

Abstract
Pain and post-herpetic neuralgia (PHN) are common and highly distressing complications of herpes zoster that remain a significant public health concern and in need of improved therapies. Zoster results from reactivation of the herpesvirus varicella zoster virus (VZV) from a neuronal latent state established at the primary infection (varicella). PHN occurs in some one fifth to one third of zoster cases with severity, incidence, and duration of pain increasing with rising patient age. While VZV reactivation and the ensuing ganglionic damage trigger the pain response, the mechanisms underlying protracted PHN are not understood, and the lack of an animal model of herpes zoster (reactivation) makes this issue more challenging. A recent preclinical rodent model has developed that opens up the potential to allow the exploration of the underlying mechanisms and treatments for VZV-induced pain. Rats inoculated with live cell-associated human VZV into the hind paw reliably demonstrate thermal hyperalgesia and mechanical allodynia for extended periods and then spontaneously recover. Dorsal root ganglia express a limited VZV gene subset, including the IE62 regulatory protein, and upregulate expression of markers suggesting a neuropathic pain state. The model has been used to investigate treatment modalities and aspects of pain signaling and is under investigation by the authors to delineate VZV genetics involved in the induction of pain. This article compares human zoster-associated pain and PHN to the pain indicators in the rat and poses important questions that, if answered, could be the basis for new treatments.
Keywords: Varicella zoster virus, Human herpesvirus 3, Post-herpetic neuralgia, Pain measurement, Animal models, Allodynia, Hyperalgesia
Varicella, latency, and zoster
Varicella zoster virus (VZV), one of the eight known human herpesviruses, causes varicella (or chickenpox) upon primary infection and herpes zoster (also called “shingles”) following reactivation from a neuronal latent state established during the primary infection. A susceptible unvaccinated population in temperate climates will largely experience varicella at early ages, often during school-based epidemics. The self-resolving disease leads to the development of an adaptive immunity that clears the infection in most and provides a protective immunity to further varicella disease. The protective immunity underlies successful development and deployment of a live attenuated varicella vaccine, which is given to most children in the USA in a two-dose regimen prior to entry to school. Its widespread use (estimated coverage rates of approximately 80% in the USA) has dramatically declined varicella cases and created a herd immunity that has reduced hospitalizations and mortality associated with varicella (Chaves et al. 2008; Reynolds et al. 2008; Seward et al. 2008).
However, most adults in the USA were infected prior to vaccine licensure (in 1995) and still harbor wild-type VZV in their ganglia with the potential to reactivate and cause zoster. VZV DNA is found in sensory and autonomic ganglia along the entire neuraxis of infected individuals in a latent state that is largely restricted to neurons. Evidence from human cadaver dorsal root ganglia (DRG) suggest that VZV latency is associated with a limited lytic gene expression repertoire, in which mRNAs from some VZV genes (including open reading frames (ORFs) 4, 21, 29, 62, 63, 66) are expressed in some neurons in human DRG (Azarkh et al. 2011; Gilden et al. 2011; Kennedy and Cohrs 2010). Protein expression from the ORF 62, 63, and 66 genes has also been described (Cohrs et al. 2003; Annunziato et al. 1998; Lungu et al. 1998; Zerboni et al. 2010b), but this is controversial because of the recent indication that many antibodies used in such analyses also cross-react with human blood group A antigens (Zerboni et al. 2011). A clear picture of the molecular protein signatures of VZV latency has not yet arisen.
Zoster is estimated to occur at lifetime risk levels of about 30% in the general population and more than 50% in those over 85 years of age. Clinically, zoster manifests as painful large vesicular skin rashes that are geographically contained, representing peripheral delivery of infectious virus by multiple neurons to the skin from one or two adjacent sensory ganglia. This establishes an intraganglionic spread of VZV prior to peripheral delivery, a phenomenon not usually seen in reactivation of herpes simplex virus (HSV)-1 and HSV-2. At the periphery, transfer of VZV from axons to skin tissue leads to replication within epidermal cells, cell–cell spread, and rash extension that is contained by innate and adaptive immune responses. Zoster can occur anywhere on the body, but the most common sites represent reactivation from the thoracic dermatomes and the cutaneous distribution of the ophthalmic branch of the trigeminal nerve. Prime factors associated with zoster are increasing age (likely reflecting natural immune senescence) and cellular immune decline from disease, its treatment, or from iatrogenic cause. Other factors include female gender, mechanical trauma, ethnicity, stress, certain diseases and underlying genetic traits, and a lack of exposure to varicella throughout life (Gershon et al. 2010; McDonald et al. 2009; Schmader et al. 1995; Thomas et al. 2004). The role of age and immune status are by far the most striking, with zoster incidence rising sharply between 50 and 60 years. People over age of 60 are at 8–10-fold increased incidence of herpes zoster compared to those under age 60. By 85, not only does lifetime incidence approach 50% (Hope-Simpson 1965) but recurrent zoster also reaches near 5–10%. A role for cellular immunity in VZV reactivation is indicated in part by the clinical trials of the zoster vaccine. When given to VZV-positive individuals, vaccine boosting of immunity results in reduced zoster incidence and less severe disease if it does occur (Oxman et al. 2005). Zoster incidence is much higher and often more severe in patients with immune suppression caused by HIV infection, cancer, organ transplantation, immune-mediated diseases, and immunosuppressive drug treatment (see Oxman 2009 for review).
Pain associated with herpes zoster
Zoster is more often associated with neurological complications and sequelae than all other human herpesviruses, with the most common being pain before, during, and after the skin manifestations. Zoster is also associated with several central nervous system (CNS) diseases of a range of severity and complexity, with some neurological disease resulting from VZV-induced vasculopathies of cranial arteries and stroke (Gilden et al. 2009, 2011; Steiner et al. 2007). Neurological disease is not only caused directly by virus replication but can result from indirect causes such as stroke (Mareedu et al. 2011; Nagel et al. 2011) or by uncontrolled innate and adaptive immune responses to the virus infections that are capable of causing significant collateral damage to normal tissue function, even after active virus clearance in the host (Gilden et al. 2011; Nagel et al. 2011).
Pain associated with zoster reaches rates as high as 90% (see recent reviews including Argoff 2011, Gilden et al. 2011, Johnson et al. 2010, Philip and Thakur 2011, and Pickering and Leplege 2011). Pain is loosely divided into zoster-associated pain (ZAP) and post-herpetic neuralgia (PHN). ZAP includes a clinical prodrome of pain encountered 2–4 days before the onset of rash, pain during rash, and pain enduring after resolution of the rash, although it is well documented that pain and neurological involvement may occur without any skin disease (termed “zoster sine herpete”) (Gilden et al. 2010). The term PHN is reserved for pain extending beyond the resolution of skin lesions, with most groups defining PHN as pain lasting longer than 3 months. PHN is the major health problem and causes much of the health burden associated with zoster, since it may last months, years, and even until death. Quality of life issues frequently come into play (Aunhachoke et al. 2011). A significant portion of zoster patients experience PHN—for example, in the shingles vaccine prevention study, PHN occurred in 31% of unvaccinated zoster patients (Oxman et al. 2005). For reasons that are not clear, PHN duration and severity increases with age, with PHN being rare in zoster patients under 40, but occurring in 21% of zoster at 60–69 and >34% over 85 years of age. While PHN patients describe several types of pain, mechanical allodynia is the most distressing and most common. This type of allodynia is pain invoked by light innocuous tactile stimulation, such as a gust of wind, which may persist long after the stimulus is gone. Allodynia is encountered by more than 70% of all PHN patients and frequently leads to impaired sleep, distress, and depression that reduce quality of life (Johnson et al. 2010).
The treatment of VZV-induced pain and PHN is greatly in need of improvement because many PHN patients do not respond to any therapy. Recent analyses of clinical treatment trials and strategies indicated that only 30–50% of patients obtain more than 50% pain relief with any treatment (Hempenstall et al. 2005), often at a cost of severe side effects. Pain during zoster may respond to antivirals that rapidly limit viral replication and spread, particularly if applied within 72 h of onset, but antivirals do not effectively reduce most PHN (Li et al. 2009; Pavan-Langston 2008). ZAP and PHN may also respond to steroids, often used in combination with antiviral cover (Tyring et al. 2000), but the benefits of steroid and antiviral treatment on PHN after the acute phase is over are debatable (Chen et al. 2010). Since PHN shows indicators of a neuropathic pain state, treatments for neuropathic pain can often be more effective than standard anti-inflammatory therapy. Indications suggest that early diagnosis and initiation of treatment strategies are more effective than if delayed (Bowsher 1997). PHN treatments can be divided into (1) antidepressants, including the tricyclic antidepressants amytryptiline, desipramine, and nortriptyline that have analgesic activities that relieve pain in some patients but not all (Hempenstall et al. 2005). These act through several mechanisms, including the inhibition of norepinephrine and serotonin uptake and blockade of sodium channels, but can have serious side effects that limit their use and prescription. (2) Anticonvulsants, of which gabapentin and the recent successor pregabalin (Jensen-Dahm et al. 2011) are the most commonly prescribed, have analgesic properties that can effectively limit PHN, with superior orobioavailability and single dosing of pregabalin allowing the minimization of side effects. (3) Opioids, the mainstay of treatment for neuropathic pain, have demonstrated effect on PHN over placebo (Haanpaa 2009; Raja et al. 2002), but the considerable side effects and addictive properties, as well as the observation that much of PHN is not responsive to opioids, have resulted in their use as second- or third-line analgesics. (4) Topical treatments that include application of a nonsteroidal anti-inflammatory drug (NSAIDS), anesthetics, lidocaine, or capsaicin patches do reduce pain in some patients, while being less effective in others. These are ideal for patients not amenable to systemic therapies and are often associated with fewer serious side effects. (5) Vaccination with the shingles vaccine reduces the burden of illness caused by zoster and PHN by 69% and the incidence of zoster by 51%. Thus, zoster and PHN in a good fraction of vaccine recipients are prevented from ever arising (Oxman et al. 2005). However, the zoster vaccine, which is based on a 14 times more infectious version of the varicella vaccine, is not yet widely used or well accepted by the elderly population in the USA. (6) More radical and rarely used treatment strategies include surgery to excise the dorsal root entry zone into the spinal cord, gangliotomy, and large-scale skin replacement. However, given that many PHN patients do not respond to any therapy, it is clear that pain and PHN associated with zoster is an unmet need for improvement.
Mechanism of VZV-induced pain
Mechanistically, pain associated with zoster and PHN are multifactorial and heterogeneous disorders of different and multiple overlapping processes and mechanisms that affect both central and peripheral processes. However, they remain poorly defined, particularly for PHN (see Delaney et al. 2009 for a recent review). The underlying initiator is VZV reactivation, lytic gene expression, virus intraganglionic spread, and virus-induced tissue damage in the ganglia. Reactivation presumably occurs initially in one reactivating sensory neuron: Models suggest that the reactivating neuron forms syncytia with and gains access to adjacent satellite cells for further replication (Reichelt et al. 2008). VZV undergoes intraganglionic spread and viral replication in adjacent neurons and non-neuronal cell types in the ganglia, reaching and infecting adjacent neurons that enable multi-axonal delivery to the periphery. The resulting host immune response to VZV replication, including lymphocytic infiltration and inflammation, most likely contributes to tissue damage. The histological analyses of Cadaver DRG from patients who died with zoster (e.g., Esiri and Tomlinson 1972; Watson et al. 1988) reveal signs of VZV replication such as intranuclear inclusions, viral antigens and particles, and ganglionic spread of virus to neurons, satellite cells and supporting fibroblasts. The ganglionic replication is associated with focal hemorrhage, demyelination, axonal degeneration, and necrosis of sensory fibers and support cells. An extensive immune infiltrate occurring after zoster has recently been characterized (Gowrishankar et al. 2010). The small fiber afferents innervating the skin show degeneration as well as impaired function, and the central system has degeneration of related motor and sensory roots and damage that may extend to the posterior nerve root and adjacent regions of the spinal cord and or brainstem. These acute changes damage the sensory and ganglionic nerves and induce abnormal afferent signals of pain that adds to the nociceptive signals that stem from the lesions themselves.
The mechanisms underlying the transition from acute to persistent long-term pain in PHN are less clear. While it has been suggested that some PHN involves persistent viral replication, this seems to be minimal, since antivirals do not routinely provide significant relief of PHN. Allodynia suggests a sensitized neuropathic hyperexcitability pain state induced by damage to the nervous system. Examination of the skin from affected dermatomes of PHN patients reveals some degeneration of the small fiber afferents infiltrating the skin and altered function of multiple neuronal types (Baron and Saguer 1993, 1995; Rowbotham et al. 1996). The CNS is also involved in some PHN patients, and it has been suggested that VZV induces a loss of inhibitory central processing interneurons that provide inhibitors and modulators to nociceptive neuron signaling. This leads to reorganization of central processing of the input and hyperexcitability of nociceptive neurons at the spinal level, giving rise to a hypersensitization state and an exaggerated response. It has also been suggested that second-order nociceptive neurons that normally respond to input from C-fibers respond instead to input from myelinated A-fibers carrying low-threshold mechanoreceptors (Bennett 1994). The hyperexcitability of neurons is due in part to molecular changes in the neurons, including changes in expression of sodium channels, changes in γ-aminobuytyric acid inhibition on nociception, and the modulation of neuropeptides and their expression patterns.
The rodent model of VZV-induced pain
Examination of the pain mechanisms in humans is clearly very restricted, and a validated VZV-induced pain model will enable exploration of mechanisms of PHN. Despite the high human specificity and host restriction of VZV, animal models of VZV pathogenesis have developed and enabled paradigm-shifting knowledge to be gained regarding primary infection. Studies in SCID-Hu mice containing skin and thymus/liver implants (Arvin et al. 2010; Moffat et al. 2007) provided the basis for a now accepted model of primary infection that involves a T cell and/or professional antigen-presenting cell-mediated viremia that distributes virus to skin from respiratory lymphoid organs such as tonsils. The model also revealed roles for innate responses in virus control in skin (Ku et al. 2005). SCID-hu mice with human ganglionic/neurological tissue (Zerboni et al. 2010a), coupled with ex vivo culture models such as human fetal ganglia explants and model neuronal cultures (Christensen et al. 2011; Gowrishankar et al. 2007; Steain et al. 2010; Markus et al. 2011; Pugazhenthi et al. 2011), will likely shed light on aspects of the VZV latent state and VZV neurotropism. However, the assessment of pain is a conscious response that requires functioning in vivo models with intact sensory enervation of both peripheral and central systems. One immune competent small animal model supporting productive VZV replication, the guinea pig, requires using cell-adapted VZV (Lowry et al. 1992; Myers and Stanberry 1991). While animal DRG contain persisting VZV DNA and virus transmits to other animals, no clinical signs of disease are apparent, and we have not observed signs of VZV-induced pain responses in this model (unpublished observations). Rats have been used to model VZV latency by several groups (Cohen 2010 for review). Animals sero-convert and VZV DNA persists in the corresponding DRG after peripheral infection, expressing mRNAs from ORFs 63, 29, and 62 and possibly the protein from ORF 63 (Cohen 2010; Cohen et al. 2004). These genes have also been reported to be expressed in human ganglia (Cohrs and Gilden 2007). Specific viral genes have been shown to be needed for efficient establishment of persistence in cotton rats, including ORF63 (Cohen et al. 2004). However, it appears that VZV does not complete a full cycle of replication in primary rat fibroblasts (unpublished observations), and the model likely relies on direct axonal infection of neurons from the periphery by the cell-associated VZV inoculum. VZV gene expression in the DRG could reflect an abortive infection in neurons in which full productive viral replication is blocked by a requirement of an essential human specific factor.
When it was shown that rats inoculated with VZV at the periphery develop prolonged behavioral indicators suggestive of an altered pain state, it opened the potential for preclinical testing and for the exploration of underlying mechanisms of pain induction. The model, developed by Fleetwood-Walker’s group (Fleetwood-Walker et al. 1999; Garry et al. 2005) and extended by others (Dalziel et al. 2004; Hasnie et al. 2007; Medhurst et al. 2008; Wallace et al. 2007), involves inoculation of high titer (we use 2 × 105 plaque-forming units (PFU) per injection) of cell-associated VZV (MeWo, CV-1 and MRC5 cells have been employed) into the glabrous region of the hind footpad. By 3–5 days following inoculation, behavioral responses to noxious thermal and innocuous mechanical stimuli initiate and remain for 6–14 weeks, and these do not develop in the controls (uninjected, uninfected cell-injected, or sham-injected footpad; Fig. 1). Animals also exhibit behavioral morbidity indicative of anxious-like behaviors in open-field paradigms (Hasnie et al. 2007) not unlike conscious behavioral changes and depression seen in many PHN patients. In the rat model, mechanical allodynia (MA) is assessed as a significant lowering of the mechanical force threshold needed to induce a paw withdrawal, applied using calibrated von Frey nylon monofilaments that apply a specific weight to the paw at bending, ranging from 1.5 to 125 g. Hasnie et al. also used a cotton bud to stimulate the footpad and measured paw withdrawal latency. The approach uses a set of timed repeated applications, and a pain positive score is to withdrawal occurring twice over ten applications. To show thermal hyperalgesia (TH), a paw withdrawal measurement is taken following application of noxious thermal stimuli applied to the plantar region of the foot. The mean latency is measured for withdrawal to rising heat applied from 30°C to 55°C, applied using apparatus such as Hargreaves apparatus that applies a consistent curve of rising heat (e.g., Harvard Apparatus, Inc.). Responses that develop are VZV-specific and do not develop in the contralateral limb or limbs injected with equal levels of uninfected cells. It has also been shown that animals show no response when injected with heat-inactivated VZV-infected cells, suggesting viable VZV is needed to infect the axons (Dalziel et al. 2004). In our hands, MeWo cells, MRC5s, or retinal pigmented epithelial lines such as ARPE provide the highest VZV titer/cell ratios, and these cells do not induce indices of pain at equivalents to VZV infection. The pain response does not develop a chronic pattern in animals inoculated with high-titer HSV-1 beyond a short-lived response (Dalziel et al. 2004).
Fig. 1.

VZV-induced pain in the rat model of PHN. Male Sprague–Dawley rats (acquired from Charles River Labs) were subjected to mechanical allodynia (MA) behavioral studies 1 week prior to the inoculation of VZV, measuring the removal of the injected (ipsilateral) and non-injected (contralateral) hind paw to non-noxious stimulation with varying size von Frey hairs to determine the minimal strength fiber that induces foot withdrawal. One day later, rats were anesthetized and injected into the ipsilateral hind paw with 100 μL containing either control cells (N=5) or cells infected with VZV1587 (N=9) expressing GFP tagged to functional ORF66, or VZV1755 (N=9) containing GFP replacing the amino terminal two third of the ORF66 coding sequence (Eisfeld et al. 2007). Virus inocula into the footpad were 2 × 105 PFU in >70% GFP-positive cell cultures. At each subsequent week post-infection, the MA behavioral analysis was repeated. Results are plotted as the MA score ratio of the injected ipsilateral hind paw to the uninjected contralateral hind paw
The level of VZV required at inoculation to induce pain is important, as pain indices are dose responsive (Garry et al. 2005; Hasnie et al. 2007). VZV infection requires injection of infected cell-associated virus, as cell-free VZV cannot be easily obtained in sufficient titer (Dalziel et al. 2004; Garry et al. 2005). The observation that treatment of rats with valacyclovir from VZV infection did not affect the onset of behavioral indices of increased pain suggests that viral replication was not needed for the chronic pain state. Rather, the initial inoculate and its ability to directly infect the axons are critical. To date, cytopathic effect has been the main gauge to assess VZV infection levels in the inoculate, and this can be difficult to standardize, resulting in VZV preparation variation from batch to batch in the number of infected to infected cells. Hasnie et al. (2007) reported only short-term TH, and it has been argued that virus titer was the cause (Delaney et al. 2009). We have improved such variability by employing green fluorescent protein (GFP) reporter fluorescent VZV, including VZV in which EGFP is either an amino terminal fusion to the VZV ORF66 protein kinase (Fig. 1, VZV1587) (Eisfeld et al. 2007) or replaces part of the ORF66 gene (Fig. 1, VZV1755). These viruses were generated using the pOka cosmids, but VZV derived from the VZV pOka BAC (Tischer et al. 2007) also induces chronic MA and TH (Fig. 2). The GFP reported in the cultures allows accurate assessment of the number of infected cells, and we routinely use cultures showing >70% GFP positivity over uninfected cells by flow cytometric analyses. The induction of pain by viruses developed from the current VZV genetic systems now sets the stage for analyses of VZV mutants for the ability to induce pain. For example, our work has now established that the VZV ORF66 protein is not required for VZV-induced pain in the model (Fig. 1).
Fig. 2.
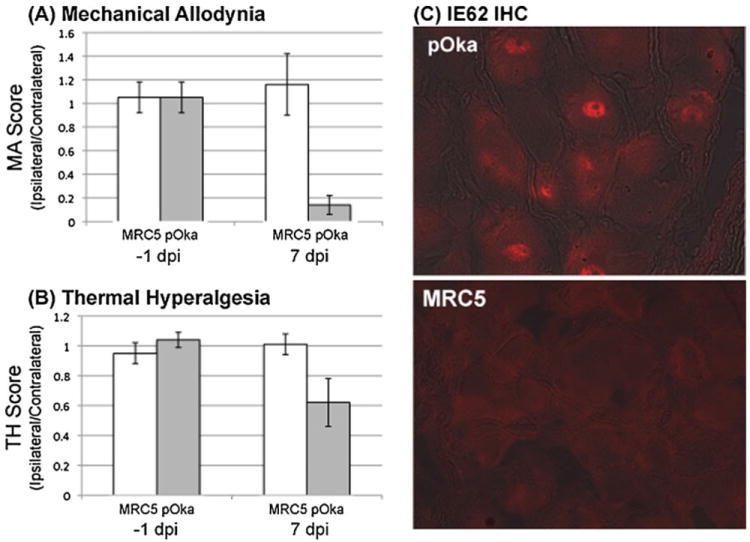
VZV pOka derived from the VZV BAC induces pain in the rat PHN model. Male rats were subjected to a mechanical allodynia (MA) and b thermal hyperalgesia (TH) behavioral testing 1 week prior to the inoculation of VZV BAC-derived pOka into the hind paw, as detailed in the legend of Fig. 1. The MA and TH levels were compared for the injected (ipsilateral) and non-injected (contralateral) hind paw to non-noxious stimulation (von Frey hairs) and heat withdrawal tests as detailed in Fig. 1. Rats were injected with control uninfected cells (N=5) or cells infected with the VZV pOka (N=9) at 2 × 105 PFU per injection. At 7 days post-infection, the MA/TH behavioral analyses were repeated. Results are plotted as the MA or TH score for the injected ipsilateral/uninjected contralateral hind paw. c Rats were then sacrificed 1–2 days later (days 8–9 post-infection), and fixed ganglia were processed for immunofluorescent probing analyses of tissue sections using rabbit anti-IE62 antibodies that were first pre-absorbed with rat tissue homogenates to remove nonspecific reactivity. Antibody was detected using Alexafluor 594-labeled anti-rabbit secondary antibody. IE62 showed nuclear staining in the DRG corresponding to the VZV pOka-injected footpad compared to the control (MRC5) cell-injected footpad DRGs (red IFA image overlaid on bright field). Two representative sections showing IE62 positivity revealed that 54 and 34 neurons showed detectable IE62 nuclear staining. IE62 staining was not detected in the DRG of uninfected cell-inoculated animals in any sections
It is not known how VZV induces prolonged pain, and why animals eventually spontaneously recover. One obvious consideration is VZV gene expression within neurons that induces the pain state and changes over time. To date, thin section immunohistochemical staining of the rat DRG has suggested IE63 protein (Fleetwood-Walker et al. 1999) and IE62 protein expression (Garry et al. 2005). Both groups reported cytoplasmic accumulation of the proteins in some neurons and more typical nuclear distribution in others. Our group has found that infection with VZV expressing GFP-tagged ORF66 does not result in levels of detectable GFP expression in the DRG: This may indicate a block to expression of the ORF66 protein, or may simply reflect very low levels of expression that is masked by an intrinsic autofluorescence of neurons in the green emission wavelength. We have found characteristic nuclear localization of IE62 in rat DRG at early times (8–10 days) post-VZV infection by immunofluorescent analyses of thin sections (Fig. 2), with multiple neurons per section staining. In two representative sections staining positive for IE62 at day 8, IE62-positive expression was found in 54 and 34 neurons. At week 4, very few neurons stained positive. We are particularly aware of reports of antibody cross-reactivity giving rise to artifacts (Zerboni et al. 2011), and we used previously detailed rabbit polyclonal antibodies (Eisfeld et al. 2006) that were pre-absorbed with uninfected rat tissue homogenates to reduce background cross-reactivities. The location of the IE62 seen in 8 days post-infection sections is reminiscent of that seen in lytic VZV infections of cultured cells, suggesting active regulation of transcription. Expression of IE62 is not seen in the contralateral ganglia or in ganglia of uninfected cell-inoculated animals. Both IE62 and IE63 proteins are known regulators of viral and host gene expression and are immediate early expressed. IE63 is involved in gene regulation (Zuranski et al. 2005), chromatin modulation (Ambagala et al. 2009; Habran et al. 2007), and the blockade of the IFN responses and signal-mediated halt to translation (Ambagala and Cohen 2007). IE62 is the principal transcriptional activator of viral gene expression (White et al. 2010) and also modulates IFN responses through cytoplasmic retention of IRF3 (Sen et al. 2010). IE62 was previously reported in multiple types of neurons, including unmyelinated C fiber afferent and myelinated αδ neurons, based on differential expression of the type III intermediate filament protein peripherin in unmyelinated sensory neurons and NF200 in larger myelinated neurons (Garry et al. 2005). While we found IE62 VZV protein expression in multiple neurons of several sections of DRG of pOka-infected rats, the levels are much less than that previously reported (Garry et al. 2005), and we have not observed the cytoplasmic forms at later times as seen by Garry et al. (2005). IE62 expression correlates with measurable MA and TH at 1 week post-infection (Fig. 2). Clearly additional studies are needed to determine if IE62 does indeed show changes in cellular localization as the model progresses into the extended pain period. The extent that other VZV proteins that are expressed in the rat DRG also needs to be addressed. Of interest is the recent suggestion that antibodies to IE62 augment brain-derived neurotrophic factor (BDNF) induced neuronal changes (Hama et al. 2010) (see below).
Expression of VZV regulatory proteins in DRG neurons suggests that VZV may influence host gene expression in the neuron to contribute to the pain state, and this has been borne out from both in intro and in vivo studies. Using an in vitro neuronal explant model, VZV infection and expression of the IE62 and IE63 regulatory proteins resulted in neurons becoming increased in sensitivity to adrenergic stimulation by norepinephrine (Kress and Fickenscher 2001). Injury to neurons changes gene expression of receptors, neurotransmitters, and signaling, and these induce multiple physiological, biochemical, and anatomical changes. Garry et al. (2005) examined expression of specific markers and peptides associated with neuropathic pain in DRG of rats showing pain indices and found upregulated expression of neuropeptide Y (NPY), galanin, ATF3, the voltage-gated calcium channel α2δ1, and the sodium channels NAv1.3 and Nav1.8 in the VZV-infected ganglia. These have all been taken to imply a neuropathic pain state is induced by VZV infection and perhaps VZV gene expression. NPY is one of several differentially expressed neuropeptides that undergo change in response to neuronal damage and is normally expressed at low levels. In DRG, it is upregulated in primary afferent DRG neurons which have been subjected to axotomy, demyelination, or injury. The increase expression of ATF-3 and galanin is likewise upregulated in neurons following peripheral nerve injuries, suggesting that the rodent host DRG has undergone some level of VZV-induced damage, as seen in human PHN patients. The increase expression of the voltage-gated calcium channel α2δ1 is consistent with PHN treatment with gabapentin, which has been proposed to exert its effect by modulating activity of this channel (Rogawski and Loscher 2004). The upregulation of the sodium channels was concluded to be further indication of neuropathic damage to neurons in rats infected by VZV. Dalziel et al. (2004) noted a lack of an obvious immune cell infiltrate, unlike that seen in the trigeminal ganglia of humans latently infected with HSV-1 (Verjans et al. 2007). However, preliminary work by us suggests the presence of a detectable VZV-induced immune infiltrate at 2 weeks following VZV infection in the footpad, in which a CD45+, CD4+, and CD8+ population was detected by flow cytometric staining of collagenase disrupted ganglia. At 4 weeks post-VZV infection (the time at which maximal pain responses develop), the levels of the infiltrate over that developing in the contralateral ganglia reach statistical significance (unpublished observations). The infiltrate appears to retract by week 8. These preliminary results suggest that an inflammatory component of the pain response may be occurring in this model. Of note is that HSV-1 latency is now well accepted to be associated with a CD8+ T cell virus-specific immune infiltrate that partly controls HSV reactivation. Its impairment by factors known to induce HSV-1 reactivation, such as stress, may well underlie recurrent HSV-1 disease (Freeman et al. 2007).
The rat model has been used as a preclinical tool to demonstrate parallel responses to known effective therapies for human PHN and to explore new drug interventions with potential to treat human PHN. Administration of gabapentin or the sodium channel inhibitors mexiletine and lamotrigine was shown by Garry et al. (2005) to provide significant short-term return to baseline indices of pain in the model when administered by gavage, using both the MA and TH formal testing. No effect of the NSAID diclofenac was found in animals. Hasnie et al. (2007) also demonstrated effectiveness of gabapentin, amytryptiline, and morphine by IP administration in reducing the pain behavior indices and further reported effective treatment by ibuprofen administration, also suggesting an inflammatory component to VZV-induced pain in the model. More recently, Medhurst et al. (2008) reported the successful and efficacious use of novel selective histamine H(3) antagonists again VZV-induced pain in the rat following oral administration, also suggesting an inflammatory component of pain in the model. Thus, the potential for new treatment modalities for relief of PHN may come to light using the model.
Perspectives and questions
The development of VZV-induced pain in the rat model allows several critical questions to be addressed that may lead to better treatment of PHN in patients. The rat model appears to mirror pain induced by VZV in human patients in that it persists for prolonged periods and shows indicators of the suspected neuropathic pain state of PHN neurons, and the pain indices respond to similar treatment strategies that are currently employed to relieve PHN. Obviously, the rat model does not fully reflect zoster (reactivated infection) and the extensive viral intraganglionic spread and virus initiated damage seen following a human VZV reactivation event. Given that pain and PHN are multifactorial and likely arise through different overlapping mechanisms and multiple pain signaling pathways, it seems likely that the model may only reflect one or some of the mechanisms underlying human PHN. Nevertheless, the model is validated for use as a preclinical model to evaluate new drugs, treatment strategies, and mechanisms (Medhurst et al. 2008; Wallace et al. 2007). Some remaining questions on the model being pursued by our group are the following:
What is the extent of VZV gene expression in the model and do changes in gene expression correlate with the pain response? It is intriguing that the MA and TH indices of pain in the rat reverse after a reproducible time, dependent on virus dose, and this reversal may correlate with cessation of expression of one or more VZV genes, or a relocalization of a regulatory protein to the cytoplasm. The rat model expressing IE62 in neurons may not be necessarily reflective of the latent-state gene expression reported by others, but perhaps more of an attempt at lytic replication that is aborted and not completed. Viral genes may be eventually silenced in the rat, resulting in return of host cell gene expression to that of normal non-neuropathic pain states. Obviously, other viral genes downstream of these VZV gene regulators may or may not be expressed—if they are, they may inflict damage on the neuron that contribute to the pain state.
Can VZV be mutated in a manner that does not induce pain indicators in this model? If so, this could form the basis of an improved and indelibly safer vaccine, as the currently licensed vaccine does reach the neuron, enter latency, and can reactivate to cause recurrent disease. A VZV that cannot access the ganglia due to inability of axonal transport or one that lacks expression of a critical gene involved in upregulating host gene expression involved in inducing the pain state would be ideal. An intriguing and quite different mechanism is that the expression of VZV IE62 protein and the intrathecal immune response to it are involved in pain. In a mouse model, antibodies to IE62 may stimulate pain hypersensitivity by cross-reacting with BDNF (Hama et al. 2010). The authors showed that antibodies reacting to IE62 region from 414 to 429 cross-reacted with the BDNF dimer and augment the expression of BDNF-related transcripts in neurons. This group showed that antibodies to IE62 peptides expressed in bacteria resulted in reduced mechanical allodynia threshold responses to von Frey hair filament stimulation in mice with nerve injury. The rat model is now open for testing of VZV recombinants that lack the IE62-BDNF cross-reacting domains that, if the model of Hama et al. (2010) is correct, should result in VZV unable to induce pain signals.
Can novel treatment strategies be used to block the pain response? Clearly the rat model not only provides a preclinical platform to evaluate new compounds but could also permit avenues for exploration of pain treatment by less conventional approaches. The development of HSV-expressing modulators of pain is one example: HSV-1 rarely causes increased nociception and enters latency in sensory neurons in ganglia. Replication-defective HSV-expressing modulators of the tonal response (such as proenkephalin) have been shown to be effective at relieving inflammatory and neuropathic pain in other models (Goss et al. 2002; Yeomans and Wilson 2009). The application of such delivery systems could be an exciting avenue of treatment that may give better relief to PHN patients and the quality of life altering consequences of it.
Acknowledgments
The authors wish to acknowledge funding and support for this work by NIH grant NS064022 from the National Institute of Neurological Diseases and Stroke, by core grant EY08098 from the National Eye Institute, and by unrestricted funds from the Eye & Ear Institute of Pittsburgh and from Research to Prevent Blindness, Inc. We also thank Mike Yee and Mingdi Zhang for excellent technical assistance.
Contributor Information
Paul R. Kinchington, Department of Ophthalmology, University of Pittsburgh, 1020 EEI Building, 203 Lothrop Street, Pittsburgh, PA 15213, USA, kinchingtonp@upmc.edu Department of Microbiology and Molecular Genetics, University of Pittsburgh, Pittsburgh, PA, USA.
William F. Goins, Department of Microbiology and Molecular Genetics, University of Pittsburgh, Pittsburgh, PA, USA
References
- Ambagala AP, Cohen JI. Varicella-zoster virus IE63, a major viral latency protein, is required to inhibit the alpha interferon-induced antiviral response. J Virol. 2007;81:7844–7851. doi: 10.1128/JVI.00325-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambagala AP, Bosma T, Ali MA, Poustovoitov M, Chen JJ, Gershon MD, Adams PD, Cohen JI. Varicella-zoster virus immediate-early 63 protein interacts with human antisilencing function 1 protein and alters its ability to bind histones h3.1 and h3.3. J Virol. 2009;83:200–209. doi: 10.1128/JVI.00645-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato P, LaRussa P, Lee P, Steinberg S, Lungu O, Gershon AA, Silverstein S. Evidence of latent varicella-zoster virus in rat dorsal root ganglia. J Infect Dis. 1998;178(Suppl 1):S48–S51. doi: 10.1086/514261. [DOI] [PubMed] [Google Scholar]
- Argoff CE. Review of current guidelines on the care of postherpetic neuralgia. Postgrad Med. 2011;123:134–142. doi: 10.3810/pgm.2011.09.2469. [DOI] [PubMed] [Google Scholar]
- Arvin AM, Moffat JF, Sommer M, Oliver S, Che X, Vleck S, Zerboni L, Ku CC. Varicella-zoster virus T cell tropism and the pathogenesis of skin infection. Curr Top Microbiol Immunol. 2010;342:189–209. doi: 10.1007/82_2010_29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aunhachoke K, Bussaratid V, Chirachanakul P, Chua-Intra B, Dhitavat J, Jaisathaporn K, Kaewkungwal J, Kampirapap K, Khuhaprema T, Pairayayutakul K, Pitisuttithum P, Sindhvananda J, Thaipisuttikul Y. Measuring herpes zoster, zoster-associated pain, postherpetic neuralgia-associated loss of quality of life, and healthcare utilization and costs in Thailand. Int J Dermatol. 2011;50:428–435. doi: 10.1111/j.1365-4632.2010.04715.x. [DOI] [PubMed] [Google Scholar]
- Azarkh Y, Dolken L, Nagel M, Gilden D, Cohrs RJ. Synthesis and decay of varicella zoster virus transcripts. J Neurovirol. 2011;17:281–287. doi: 10.1007/s13365-011-0029-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron R, Saguer M. Postherpetic neuralgia. Are C-nociceptors involved in signalling and maintenance of tactile allodynia? Brain. 1993;116(Pt 6):1477–1496. doi: 10.1093/brain/116.6.1477. [DOI] [PubMed] [Google Scholar]
- Baron R, Saguer M. Mechanical allodynia in postherpetic neuralgia: evidence for central mechanisms depending on nociceptive C-fiber degeneration. Neurology. 1995;45:S63–S65. doi: 10.1212/wnl.45.12_suppl_8.s63. [DOI] [PubMed] [Google Scholar]
- Bennett GJ. Hypotheses on the pathogenesis of herpes zoster-associated pain. Ann Neurol. 1994;35(Suppl):S38–S41. doi: 10.1002/ana.410350712. [DOI] [PubMed] [Google Scholar]
- Bowsher D. The effects of pre-emptive treatment of postherpetic neuralgia with amitriptyline: a randomized, double-blind, placebo-controlled trial. J Pain Symptom Manage. 1997;13:327–331. doi: 10.1016/s0885-3924(97)00077-8. [DOI] [PubMed] [Google Scholar]
- Chaves SS, Haber P, Walton K, Wise RP, Izurieta HS, Schmid DS, Seward JF. Safety of varicella vaccine after licensure in the United States: experience from reports to the vaccine adverse event reporting system, 1995–2005. J Infect Dis. 2008;197(Suppl 2):S170–S177. doi: 10.1086/522161. [DOI] [PubMed] [Google Scholar]
- Chen N, Yang M, He L, Zhang D, Zhou M, Zhu C. Corticosteroids for preventing postherpetic neuralgia. Cochrane Database Syst Rev. 2010 doi: 10.1002/14651858.CD005582.pub3. CD005582. [DOI] [PubMed] [Google Scholar]
- Christensen J, Steain M, Slobedman B, Abendroth A. Differentiated neuroblastoma cells provide a highly efficient model for studies of productive varicella-zoster virus infection of neuronal cells. J Virol. 2011;85:8436–8442. doi: 10.1128/JVI.00515-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JI. Rodent models of varicella-zoster virus neurotropism. Curr Top Microbiol Immunol. 2010;342:277–289. doi: 10.1007/82_2010_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JI, Cox E, Pesnicak L, Srinivas S, Krogmann T. The varicella-zoster virus open reading frame 63 latency-associated protein is critical for establishment of latency. J Virol. 2004;78:11833–11840. doi: 10.1128/JVI.78.21.11833-11840.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohrs RJ, Gilden DH, Kinchington PR, Grinfeld E, Kennedy PG. Varicella-zoster virus gene 66 transcription and translation in latently infected human Ganglia. J Virol. 2003;77(12):6660–6665. doi: 10.1128/JVI.77.12.6660-6665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohrs RJ, Gilden DH. Prevalence and abundance of latently transcribed varicella-zoster virus genes in human ganglia. J Virol. 2007;81:2950–2956. doi: 10.1128/JVI.02745-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalziel RG, Bingham S, Sutton D, Grant D, Champion JM, Dennis SA, Quinn JP, Bountra C, Mark MA. Allodynia in rats infected with varicella zoster virus—a small animal model for post-herpetic neuralgia. Brain Res Brain Res Rev. 2004;46:234–242. doi: 10.1016/j.brainresrev.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Delaney A, Colvin LA, Fallon MT, Dalziel RG, Mitchell R, Fleetwood-Walker SM. Postherpetic neuralgia: from preclinical models to the clinic. Neurotherapeutics. 2009;6:630–637. doi: 10.1016/j.nurt.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisfeld AJ, Turse SE, Jackson SA, Lerner EC, Kinchington PR. Phosphorylation of the varicella-zoster virus (VZV) major transcriptional regulatory protein IE62 by the VZV open reading frame 66 protein kinase. J Virol. 2006;80:1710–1723. doi: 10.1128/JVI.80.4.1710-1723.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisfeld AJ, Yee MB, Erazo A, Abendroth A, Kinchington PR. Downregulation of class I major histocompatibility complex surface expression by varicella-zoster virus involves open reading frame 66 protein kinase-dependent and -independent mechanisms. J Virol. 2007;81:9034–9049. doi: 10.1128/JVI.00711-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esiri MM, Tomlinson AH. Herpes zoster. Demonstration of virus in trigeminal nerve and ganglion by immunofluorescence and electron microscopy. J Neurol Sci. 1972;15:35–48. doi: 10.1016/0022-510x(72)90120-7. [DOI] [PubMed] [Google Scholar]
- Fleetwood-Walker SM, Quinn JP, Wallace C, Blackburn-Munro G, Kelly BG, Fiskerstrand CE, Nash AA, Dalziel RG. Behavioural changes in the rat following infection with varicella-zoster virus. J Gen Virol. 1999;80(Pt 9):2433–2436. doi: 10.1099/0022-1317-80-9-2433. [DOI] [PubMed] [Google Scholar]
- Freeman ML, Sheridan BS, Bonneau RH, Hendricks RL. Psychological stress compromises CD8+ T cell control of latent herpes simplex virus type 1 infections. J Immunol. 2007;179:322–328. doi: 10.4049/jimmunol.179.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garry EM, Delaney A, Anderson HA, Sirinathsinghji EC, Clapp RH, Martin WJ, Kinchington PR, Krah DL, Abbadie C, Fleetwood-Walker SM. Varicella zoster virus induces neuropathic changes in rat dorsal root ganglia and behavioral reflex sensitisation that is attenuated by gabapentin or sodium channel blocking drugs. Pain. 2005;118:97–111. doi: 10.1016/j.pain.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Gershon AA, Gershon MD, Breuer J, Levin MJ, Oaklander AL, Griffiths PD. Advances in the understanding of the pathogenesis and epidemiology of herpes zoster. J Clin Virol. 2010;48(Suppl 1):S2–S7. doi: 10.1016/S1386-6532(10)70002-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilden D, Cohrs RJ, Mahalingam R, Nagel MA. Varicella zoster virus vasculopathies: diverse clinical manifestations, laboratory features, pathogenesis, and treatment. Lancet Neurol. 2009;8:731–740. doi: 10.1016/S1474-4422(09)70134-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilden D, Cohrs RJ, Mahalingam R, Nagel MA. Neurological disease produced by varicella zoster virus reactivation without rash. Curr Top Microbiol Immunol. 2010;342:243–253. doi: 10.1007/82_2009_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilden D, Mahalingam R, Nagel MA, Pugazhenthi S, Cohrs RJ. Review: the neurobiology of varicella zoster virus infection. Neuropathol Appl Neurobiol. 2011;37:441–463. doi: 10.1111/j.1365-2990.2011.01167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss JR, Harley CF, Mata M, O’Malley ME, Goins WF, Hu X, Glorioso JC, Fink DJ. Herpes vector-mediated expression of proenkephalin reduces bone cancer pain. Ann Neurol. 2002;52:662–665. doi: 10.1002/ana.10343. [DOI] [PubMed] [Google Scholar]
- Gowrishankar K, Slobedman B, Cunningham AL, Miranda-Saksena M, Boadle RA, Abendroth A. Productive varicella-zoster virus infection of cultured intact human ganglia. J Virol. 2007;81:6752–6756. doi: 10.1128/JVI.02793-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar K, Steain M, Cunningham AL, Rodriguez M, Blumbergs P, Slobedman B, Abendroth A. Characterization of the host immune response in human ganglia after herpes zoster. J Virol. 2010;84:8861–8870. doi: 10.1128/JVI.01020-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haanpaa M. Controlled release oxycodone—an evidence-based treatment for pain in acute herpes zoster. Pain. 2009;142:175–176. doi: 10.1016/j.pain.2009.01.025. [DOI] [PubMed] [Google Scholar]
- Habran L, El MN, Di Valentin E, Sadzot-Delvaux C, Bontems S, Piette J. The varicella-zoster virus immediate-early 63 protein affects chromatin-controlled gene transcription in a cell-type dependent manner. BMC Mol Biol. 2007;8:99. doi: 10.1186/1471-2199-8-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hama Y, Shiraki K, Yoshida Y, Maruyama A, Yasuda M, Tsuda M, Honda M, Takahashi M, Higuchi H, Takasaki I, Daikoku T, Tsumoto T. Antibody to varicella-zoster virus immediate-early protein 62 augments allodynia in zoster via brain-derived neurotrophic factor. J Virol. 2010;84:1616–1624. doi: 10.1128/JVI.02061-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasnie FS, Breuer J, Parker S, Wallace V, Blackbeard J, Lever I, Kinchington PR, Dickenson AH, Pheby T, Rice AS. Further characterization of a rat model of varicella zoster virus-associated pain: relationship between mechanical hypersensitivity and anxiety-related behavior, and the influence of analgesic drugs. Neuroscience. 2007;144:1495–1508. doi: 10.1016/j.neuroscience.2006.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempenstall K, Nurmikko TJ, Johnson RW, A’Hern RP, Rice AS. Analgesic therapy in postherpetic neuralgia: a quantitative systematic review. PLoS Med. 2005;2:e164. doi: 10.1371/journal.pmed.0020164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope-Simpson RE. The nature of herpes zoster: a long-term study and a new hypothesis. Proc R Soc Med. 1965;58:9–20. [PMC free article] [PubMed] [Google Scholar]
- Jensen-Dahm C, Rowbotham MC, Reda H, Petersen KL. Effect of a single dose of pregabalin on herpes zoster pain. Trials. 2011;12:55. doi: 10.1186/1745-6215-12-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RW, Bouhassira D, Kassianos G, Leplege A, Schmader KE, Weinke T. The impact of herpes zoster and post-herpetic neuralgia on quality-of-life. BMC Med. 2010;8:37. doi: 10.1186/1741-7015-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy PG, Cohrs RJ. Varicella-zoster virus human ganglionic latency: a current summary. J Neurovirol. 2010;16:411–418. doi: 10.3109/13550284.2010.515652. [DOI] [PubMed] [Google Scholar]
- Kress M, Fickenscher H. Infection by human varicella-zoster virus confers norepinephrine sensitivity to sensory neurons from rat dorsal root ganglia. FASEB J. 2001;15:1037–1043. doi: 10.1096/fj.00-0440com. [DOI] [PubMed] [Google Scholar]
- Ku CC, Besser J, Abendroth A, Grose C, Arvin AM. Varicella-zoster virus pathogenesis and immunobiology: new concepts emerging from investigations with the SCIDhu mouse model. J Virol. 2005;79:2651–2658. doi: 10.1128/JVI.79.5.2651-2658.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Chen N, Yang J, Zhou M, Zhou D, Zhang Q, He L. Antiviral treatment for preventing postherpetic neuralgia. Cochrane Database Syst Rev. 2009 doi: 10.1002/14651858.CD006866.pub2. CD006866. [DOI] [PubMed] [Google Scholar]
- Lowry PW, Solem S, Watson BN, Koropchak CM, Thackray HM, Kinchington PR, Ruyechan WT, Ling P, Hay J, Arvin AM. Immunity in strain 2 guinea-pigs inoculated with vaccinia virus recombinants expressing varicella-zoster virus glycoproteins I, IV, V or the protein product of the immediate early gene 62. J Gen Virol. 1992;73(Pt 4):811–819. doi: 10.1099/0022-1317-73-4-811. [DOI] [PubMed] [Google Scholar]
- Lungu O, Panagiotidis CA, Annunziato PW, Gershon AA, Silverstein SJ. Aberrant intracellular localization of varicella-zoster virus regulatory proteins during latency. Proc Natl Acad Sci U S A. 1998;95:7080–7085. doi: 10.1073/pnas.95.12.7080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareedu J, Hanumaiah RG, Hale E, Habte-Gabr E. Varicella zoster vasculopathy. J Int Assoc Physicians AIDS Care (Chic) 2011;10:144–145. doi: 10.1177/1545109710397366. [DOI] [PubMed] [Google Scholar]
- Markus A, Grigoryan S, Sloutskin A, Yee MB, Zhu H, Yang IH, Thakor NV, Sarid R, Kinchington PR, Goldstein RS. Varicella-zoster virus (VZV) infection of neurons derived from human embryonic stem cells: direct demonstration of axonal infection, transport of VZV, and productive neuronal infection. J Virol. 2011;85:6220–6233. doi: 10.1128/JVI.02396-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JR, Zeringue AL, Caplan L, Ranganathan P, Xian H, Burroughs TE, Fraser VJ, Cunningham F, Eisen SA. Herpes zoster risk factors in a national cohort of veterans with rheumatoid arthritis. Clin Infect Dis. 2009;48:1364–1371. doi: 10.1086/598331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medhurst SJ, Collins SD, Billinton A, Bingham S, Dalziel RG, Brass A, Roberts JC, Medhurst AD, Chessell IP. Novel histamine H3 receptor antagonists GSK189254 and GSK334429 are efficacious in surgically-induced and virally-induced rat models of neuropathic pain. Pain. 2008;138:61–69. doi: 10.1016/j.pain.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Moffat J, Ku CC, Zerboni L, Sommer M, Arvin A. VZV: pathogenesis and the disease consequences of primary infection. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K, editors. Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press; Cambridge: 2007. [PubMed] [Google Scholar]
- Myers MG, Stanberry LR. Drug testing for activity against varicella-zoster virus in hairless guinea pigs. Antiviral Res. 1991;15:341–344. doi: 10.1016/0166-3542(91)90015-j. [DOI] [PubMed] [Google Scholar]
- Nagel MA, Traktinskiy I, Azarkh Y, Kleinschmidt-DeMasters B, Hedley-Whyte T, Russman A, VanEgmond EM, Stenmark K, Frid M, Mahalingam R, Wellish M, Choe A, Cordery-Cotter R, Cohrs RJ, Gilden D. Varicella zoster virus vasculopathy: analysis of virus-infected arteries. Neurology. 2011;77:364–370. doi: 10.1212/WNL.0b013e3182267bfa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxman MN. Herpes zoster pathogenesis and cell-mediated immunity and immunosenescence. J Am Osteopath Assoc. 2009;109:S13–S17. [PubMed] [Google Scholar]
- Oxman MN, Levin MJ, Johnson GR, Schmader KE, Straus SE, Gelb LD, Arbeit RD, Simberkoff MS, Gershon AA, Davis LE, Weinberg A, Boardman KD, Williams HM, Zhang JH, Peduzzi PN, Beisel CE, Morrison VA, Guatelli JC, Brooks PA, Kauffman CA, Pachucki CT, Neuzil KM, Betts RF, Wright PF, Griffin MR, Brunell P, Soto NE, Marques AR, Keay SK, Goodman RP, Cotton DJ, Gnann JW, Jr, Loutit J, Holodniy M, Keitel WA, Crawford GE, Yeh SS, Lobo Z, Toney JF, Greenberg RN, Keller PM, Harbecke R, Hayward AR, Irwin MR, Kyriakides TC, Chan CY, Chan IS, Wang WW, Annunziato PW, Silber JL. Avaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271–2284. doi: 10.1056/NEJMoa051016. [DOI] [PubMed] [Google Scholar]
- Pavan-Langston D. Herpes zoster antivirals and pain management. Ophthalmology. 2008;115:S13–S20. doi: 10.1016/j.ophtha.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Philip A, Thakur R. Post herpetic neuralgia. J Palliat Med. 2011;14:765–773. doi: 10.1089/jpm.2011.9685. [DOI] [PubMed] [Google Scholar]
- Pickering G, Leplege A. Herpes zoster pain, postherpetic neuralgia, and quality of life in the elderly. Pain Pract. 2011;11:397–402. doi: 10.1111/j.1533-2500.2010.00432.x. [DOI] [PubMed] [Google Scholar]
- Pugazhenthi S, Nair S, Velmurugan K, Liang Q, Mahalingam R, Cohrs RJ, Nagel MA, Gilden D. Varicella-zoster virus infection of differentiated human neural stem cells. J Virol. 2011;85:6678–6686. doi: 10.1128/JVI.00445-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja SN, Haythornthwaite JA, Pappagallo M, Clark MR, Travison TG, Sabeen S, Royall RM, Max MB. Opioids versus antidepressants in postherpetic neuralgia: a randomized, placebo-controlled trial. Neurology. 2002;59:1015–1021. doi: 10.1212/wnl.59.7.1015. [DOI] [PubMed] [Google Scholar]
- Reichelt M, Zerboni L, Arvin AM. Mechanisms of varicella-zoster virus neuropathogenesis in human dorsal root ganglia. J Virol. 2008;82:3971–3983. doi: 10.1128/JVI.02592-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds MA, Chaves SS, Harpaz R, Lopez AS, Seward JF. The impact of the varicella vaccination program on herpes zoster epidemiology in the United States: a review. J Infect Dis. 2008;197(Suppl 2):S224–S227. doi: 10.1086/522162. [DOI] [PubMed] [Google Scholar]
- Rogawski MA, Loscher W. The neurobiology of antiepileptic drugs for the treatment of nonepileptic conditions. Nat Med. 2004;10:685–692. doi: 10.1038/nm1074. [DOI] [PubMed] [Google Scholar]
- Rowbotham MC, Yosipovitch G, Connolly MK, Finlay D, Forde G, Fields HL. Cutaneous innervation density in the allodynic form of postherpetic neuralgia. Neurobiol Dis. 1996;3:205–214. doi: 10.1006/nbdi.1996.0021. [DOI] [PubMed] [Google Scholar]
- Schmader K, George LK, Burchett BM, Pieper CF, Hamilton JD. Racial differences in the occurrence of herpes zoster. J Infect Dis. 1995;171:701–704. doi: 10.1093/infdis/171.3.701. [DOI] [PubMed] [Google Scholar]
- Sen N, Sommer M, Che X, White K, Ruyechan WT, Arvin AM. Varicella-zoster virus immediate-early protein 62 blocks interferon regulatory factor 3 (IRF3) phosphorylation at key serine residues: a novel mechanism of IRF3 inhibition among herpesviruses. J Virol. 2010;84:9240–9253. doi: 10.1128/JVI.01147-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seward JF, Marin M, Vazquez M. Varicella vaccine effectiveness in the US vaccination program: a review. J Infect Dis. 2008;197(Suppl 2):S82–S89. doi: 10.1086/522145. [DOI] [PubMed] [Google Scholar]
- Steain M, Slobedman B, Abendroth A. Experimental models to study varicella-zoster virus infection of neurons. Curr Top Microbiol Immunol. 2010;342:211–228. doi: 10.1007/82_2010_15. [DOI] [PubMed] [Google Scholar]
- Steiner I, Kennedy PG, Pachner AR. The neurotropic herpes viruses: herpes simplex and varicella-zoster. Lancet Neurol. 2007;6:1015–1028. doi: 10.1016/S1474-4422(07)70267-3. [DOI] [PubMed] [Google Scholar]
- Thomas SL, Wheeler JG, Hall AJ. Case–control study of the effect of mechanical trauma on the risk of herpes zoster. BMJ. 2004;328:439. doi: 10.1136/bmj.37991.511829.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischer BK, Kaufer BB, Sommer M, Wussow F, Arvin AM, Osterrieder N. A self-excisable infectious bacterial artificial chromosome clone of varicella-zoster virus allows analysis of the essential tegument protein encoded by ORF9. J Virol. 2007;81(23):13200–13208. doi: 10.1128/JVI.01148-07. Epub 2007 Oct 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyring SK, Beutner KR, Tucker BA, Anderson WC, Crooks RJ. Antiviral therapy for herpes zoster: randomized, controlled clinical trial of valacyclovir and famciclovir therapy in immunocompetent patients 50 years and older. Arch Fam Med. 2000;9:863–869. doi: 10.1001/archfami.9.9.863. [DOI] [PubMed] [Google Scholar]
- Verjans GM, Hintzen RQ, van Dun JM, Poot A, Milikan JC, Laman JD, Langerak AW, Kinchington PR, Osterhaus AD. Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc Natl Acad Sci U S A. 2007;104:3496–3501. doi: 10.1073/pnas.0610847104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace VC, Segerdahl AR, Lambert DM, Vandevoorde S, Blackbeard J, Pheby T, Hasnie F, Rice AS. The effect of the palmitoylethanolamide analogue, palmitoylallylamide (L-29) on pain behaviour in rodent models of neuropathy. Br J Pharmacol. 2007;151:1117–1128. doi: 10.1038/sj.bjp.0707326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson CP, Morshead C, Van der Kooy D, Deck J, Evans RJ. Postherpetic neuralgia: post-mortem analysis of a case. Pain. 1988;34:129–138. doi: 10.1016/0304-3959(88)90158-3. [DOI] [PubMed] [Google Scholar]
- White K, Peng H, Hay J, Ruyechan WT. Role of the IE62 consensus binding site in transactivation by the varicella-zoster virus IE62 protein. J Virol. 2010;84:3767–3779. doi: 10.1128/JVI.02522-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeomans DC, Wilson SP. Herpes virus-based recombinant herpes vectors: gene therapy for pain and molecular tool for pain science. Gene Ther. 2009;16:502–508. doi: 10.1038/gt.2009.25. [DOI] [PubMed] [Google Scholar]
- Zerboni L, Reichelt M, Arvin A. Varicella-zoster virus neurotropism in SCID mouse–human dorsal root ganglia xenografts. Curr Top Microbiol Immunol. 2010a;342:255–276. doi: 10.1007/82_2009_8. [DOI] [PubMed] [Google Scholar]
- Zerboni L, Sobel RA, Ramachandran V, Rajamani J, Ruyechan W, Abendroth A, Arvin A. The expression of varicellazoster virus immediate early regulatory protein IE63 in neurons of latently infected human sensory ganglia. J Virol. 2010b doi: 10.1128/JVI.02416-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerboni L, Sobel RA, Lai M, Triglia R, Steain M, Abendroth A, Arvin A. Apparent expression of varicella-zoster virus proteins in latency resulting from reactivity of murine and rabbit antibodies with human blood group A determinants in sensory neurons. J Virol. 2011 doi: 10.1128/JVI.05950-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuranski T, Nawar H, Czechowski D, Lynch JM, Arvin A, Hay J, Ruyechan WT. Cell-type-dependent activation of the cellular EF-1alpha promoter by the varicella-zoster virus IE63 protein. Virology. 2005;338:35–42. doi: 10.1016/j.virol.2005.05.005. [DOI] [PubMed] [Google Scholar]