

Abstract
Neurodegenerative diseases such as Alzheimer's (AD) are characterized by an abnormal aggregation of misfolded beta-sheet rich proteins such as β-amyloid (Aβ). Various ubiquitously-expressed molecular chaperones control the correct folding of cellular proteins and prevent the accumulation of harmful species. We here describe a novel anti-aggregant chaperone function for the neuroendocrine protein proSAAS, an abundant secretory polypeptide that is widely expressed within neural and endocrine tissues and which has previously been associated with neurodegenerative disease in various proteomics studies. In the brains of 12-month old APdE9 mice, and in the cortex of a human AD-affected brain, proSAAS immunoreactivity was highly colocalized with amyloid pathology. Immunoreactive proSAAS co-immunoprecipitated with Aβ immunoreactivity in lysates from APdE9 mouse brains. In vitro, proSAAS efficiently prevented the fibrillation of Aβ1-42 at molar ratios of 1:10, and this anti-aggregation effect was dose-dependent. Structure-function studies showed that residues 97-180 were sufficient for the anti-aggregation function against Aβ. Finally, inclusion of recombinant proSAAS in the medium of Neuro2a cells, as well as lentiviral-mediated proSAAS overexpression, blocked the neurocytotoxic effect of Aβ1-42 in Neuro2a cells. Taken together, our results suggest that proSAAS may play a role in Alzheimer's disease pathology.
Keywords: proSAAS, protein aggregation, Alzheimer's disease, Parkinson's disease, chaperone
Introduction
Many neurodegenerative diseases share the common feature of abnormal protein aggregates in regions of neuronal loss and dysfunction. For example, Alzheimer's disease (AD) (the leading cause of dementia) is characterized by neuronal loss in the hippocampus and in the cortex, in close proximity to extracellular plaques consisting of aberrantly aggregated Aβ1-42 peptides and intracellular neurofibrillary tangles composed of hyperphosphorylated tau. Identifying drugs or molecular chaperones which block the aggregation of Aβ1-42, tau, or α-synuclein may represent a viable strategy for slowing down the progression of neurodegenerative diseases.
The secretory protein proSAAS is expressed in neurons throughout the brain at high levels (Lanoue & Day 2001),(Morgan et al. 2005). It is partially proteolytically processed within the regulated secretory pathway (Sayah et al. 2001), where it has been well-characterized as a potent and specific inhibitor of prohormone convertase 1/3 (PC1/3) (Fricker et al. 2000), (Qian et al. 2000), (Cameron et al. 2000). However, proSAAS is also expressed in many non-PC1/3-expressing cells, raising the possibility of additional functions (Feng et al. 2001, Lanoue & Day 2001). Indeed, recent studies have now shown that various proSAAS-derived peptides participate in a number of physiologically important systems, including circadian rhythm (Atkins et al. 2010, Hatcher et al. 2008), food intake (Wardman et al. 2011), energy balance (Morgan et al. 2010), and fetal neuropeptide processing (Morgan et al. 2010). Furthermore, the expression of PC1/3 and proSAAS is not always co-regulated. Although proSAAS acts as an endogenous inhibitor of PC1/3, long-term treatment of AtT-20 cells with secretagogues increases PC1/3 mRNA levels without affecting proSAAS mRNA (Mzhavia et al. 2002). These differences between the expression and regulation of PC1/3 and proSAAS support the hypothesis that proSAAS may have functions unrelated to PC1/3.
Interestingly, in the decade since its discovery, proSAAS has been repeatedly implicated in various neurodegenerative diseases. ProSAAS immunoreactivity has been found in neurofibrillary tangles and neuritic plaques of brain tissues from patients with AD, parkinsonism-dementia complex, and Pick's disease, implying a possible involvement of proSAAS in the pathophysiology of general tauopathies (Kikuchi et al. 2003, Wada et al. 2004). In addition, four independent proteomic studies have identified proSAAS as a candidate biomarker in both AD and frontotemporal dementia, with significant reduction in the levels of proSAAS-derived peptides in patient cerebrospinal fluid (CSF) (Abdi et al. 2006, Jahn et al. 2011, Davidsson et al. 2002, Finehout et al. 2007). Finally, CSF proSAAS levels are reduced in patients with a spinal nerve root injury from lumbar disk herniation (Liu et al. 2006).
7B2, a small secretory protein that serves as a convertase binding protein (Braks & Martens 1994), has also been reported as a possible protein chaperone (Helwig et al. 2012). Like proSAAS, 7B2 is found in neurons lacking convertase expression, suggesting alternative functions. Indeed, others have shown that 7B2 blocks the aggregation of several unrelated secretory proteins, including insulin-like growth factor 1 (Chaudhuri et al. 1995); proPC2 (Lee & Lindberg 2008); Aβ1-42; and α-synuclein (Helwig et al. 2012). Based on these studies, and the structural similarity of proSAAS to 7B2, we hypothesized that proSAAS might function as an anti-aggregant chaperone in AD. In the study presented here, we have used mouse models of AD, as well as human post-mortem tissues of AD patients, to show that proSAAS co-localizes with proteins involved in AD. Further, we have used in vitro aggregation assays to demonstrate a potential function for proSAAS as an anti-aggregant, and neurotoxicity assays to show effects of endogenous as well as exogenous proSAAS in the blockade of Aβ1-42-mediated neurotoxicity.
Materials and Methods
Immunofluorescent labeling of human brain tissues for proSAAS and AD markers
A hippocampal tissue sample from a 73-year old donor with AD was obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland-Baltimore, MD. The tissue was formalin-fixed, cryoembedded and sectioned at 16 μm. For immunohistochemistry, tissue sections were blocked for 1 h in blocking solution (phosphate-buffered saline; PBS) containing 3% bovine serum albumin (BSA) and 0.5% Triton X-100 before incubation with rabbit anti-proSAAS (LS45, 1:50) and monoclonal mouse antibody raised against Aβ17-26 (clone 4G8, 1:1000, Cell Sciences, Canton, MA) in blocking solution overnight at 4 C. The proSAAS antiserum was raised in rabbits against recombinant His-tagged 21 kDa proSAAS (Fortenberry et al. 2002) and has previously been used to image proSAAS in pancreatic tissues (Guest et al. 2002). Sections were rinsed, incubated with Cy3-conjugated goat anti-rabbit (1:200, A10520, Invitrogen, Carlsbad, CA) and/or Cy2-conjugated donkey anti-mouse (1:250, AP124J, Millipore, Billercia, MA) in blocking solution containing Hoechst 33342 (1:10,000, ALX-620-050, Axxora LLC, San Diego, CA) for 2 h at room temperature. Slides were rinsed in PBS, coverslipped with Fluoromount G (Electron Microscopy Sciences, Hatfield, PA) and visualized using a confocal Olympus BX61 (Olympus, Tokyo, Japan) and an epifluorescence Nikon Eclipse TE2000-E microscope (Nikon, Tokyo, Japan). Images were merged using processing software (Olympus FluoView, Nikon MetaView). Anatomical localization of immunoreactivity within the brain was annotated according to the Allen Human Brain Atlas and Gray's Anatomy of the Human Body (30th edition).
Animal Models
For the examination of amyloid plaques, 12-month old male APP695/PSEN1dE9 mice (APdE9; B6C3-Tg(APPswe,PSEN1dE9)85Dbo/Mmjax; Jackson Laboratory) were sacrificed and their brains were fixed with Accustain (Sigma Aldrich, St. Louis, MO) and subjected to paraffin processing. All studies were conducted under the approval of Institutional Animal Care and Use Committee (IACUC) at the University of Houston.
Immunofluorescence of mouse brain tissue for proSAAS and AD markers
Ten μm brain sections were treated with Aqua DePar and Reveal antigen retrieval solutions in a Decloaking Chamber system (Biocare Medical, Concord, CA). The sections were then incubated with avidin/biotin blocking kit (Vector Laboratories, Burlingame, CA), blocked in 5% normal goat serum in Tris-buffered saline (TBS) containing 0.5% Triton X-100 for 20 min, followed by an incubation with polyclonal rabbit anti-proSAAS (#45, 1:50) for one hour. The sections were washed, incubated with biotinylated goat anti-rabbit antibody (Vector Laboratories) for 30 min, washed, then incubated with Texas Red Avidin DCS (Vector laboratories) for 10 min, and washed again. For co-localization studies, sections were subjected to another round of re-blocking and then stained with a pan-Aβ monoclonal mouse antibody (4G8, 1:250, Covance) for amyloid pathology. The sections were washed, incubated with biotinylated goat anti-mouse antibody (Vector Laboratories) for 30 min, and washed again before incubating with Fluorescein-Avidin for 10 min. The sections were then washed extensively, mounted using Vectashield media, and viewed under a confocal microscope (Olympus IX6a DSU). The Neurolucida program was used to process the images (Microbrightfield, Inc, Willston, VT). Dense core plaques were stained with methoxy-X04 (1 μM) and then washed extensively before imaging.
Preparation of recombinant full-length and N-terminally truncated His-tagged proSAAS
A 21 kDa mouse proSAAS (1-180) plasmid was prepared as described previously (Fortenberry et al. 2002). Plasmids encoding N-terminally truncated His-tagged proSAAS fragments were generated using the pQE30 21 kDa proSAAS plasmid (Fortenberry et al. 2002) as a template. PCR was performed using the GC-Rich PCR system (Roche Applied Science, Indianapolis, IN) and the following primers: a common 3′ primer (5′-TAG GAA GCT TTT ACG GGG CAG GAG CAG CCT C-3′) and the 5′ primers (5′-CGC GCA TGC GCG CAG GAG GCT GAG GAT CAG CA-5′) for proSAAS62-180 and (5′-CGC GCA TGC GAC GCT CCA GCT GCA CAG CTC GC-3′) for proSAAS97-180. The PCR products were cloned into pQE30 (Qiagen, Valencia, CA) at the HindIII and SphI sites and constructs were verified by sequencing. Plasmids were expressed in E. coli XL-1 Blue cells (Stratagene, La Jolla, CA), induced with 1 mM (final concentration) IPTG, and protein purified using the native BugBuster method (Novagen, Gibbstown, NJ). Briefly, cells were lysed in Bug Buster Protein Extraction Reagent containing 1 mM phenylmethanesulfonylfluoride, 15 μl of Benzonase nuclease solution and 15 kU of rLysozyme (Novagen) for 20 min at room temperature, centrifuged at 16,000g for 20 min, and loaded onto a Ni-NTA resin previously equilibrated with binding buffer (0.5 M NaCl, 20 mM Tris-HCl, 5 mM imidazole, pH 7.9). The column was washed with binding buffer, followed by wash buffer (0.5 M NaCl, 20 mM Tris-HCl, 60 mM imidazole, pH 7.9), and then eluted with elution buffer (0.5 M NaCl, 20 mM Tris-HCl, 1 M imidazole, pH 7.9). Peak fractions were subjected to buffer exchange at 4 C using a Superdex 200 10/300GL gel filtration column equilibrated with 5 mM acetic acid; proteins were concentrated by lyophilization and resuspended in 5 mM acetic acid. ProSAAS138-180 and proSAAS97-137 were synthesized at more than 85% purity at the University of Maryland-Baltimore, Biopolymer Core Facility.
Peptide/protein preparation
Aβ1-42 (Biopeptide, San Diego, CA or California Peptide, Napa, CA) was treated at a final concentration of 1 mM for 1 h with the denaturant hexafluoroisopropanol (99%), lyophilized in aliquots, and stored at -80 C until use.
In vitro fibrillation assays
Aβ1-42 (22 μM) was fibrillated in 96-well plates in Tris-HCl buffer, pH 7.4, in the presence or absence of a) full-length and N-terminally truncated proSAAS; b) proSAAS sequences from different species; or c) either BSA or carbonic anhydrase as negative controls, at the final concentrations indicated in the figures. The plates were incubated at 37 C and agitated (setting 30) in the presence of 10 μM thioflavin T (ThT). Fibrillation was measured as an increase in ThT fluorescence (Ex 444 nm, Em 485 nm) upon binding to fibrils, measured at the times indicated. The data were then normalized: 0% was set as the lowest ThT fluorescence value for each condition (time 0) and 100% was set as the highest ThT fluorescence value for the assay.
Electron microscopy
Aβ1-42 (22 μM) was fibrillated in the presence or absence of 21 kDa proSAAS (2 μM) as described above but lacking ThT. After 72 h, the vehicle-treated sample (diluted 1000 times with water) and proSAAS-treated Aβ1-42 samples (undiluted) were adsorbed onto Formvar-coated 400 mesh copper grids and negatively stained with 2% phosphotungstic acid, pH 7. Excess solution was wicked off and the grids were examined under a transmission electron microscope (TEM) (Tecnai T12, FEI) operated at 80 kV. Images were acquired using an AMT bottom mount CCD camera and AMT600 software, at 30,000–52,000x magnification.
Dot blot analysis
Aβ1-42 was fibrillated as described above in the presence or absence of 21 kDa proSAAS. After 48 h, one-third of each reaction was reserved (“total”), and the remainder of the samples was centrifuged for 30 min at 20,000g at 4 C. An appropriate volume of PBS was added to the supernatant and pellet samples to bring all of the final volumes to 100 μl. Ten μl aliquots of the reactions were then spotted onto a 0.2 μM nitrocellulose membrane (Bio-Rad, Hercules, CA), air-dried, and blocked for 30 min in TBS containing 0.5% BSA, 0.2% goat serum, 0.3% Triton X-100 before incubating with monoclonal anti-β-amyloid antibody (6E10, 1:1000, Covance, Princeton, NJ) overnight at 4 C. The following day, the membrane was washed and then incubated with anti-mouse antiserum conjugated to horseradish peroxidase for 1.5 h. The membranes were washed before developing with a chemiluminescent substrate (Supersignal West Pico; Pierce, Rockford, IL) on HyBlot CL autoradiography film (Denville Scientific, Inc.). Supernatant:pellet dot intensity ratios were calculated using Image J software (NIH, Bethesda, MD).
Cell culture
Neuro2a cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and were maintained in DMEM high glucose: Opti-MEM (1:1) medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Invitrogen) at 37 C in a humidified atmosphere containing 5% CO2.
Aβ1-42 oligomer preparation
Dried Aβ1-42 peptide films were resuspended in DMSO at a concentration of 5 mM, sonicated for 10 min in a water bath sonicator, and diluted to a final concentration of 100 μM with phenol red-free Ham's F12 (Biosource, CA). The aliquots were then briefly vortexed, and centrifuged before incubation at 4 C for 24 h to form Aβ1-42 oligomers (Stine et al. 2011).
Co-immunoprecipitation
12-month old APP695/PSdE9 mouse brain was homogenized in TPER (Pierce Biotechnology) solution with HALT Protease Inhibitor cocktail (Pierce Biotechnology). Samples were normalized to 2 μg/μl protein in TPER and HALT, cleared by centrifugation for 10 min at 14,000g to eliminate insoluble material, and the supernatant was aliquoted and stored at -80 C prior to use. For co-immunoprecipitation studies, aliquots were thawed on ice at 4 C, briefly vortexed, and then centrifuged for 10 min at 14,000g. Fifty 슰 l of Protein G Dynabeads (Invitrogen) were washed with PBS containing 0.02% Tween-20 and then bound either to monoclonal anti-GAPDH (10 μg; Invitrogen) or to anti-Abeta 6E10 (10 μg; Covance) and washed again. The Protein G Dynabeads were then resuspended in 200 μl PBS containing 0.02% Tween-20, and 75 μg of protein lysate were then added to the suspension. Samples were incubated for 30 min at room temperature with constant rotation. Following incubation, samples were placed on a magnet to allow removal of the supernatant. Beads were washed three times with gentle resuspension in PBS containing 0.02% Tween-20. Bound proteins were eluted from the beads under denaturing conditions with 20 μl of 50 mM glycine, pH 2.8, and then resuspended in 2X Laemmli sample buffer (Bio-Rad) containing 5% beta-mercaptoethanol. These samples were electrophoresed on a 4-15% TGX gel (Bio-Rad), transferred to a nitrocellulose membrane, and then probed for proSAAS, followed by anti-rabbit HRP secondary at 1:10,000 (Jackson Immunoresearch, West Grove, PA). Bands were visualized using ECL Plus Western Blotting Detection Reagent (GE Healthcare).
Cytotoxicity assay
Neuro2a cells were plated in 96-well plates at a density of 5 × 103 cells/well. On the following day, cells were washed with serum-free medium (DMEM) and treated with either vehicle, Aβ1-42 oligomers (10 μM final concentration), prepared as described above, and/or 21 kDa proSAAS (at the concentrations indicated) or α-lactalbumin/carbonic anhydrase as negative controls (6 μM) for 48 h. Cell viability was measured using the WST-1 cell proliferation reagent (Roche, Mannheim) and absorption at 450 nm was measured every 30 min. The value for vehicle-treated cells was set as 100%. Cell viability was assessed by labeling the cells with calcein AM (Invitrogen), and representative images of these cells were taken using a Nikon Eclipse TE2000-E fluorescent microscope.
Lentiviral overexpression and siRNA knockdown of proSAAS in Neuro2a cells
Lentiviral particles encoding full-length proSAAS were commercially synthesized using a pReceiver-Lv105 vector system (GeneCopoeia, Rockville, MD; LP-U0612-Lv105-0200-S). Neuro2a cells were seeded at 1 × 103 cells/well into 96-well plates. The following day, cells were washed twice with PBS, and incubated with proSAAS or control lentivirus (negative control particles, LP-NEG-LV105, GeneCopoeia) diluted in 50 μl of PBS containing 8 (μg/mL Polybrene, at a multiplicity of infection of 1. After 30 min, 50 μl of high-glucose DMEM containing 2% FBS were added to each well, and cells were incubated for 36 h at 37 C. The medium was then changed to DMEM containing 10 μM Aβ1-42 for an additional 48 h.
For the siRNA knockdown experiment, three different specific sequences of stealth siRNA (Invitrogen) were designed to target the murine proSAAS mRNA sequence (MSS282573, MSS282574, MSS282575), and the most effective siRNA, MSS282573, was employed for subsequent experiments. A control scrambled sequence was designed to have the same GC content (46-2000; Invitrogen). Neuro2a cells grown in 96-well plates were transfected sequentially with 100 nM of the respective siRNA on the first day and 200 nM on the second day using Lipofectamine 2000 (Invitrogen). On the third day, the medium was changed to DMEM containing 10 μM Aβ1-42 for 48h. Cell viability was assessed using the WST-1 cell proliferation assay and proSAAS was measured by radioimmunoassay (Sayah et al. 2001).
Statistical analysis
Either the Student's unpaired t-test, or a one-way ANOVA followed by Newman-Keuls multiple comparison was used to assess significance, as indicated in each figure; p-values with a value of p<0.05 were taken as statistically significant.
Results
ProSAAS co-localizes with amyloid deposits in a human AD patient and in mouse models of AD
Previous work in our laboratory has shown that another convertase chaperone, 7B2, colocalizes with amyloid plaques (Helwig et al. 2012). We therefore decided to investigate the possibility that proSAAS, which is much more abundant in the brain than 7B2, might also colocalize with amyloid plaques. Coronal sections of the hippocampus from a human AD brain (Figure 1A-D) and a control brain (Figure 1E-H) were stained with our polyclonal antisera to 21 kDa rodent proSAAS (Figure 1B, F) (described in (Guest et al. 2002)) or a pan-Aβ monoclonal antibody raised against Aβ17-24 (Figure 1C, G). We found strong co-localization of proSAAS with extracellular Aβ deposits in the AD brain (Figure 1D). In the control brain, a similar level of proSAAS was detected, but no Aβ immunoreactivity was observed (Figure 1H).
Figure 1. proSAAS co-localizes with Aβ1-42 in human AD brain.
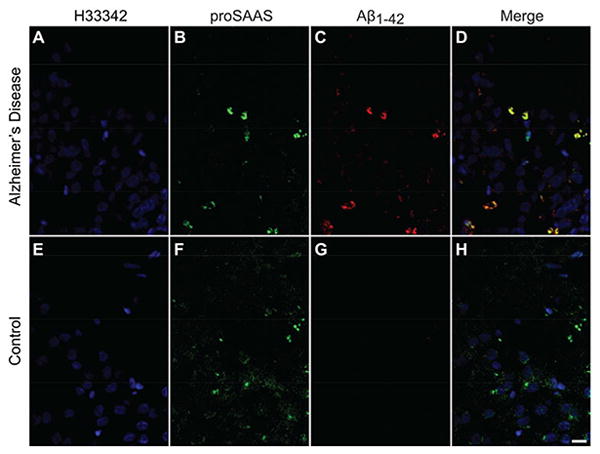
Coronal sections of human hippocampus from an AD patient (A-D) and a healthy control (E-H) were stained for proSAAS (B, F) and Aβ1-42 (C, G). Aβ1-42 staining was only observed in the hippocampus of the AD patient. The merged images show co-localization of Aβ1-42 and proSAAS (D, H). Scale bar: 50 μm.
We also performed immunohistochemistry on hippocampal and cortical slices from 12-month-old APdE9 mice (Figure 2). This mouse AD model contains mutations in both the amyloid precursor protein (APP) and presenilin 1 genes that are known to cause familial AD (Haass et al. 1995), and result in high amyloid plaque burden in mice by 9 months of age (Jankowsky et al. 2004). Indeed, we found multiple methoxy-X04 positive plaques (Figure 2B) in our sections, and they were highly co-localized with proSAAS (Figure 2A, C). Higher magnification images depict examples of plaques in the hippocampus (Figure 2D-G) and cortex (Figure 2H-K); in both tissues there is clear co-localization of β-amyloid and proSAAS immunoreactivity. Interestingly, proSAAS and amyloid co-localization occurred in both dense core plaques (4G8+/methoxy-X04+) and in diffuse plaques (only 4G8+). One example of proSAAS immunoreactivity in diffuse cortical plaques is shown in panel H, where proSAAS and 4G8 co-localization is clear; however, there is a paucity of methoxy-X04 staining (denoted by the arrowhead). Taken together, these results suggest a possible physiological role for proSAAS in β-amyloid pathology in AD.
Figure 2. proSAAS co-localizes with Aβ1-42 and methoxy-X04 positive plaques in the hippocampus and cortex of aged APdE9 mice.
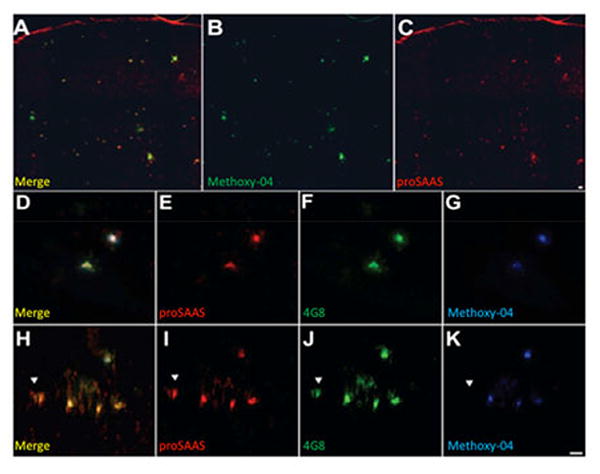
Tissues from 12-month old APdE9 mice were stained for proSAAS, Aβ1-42, and methoxy-X04 for dense core plaques. A. Merged image of methoxy-X04 stain (B) and proSAAS (C). D-G are images of hippocampal slices stained with antisera to proSAAS (E), β- amyloid (4G8) (F), or methoxy-X04 (G). Cortical sections were also stained with proSAAS (I), beta-amyloid (4G8) (J), or methoxy-X04 (K). The left panels (D, H) are merged images showing co-localization of proSAAS immunoreactivity with both diffuse and dense core plaques. Scale bar: 100 μm.
ProSAAS co-immunoprecipitates with Aβ
To determine whether proSAAS directly interacts with β-amyloid in vivo, a co-immunoprecipitation experiment was conducted. β-amyloid was immunoprecipitated from an aged (12-month old) APP/PSdE9 mouse brain lysate using the 6E10 Aβ antiserum. The immunoprecipitates were then blotted with antiserum directed against proSAAS, raised in rabbits against recombinant His-tagged 21 kDa mouse proSAAS (Fortenberry et al. 2002). Controls included the use of GAPDH IgG rather than Abeta IgG; and beads lacking IgG. The results (Figure 3) clearly indicate the presence of an immunoreactive band consistent with that of proSAAS 1-180 (Fortenberry et al. 2002).
Figure 3. ProSAAS co-immunoprecipitates with Aβ.
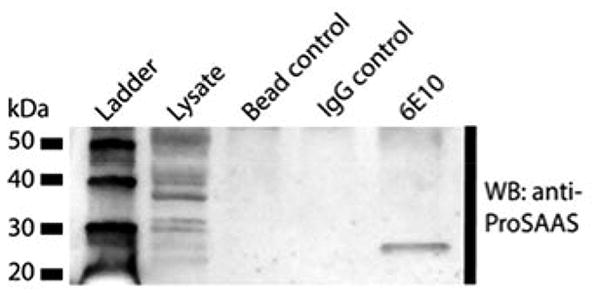
Aβ was immunoprecipitated from mouse brain lysates with 6E10 Aβ antibody and immunoprecipitated proteins were then blotted for immunoreactive proSAAS. Immunoprecipitation with the 6E10 Aβ antibody resulted in a single proSAAS-ir co-precipitating band with a molecular mass corresponding to that of proSAAS 1-180, while control immunoprecipitations using beads only or GAPDH IgG showed no detectable bound proSAAS-ir. Data are representative of three independent experiments.
proSAAS prevents fibrillation of Aβ1-42 in vitro
Aggregates of misfolded proteins found in brains of patients afflicted with neurodegenerative diseases often consist of detergent-insoluble, β-sheet rich fibrils that bind to dyes such as ThT. We therefore tested whether proSAAS could block the formation of ThT-binding Aβ1-42 fibrils. We found that the addition of recombinant 21 kDa mproSAAS (N-terminal domain) was able to prevent Aβ1-42 fibrillation in a dose-dependent manner (Figure 4A). ProSAAS was a highly potent inhibitor of fibrillation; Aβ1-42 fibrillation was blocked by 50% even at the low molar ratio of 37:1 (Aβ1-42: proSAAS). Transmission electron microscopy (TEM) further confirmed the effect of proSAAS on Aβ1-42 fibrils: 72 h after the initiation of fibrillation, samples were processed for TEM (Figure 4B). Aβ1-42 alone formed fibrils that were on average 575 ± 302 nm (n=10, mean ± SD) in length, while the length of Aβ1-42 fibrils formed in the presence of 2 μM proSAAS was about 75% shorter, averaging 143 nm ± 113 nm (n=10, mean ± SD, p<0.0001). The TEM data support the finding that proSAAS blocks the fibrillation of Aβ1-42, and indicate that the decrease in ThT fluorescence observed in the presence of proSAAS is not an artifact due to fluorescence quenching. In addition, a dot blot analysis showed that the majority of the Aβ1-42 was insoluble (i.e. pelletable) following the fibrillation assay. Samples incubated with proSAAS had more Aβ1-42 in the supernatant and a lesser amount of pelleted Aβ1-42, suggesting that the generation of insoluble Aβ1-42 species was inhibited in the presence of proSAAS (Figure 4C). We were not able to deaggregate pre-formed fibrils with later addition of proSAAS (Figure 4D).
Figure 4. ProSAAS blocks fibrillation of Aβ1-42 in a dose- and time-dependent manner.
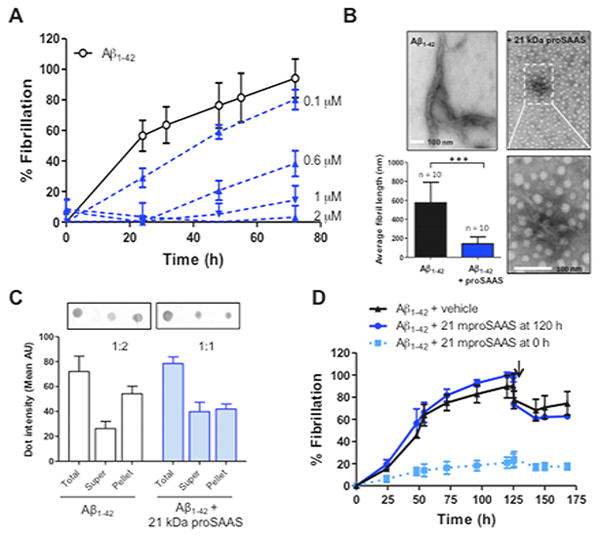
A. Aβ1-42 (22 μM) was fibrillated in the presence of increasing concentrations of 21 kDa proSAAS (0–2 μM). Each fibrillation point represents the mean ± SD, N=4. B. After 72 h, fibril formation of Aβ1-42 in the presence and absence of 21 kDa proSAAS was confirmed by TEM (upper panels). The fibrils in the sample containing Aβ1-42 and proSAAS are shown at a higher magnification (bottom right panel). In the presence of 21 kDa proSAAS, fibril length was significantly decreased (bottom left panel). Each bar represents the mean ± SD, N=10. ***=p<0.05, Student's t-test. C. Following fibrillation, samples were subjected to a dot blot analysis. Quantification of the dot intensities revealed that the distribution of Aβ1-42 shifted from the pellet (insoluble Aβ1-42) to the supernatant (soluble Aβ1-42) in the presence of 21 kDa proSAAS. D proSAAS cannot break pre-formed Aβ1-42 fibrils. Vehicle (black triangles) or 2 μM 21 kDa proSAAS (blue circles) was added to Aβ1-42 120 h after the initiation of fibrillation, as indicated by the arrow. The blue squares represent Aβ1-42 fibrillated in the presence of 21 kDa proSAAS (2 μM) for the entire duration of the assay. Each fibrillation point represents the mean ± SD, N=3.
Residues 97-180 are sufficient to block fibrillation of Aβ1-42
We next attempted to identify the region within proSAAS responsible for the anti-fibrillation effect; Figure 5A illustrates the proSAAS constructs and synthetic peptides used for this study. Secondary structure programs predict that the 21 kDa N-terminal domain of proSAAS (referred to here as #2) contains three α-helices; N-terminally truncated constructs were designed to test whether these helices played a role in anti-fibrillation. Constructs #1-4 were able to efficiently prevent fibrillation of Aβ1-42, while construct #5 (residues 138-180), representing putative α-helix III, was not able to block fibrillation (Figure 5B). We then tested construct #6 (residues 97-137) to determine whether putative α-helix II was sufficient to prevent fibrillation, or whether both α- helices II and III were required. Since construct #6 was inactive, these results suggest that both α-helices II and III are required. Further, the expression of a protein segment encoding both of the two α-helices may be necessary for proper folding because simultaneous addition of constructs #5 and #6 to Aβ1-42 had no effect on fibrillation (data not shown). Interestingly, addition of full-length proSAAS from Xenopus and zebrafish also prevented fibrillation of Aβ1-42, suggesting conservation of function despite limited sequence homology (Figure 5C). We conclude that residues 97-180 within the N-terminal domain are sufficient to function as an anti-aggregation chaperone for Aβ1-42.
Figure 5. Structure-function analysis: proSAAS residues 97-180 are sufficient to block Aβ1-42 fibrillation in vitro.
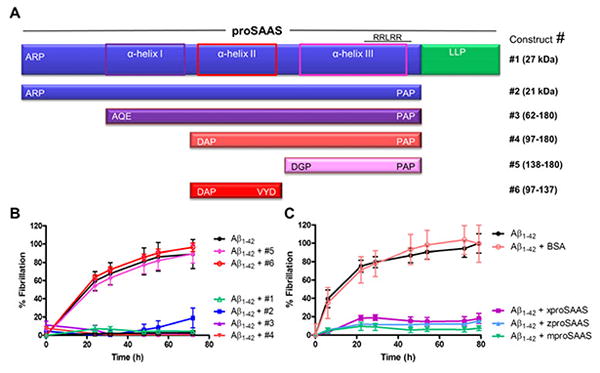
A. Domain structure of proSAAS and a schematic representation of predicted α-helical structures (in purple/pink boxes), and the N-terminally truncated proSAAS constructs. B. Aβ1-42 (22 μM) was fibrillated in the presence of vehicle or 2 μM proSAAS constructs #1–6. Constructs 1–4 were able to efficiently block fibrillation, whereas constructs #5 and #6 had no effect. C. Aβ1-42 (22 μM) was fibrillated in the presence of vehicle or full-length proSAAS sequences from 3 different species: Xenopus, zebrafish, and mouse. ProSAAS proteins from all species were able to completely block Aβ1-42 fibrillation. Aβ1-42 was also fibrillated in the presence of BSA as a negative control. Each fibrillation point represents the mean ± SD, N=4.
21 kDa proSAAS prevents Aβ1-42 mediated cytotoxicity
Aβ1-42 oligomers have been proposed to be the actual cytotoxic species, rather than fibrils, (reviewed in (Benilova et al. 2012)). Therefore, we treated Neuro2a cells with Aβ1-42 oligomers and tested whether recombinant proSAAS could exhibit cytoprotective effects. Our oligomerization protocol produced an amyloid sample containing a mixture of monomeric, tetrameric, and aggregated Aβ1-42 species (data not shown). Neuro2a cells were treated with 10 μM of these Aβ1-42 oligomers in the presence or absence of recombinant 21 kDa proSAAS for 48 h. We observed 50% cell death when Neuro2a cells were treated with Aβ1-42; however, in the presence of proSAAS, Aβ1-42-induced cytotoxicity was inhibited (Figure 6A). The effect of proSAAS was dose-dependent and reached full protection at a concentration of 3 μM. Similar concentrations of α-lactalbumin and carbonic anhydrase were also added to Aβ1-42-treated Neuro2a cells as negative controls, but were unable to block cytotoxicity. A parallel set of cells was treated with vehicle or with Aβ1-42 in the presence of vehicle, 21 kDa proSAAS, or α-lactalbumin, and stained with calcein AM; these data confirmed proSAAS-induced protection from amyloid cytotoxicity (Figure 6B). We also attempted to test whether the specific N-terminally truncated proSAAS constructs that showed anti-fibrillation effects (constructs #3 and #4) would also exhibit neuroprotection against Aβ1-42-induced cytotoxicity, but neither of these smaller constructs, nor the peptides (#5 and #6) were neuroprotective when added exogenously (data not shown.)
Figure 6. ProSAAS prevents Aβ1-42 -induced cytotoxicity in Neuro2a cells.
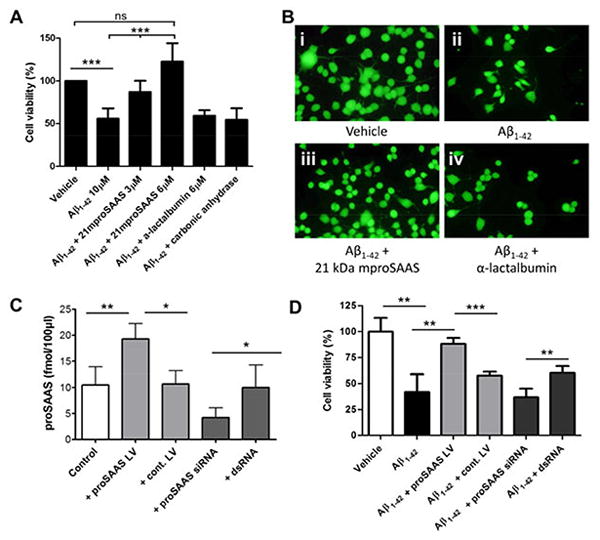
A. Neuro2a cells were incubated for 48 h with a 10 μM Aβ1-42 oligomer mixture, which resulted in about 50% cytotoxicity. The addition of 21 kDa mproSAAS to the medium during the 48 h treatment significantly prevented Aβ1-42-mediated cell death in a dose-dependent manner, as assessed by WST-1 viability assay. α-lactalbumin (6 μM) and carbonic anhydrase (6 μM) were added as negative controls in parallel sets of Aβ1-42-treated Neuro2a cells. Results represent the mean ± SD, N=4. B. Representative images of Neuro2a cells treated with vehicle (i), Aβ1-42 (ii), Aβ1-42 + 21 kDa proSAAS (3 μM) (iii), and Aβ1-42 + α-lactalbumin (3 μM) (iv), stained with calcein AM. C. In order to validate the proSAAS siRNA and lentivirus, endogenous proSAAS levels in a 6-well plate of Neuro2a cells were measured by radioimmunoassay after treatment with either proSAAS-encoding lentivirus, no lentivirus, proSAAS siRNA, or control dsRNA. Control lentivirus and control dsRNA had no effect on the endogenous level of proSAAS. D. Lentiviral-mediated overexpression of proSAAS rescued Neuro2a cells from Aβ1-42-induced cytotoxicity, as measured in the 96-well plate cytotoxicity assay. siRNA mediated knockdown of proSAAS resulted in reduced cell viability compared to Neuro2a cells treated with Aβ1-42 and a control dsRNA. One-way ANOVA or t-test was used to determine statistical significance. * = p<0.05, ** = p<0.01, and *** = p<0.001.
We then tested whether endogenously-expressed proSAAS could protect Neuro2a cells from Aβ1-42-induced cytotoxicity. We first used a lentivirus construct to overexpress proSAAS and proSAAS siRNA to knockdown endogenous proSAAS in a test experiment; the result of these manipulations of proSAAS expression is shown in Figure 6, panel C. The addition of proSAAS-encoding virus nearly doubled the cellular proSAAS levels as compared to a control irrelevant virus in this expression assay. When this same proSAAS lentivirus was used in the standard Neuro2A Aβ1-42-induced cytotoxicity test, it greatly increased the number of viable cells (p<0.001). Since the majority of secreted molecules will accumulate in the medium in this cell line, the cytoprotective effect of the proSAAS lentivirus is likely to be achieved via released proSAAS.
We achieved about a 40% drop in the level of intracellular proSAAS with siRNA (Figure 6, panel C); the effect of this reduction on secreted proSAAS levels is difficult to ascertain, and the effect on enhancement of cytotoxicity was small. Decreased expression led to increased cell death when proSAAS-siRNA-treated cells were compared to irrelevant ds-RNA-treated cells (p<0.01), but significance was not achieved when proSAAS siRNA was compared to non-RNA treated cells (Figure 6D).
Taken together, these data show that increases in both exogenously added recombinant proSAAS as well as endogenously synthesized proSAAS result in effective cytoprotection from Aβ1-42.
Discussion
The much broader distribution pattern of proSAAS with respect to PC1/3 led us to hypothesize that proSAAS may perform other functions in addition to prohormone convertase inhibition, specifically in relation to neurodegenerative diseases. We found a high incidence of co-localization between proSAAS and Aβ1-42-positive plaques in human AD brain tissues, as well as in the brains of AD mouse models. Whether this co-localization is functional or nonspecific is not clear from these data alone. It is interesting to note that very strong proSAAS localization was observed mainly in dense core pathology, but Figure 2H also shows accumulation in early stage diffuse plaques, suggesting that proSAAS might modulate plaque formation at different stages. These studies lend support to the idea of physical association of proSAAS and the amyloidogenic peptide in vivo, and are experimentally supported by our finding that Abeta and proSAAS are co-immunoprecipitated from brain extracts of APP-overexpressing mice.
It was somewhat puzzling to find proSAAS-immunoreactivity within amyloid plaques given the knowledge that proSAAS is an effective inhibitor of Aβ1-42 fibrillation in vitro (Figure 4A). We speculate that in certain brain areas, the local concentration of Aβ1-42 might exceed the ability of proSAAS to block Aβ1-42 plaque formation. Alternatively, if secreted proSAAS becomes trapped within plaques concomitantly with disease progression, this may leave reduced levels of available proSAAS to block Aβ fibrillation and/or cytotoxicity, contributing to pathophysiology. This latter explanation would be consistent with the repeated proteomics observation that patients with AD exhibit reduced CSF levels of proSAAS (Abdi et al. 2006, Jahn et al. 2011, Davidsson et al. 2002, Finehout et al. 2007).
While the mechanism of protein fibrillation is still unclear, inappropriate hydrophobic interactions are thought to contribute to fibril formation (Lin et al. 2008). We localized the anti-fibrillation sequence to a 97-180 residue segment within the N-terminal domain of proSAAS; this sequence may block Aβ1-42 fibrillation by preventing these inappropriate hydrophobic interactions. A loss of α-helical structure might eliminate the effect of proSAAS on fibril formation; mutagenesis will be required to test this hypothesis. Interestingly, when proSAAS was added at a time when Aβ1-42 peptides had already formed fibrils, ThT fluorescence did not decrease, indicating that proSAAS cannot disrupt pre-formed fibrils (Figure 4D). Moreover, we found that the chaperone action of proSAAS did not extend to an ability to refold and reactivate inactive luciferase (unpublished data). We conclude that proSAAS most likely acts by blocking inappropriate hydrophobic interactions that lead to protein aggregation, and not by classical chaperone refolding mechanisms. We were unable to correlate the structure-function results observed in vitro with structure-function results observed in vivo; proSAAS constructs smaller than 21 kDa proSAAS did not confer protection against Aβ cytotoxicity in Neuro2A cells. Possible reasons for this discrepancy include differences in sensitivity, solubility or protein conformation in the two types of assays which result in the need for additional N-terminal sequence in the cytotoxicity assays.
In addition to insoluble fibrils, other types of soluble Aβ populations have been reported to contribute to the pathophysiology of AD (Walsh et al. 1997, Harper et al. 1997, Lambert et al. 1998). Soluble Aβ1-42 oligomers are believed to be significantly more cytotoxic than fibrils; the formation of amyloid plaques might represent a mechanism for cellular sequestration of these oligomers (reviewed in (Benilova et al. 2012)). This view is supported by postmortem studies that identified only weak correlation between plaque density and neuropsychological tests (Terry et al. 1991). In our studies, proSAAS exhibited neuroprotection against cytotoxicity induced by oligomeric Aβ1-42 species, and this effect could be obtained either by increasing exogenous levels of proSAAS or by increasing endogenously synthesized and secreted proSAAS. Combined with our in vitro fibrillation data, these observations suggest that proSAAS may act on multiple Aβ1-42 species to influence AD pathophysiology.
Chaperones play an important role in maintaining neuronal protein homeostasis, and chaperone dysfunction has previously been implicated in the pathogenesis of AD as well as Parkinson's disease (reviewed in (Ali et al. 2010)). An important feature of chaperones is that they bind to multiple substrates/clients. For example, heat shock proteins (HSPs), the most common type of molecular chaperone, as well as two non-HSPs, αB-crystallin and clusterin, can block both Aβ1-42 and α-synuclein fibrillation in vitro (reviewed in (Ali et al. 2010); (Shammas et al. 2011, Rekas et al. 2004, Yerbury et al. 2007). A recent study shows that addition of several known secreted chaperones, including clusterin, haptoglobulin and -2 macroglobulin protects SH-SY5Y cells against toxicity from oligomers of Aβ1-42, islet amyloid polypeptide, and a bacterial chaperone, HypF-N; this occurs by direct binding of chaperone proteins to oligomers, reducing their cytotoxic potential (Mannini et al. 2012). A similar mechanism may be operational here, since our co-immunoprecipitation data support the idea of direct A -proSAAS binding; however, additional experiments are required to show that proSAAS binds directly to oligomers.
Aβ1-42 plaques are generally found extracellularly; however, proSAAS immunoreactivity was co-localized with both extracellular and intracellular protein deposits. Given that Aβ1-42 is secreted and can be internalized from the extracellular space (reviewed in (Mohamed & Posse de Chaves 2011);(Takuma et al. 2009, Emmanouilidou et al. 2010, Volpicelli-Daley et al. 2011), we speculate that proSAAS can interact with Aβ1-42, either following joint secretion into the extracellular space or following reuptake. While the majority of Aβ1-42/proSAAS co-localization clearly occurs extracellularly, the latter possibility is intriguing since it has been shown that Aβ1-42 accumulates in acidic vesicles where it becomes concentrated and begins to aggregate (Hu et al. 2009). Additional immunocytochemical studies at a higher resolution may reveal the extent of subcellular colocalization of proSAAS with Aβ1-42 within cells.
In summary, we have identified a molecular chaperone-like activity for the N-terminal domain of proSAAS. We used ThT assays and TEM studies to demonstrate that proSAAS can block the fibrillation of Aβ1-42 in vitro, and show that addition of recombinant proSAAS to Neuro2a cells results in protection from Aβ1-42-induced cytotoxicity. These data are in agreement with our immunohistochemistry data which illustrate co-localization of proSAAS and amyloid plaques deposits in vivo. Collectively, these data suggest that proSAAS may play a role in the pathogenesis of Alzheimer's disease, and highlight the therapeutic potential of drugs targeting this mechanism for prevention of neuronal cytotoxicity.
Acknowledgments
This work was funded by NIH DK49703 to IL and NIH AG039008 and Alzheimer's Association NIRG-08-92033 to JE. MH was supported by the German Academy of Sciences Leopoldina Foundation (LPDS 2009-33). The authors would also like to acknowledge the NICHD Brain and Tissue Bank for Developmental Disorders of the University of Maryland-Baltimore for the human brain samples; UMB Biopolymer Core for peptide synthesis; and the UMB Core Imaging Facility for EM expertise. The authors declare that they have no conflict of interest in publishing this work.
Abbreviations
- AD
Alzheimer's disease
- PD
Parkinson's disease
- PC1/3
prohormone convertase 1/3
- CSF
cerebrospinal fluid
- NICHD
National Institute of Child Health and Human Development
- PBS
phosphate-buffered saline
- BSA
bovine serum albumin
- TBS
Tris-buffered saline
- ThT
Thioflavin T
- TEM
transmission electron microscopy
- FBS
fetal bovine serum
- APP
amyloid precursor protein
Reference List
- Abdi F, Quinn JF, Jankovic J, et al. Detection of biomarkers with a multiplex quantitative proteomic platform in cerebrospinal fluid of patients with neurodegenerative disorders. J Alzheimers Dis. 2006;9:293–348. doi: 10.3233/jad-2006-9309. [DOI] [PubMed] [Google Scholar]
- Ali YO, Kitay BM, Zhai RG. Dealing with misfolded proteins: examining the neuroprotective role of molecular chaperones in neurodegeneration. Molecules. 2010;15:6859–6887. doi: 10.3390/molecules15106859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins N, Jr, Mitchell JW, Romanova EV, Morgan DJ, Cominski TP, Ecker JL, Pintar JE, Sweedler JV, Gillette MU. Circadian integration of glutamatergic signals by little SAAS in novel suprachiasmatic circuits. PLoS One. 2010;5:e12612. doi: 10.1371/journal.pone.0012612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci. 2012 doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- Braks JAM, Martens GJM. 7B2 is a neuroendocrine chaperone that transiently interacts with prohormone convertase PC2 in the secretory pathway. Cell. 1994;78:263–273. doi: 10.1016/0092-8674(94)90296-8. [DOI] [PubMed] [Google Scholar]
- Cameron A, Fortenberry Y, Lindberg I. The SAAS granin exhibits structural and functional homology to 7B2 and contains a highly potent hexapeptide inhibitor of PC1. FEBS Lett. 2000;473:135–138. doi: 10.1016/s0014-5793(00)01511-8. [DOI] [PubMed] [Google Scholar]
- Chaudhuri B, Stephen C, Huijbregts R, Martens G. The neuroendocrine protein 7B2 acts as a molecular chaperone in the in vitro folding of human insulin-like growth factor-1 secreted from yeast. Biochem Biophys Res Commun. 1995;211:417–425. doi: 10.1006/bbrc.1995.1830. [DOI] [PubMed] [Google Scholar]
- Davidsson P, Sjogren M, Andreasen N, Lindbjer M, Nilsson CL, Westman-Brinkmalm A, Blennow K. Studies of the pathophysiological mechanisms in frontotemporal dementia by proteome analysis of CSF proteins. Brain Res Mol Brain Res. 2002;109:128–133. doi: 10.1016/s0169-328x(02)00549-1. [DOI] [PubMed] [Google Scholar]
- Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. Journal of Neuroscience. 2010;30:6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Reznik SE, Fricker LD. Distribution of proSAAS-derived peptides in rat neuroendocrine tissues. Neuroscience. 2001;105:469–478. doi: 10.1016/s0306-4522(01)00200-7. [DOI] [PubMed] [Google Scholar]
- Finehout EJ, Franck Z, Choe LH, Relkin N, Lee KH. Cerebrospinal fluid proteomic biomarkers for Alzheimer's disease. Ann Neurol. 2007;61:120–129. doi: 10.1002/ana.21038. [DOI] [PubMed] [Google Scholar]
- Fortenberry Y, Hwang JR, Apletalina EV, Lindberg I. Functional characterization of ProSAAS: similarities and differences with 7B2. J Biol Chem. 2002;277:5175–5186. doi: 10.1074/jbc.M104531200. [DOI] [PubMed] [Google Scholar]
- Fricker LD, McKinzie AA, Sun J, et al. Identification and characterization of proSAAS, a granin-like neuroendocrine peptide precursor that inhibits prohormone processing. J Neurosci. 2000;20:639–648. doi: 10.1523/JNEUROSCI.20-02-00639.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guest PC, Abdel-Halim SM, Gross DJ, Clark A, Poitout V, Amaria R, Ostenson C, Hutton JC. Proinsulin processing in the diabetic Goto-Kakizaki rat. J Endocrinol. 2002;175:637–647. doi: 10.1677/joe.0.1750637. [DOI] [PubMed] [Google Scholar]
- Haass C, Lemere CA, Capell A, Citron M, Seubert P, Schenk D, Lannfelt L, Selkoe DJ. The Swedish mutation causes early-onset Alzheimer's disease by beta-secretase cleavage within the secretory pathway. Nat Med. 1995;1:1291–1296. doi: 10.1038/nm1295-1291. [DOI] [PubMed] [Google Scholar]
- Harper JD, Lieber CM, Lansbury PT., Jr Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer's disease amyloid-beta protein. Chem Biol. 1997;4:951–959. doi: 10.1016/s1074-5521(97)90303-3. [DOI] [PubMed] [Google Scholar]
- Hatcher NG, Atkins NJ, Annangudi SP, Forbes AJ, Kelleher NL, Gillette MU, Sweedler JV. Mass spectrometry-based discovery of circadian peptides. Proc Natl Acad Sci U S A. 2008;105:12527–12532. doi: 10.1073/pnas.0804340105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helwig M, Hoshino A, Berridge C, Lee SN, Lorenzen N, Otzen DE, Eriksen JL, Lindberg I. The neuroendocrine protein 7B2 suppresses the aggregation of neurodegenerative disease-related proteins. J Biol Chem. 2012;288:1114–1124. doi: 10.1074/jbc.M112.417071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Crick SL, Bu G, Frieden C, Pappu RV, Lee JM. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20324–20329. doi: 10.1073/pnas.0911281106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn H, Wittke S, Zurbig P, et al. Peptide fingerprinting of Alzheimer's disease in cerebrospinal fluid: identification and prospective evaluation of new synaptic biomarkers. PLoS One. 2011;6:e26540. doi: 10.1371/journal.pone.0026540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Kikuchi K, Arawaka S, Koyama S, et al. An N-terminal fragment of ProSAAS (a granin-like neuroendocrine peptide precursor) is associated with tau inclusions in Pick's disease. Biochem Biophys Res Commun. 2003;308:646–654. doi: 10.1016/s0006-291x(03)01391-3. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanoue E, Day R. Coexpression of proprotein convertase SPC3 and the neuroendocrine precursor proSAAS. Endocrinology. 2001;142:4141–4149. doi: 10.1210/endo.142.9.8386. [DOI] [PubMed] [Google Scholar]
- Lee SN, Lindberg I. 7B2 prevents unfolding and aggregation of prohormone convertase 2. Endocrinology. 2008;149:4116–4127. doi: 10.1210/en.2008-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MS, Chen LY, Tsai HT, Wang SS, Chang Y, Higuchi A, Chen WY. Investigation of the mechanism of beta-amyloid fibril formation by kinetic and thermodynamic analyses. Langmuir. 2008;24:5802–5808. doi: 10.1021/la703369b. [DOI] [PubMed] [Google Scholar]
- Liu XD, Zeng BF, Xu JG, Zhu HB, Xia QC. Proteomic analysis of the cerebrospinal fluid of patients with lumbar disk herniation. Proteomics. 2006;6:1019–1028. doi: 10.1002/pmic.200500247. [DOI] [PubMed] [Google Scholar]
- Mannini B, Cascella R, Zampagni M, et al. Molecular mechanisms used by chaperones to reduce the toxicity of aberrant protein oligomers. Proc Natl Acad Sci U S A. 2012;109:12479–12484. doi: 10.1073/pnas.1117799109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed A, Posse de Chaves E. Abeta internalization by neurons and glia. Int J Alzheimers Dis. 2011;2011:127984. doi: 10.4061/2011/127984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DJ, Mzhavia N, Peng B, Pan H, Devi LA, Pintar JE. Embryonic gene expression and pro-protein processing of proSAAS during rodent development. J Neurochem. 2005;93:1454–1462. doi: 10.1111/j.1471-4159.2005.03138.x. [DOI] [PubMed] [Google Scholar]
- Morgan DJ, Wei S, Gomes I, Czyzyk T, Mzhavia N, Pan H, Devi LA, Fricker LD, Pintar JE. The propeptide precursor proSAAS is involved in fetal neuropeptide processing and body weight regulation. J Neurochem. 2010;113:1275–1284. doi: 10.1111/j.1471-4159.2010.06706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mzhavia N, Qian Y, Feng Y, Che FY, Devi LA, Fricker LD. Processing of proSAAS in neuroendocrine cell lines. Biochem J. 2002;361:67–76. doi: 10.1042/0264-6021:3610067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Devi LA, Mzhavia N, Munzer S, Seidah NG, Fricker LD. The C-terminal region of proSAAS is a potent inhibitor of prohormone convertase 1. J Biol Chem. 2000;275:23596–23601. doi: 10.1074/jbc.M001583200. [DOI] [PubMed] [Google Scholar]
- Rekas A, Adda CG, Andrew Aquilina J, et al. Interaction of the molecular chaperone alphaB-crystallin with alpha-synuclein: effects on amyloid fibril formation and chaperone activity. J Mol Biol. 2004;340:1167–1183. doi: 10.1016/j.jmb.2004.05.054. [DOI] [PubMed] [Google Scholar]
- Sayah M, Fortenberry Y, Cameron A, Lindberg I. Tissue distribution and processing of proSAAS by proprotein convertases. J Neurochem. 2001;76:1833–1841. doi: 10.1046/j.1471-4159.2001.00165.x. [DOI] [PubMed] [Google Scholar]
- Shammas SL, Waudby CA, Wang S, et al. Binding of the molecular chaperone alphaB-crystallin to Abeta amyloid fibrils inhibits fibril elongation. Biophys J. 2011;101:1681–1689. doi: 10.1016/j.bpj.2011.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine WB, Jungbauer L, Yu C, LaDu MJ. Preparing synthetic Abeta in different aggregation states. Methods in molecular biology. 2011;670:13–32. doi: 10.1007/978-1-60761-744-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuma K, Fang F, Zhang W, et al. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20021–20026. doi: 10.1073/pnas.0905686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada M, Ren CH, Koyama S, et al. A human granin-like neuroendocrine peptide precursor (proSAAS) immunoreactivity in tau inclusions of Alzheimer's disease and parkinsonism-dementia complex on Guam. Neurosci Lett. 2004;356:49–52. doi: 10.1016/j.neulet.2003.11.028. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate. J Biol Chem. 1997;272:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- Wardman JH, Berezniuk I, Di S, Tasker JG, Fricker LD. ProSAAS-derived peptides are colocalized with neuropeptide Y and function as neuropeptides in the regulation of food intake. PLoS One. 2011;6:e28152. doi: 10.1371/journal.pone.0028152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yerbury JJ, Poon S, Meehan S, Thompson B, Kumita JR, Dobson CM, Wilson MR. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 2007;21:2312–2322. doi: 10.1096/fj.06-7986com. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]