

Abstract
The use of proteomics for direct detection of expressed pathways producing natural products has yielded many new compounds, even when used in a screening mode without a bacterial genome sequence available. Here we quantify the advantages of having draft DNA-sequence available for strain-specific proteomics using the latest in ultrahigh-resolution mass spectrometry (MS) for both proteins and the small molecules they generate. Using the draft sequence of Streptomyces lilacinus NRRL B-1968, we show a >10-fold increase in the number of peptide identifications vs. using publicly available databases. Detected in this strain were six expressed gene clusters with varying homology to those known. To date, we have identified three of these clusters as encoding for the production of griseobactin (known), rakicidin D (an orphan NRPS/PKS hybrid cluster), and a putative thr and DHB-containing siderophore produced by a new non-ribosomal peptide sythetase gene cluster. The remaining three clusters show lower homology to those known, and likely encode enzymes for production of novel compounds. Using an interpreted strain-specific DNA sequence enables deep proteomics for the detection of multiple pathways and their encoded natural products in a single cultured bacterium.
Keywords: Proteomics, Natural Products, Mass Spectrometry, Metabolomics, Genome Mining
Introduction
Natural products have historically been a rich source of drug leads for the pharmaceutical industry [19]. In particular, natural products or their derivatives make up a large number of FDA approved antibiotic and antiproliferative drugs. Many natural products with clinical utility are synthesized by polyketide synthases (PKSs) or nonribosomal peptide synthetases (NRPSs) [25]. Both of these enzyme families create structurally and functionally diverse compound scaffolds. NRPSs and PKSs both contain numerous domains and synthesize natural products via multi-modular assembly lines. They have been reviewed at length elsewhere [13].
Traditional approaches to natural product discovery have relied primarily on bioassay-guided approaches with repeated fractionation to isolate the compound of interest. This approach is constrained by the bioactivity used in the screen and is often frustrated by the observation that many different bacteria produce the same compounds. The advent of genome sequencing led to the recognition that even well-studied microorganisms possess a significantly greater potential for the production of natural products than has been realized to date [26]. Genome-mining approaches were developed to capitalize on this apparent wealth of biosynthetic potential—and with some successes [5]. However, a paradigm arose that held that some pathways were simply “cryptic” and were not expressed to detectable levels under laboratory conditions [22,24].
Several research groups, including our own, turned to proteomics to complement genome mining and bioassay guided methodologies (Table 1). Our lab developed a “protein-first” strategy termed PrISM (short for Proteomic Investigation of Secondary Metabolism) for identification of natural products via their biosynthetic pathways. By identifying biosynthetic proteins first, cryptic gene clusters are avoided, and natural products are found in a “structure-based” approach independent of bioactivity. By focusing on a characteristic ion associated with the phosphopantetheine post-translational modification that is essential for the function of thiotemplated systems, active site peptides could be identified, enabling the sequencing and characterization of whole biosynthetic clusters [2]. This approach was successfully employed in the discovery of several new natural products including koranimine [12], flavopeptin [6], and gobichelins A and B [8].
Table 1.
Summary of published work using microbial proteomics to interrogate the expression of genes and gene clusters encoding enzymes responsible for the biosynthesis of secondary metabolites and natural products.
| Organism(s) Studied | Instrumentation | Biosynthetic Pathways Studied | Reference |
|---|---|---|---|
| Bacillus subtilis, Streptomyces sp. | LTQ-FTICR (12T) | gramicidin S, kurstakins, zwittermicins | 2 |
| Bacillus subtilis | LTQ-Ion Trap | surfactin, bacillibactin, bacillaene, plipastatin | 17 |
| Bacillus subtilis | LTQ-Ion Trap | surfactin, bacillibactin, plipastatin | 16 |
| Streptomyces sp. | LTQ-FTICR (7T) | flavopeptin | 6 |
| Bacillus subtilis | FTICR | nikkomycin, pyoluteorin, prodigiosin, gramicidin, mycosubtilin, enterobactin | 11 |
| Streptococcus agalactiae | LTQ | putative pks | 18 |
| Streptomyces sp. | LTQ-Orbitrap | gobichelin | 8 |
| Actinobacteria | LTQ-FTICR (7T) | foroxymithine, antibiotic S213 L, actinomycin, etc. | 7 |
| Bacillus sp. | LTQ-FTICR (7T) | koranimine | 12 |
| Frankia sp. | LTQ | 25+ individual pathways | 23 |
As technology has improved over the past decade, mass spectrometry-based proteomics has played a growing role in both the discovery and characterization of these proteins and their biosynthetic products even when genome-predicted ORFs from related strains were used as protein databases. Table 1 highlights the diversity of organisms studied using proteomics and the diversity of chemical structures identified. A variety of MS instrumentation has been employed, including standalone ion traps like the linear trap quadrupole (LTQ) which are relatively inexpensive bench-top mass spectrometers. As such, many labs have access to these types of instruments either in their own laboratory or in university-run cores. Thus, while original publications used expensive, Fourier-Transform mass spectrometers, this requirement has eased with the availability of bench-top FTMS instruments. High quality genome sequence can increase the accessibility of this approach even further by compensating somewhat for lower quality mass spectrometry data. In particular, the necessity of high mass accuracy is reduced when searching a highly targeted database as compared to a large, comprehensive database such as NCBInr.
Recently, we began using sequenced organisms and searching against strain-specific databases created from protein sequences predicted from the draft genome sequence of the exact strain being studied. We term this approach “Genome-Enabled” PrISM (GE-PrISM) and the work and data flows that comprise it are shown in Fig. 1. Here we use a single actinomycete to quantify the advantages of this approach and offer a look ahead at the intersection of genomics-, proteomics-, and metabolomics-based tool sets for natural products discovery from the microbial world.
Fig. 1.

Workflow of “genome-enabled” PrISM, an extended form of a proteomics-based approach to discover expressed gene clusters in microbial systems. From left to right, the workflow entails microbial culture, striation of the proteome according to mass by SDS-PAGE, in-gel digestion followed by LC-MS/MS and searching of peptide fragment ion data against the assembled genome of a specific microbial strain. The structural elements of a molecule are determined using the predicted substrates for domains in the assembly line and the rules of co-linear biosynthesis. This informs a targeted metabolomics-based approach to identify the small molecule being produced.
Materials and Methods
Strain Acquisition and Sequencing
Streptomyces lilacinus NRRL B-1968T was acquired from the Agricultural Research Service (ARS) collection. DNA isolation was performed using the MoBio UltraClean Microbial DNA Isolation kit. Library preparation was performed using Nextera version 1 kits and protocols. Illumina sequencing was performed at the UIUC genomic sequencing facility on a HiSeq 2000 for 2x100 cycles and resulted in 3,297,810 paired reads. This corresponds to 96x coverage for this strain. Genome assembly and gene prediction were performed using previously described methods [10].
Proteomics
S. lilacinus was grown in 5 mL of ISP2 medium at 30 degrees Celsius for 2 days with shaking (200 rpm). Cell pellets were collected via centrifugation at 14,000 rpm for 10 minutes. Cell lysis was performed using previously described methods [8]. The resulting lysate was analyzed via SDS-PAGE. High molecular weight proteins (HMWPs) (>200 kDa) were excised and subjected to in-gel trypsin digestion. The resultant peptides were subjected to nanocapillary LC-MS/MS analysis on either a hybrid Velos 12 Tesla FTICR system or a Velos Orbitrap Elite (Thermo Fisher Scientific, Waltham, MA).
Analysis of Proteomics Data
For initial experiments, mass spectrometry data were searched against large publicly available bacterial databases (SwissProt or NCBInr) to look for the presence of proteins involved in the biosynthesis of secondary metabolites, primarily PKS- and NRPS-derived compounds. The search process was slow and required a non-trivial amount of computing capacity. Recently, we have begun searching against strain-specific databases created from ORF predictions acquired from the genome sequence of the specific strain being interrogated.
Small Molecule Mass Spectrometry
Cultures for small molecule analysis were grown following the procedures described above. Culture supernatant was collected and extracted using Oasis HLB solid phase extraction columns (Waters Corp, Milford, MA) according to manufacturer’s instructions and eluted with 80% ACN. The resultant extract was reduced to dryness. Samples were resuspended in 95% H2O/5% ACN (with 0.2% formic acid) to a final concentration of 2 mg/mL. Forty μg of sample was loaded onto a 5 μm Luna C18 column (2 mm i.d.; 150 mm) (Phenomenex, Torrance, CA). A 60 minute LC gradient was employed at a flow rate of 200 μL/min on an Agilent 1150 LC system (Agilent, Santa Clara, CA). Mass spectrometry was performed on a Q-Exactive mass spectrometer (Thermo Fisher Scientific, Waltham, MA). Intact MS spectra were acquired at a resolution of 35,000. The top 5 most intense ions were selected for fragmentation in a data-dependent acquisition mode. MS/MS spectra were acquired at a resolution of 17,500.
Small Molecule Data Analysis
The SIEVE software platform (Thermo Fisher Scientific, Waltham, MA) was used for the analysis of metabolite-level mass spectrometry data. Automated component detection was performed at the intact mass (MS1) level. Deisotoping and collapsing of numerous adduct forms to a single component were performed to reduce data redundancy and allow accurate calculation of a neutral mass for each component. To rapidly identify any known natural products, all components were searched against a custom accurate mass database consisting of 11,413 known bacterial metabolites using a mass tolerance of 3 ppm. The database was prepared using Antibase [1], Dictionary of Natural Products [9], Norine [3], and additional bacterial natural products identified in the literature.
Stable Isotope Labeling
NRPS proteins identified during proteomic analysis were analyzed for the presence of adenylation domains. Adenylation domains were analyzed using NRPSPredictor to predict substrate specificity [21]. When confident A-domain predictions were available, stable isotope labeling was performed using the predicted monomer(s) likely to be incorporated into the final secondary metabolite of interest. Isotopically labeled forms of these monomers were purchased from Cambridge Isotope Laboratories (Andover, MA). Labeled amino acids were dissolved in ultrapure water and sterile filtered (0.2 μm) into previously autoclaved media to a final concentration of 1 mM immediately prior to inoculation. Following the addition of labeled monomer, cultures were grown as previously described. Unlabeled control samples were grown in parallel.
Results
Genome Sequence of B-1968
The draft genome of S. lilacinus B-1968 was assembled into 351 scaffolds comprised of 2963 contigs, covering 6,865,648 bases with a GC content of 72.1%. ORF prediction was performed with Prodigal and resulted in 7228 predicted ORFs. Profile hidden Markov models were used to find 67 natural product gene clusters, although 44 of these clusters are partial due to breaks in the assembly. Thus, the strain could contain a maximum of 67 natural product gene clusters. The actual number is likely to be lower due to the possibility of a fragmented cluster being present on more than one contig. The tally of natural product gene clusters is as follows: type I PKS, three complete, 41 partial; NRPS, three complete, two contigs containing partial clusters; NRPS/PKS hybrid, one partial; microcins, one complete; terpene synthase and isoprenoid modification clusters, eight complete; lantipeptide, two complete; NRPS-independent siderophores (aerobactin-like), one complete; type II PKS, two complete; type III PKS, one complete; indigoidine, one complete; phosphonate, one complete.
Proteomics based on the interpreted ORFs from the sequence contigs revealed the results shown in Table 2. Comparison of database search metrics for traditional PrISM vs. GE-PrISM show a >20 fold speed improvement for searching and a very significant increase of >15 fold in the number of PKS and NRPS peptides identified (Table 2). Using the traditional PrISM approach and searching the LC-MS/MS data against the publicly available NCBInr database, only 11 PKS- and 6 NRPS-derived peptides were identified. Using the same data and searching against the genome-enabled strain specific database identified 219 PKS- and 103 NRPS-derived peptides. Additionally, the search time was decreased from 4 hours per search to approximately 12 minutes per search. Note that for several proteins in these database hits, the sequence coverage rose from <2% to over 60%. These proteins mapped onto 9 sequence contigs. Overall, evidence for expression of 6 gene clusters could be determined (vide infra). The “deep” proteomics enabled by having the precise genome of the strain being interrogated identified 43 peptides from a novel gene cluster predicted to produce a PKS product. Despite multiple labeling attempts and MS-based searching of the exported metabolome, the corresponding product could not be identified to date. Further searching in the intracellular space and membrane-bound fractions are ongoing for this target.
Table 2.
Comparison of the traditional PrISM workflow with “genome-enabled” version employing strain-specific protein databases generated from assembled DNA-sequences. A >15-fold increase in the number of detected peptides corresponding to enzymes involved in secondary metabolite biosynthesis is coupled with a significant reduction in search time.
| Method | Database | Average Search Time | PKS Peptides Identified | PKS Avg Peptides per ORF | NRPS Peptides Identified | NRPS Avg Peptides per ORF |
|---|---|---|---|---|---|---|
| Traditional PrISM | NCBInr | 4 hours | 11 | 1.1 | 6 | 1.2 |
| Genome-Enabled PrISM | S. lilacinus Strain-Specific Database | 12 minutes | 219 | 8.4 | 103 | 9.4 |
Case Study 1: Griseobactin
The genome of S. lilacinus contained a gene cluster with a high degree of similarity to the biosynthetic genes proposed by Patzer et al. to be responsible for the production of griseobactin [20]. Proteomics experiments identified 23 peptides from 3 ORFs encoded by genes in this cluster (Table S1). With GE-PrISM, sequence coverage >75% was often observed, even for several large (>100 kDa) PKS and NRPS proteins. This high degree of sequence coverage allowed us to quickly and confidently identify that a given gene cluster was indeed being expressed. At the metabolite level, HRMS analysis of the culture supernatant successfully identified griseobactin via the automated dereplication process (Fig. S1 and S2). In addition to the protonated form, low levels of a compound with a mass consistent with Al3+-adducted griseobactin were also observed (data not shown). This is consistent with the original report [20]. Our analysis detected both the singly and doubly charged forms of the aluminum complexed griseobactin species, in contrast to the original paper which detected only the doubly charged form.
Case Study 2: A New Threonine-Containing Siderophore-Like Compound and Its Associated Cluster
The identification of additional NRPS-derived peptides afforded to us through the use of the genome-specific database allowed us to identify an additional NRPS cluster expressed under these conditions. A-domain analysis of the proteomics data predicted that threonine would be incorporated into the final product. Based on this information, isotopically labeled threonine was added to the culture medium in a targeted approach to uncover the metabolite produced by this cluster. Small molecule mass spectrometry identified a putative compound consistent with the expected mass shift due to the incorporation of the labeled threonine (Fig. 2). The isotopic distribution suggested that the compound incorporated three threonine residues, which was confirmed using tandem mass spectrometry (data not shown). Further analysis of the tandem mass spectrometry data showed the compound to be a linear peptide consisting of a tri-threonine core terminated by a dihydroxy-benzoic acid at each terminus (Fig. 3). The structure suggests that this compound acts as a catecholate siderophore, as dihydroxy benzoic acid monomers are characteristic of this class of compounds. The monomer composition and arrangement of this compound are notably similar to the previously described griseobactin compound. Interestingly, however, the data suggest that these two compounds are indeed produced by two distinct gene clusters, both of which are present in this strain. This was confirmed at both the genome and proteome level, as highly similar but distinct peptides were detected for each gene cluster (i.e. peptides from the thr adenylation domains for both the griseobactin cluster and the novel compound cluster were observed in the proteomics data). This is consistent with the genomics data, which upon interpretation yields two separate clusters as well.
Fig. 2.
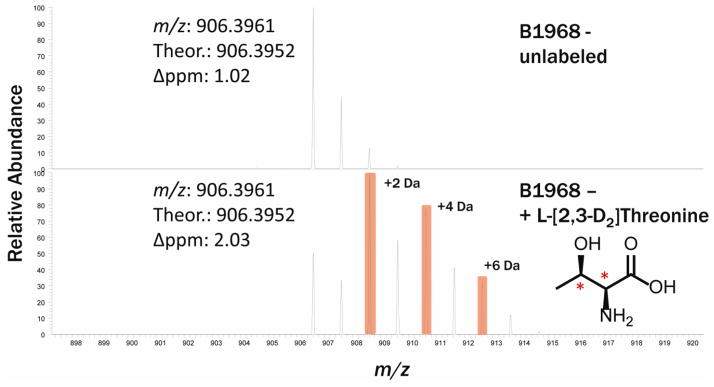
Intact mass spectrum of novel NRPS compound without (top) and with (bottom) the addition of 2,3-d2 labeled threonine (inset). The isotope pattern is consistent with the addition of three threonine monomers.
Fig. 3.

Draft structure of a new catecholate siderophore and its high-resolution MS2 analysis. Peaks highlighted in red are consistent with the predicted fragmentation of this compound. The table below shows the explanation given for the most abundant signals in the tandem mass spectrum.
Case Study 3: The Gene Cluster Producing Rakicidin D
Further proteomic analysis of the same strain identified additional NRPS- and PKS-derived peptides that were distinct from the gene clusters mentioned above. Using our GE-PrISM methodology, we putatively identified the compound produced by this strain as rakicidin D based on accurate intact mass measurements and confirmed this identification with tandem mass spectrometry.
Rakicidin D is a member of a family of cyclic depsipeptide compounds with cytotoxic properties that includes vinylamicin [14], microtermolide A [4], and the antibiotic BE-43547 [15]. Rakicidin A has uniquely demonstrated enhanced cytotoxicity under hypoxic conditions [27]. To date, no biosynthetic proteins have been associated with the production of rakicidin D or any of the members of this class of cyclic depsipeptides. The proteomics data we acquired coupled with the genome-specific database allowed us to rapidly assign a hybrid NRPS/PKS-containing region of the genome as the cluster responsible for rakicidin biosynthesis (Fig. 5). Preliminary analysis of the genomics data identified the presence of only one NRPS/PKS hybrid cluster in this strain. Proteomic analysis identified peptides matching to 9 ORFs present on this contig. Further supporting the assignment, A-domain predictions for this cluster, where available, were consistent with the amino acid monomers present in rakicidin D. Additionally, genes encoding for the biosythentic enzymes required to produce the modified amino acids in rakicidin D are represented in the cluster.
Fig. 5.

A) Map of contig containing putative biosynthetic genes of rakicidin D. NRPS and PKS genes are shown in blue, while transport genes are shown in orange. rakG and rakD, which correspond with the most striking features of rakicidin D, are colored in green and red, respectively. B) Bioinformatic prediction of the domain architecture of NRPS and PKS genes in the biosynthetic cluster. Proteins identified using MS-based proteomics are highlighted in red. C) Structure of rakicidin D. The methyl group of the sarcosine substituent is circled in green, corresponding to the NRPS protein RakG which contains an A-domain specific to glycine as well as an N-methyltransferase domain. The hydroxyl group of the hydroxyasparagine monomer is circled in red, corresponding to RakO, a putative asparagine oxygenase.
Discussion and Future Prospects
While the original PrISM platform has led to the identification of dozens of known natural product gene clusters and numerous novel secondary metabolites, the introduction of genome-specific databases has led to significant improvements in the platform. Studying natural products biosynthesis at the protein level offers the advantage of selecting for only expressed gene clusters. Annotated proteins in databases such as NCBInr are necessarily biased toward known systems. The initial PrISM pipeline was thus prone to identifying expressed gene clusters similar to known clusters. This limited our ability to detect truly novel compounds with new chemical scaffolds.
Originally, the PrISM platform was limited by a self-imposed bias of looking only at high molecular weight proteins. This was done as a way to eliminate highly abundant so-called “house-keeping” proteins by taking advantage of the fact that many NRPS and PKS proteins are significantly larger in size than most of the highly expressed enzymes involved in primary metabolism. This is not true for all secondary metabolism enzymes, however, including those responsible for production of many critical antibiotics of the aminoglycoside family and others. By dramatically reducing the required search space, GE-PrISM has allowed us to bypass the gel screening step entirely. Instead, cells from a bacterial culture are lysed and the entire proteome content of the organism is digested directly and analyzed en masse in a single LC-MS/MS run. Initial results have identified up to 1,000 proteins in a single LC-MS/MS injection. This included the identification of NRPS- and PKS-derived peptides consistent with what we have accomplished using our gel-based platform. Additionally, aminoglycoside and terpenoid biosynthetic proteins were detected, significantly expanding our coverage of the space populated by natural products. Significantly, these results were obtained using draft-quality genomes comprised of >15 contigs. Scaffolds of genome sequences containing fragmented clusters do not hinder MS-based proteomics, but do create the need for contig-end PCR or additional informatics to identify all the contigs containing a specific gene cluster. Targeted genome-specific databases are clearly advantageous for maximizing protein and proteome coverage in a high-throughput paradigm.
Genome Enabled PrISM has increased our hit rate by allowing us to identify peptides with low homology to genes found in publicly available databases. In addition to increasing the number of peptides identified, this also allows us to identify clusters with low homology to known clusters, ultimately providing a pathway for the high-throughput discovery of compounds with completely new structural scaffolds. This would open up new chemical space to help address the ever-growing problem of antibiotic resistant pathogens. The introduction of high quality genome data also aids significantly in the structure elucidation phase of the PrISM workflow. The addition of an even greater number of sequenced microbes increases the accuracy of prediction algorithms such as NRPSPredictor and others, further accelerating the path from gene discovery to small molecule discovery.
Conclusions
The advent of high throughput, low-cost genomic sequencing has made it readily accessible to many labs, many of which would not consider themselves “genomicists”. The combination of genome sequence data with both proteomics and metabolomics data (all using low ppm mass accuracy) allows for the rapid and confident identification of natural products and the genes encoding their biosynthesis. The potential of this technology has yet to be realized by the community at large, and still requires some software development to operate in an automated mode at the thousands of strains scale. The availability of the S. lilacinus genome sequence increased the number of secondary metabolism biosynthetic proteins identified owing to the far higher quality, “deep” proteomic data (along with a significant time savings for database searching). GE-PrISM has removed the main bottlenecks that existed in our previous PrISM workflow and helps identify the small molecule being produced by identification of the corresponding gene cluster. This small pilot experiment culminated in the assignment of multiple orphan gene clusters with the secondary metabolite(s) they produce – even in a single bacterial strain.
Genomics thus enables a more thorough analysis of expressed proteins, which in turn propels novel insights into the microbial world by rapidly identifying which small molecules are being actively produced. Similarly, metabolomics data both informs and is informed by gene- and protein-level data. While genomics and proteomics data aid greatly in the elucidation of natural product structures, secondary metabolites themselves such as quorum sensors can also feedback to the gene and protein levels. The intersection of these omics-based technologies (two of which rely on mass spectrometry) offers a promising path toward a much-anticipated resurgence in natural products research.
Supplementary Material
Fig. 4.

High-resolution tandem mass spectrum confirming the presence of rakicidin D. Peaks highlighted in red are consistent with the expected fragment ions from rakicidin D.
Acknowledgments
The Department of Chemistry at Northwestern University and these grants from the National Institutes of Health supported this work: GM 067725 from NIGMS (NLK) and GM 077596 from NIGMS (WWM). We also acknowledge support from the Institute for Genomic Biology IGB Fellows Program at UIUC (JRD). The authors would also like to thank Claudia K. Jones for her scholarly work.
References
- 1.Antibase: the natural compound identifier. Wiley VCH; 2011. [Google Scholar]
- 2.Bumpus SB, Evans BS, Thomas PM, Ntai I, Kelleher NL. A proteomics approach to discovering natural products and their biosynthetic pathways. Nat Biotechnol. 2009;27 (10):951–956. doi: 10.1038/nbt.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caboche S, Pupin M, Leclere V, Fontaine A, Jacques P, Kucherov G. NORINE: a database of nonribosomal peptides. Nucleic Acids Res. 2008;36:D326–331. doi: 10.1093/nar/gkm792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carr G, Poulsen M, Klassen JL, Hou Y, Wyche TP, Bugni TS, Currie CR, Clardy J. Microtermolides A and B from termite-associated Streptomyces sp. and structural revision of vinylamycin. Org Lett. 2012;14 (11):2822–2825. doi: 10.1021/ol301043p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Challis GL. Genome mining for novel natural product discovery. J Med Chem. 2008;51 (9):2618–2628. doi: 10.1021/jm700948z. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, McClure RA, Zheng Y, Thomson RJ, Kelleher NL. Proteomics guided discovery of flavopeptins: anti-proliferative aldehydes synthesized by a reductase domain-containing non-ribosomal peptide synthetase. J Am Chem Soc. 2013;135 (28):10449–10456. doi: 10.1021/ja4031193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Y, Ntai I, Ju KS, Unger M, Zamdborg L, Robinson SJ, Doroghazi JR, Labeda DP, Metcalf WW, Kelleher NL. A proteomic survey of nonribosomal peptide and polyketide biosynthesis in actinobacteria. J Proteome Res. 2012;11 (1):85–94. doi: 10.1021/pr2009115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Unger M, Ntai I, McClure RA, Albright JC, Thomson RJ, Kelleher NL. Gobichelin A and B: mixed-ligand siderophores discovered using proteomics. Med Chem Comm. 2013;4 (1):233–238. doi: 10.1039/C2MD20232H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dictionary of Natural Products. CRC Press; 2013. [Google Scholar]
- 10.Doroghazi JR, Ju KS, Brown DW, Labeda DP, Deng Z, Metcalf WW, Chen W, Price NP. Genome sequences of three tunicamycin-producing Streptomyces strains, S. chartreusis NRRL 12338, S. chartreusis NRRL 3882, and S. lysosuperificus ATCC 31396. J Bacteriol. 2011;193 (24):7021–7022. doi: 10.1128/JB.06262-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dorrestein PC, Blackhall J, Straight PD, Fischbach MA, Garneau-Tsodikova S, Edwards DJ, McLaughlin S, Lin M, Gerwick WH, Kolter R, Walsh CT, Kelleher NL. Activity screening of carrier domains within nonribosomal peptide synthetases using complex substrate mixtures and large molecule mass spectrometry. Biochemistry-US. 2006;45 (6):1537–1546. doi: 10.1021/bi052333k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans BS, Ntai I, Chen YQ, Robinson SJ, Kelleher NL. Proteomics-based discovery of koranimine, a cyclic imine natural product. J Am Chem Soc. 2011;133 (19):7316–7319. doi: 10.1021/ja2015795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem Rev. 2006;106 (8):3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 14.Igarashi M, Shida T, Sasaki Y, Kinoshita N, Naganawa H, Hamada M, Takeuchi T. Vinylamycin, a new depsipeptide antibiotic, from Streptomyces sp. Jpn J Antibiot. 1999;52 (10):873–879. doi: 10.7164/antibiotics.52.873. [DOI] [PubMed] [Google Scholar]
- 15.Igarishi Y, Shimasaki R, Miyanaga S, Oku N, Onaka H, Sakurai H, Saiki I, Kitani S, Nihara T, Wimonsiravude W, Panbangred W. Rakicidin D, an inhibitor of tumor cell invasion from marine-derived Streptomyces sp. Nature. 2010;63:563–5654. doi: 10.1038/ja.2010.70. [DOI] [PubMed] [Google Scholar]
- 16.Meier JL, Niessen S, Hoover HS, Foley TL, Cravatt BF, Burkart MD. An orthogonal active site identification system (OASIS) for proteomic profiling of natural product biosynthesis. ACS Chem Biol. 2009;4 (11):948–957. doi: 10.1021/cb9002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meier JL, Patel AD, Niessen S, Meehan M, Kersten R, Yang JY, Rothmann M, Cravatt BF, Dorrestein PC, Burkart MD, Bafna V. Practical 4′-phosphopantetheine active site discovery from proteomic samples. J Proteome Res. 2011;10 (1):320–329. doi: 10.1021/pr100953b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meluzzi D, Zheng WH, Hensler M, Nizet V, Dorrestein PC. Top-down mass spectrometry on low-resolution instruments: characterization of phosphopantetheinylated carrier domains in polyketide and non-ribosomal biosynthetic pathways. Bioorg Med Chem Lett. 2008;18 (10):3107–3111. doi: 10.1016/j.bmcl.2007.10.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. J Nat Prod. 2007;70 (3):461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 20.Patzer SI, Braun V. Gene cluster involved in the biosynthesis of griseobactin, a catechol-peptide siderophore of Streptomyces sp ATCC 700974. J Bacteriol. 2010;192 (2):426–435. doi: 10.1128/JB.01250-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rottig M, Medema MH, Blin K, Weber T, Rausch C, Kohlbacher O. NRPSpredictor2-a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 2011;39:W362–W367. doi: 10.1093/nar/gkr323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scherlach K, Hertweck C. Triggering cryptic natural product biosynthesis in microorganisms. Org Biomol Chem. 2009;7 (9):1753–1760. doi: 10.1039/b821578b. [DOI] [PubMed] [Google Scholar]
- 23.Udwary DW, Gontang EA, Jones AC, Jones CS, Schultz AW, Winter JM, Yang JY, Beauchemin N, Capson TL, Clark BR, Esquenazi E, Eustaquio AS, Freel K, Gerwick L, Gerwick WH, Gonzalez D, Liu WT, Malloy KL, Maloney KN, Nett M, Nunnery JK, Penn K, Prieto-Davo A, Simmons TL, Weitz S, Wilson MC, Tisa LS, Dorrestein PC, Moore BS. Significant natural product biosynthetic potential of actinorhizal symbionts of the genus Frankia, as revealed by comparative genomic and proteomic analyses. Appl Environ Microb. 2011;77 (11):3617–3625. doi: 10.1128/AEM.00038-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Wezel GP, McDowall KJ. The regulation of the secondary metabolism of Streptomyces: new links and experimental advances. Nat Prod Rep. 2011;28 (7):1311–1333. doi: 10.1039/c1np00003a. [DOI] [PubMed] [Google Scholar]
- 25.Walsh CT. Polyketide and nonribosomal peptide antibiotics. Science. 2004;303:1805–1810. doi: 10.1126/science.1094318. [DOI] [PubMed] [Google Scholar]
- 26.Walsh CT, Fischbach MA. Natural products version 2.0: connecting genes to molecules. J Am Chem Soc. 2010;132 (8):2469–2493. doi: 10.1021/ja909118a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamazaki Y, Kunimoto S, Ikeda D. Rakicidin A: a hypoxia-selective cytotoxin. Biol Pharm Bull. 2007;30 (2):261–265. doi: 10.1248/bpb.30.261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.