optic nerve head surface change in experimental glaucoma. Invest Ophthalmol Vis Sci. 2012;53:3939–3950. doi: 10.1167/iovs.12-9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weber AJ, Harman CD, Viswanathan S. Effects of optic nerve injury, glaucoma, and neuroprotection on the survival, structure, and function of ganglion cells in the mammalian retina. J Physiol. 2008;586:4393–4400. doi: 10.1113/jphysiol.2008.156729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stevens SL, Ciesielski TM, Marsh BJ, Yang T, Homen DS, Boule JL, et al. Toll-like receptor 9: a new target of ischemic preconditioning in the brain. J Cereb Blood Flow Metab. 2008;28:1040–1047. doi: 10.1038/sj.jcbfm.9600606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Porciatti V, Nagaraju M. Head-up tilt lowers IOP and improves RGC dysfunction in glaucomatous DBA/2J mice. Exp Eye Res. 2010;90:452–460. doi: 10.1016/j.exer.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kong YX, van Bergen N, Bui BV, Chrysostomou V, Vingrys AJ, Trounce IA, et al. Impact of aging and diet restriction on retinal function during and after acute intraocular pressure injury. Neurobiol Aging. 2012;33:1126, e15–25. doi: 10.1016/j.neurobiolaging.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 52.Bui BV, He Z, Vingrys AJ, Nguyen CT, Wong VH, Fortune B. Using the electroretinogram to understand how intraocular pressure elevation affects the rat retina. J Ophthalmol. 2013;2013:262467. doi: 10.1155/2013/262467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Banitt MR, Ventura LM, Feuer WJ, Savatovsky E, Luna G, Shif O, et al. Progressive loss of retinal ganglion cell function precedes structural loss by several years in glaucoma suspects. Invest Ophthalmol Vis Sci. 2013;54:2346–2352. doi: 10.1167/iovs.12-11026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weber AJ, Kaufman PL, Hubbard WC. Morphology of single ganglion cells in the glaucomatous primate retina. Invest Ophthalmol Vis Sci. 1998;39:2304–2320. [PubMed] [Google Scholar]
- 55.Eisenlohr JE, Langham ME, Maumenee AE. Manometric studies of the pressure-volume relationship in living and enucleated eyes of individual human subjects. Br J Ophthalmol. 1962;46:536–548. doi: 10.1136/bjo.46.9.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dastiridou AI, Ginis HS, De Brouwere D, Tsilimbaris MK, Pallikaris IG. Ocular rigidity, ocular pulse amplitude, and pulsatile ocular blood flow: the effect of intraocular pressure. Invest Ophthalmol Vis Sci. 2009;50:5718–5722. doi: 10.1167/iovs.09-3760. [DOI] [PubMed] [Google Scholar]
- 57.Pallikaris IG, Kymionis GD, Ginis HS, Kounis GA, Tsilimbaris MK. Ocular rigidity in living human eyes. Invest Ophthalmol Vis Sci. 2005;46:409–414. doi: 10.1167/iovs.04-0162. [DOI] [PubMed] [Google Scholar]
- 58.Silver DM, Geyer O. Pressure-volume relation for the living human eye. Curr Eye Res. 2000;20:115–120. [PubMed] [Google Scholar]
- 59.Wang J, Freeman EE, Descovich D, Harasymowycz PJ, Kamdeu Fansi A, Li G, et al. Estimation of ocular rigidity in glaucoma using ocular pulse amplitude and pulsatile choroidal blood flow. Invest Ophthalmol Vis Sci. 2013;54:1706–1711. doi: 10.1167/iovs.12-9841. [DOI] [PubMed] [Google Scholar]
- 60.Markin VS, Sachs F. Thermodynamics of mechanosensitivity. Phys Biol. 2004;1:110–124. doi: 10.1088/1478-3967/1/2/007. [DOI] [PubMed] [Google Scholar]
- 61.Wiggins P, Phillips R. Membrane-protein interactions in mechanosensitive channels. Biophys J. 2005;88:880–902. doi: 10.1529/biophysj.104.047431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Niittykoski M, Kalesnykas G, Larsson KP, Kaarniranta K, Akerman KE, Uusitalo H. Altered calcium signaling in an experimental model of glaucoma. Invest Ophthalmol Vis Sci. 2010;51:6387–6393. doi: 10.1167/iovs.09-3816. [DOI] [PubMed] [Google Scholar]
- 63.Agar A, Li S, Agarwal N, Coroneo MT, Hill MA. Retinal ganglion cell line apoptosis induced by hydrostatic pressure. Brain Res. 2006;1086:191–200. doi: 10.1016/j.brainres.2006.02.061. [DOI] [PubMed] [Google Scholar]
- 64.Sappington RM, Carlson BJ, Crish SD, Calkins DJ. The microbead occlusion model: a paradigm for induced ocular hypertension in rats and mice. Invest Ophthalmol Vis Sci. 2010;51:207–216. doi: 10.1167/iovs.09-3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pang IH, Clark AF. Rodent models for glaucoma retinopathy and optic neuropathy. J Glaucoma. 2007;16:483–505. doi: 10.1097/IJG.0b013e3181405d4f. [DOI] [PubMed] [Google Scholar]
- 66.Ryskamp DA, Witkovsky P, Barabas P, Huang W, Koehler C, Akimov NP, et al. The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells. J Neurosci. 2011;31:7089–7101. doi: 10.1523/JNEUROSCI.0359-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xia J, Lim JC, Lu W, Beckel JM, Macarak EJ, Laties AM, et al. Neurons respond directly to mechanical deformation with pannexin-mediated ATP release and autostimulation of P2X7 receptors. J Physiol. 2012;590:2285–2304. doi: 10.1113/jphysiol.2012.227983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Resta V, Novelli E, Vozzi G, Scarpa C, Caleo M, Ahluwalia A, et al. Acute retinal ganglion cell injury caused by intraocular pressure spikes is mediated by endogenous extracellular ATP. Eur J Neurosci. 2007;25:2741–2754. doi: 10.1111/j.1460-9568.2007.05528.x. [DOI] [PubMed] [Google Scholar]
- 69.Balaratnasingam C, Morgan WH, Bass L, Ye L, McKnight C, Cringle SJ, et al. Elevated pressure induced astrocyte damage in the optic nerve. Brain Res. 2008;1244:142–154. doi: 10.1016/j.brainres.2008.09.044. [DOI] [PubMed] [Google Scholar]
- 70.Fu CT, Tran T, Sretavan D. Axonal/glial upregulation of EphB/ephrin-B signaling in mouse experimental ocular hypertension. Invest Ophthalmol Vis Sci. 2010;51:991–1001. doi: 10.1167/iovs.09-3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Molnar T, Barabas P, Birnbaumer L, Punzo C, Kefalov V, Krizaj D. Store-operated channels regulate intracellular calcium in mammalian rods. J Physiol. 2012;590:3465–3481. doi: 10.1113/jphysiol.2012.234641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xue T, Do MT, Riccio A, Jiang Z, Hsieh J, Wang HC, et al. Melanopsin signalling in mammalian iris and retina. Nature. 2011;479:67–73. doi: 10.1038/nature10567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sappington RM, Sidorova T, Long DJ, Calkins DJ. TRPV1: contribution to retinal ganglion cell apoptosis and increased intracellular Ca2+ with exposure to hydrostatic pressure. Invest Ophthalmol Vis Sci. 2009;50:717–728. doi: 10.1167/iovs.08-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Frye A, Ryskamp D, Krizžaj D. IOVS Abstr. Ft. Lauderdale, FL: 2012. Overstimulation of TRPV4 in vivo induces selective apoptosis of retinal ganglion cells. An acute in vivo experimental model for glaucoma. [Google Scholar]
- 75.Loukin S, Zhou X, Su Z, Saimi Y, Kung C. Wild-type and brachyolmia-causing mutant TRPV4 channels respond directly to stretch force. J Biol Chem. 2010;285:27176–27181. doi: 10.1074/jbc.M110.143370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–438. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- 77.Matthews BD, Thodeti CK, Tytell JD, Mammoto A, Overby DR, Ingber DE. Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface beta1 integrins. Integr Biol (Camb) 2010;2:435–442. doi: 10.1039/c0ib00034e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tomita G. The optic nerve head in normal-tension glaucoma. Curr Opin Ophthalmol. 2000;11:116–120. doi: 10.1097/00055735-200004000-00009. [DOI] [PubMed] [Google Scholar]
- 79.Nickells RW, Howell GR, Soto I, John SW. Under pressure: cellular and molecular responses during glaucoma, a common neurodegeneration with axonopathy. Annu Rev Neurosci. 2012;35:153–179. doi: 10.1146/annurev.neuro.051508.135728. [DOI] [PubMed] [Google Scholar]
- 80.Wang SY, Singh K, Lin SC. The association between glaucoma prevalence and supplementation with the oxidants calcium and iron. Invest Ophthalmol Vis Sci. 2012;53:725–731. doi: 10.1167/iovs.11-9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huang W, Fileta J, Rawe I, Qu J, Grosskreutz CL. Calpain activation in experimental glaucoma. Invest Ophthalmol Vis Sci. 2010;51:3049–3054. doi: 10.1167/iovs.09-4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Garcia-Valenzuela E, Shareef S, Walsh J, Sharma SC. Programmed cell death of retinal ganglion cells during experimental glaucoma. Exp Eye Res. 1995;61:33–44. doi: 10.1016/s0014-4835(95)80056-5. [DOI] [PubMed] [Google Scholar]
- 83.Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150:887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.de Rivero Vaccari JP, Lotocki G, Alonso OF, Bramlett HM, Dietrich WD, Keane RW. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J Cereb Blood Flow Metab. 2009;29:1251–1261. doi: 10.1038/jcbfm.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Abulafia DP, de Rivero Vaccari JP, Lozano JD, Lotocki G, Keane RW, Dietrich WD. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J Cereb Blood Flow Metab. 2009;29:534–544. doi: 10.1038/jcbfm.2008.143. [DOI] [PubMed] [Google Scholar]
- 86.Kanneganti TD, Lamkanfi M, Kim YG, Chen G, Park JH, Franchi L, et al. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity. 2007;26:433–443. doi: 10.1016/j.immuni.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 87.Clarke TC, Williams OJ, Martin PE, Evans WH. ATP release by cardiac myocytes in a simulated ischaemia model: inhibition by a connexin mimetic and enhancement by an antiarrhythmic peptide. Eur J Pharmacol. 2009;605:9–14. doi: 10.1016/j.ejphar.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 88.Seminario-Vidal L, Kreda S, Jones L, O’Neal W, Trejo J, Boucher RC, et al. Thrombin promotes release of ATP from lung epithelial cells through coordinated activation of rho-and Ca2+-dependent signaling pathways. J Biol Chem. 2009;284:20638–20648. doi: 10.1074/jbc.M109.004762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ingber DE. From cellular mechanotransduction to biologically inspired engineering: 2009 Pritzker Award Lecture, BMES Annual Meeting October 10, 2009. Ann Biomed Eng. 2010;38:1148–1161. doi: 10.1007/s10439-010-9946-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Burnstock G. Release of vasoactive substances from endothelial cells by shear stress and purinergic mechanosensory transduction. J Anat. 1999;194:335–342. doi: 10.1046/j.1469-7580.1999.19430335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sadananda P, Shang F, Liu L, Mansfield KJ, Burcher E. Release of ATP from rat urinary bladder mucosa: role of acid, vanilloids and stretch. Br J Pharmacol. 2009;158:1655–1662. doi: 10.1111/j.1476-5381.2009.00431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Winters SL, Davis CW, Boucher RC. Mechanosensitivity of mouse tracheal ciliary beat frequency: roles for Ca2+, purinergic signaling, tonicity, and viscosity. Am J Physiol Cell Physiol. 2007;292:L614–L624. doi: 10.1152/ajplung.00288.2005. [DOI] [PubMed] [Google Scholar]
- 93.Wheeler-Schilling TH, Marquordt K, Kohler K, Guenther E, Jabs R. Identification of purinergic receptors in retinal ganglion cells. Brain Res Mol Brain Res. 2001;92:177–180. doi: 10.1016/s0169-328x(01)00160-7. [DOI] [PubMed] [Google Scholar]
- 94.Zhang X, Li A, Ge J, Reigada D, Laties AM, Mitchell CH. Acute increase of intraocular pressure releases ATP into the anterior chamber. Exp Eye Res. 2007;85:637–643. doi: 10.1016/j.exer.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 95.Li A, Zhang X, Zheng D, Ge J, Laties AM, Mitchell CH. Sustained elevation of extracellular ATP in aqueous humor from humans with primary chronic angle-closure glaucoma. Exp Eye Res. 2011;93:528–533. doi: 10.1016/j.exer.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Reigada D, Lu W, Zhang M, Mitchell CH. Elevated pressure triggers a physiological release of ATP from the retina: possible role for pannexin hemichannels. Neurosci. 2008;157:396–404. doi: 10.1016/j.neuroscience.2008.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lu W, Rasmussen C, Gabelt B, Hennes B, Kaufman P, Laties AM, et al. Upregulation of NTPDase 1 in an experimental monkey glaucoma model. Invest Ophthalmol Vis Sci. 2007;48:4804. (Abstract) [Google Scholar]
- 98.Lu W, Hu H, Laties AM, Sevigney J, Mitchell CH. Upregulation of Retinal NTPDase 1 and vitreal ATP levels in an experimental rat glaucoma model. Invest Ophthalmol Vis Sci. 2008;49 ARVO E abstract:869. [Google Scholar]
- 99.Gulbransen BD, Bashashati M, Hirota SA, Gui X, Roberts JA, MacDonald JA, et al. Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat Med. 2012;18:600–604. doi: 10.1038/nm.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Clark AK, Staniland AA, Marchand F, Kaan TK, McMahon SB, Malcangio M. P2X7-dependent release of interleukin-1beta and nociception in the spinal cord following lipopolysaccharide. J Neurosci. 2010;30:573–582. doi: 10.1523/JNEUROSCI.3295-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang X, Zhang M, Laties AM, Mitchell CH. Stimulation of P2X7 receptors elevates Ca2+ and kills retinal ganglion cells. Invest Ophthalmol Vis Sci. 2005;46:2183–2191. doi: 10.1167/iovs.05-0052. [DOI] [PubMed] [Google Scholar]
- 102.Hu H, Lu W, Zhang M, Zhang X, Argall AJ, Patel S, et al. Stimulation of the P2X7 receptor kills rat retinal ganglion cells in vivo. Exp Eye Res. 2010;91:425–432. doi: 10.1016/j.exer.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mitchell CH, Lu W. Retinal ganglion cells and glaucoma: traditional patterns and new possibilities. Curr Topics Membr. 2008;62:301–322. [Google Scholar]
- 104.Mitchell CH, Lu W, Hu H, Zhang X, Reigada D, Zhang M. The P2X(7) receptor in retinal ganglion cells: a neuronal model of pressure-induced damage and protection by a shifting purinergic balance. Purinergic Signal. 2009;5:241–249. doi: 10.1007/s11302-009-9142-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Newman EA. Propagation of intercellular calcium waves in retinal astrocytes and Muller cells. J Neurosci. 2001;21:2215–2223. doi: 10.1523/JNEUROSCI.21-07-02215.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Newman EA. Glial cell inhibition of neurons by release of ATP. J Neurosci. 2003;23:1659–1666. doi: 10.1523/JNEUROSCI.23-05-01659.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82:1013–1067. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- 108.Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bunse S, Locovei S, Schmidt M, Qiu F, Zoidl G, Dahl G, et al. The potassium channel subunit Kvbeta3 interacts with pannexin 1 and attenuates its sensitivity to changes in redox potentials. Febs J. 2009;276:6258–6270. doi: 10.1111/j.1742-4658.2009.07334.x. [DOI] [PubMed] [Google Scholar]
- 110.Dubyak GR. Both sides now: multiple interactions of ATP with pannexin-1 hemichannels. Focus on “A permeant regulating its permeation pore: inhibition of pannexin 1 channels by ATP”. Am J Physiol Cell Physiol. 2009;296:C235–C241. doi: 10.1152/ajpcell.00639.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bao L, Locovei S, Dahl G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Letts. 2004;572:65–68. doi: 10.1016/j.febslet.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 112.Seminario-Vidal L, Okada SF, Sesma JI, Kreda SM, van Heusden CA, Zhu Y, et al. Rho signaling regulates pannexin 1-mediated ATP release from airway epithelia. J Biol Chem. 2011;286:26277–26286. doi: 10.1074/jbc.M111.260562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xia J, Lim JC, Lu W, Beckel JM, Macarak EJ, Laties AM, et al. Neurons respond directly to mechanical deformation with pannexin-mediated ATP release and autostimulation of P2X7 receptors. J Physiol. 2012;590.10:2285–2304. doi: 10.1113/jphysiol.2012.227983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dvoriantchikova G, Ivanov D, Panchin Y, Shestopalov VI. Expression of pannexin family of proteins in the retina. FEBS Letts. 2006;580:2178–2182. doi: 10.1016/j.febslet.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 115.Franke H, Klimke K, Brinckmann U, Grosche J, Francke M, Sperlagh B, et al. P2X(7) receptor-mRNA and -protein in the mouse retina; changes during retinal degeneration in BALBCrds mice. Neurochem Int. 2005;47:235–242. doi: 10.1016/j.neuint.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 116.Vessey KA, Fletcher EL. Rod and cone pathway signalling is altered in the P2X7 receptor knock out mouse. Plos One. 2012;7:290–305. doi: 10.1371/journal.pone.0029990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yang H, Thompson H, Roberts MD, Sigal IA, Downs JC, Burgoyne CF. Deformation of the early glaucomatous monkey optic nerve head connective tissue after acute IOP elevation in 3-D histomorphometric reconstructions. Invest Ophthalmol Vis Sci. 2011;52:345–363. doi: 10.1167/iovs.09-5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vanden Abeele F, Bidaux G, Gordienko D, Beck B, Panchin YV, Baranova AV, et al. Functional implications of calcium permeability of the channel formed by pannexin 1. J Cell Biol. 2006;174:535–546. doi: 10.1083/jcb.200601115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Locovei S, Wang J, Dahl G. Activation of pannexin 1 channels by ATP through P2Y receptors and by cytoplasmic calcium. FEBS Lett. 2006;580:239–244. doi: 10.1016/j.febslet.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 120.Poornima V, Madhupriya M, Kootar S, Sujatha G, Kumar A, Bera AK. P2X7 receptor-pannexin 1 hemichannel association: effect of extracellular calcium on membrane permeabilization. J Mol Neurosci. 2012;46:585–594. doi: 10.1007/s12031-011-9646-8. [DOI] [PubMed] [Google Scholar]
- 121.Pelegrin P, Barroso-Gutierrez C, Surprenant A. P2X7 receptor differentially couples to distinct release pathways for IL-1beta in mouse macrophage. J Immunol. 2008;180:7147–7157. doi: 10.4049/jimmunol.180.11.7147. [DOI] [PubMed] [Google Scholar]
- 122.Silverman WR, de Rivero Vaccari JP, Locovei S, Qiu F, Carlsson SK, Scemes E, et al. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J Biol Chem. 2009;284:18143–18151. doi: 10.1074/jbc.M109.004804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Striedinger K, Petrasch-Parwez E, Zoidl G, Napirei M, Meier C, Eysel UT, et al. Loss of connexin36 increases retinal cell vulnerability to secondary cell loss. Eur J Neurosci. 2005;22:605–616. doi: 10.1111/j.1460-9568.2005.04228.x. [DOI] [PubMed] [Google Scholar]
- 124.Orellana JA, Saez PJ, Shoji KF, Schalper KA, Palacios-Prado N, Velarde V, et al. Modulation of brain hemi-channels and gap junction channels by pro-inflammatory agents and their possible role in neurodegeneration. Antioxid Redox Signal. 2009;11:369–399. doi: 10.1089/ars.2008.2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Contreras JE, Sanchez HA, Eugenin EA, Speidel D, Theis M, Willecke K, et al. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc Natl Acad Sci USA. 2002;99:495–500. doi: 10.1073/pnas.012589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bao L, Locovei S, Dahl G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 2004;572:65–68. doi: 10.1016/j.febslet.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 127.Barbe MT, Monyer H, Bruzzone R. Cell-cell communication beyond connexins: the pannexin channels. Physiology (Bethesda) 2006;21:103–114. doi: 10.1152/physiol.00048.2005. [DOI] [PubMed] [Google Scholar]
- 128.Brough D, Pelegrin P, Rothwell NJ. Pannexin-1-dependent caspase-1 activation and secretion of IL-1beta is regulated by zinc. Eur J Immunol. 2009;39:352–358. doi: 10.1002/eji.200838843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Scemes E, Spray DC. Extracellular K(+) and astrocyte signaling via connexin and pannexin channels. Neurochem Res. 2012;37:2310–2316. doi: 10.1007/s11064-012-0759-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dvoriantchikova G, Ivanov D, Barakat D, Grinberg A, Wen R, Slepak VZ, Shestopalov VI. Genetic ablation of Pannexin1 protects retinal neurons from ischemic injury. PlosOne. 2012;7:e31991. doi: 10.1371/journal.pone.0031991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bargiotas P, Krenz A, Hormuzdi SG, Ridder DA, Herb A, Barakat W, et al. Pannexins in ischemia-induced neuro-degeneration. Proc Natl Acad Sci USA. 2011;108:20772–20777. doi: 10.1073/pnas.1018262108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang L, Deng T, Sun Y, Liu K, Yang Y, Zheng X. Role for nitric oxide in permeability of hippocampal neuronal hemichannels during oxygen glucose deprivation. J Neurosci Res. 2008;86:2281–2291. doi: 10.1002/jnr.21675. [DOI] [PubMed] [Google Scholar]
- 133.Orellana JA, Hernandez DE, Ezan P, Velarde V, Bennett MV, Giaume C, et al. Hypoxia in high glucose followed by reoxygenation in normal glucose reduces the viability of cortical astrocytes through increased permeability of connexin 43 hemichannels. Glia. 2010;58:329–343. doi: 10.1002/glia.20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Domercq M, Perez-Samartin A, Aparicio D, Alberdi E, Pampliega O, Matute C. P2X7 receptors mediate ischemic damage to oligodendrocytes. Glia. 2009;58:730–740. doi: 10.1002/glia.20958. [DOI] [PubMed] [Google Scholar]
- 135.Bargiotas P, Monyer H, Schwaninger M. Hemichannels in cerebral ischemia. Curr Mol Med. 2009;9:186–194. doi: 10.2174/156652409787581646. [DOI] [PubMed] [Google Scholar]
- 136.Thompson RJ, Zhou N, MacVicar BA. Ischemia opens neuronal gap junction hemichannels. Science. 2006;312:924–927. doi: 10.1126/science.1126241. [DOI] [PubMed] [Google Scholar]
- 137.Pelegrin P, Surprenant A. The P2X(7) receptor-pannexin connection to dye uptake and IL-1beta release. Purinergic Signal. 2009;5:129–137. doi: 10.1007/s11302-009-9141-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Locovei S, Scemes E, Qiu F, Spray DC, Dahl G. Pannexin1 is part of the pore forming unit of the P2X(7) receptor death complex. FEBS Lett. 2007;581:483–488. doi: 10.1016/j.febslet.2006.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Iglesias R, Locovei S, Roque A, Alberto AP, Dahl G, Spray DC, et al. P2X7 receptor-Pannexin1 complex: pharmacology and signaling. Am J Physiol Cell Physiol. 2008;295:C752–C760. doi: 10.1152/ajpcell.00228.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Orellana JA, Froger N, Ezan P, Jiang JX, Bennett MV, Naus CC, et al. ATP and glutamate released via astroglial connexin43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J Neurochem. 2011;118:826–840. doi: 10.1111/j.1471-4159.2011.07210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Pelegrin P, Surprenant A. Pannexin-1 couples to maitotoxin- and nigericin-induced interleukin-1beta release through a dye uptake-independent pathway. J Biol Chem. 2007;282:2386–2394. doi: 10.1074/jbc.M610351200. [DOI] [PubMed] [Google Scholar]
- 142.Ziegler G, Harhausen D, Schepers C, Hoffmann O, Rohr C, Prinz V, et al. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem Biophys Res Commun. 2007;359:574–579. doi: 10.1016/j.bbrc.2007.05.157. [DOI] [PubMed] [Google Scholar]
- 143.Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci USA. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shibuya E, Meguro A, Ota M, Kashiwagi K, Mabuchi F, Iijima H, et al. Association of Toll-like receptor 4 gene polymorphisms with normal tension glaucoma. Invest Ophthalmol Vis Sci. 2008;49:4453–4457. doi: 10.1167/iovs.07-1575. [DOI] [PubMed] [Google Scholar]
- 145.Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 146.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 147.Zhang Z, Trautmann K, Schluesener HJ. Microglia activation in rat spinal cord by systemic injection of TLR3 and TLR7/8 agonists. J Neuroimmunol. 2005;164:154–160. doi: 10.1016/j.jneuroim.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 148.Shiose S, Chen Y, Okano K, Roy S, Kohno H, Tang J, et al. Toll-like receptor 3 is required for development of retinopathy caused by impaired all-trans-retinal clearance in mice. J Biol Chem. 2011;286:15543–15555. doi: 10.1074/jbc.M111.228551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Luo C, Yang X, Kain AD, Powell DW, Kuehn MH, Tezel G. Glaucomatous tissue stress and the regulation of immune response through glial Toll-like receptor signaling. Invest Ophthalmol Vis Sci. 2010;51:5697–5707. doi: 10.1167/iovs.10-5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ayna G, Krysko DV, Kaczmarek A, Petrovski G, Vandenabeele P, Fesus L. ATP release from dying autophagic cells and their phagocytosis are crucial for inflammasome activation in macrophages. PLoS One. 2012;7:e40069. doi: 10.1371/journal.pone.0040069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Riteau N, Gasse P, Fauconnier L, Gombault A, Couegnat M, Fick L, et al. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am J Respir Crit Care Med. 2010;182:774–783. doi: 10.1164/rccm.201003-0359OC. [DOI] [PubMed] [Google Scholar]
- 152.Pelegrin P. Targeting interleukin-1 signaling in chronic inflammation: focus on P2X(7) receptor and Pannexin-1. Drug News Perspect. 2008;21:424–433. doi: 10.1358/dnp.2008.21.8.1265800. [DOI] [PubMed] [Google Scholar]
- 153.de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW. A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci. 2008;28:3404–3414. doi: 10.1523/JNEUROSCI.0157-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tezel G, Yang X, Yang J, Wax MB. Role of tumor necrosis factor receptor-1 in the death of retinal ganglion cells following optic nerve crush injury in mice. Brain Res. 2004;996:202–212. doi: 10.1016/j.brainres.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 155.Nakazawa T, Nakazawa C, Matsubara A, Noda K, Hisatomi T, She H, et al. Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J Neurosci. 2006;26:12633–12641. doi: 10.1523/JNEUROSCI.2801-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hyakkoku K, Hamanaka J, Tsuruma K, Shimazawa M, Tanaka H, Uematsu S, et al. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience. 2010;171:258–267. doi: 10.1016/j.neuroscience.2010.08.054. [DOI] [PubMed] [Google Scholar]
- 157.Dvoriantchikova G, Barakat DJ, Hernandez E, Shestopalov VI, Ivanov D. Toll-like receptor 4 contributes to retinal ischemia/reperfusion injury. Mol Vis. 2010;16:1907–1912. [PMC free article] [PubMed] [Google Scholar]
- 158.Barakat DJ, Dvoriantchikova G, Ivanov D, Shestopalov VI. Astroglial NF-kappaB mediates oxidative stress by regulation of NADPH oxidase in a model of retinal ischemia reperfusion injury. J Neurochem. 2012;120:586–597. doi: 10.1111/j.1471-4159.2011.07595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Murphy N, Cowley TR, Richardson JC, Virley D, Upton N, Walter D, Lynch MA. The Neuroprotective effect of a specific P2X(7) receptor antagonist derives from its ability to inhibit assembly of the NLRP3 inflammasome in glial cells. Brain Pathol. 2011;22:295–306. doi: 10.1111/j.1750-3639.2011.00531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tezel G, Wax MB. Increased production of tumor necrosis factor-alpha by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J Neurosci. 2000;20:8693–8700. doi: 10.1523/JNEUROSCI.20-23-08693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yang X, Luo C, Cai J, Powell DW, Yu D, Kuehn MH, et al. Neurodegenerative and inflammatory pathway components linked to TNF-alpha/TNFR1 signaling in the glaucomatous human retina. Invest Ophthalmol Vis Sci. 2011;52:8442–8454. doi: 10.1167/iovs.11-8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tezel G, Yang X, Luo C, Cai J, Powell DW. An astrocyte-specific proteomic approach to inflammatory responses in experimental rat glaucoma. Invest Ophthalmol Vis Sci. 2012;53:4220–4233. doi: 10.1167/iovs.11-9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cabal-Hierro L, Lazo PS. Signal transduction by tumor necrosis factor receptors. Cell Signal. 2012;24:1297–1305. doi: 10.1016/j.cellsig.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 164.Blander JM, Sander LE. Beyond pattern recognition: five immune checkpoints for scaling the microbial threat. Nat Rev Immunol. 2012;12:215–225. doi: 10.1038/nri3167. [DOI] [PubMed] [Google Scholar]
- 165.Ogura Y, Sutterwala FS, Flavell RA. The inflammasome: first line of the immune response to cell stress. Cell. 2006;126:659–662. doi: 10.1016/j.cell.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 166.Creagh EM, O’Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006;27:352–357. doi: 10.1016/j.it.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 167.Harari OA, Liao JK. NF-kappaB and innate immunity in ischemic stroke. Ann N Y Acad Sci. 2010;1207:32–40. doi: 10.1111/j.1749-6632.2010.05735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Takahashi Y, Katai N, Murata T, Taniguchi SI, Hayashi T. Development of spontaneous optic neuropathy in NF-kappaBetap50-deficient mice: requirement for NF-kappaBetap50 in ganglion cell survival. Neuropathol Appl Neurobiol. 2007;33:692–705. doi: 10.1111/j.1365-2990.2007.00862.x. [DOI] [PubMed] [Google Scholar]
- 169.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 170.Hanamsagar R, Hanke ML, Kielian T. Toll-like receptor (TLR) and inflammasome actions in the central nervous system. Trends Immunol. 2012;33:333–342. doi: 10.1016/j.it.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tezel G. Immune regulation toward immunomodulation for neuroprotection in glaucoma. Curr Opin Pharmacol. 2013;13:23–31. doi: 10.1016/j.coph.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Baudouin SJ, Angibaud J, Loussouarn G, Bonnamain V, Matsuura A, Kinebuchi M, et al. The signaling adaptor protein CD3zeta is a negative regulator of dendrite development in young neurons. Mol Biol Cell. 2008;19:2444–2456. doi: 10.1091/mbc.E07-09-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Corriveau RA, Huh GS, Shatz CJ. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron. 1998;21:505–520. doi: 10.1016/s0896-6273(00)80562-0. [DOI] [PubMed] [Google Scholar]
- 174.Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000;290:2155–2159. doi: 10.1126/science.290.5499.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ishii T, Hirota J, Mombaerts P. Combinatorial coexpression of neural and immune multigene families in mouse vomeronasal sensory neurons. Curr Biol. 2003;13:394–400. doi: 10.1016/s0960-9822(03)00092-7. [DOI] [PubMed] [Google Scholar]
- 176.Syken J, Shatz CJ. Expression of T cell receptor beta locus in central nervous system neurons. Proc Natl Acad Sci USA. 2003;100:13048–13053. doi: 10.1073/pnas.1735415100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Syken J, Grandpre T, Kanold PO, Shatz CJ. PirB restricts ocular-dominance plasticity in visual cortex. Science. 2006;313:1795–1800. doi: 10.1126/science.1128232. [DOI] [PubMed] [Google Scholar]
- 178.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 180.Wu ZP, Washburn L, Bilousova TV, Boudzinskaia M, Escande-Beillard N, Querubin J, et al. Enhanced neuronal expression of major histocompatibility complex class I leads to aberrations in neurodevelopment and neurorepair. J Neuroimmunol. 2011;232:8–16. doi: 10.1016/j.jneuroim.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Joseph MS, Bilousova T, Zdunowski S, Wu ZP, Middleton B, Boudzinskaia M, et al. Transgenic mice with enhanced neuronal major histocompatibility complex class I expression recover locomotor function better after spinal cord injury. J Neurosci Res. 2011;89:365–372. doi: 10.1002/jnr.22557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Boulanger LM, Huh GS, Shatz CJ. Neuronal plasticity and cellular immunity: shared molecular mechanisms. Curr Opin Neurobiol. 2001;11:568–578. doi: 10.1016/s0959-4388(00)00251-8. [DOI] [PubMed] [Google Scholar]
- 183.Fourgeaud L, Boulanger LM. Synapse remodeling, compliments of the complement system. Cell. 2007;131:1034–1036. doi: 10.1016/j.cell.2007.11.031. [DOI] [PubMed] [Google Scholar]
- 184.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kiefer F, Vogel WF, Arnold R. Signal transduction and co-stimulatory pathways. Transpl Immunol. 2002;9:69–82. doi: 10.1016/s0966-3274(02)00009-6. [DOI] [PubMed] [Google Scholar]
- 186.Baniyash M. TCR zeta-chain downregulation: curtailing an excessive inflammatory immune response. Nat Rev Immunol. 2004;4:675–687. doi: 10.1038/nri1434. [DOI] [PubMed] [Google Scholar]
- 187.Shatz CJ. MHC class I: an unexpected role in neuronal plasticity. Neuron. 2009;64:40–45. doi: 10.1016/j.neuron.2009.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bosco A, Crish SD, Steele MR, Romero CO, Inman DM, Horner PJ, et al. Early reduction of microglia activation by irradiation in a model of chronic glaucoma. Plos One. 2012;7:e43602. doi: 10.1371/journal.pone.0043602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Bosco A, Inman DM, Steele MR, Wu G, Soto I, Marsh-Armstrong N, et al. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2008;49:1437–1446. doi: 10.1167/iovs.07-1337. [DOI] [PubMed] [Google Scholar]
- 190.Bosco A, Steele MR, Vetter ML. Early microglia activation in a mouse model of chronic glaucoma. J Comp Neurol. 2011;519:599–620. doi: 10.1002/cne.22516. [DOI] [PMC free article] [PubMed] [Google Scholar]