

Abstract
Although acute lung injury (ALI) contributes significantly to critical illness, resolution often occurs spontaneously through endogenous pathways. We recently found that mechanical ventilation increases levels of pulmonary adenosine, a signaling molecule known to attenuate lung inflammation. Here, we hypothesized a contribution of transcriptionally controlled pathways to pulmonary adenosine receptor signaling during ALI. We gained initial insight from microarray analysis of pulmonary epithelia exposed to conditions of cyclic mechanical stretch - a mimic for ventilation-induced lung disease. Surprisingly, these studies revealed a selective induction of the ADORA2B. Utilizing real-time RT-PCR and western blotting, we confirmed an up to 9-fold induction of the ADORA2B following cyclic mechanical stretch (A549, Calu-3 or HPAEpiC). Studies utilizing ADORA2B promoter constructs identified a prominent region within the ADORA2B promoter conveying stretch responsiveness. This region of the promoter contained a binding site for the transcription factor hypoxia-inducible factor (HIF)-1. Additional studies utilizing site-directed mutagenesis or transcription factor binding assays demonstrated a functional role for HIF1 in stretch-induced increases of ADORA2B expression. Moreover, studies of ventilator induced lung injury revealed induction of the ADORA2B during ALI in vivo that was abolished following HIF-inhibition or genetic deletion of Hif1a. Together, these studies implicate HIF in the transcriptional control of pulmonary adenosine signaling during ALI.
Keywords: hypoxia-inducible factor, adenosine receptors, A2B, Adora2b, A1, A2A, A3, stretch, ventilator induced lung injury, transcription, inflammation, metabolism
Introduction
Acute lung injury (ALI) is a syndrome consisting of acute hypoxemic respiratory failure with bilateral pulmonary infiltrates, not attributable to left heart failure (1, 2). While there is currently no specific therapy available, management consists of aggressive treatment of the initiating cause, vigilant supportive care, and the prevention of nosocomial infections. Despite optimal management, mortality ranges between 35 and 60% (2). In fact, approximately 200,000 patients develop ALI annually in the USA, leading to 75,000 deaths and accounting for up to 3.6 million hospital days (3). Among the hallmarks of acute lung injury is a massive accumulation of inflammatory cells into different compartments of the lungs, in conjunction with cytokine release and inflammatory activation of recruited or resident cells. Other characteristics include a disruption of the alveolar-capillary barrier function, resulting in extensive pulmonary edema and attenuated gas exchange. In such settings, several factors contribute to pulmonary tissue hypoxia. First, ALI is frequently associated with obstruction of airflow into the distal airways, resulting in attenuated oxygen supply into atelectatic areas of the lungs. Secondly, pulmonary edema causing interstitial and intra-alveolar fluid accumulation results in attenuated gas exchange and increased alveolar-arterial oxygen gradient. Finally, mechanical ventilation and inflammatory cell accumulation and activation are associated with dramatic increases of metabolic requirements, including oxygen, thereby causing additional shifts in pulmonary oxygen supply and demand ratio and resulting in pulmonary hypoxia (4–7).
Despite the large impact of ALI on morbidity and mortality in critically ill patients (2), many episodes of ALI are self-limiting and resolve spontaneously through unknown mechanisms. For example, patients undergoing major thoracic surgery for lung cancer have an overall ALI incidence of less than 5%, open heart surgery with cardiopulmonary bypass less than 0.5% or kidney transplantation of less than 0.2% (8). These clinical observations have inspired many studies of endogenous pathways that dampen acute increases in the capillary-alveolar barrier and lung inflammation in different models of ALI, including hypoxia- (9–14), chemical-(15, 16) or endotoxin- induced (5, 17, 18) forms of lung injury.
Previous studies have identified the extracellular signaling molecule adenosine in endogenous lung protection during ALI (8, 19). Mice deficient in the production of extracellular adenosine experience enhanced lung inflammation or pulmonary edema during ALI induced by mechanical ventilation or endotoxin (8, 20). Extracellular adenosine can signal through four distinct adenosine receptors (ADORs); ADORA1, ADORA2, ADORA2B, ADORA3 (21–23). The ADORA2 and ADORA2B have specifically been implicated in dampening acute inflammation, including lung protection during ALI (5, 17, 24–28). While previous studies have shown increased pulmonary adenosine levels and signaling during ALI (8, 17, 20, 27), regulatory mechanisms controlling pulmonary adenosine signaling are only poorly understood. In the present study, we hypothesized a contribution of transcriptionally regulated pathways in the endogenous control of pulmonary adenosine signaling events during ALI. To address this hypothesis, we combined in vitro studies of cyclic mechanical stretch of pulmonary epithelia, with in vivo studies of ALI induced by mechanical ventilation. Serendipitously, these studies identified a role for HIF-1 signaling pathways (6, 29) in the transcriptional control of pulmonary ADORA2B signaling during ALI.
Material and Methods
Cell culture
Calu-3 human airway epithelial cells, human microvascular endothelial cells (HMEC-1 – used for promoter studies) or cultured pulmonary epithelial A549 cells, were cultured as described previously (10, 30–32). Primary pulmonary epithelial cells (HPAEpiC) (ScienCell Research Laboratories, CA, USA) were cultured according to supplier's instructions.
In vitro stretch model
To study the consequences of cyclic mechanical stretch on adenosine receptor transcription, we adopted a previously described in vitro model resembling mechanical ventilation by applying cyclic mechanical stretch (3). In short, Calu-3, A549, HMEC-1 or HPAEpiC were plated on BioFlex culture plates-collagen type I (BF-3001C; FlexCell International) and allowed to attach and grow to ~80% confluence. All cells were cultured in 4ml media: Calu-3 cells were grown in Advanced MEM (GIBCO), A549 cells were grown in DME-12 (GIBCO), both cell lines with 10% FBS, 0.02% L-Glutamine, HMEC-1 were grown in MCDB131 (GIBCO) 131, 0.2% hydrocortisone, 0.02% EGF and 10% FBS, and HPAEpiC were grown in AEpiCM (ScienCell). For promoter analyses, HMECs were transfected in Opti-Mem plus GlutaMAX (Gibco) media. Plates were then placed on a FlexCell FX-4000T Tension Plus System and stretched at % stretch indicated, 30% maximum, 0.7% stretch minimum, sine wave 5s on, 5s off. Cells were collected at indicated time points from duplicate wells, flash-frozen and stored at −80°C for further analysis. For control, cells were cultured under similar conditions at rest (no cyclic mechanical stretch). Transcriptional levels of ADORs were determined as described previously (11).
Gene arrays
The transcriptional profile of adenosine receptor expression in Calu-3 cells subjected to 30% stretch for 24h was assessed from total RNA (isolated using Qiagen RNeasy kit) using quantitative GeneChip arrays (Human genome U133plus 2.0 Array). Analyses were done by the Microarray Core at UC Denver.
ADORA2B promoter constructs
We used previously designed constructs (pGL3 basic luciferase expression vector) (33). Reporter construct `1095bp' is the full length promoter containing a binding site for the transcription factor hypoxia-inducible factor (HIF; hypoxia responsive element, HRE) and reporter construct `477bp' is truncated and lacking the HRE. In a subset of experiments, constructs were utilized where a HIF-1α binding site mutation has been introduced; here the original sequence ACGTG was altered to AATCG.
ADORA2B reporter assay
To measure the transcriptional activity of the ADORA2B, we used HMEC-1 cells as an easily transfectable cellular model. HMEC-1 were plated in 6-well plates at a density of 1 × 106 cell/well and left to adhere overnight. Monolayers were then transfected with 2μg of either ADORA2B-luc promoter reporter or control pGL3 vector, and co-transfected with 0.02μg Renilla reporter vector for 24h using Fugene 6 transfection reagent (Roche, in accordance with the manufacturer's instructions). Cells were exposed to cyclic mechanical stretch at 30% for specified time points up to 24h before being washed in ice-cold PBS and collected in 200μl 1x passive lysis buffer (Promega). The Dual Luciferase Reporter Assay (Promega) was carried out according to manufacturer's instructions. All activity was normalized to constitutively expressed Renilla-luciferase.
Chromatin immunoprecipitation (ChIP) assay
Chromatin immunoprecipitation was performed as described previously using an antibody against HIF-1α (9, 34). HIF-1 binding to A2B promoter DNA in stretch was quantified by standard PCR using primers (forward: 5 – CAGGGT GTC GGC AAA CTT CC- 3 and reverse: 5 – CTT GTT GGA TTT GGG GGC A -3) designed to amplify a 374bp region of the A2B promoter. Chromatin incubated with IgG antibody was used to control for nonspecific binding of DNA.
Inhibition of HIF1A in vitro and in vivo
Echinomycin (Sigma-Aldrich, USA) was administered to Calu-3 cells at concentrations of 3, 30 and 300 nM and cells were exposed to cyclic mechanical stretch at 30%. In vivo, 30 μg Echinomycin was administered to experimental mice i.p. 1h before induction of anesthesia and subsequent ventilation (35).
Murine mechanical ventilation
All animal protocols were in accordance with the guidelines of the National Institute for Health for the use of laboratory animals and approved by the Institutional Animal Care and Use Committee of the University of Colorado. C57BL6/J mice (Charles River Laboratories) were matched according to sex, age and weight. For Hif1a tissue specific knockout in the alveolar epithelium, triple transgenic mice (Hif1af/f SPC-rtA Tet-O-Cre [Hif1af/fSurfactantCre+]) were induced by Doxycyclin therapy over 5 days i.p and p.o. as described (35). Ventilator induced lung injury was induced as described previously (36).
Mouse and human cell transcriptional analysis
Transcriptional studies of adenosine receptors during murine ALI were carried out as described previously (27).
Human and mouse protein analysis
Western blotting studies for murine or human ADORA2B were carried out as described previously (11, 27). In studies for HIF1A Western blotting, we used Anti-HIF1 alpha [H1alpha67] antibody (ab1) (35).
Statistical Analysis
Data are presented as mean ± SEM. Statistical analysis was performed using ANOVA and the Student t-test (two sided, < 0.05). A value of P<0.05 or P<0.001 was considered statistically significant.
Results
Selective induction of the ADORA2B during cyclic mechanical stretch of pulmonary epithelia
Previous studies have identified extracellular adenosine signaling events in endogenous protection from pathologic lung inflammation during ALI (14, 17, 20, 27, 37). However, transcriptional mechanisms that control adenosine receptor expression during ALI are only poorly understood. To gain insight into the transcriptional control of pulmonary adenosine signaling events during ALI, we utilized cyclic mechanical stretch exposure of pulmonary epithelia as a well-defined in vitro model (8). As such, we performed a screening experiment wherein Calu-3 cells were exposed to cyclic mechanical stretch at 30% over 24h and microarray analysis used to study the transcriptional response of ADORs (microarray data were accepted and published on GEO NCBI, submission number GSE27128: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE27128). Consistent with previous studies of pulmonary AR expression (38), we observed highest basal expression levels of the ADORA2B subtype in untreated Calu-3 cells. Moreover, ADORA2B transcript levels were selectively induced following stretch exposure (Fig. 1). Taken together, these studies reveal for the first time a selective induction of the ADORA2B during cyclic mechanical stretch. Furthermore, these findings closely correlate with other studies that have shown a selective induction of the ADORA2B during in vivo exposure of mice to ALI (17, 27).
Figure 1. Transcriptional responses of adenosine receptors during cyclic mechanical stretch of pulmonary epithelial cells.

Confluent Calu-3 cells were plated on collagen coated BioFlex plates and underwent cyclic mechanical stretch for 24h. RNA was extracted using Trizol and microarray technology was used to assess relative fluorescence in A1A, A2A, A2B and A3 adenosine receptors (AR) in stretch vs. control conditions. Results are presented as mean ± SEM (P<0.001, n = 3).
ADORA2B induction is time- and stretch-dependent
Based on the above micro-array studies showing selective induction of ADORA2B transcript, we next utilized different stretch conditions and examined ADORA2B transcript and protein levels. For this purpose, Calu-3 cells were exposed to 0, 10, 20 or 30% cyclical stretch, and protein and RNA analyses used to observe changes in expression (Fig. 2A and B). We observed that 30% stretch exposure was associated with the most profound induction of ADORA2B transcript and protein levels. Therefore, we performed cyclic mechanical stretch of Calu-3 epithelia at 30% over different time periods (2, 4, 8, and 24 h; Fig. 2C and 2D). Moreover, we also exposed human primary alveolar epithelial cells (HPAEpiC) to a time course of cyclic mechanical stretch at 30% stretch. Consistently, these studies revealed induction of the ADORA2B with cyclic mechanical stretch exposure.
Figure 2. Regulation of the ADORA2B by cyclic mechanical stretch of pulmonary epithelial cells.

To measure ADORA2B expression in pulmonary epithelial cells, Calu-3's were exposed to cyclic mechanical stretch for indicated time periods and stretch intensity. Transcript levels were determined by real-time quantitative PCR (O-PCR). Data was calculated relative to β-actin and expressed as fold change relative to control. β-actin was also used to control for protein loading. A) Calu-3's were exposed to 0, 10, 20 and 30% stretch. B) Protein change was determined following 0, 10, 20 and 30% stretch. C) Induction of ADORA2B mRNA and D) ADORA2B protein in Calu-3 cells following cyclic mechanical stretch at 30% for 2, 4, 8, and 24h vs. control at 0h. E) ADORA2B transcript and F) representative Western blots are shown following 30% stretch in HPAEpiC. Data are mean ± SEM (n = 3).
Identification of a functional HIF-1 binding site that regulates ADORA2B promoter activity during cyclic mechanical stretch
Having shown that ADORA2B transcript and protein levels are induced during cyclic mechanical stretch of pulmonary epithelia, we next went on to study transcriptional pathways that could represent a regulatory component for the ADORA2B during stretch conditions. Analysis of the human ADORA2B promoter region (33) revealed several transcription factor binding sites, including a hypoxia-responsive element (HRE; HIF-binding site; 5-CACGTGG-3) at position −642 to −647bp relative to the transcription start site in combination with an ancillary HIF site (HAS; 5-CGGGAG-3 at −546 to −541), and binding sites for SP1 (−528 to −534bp), E2F (−402 to −406bp) and AP2 (−223 to −225bp) (Fig. 3A). To address the functional relevance of these transcription factor binding sites, we transfected a full-length (1095bp) and a truncated promoter-reporter construct (477bp) into HMEC-1 (Fig 3B), exposed the cells to 30% cyclic mechanical stretch over 24h and analyzed promoter activity. These studies revealed robust increases of luciferase activity with stretch-exposure of the full-length promoter (1095bp), which was completely abolished in the truncated construct (477bp; Fig. 3C), indicating an area of particular relevance for stretch-inducibility located between 477bp and 1095bp of the ADORA2B promoter. In conjunction with previous studies showing that the ADORA2B is induced during ambient hypoxia (10, 11, 33), along with the fact that this defined regulatory region within the ADORA2B promoter contains a prominent HRE/HAS site, we next performed site-directed mutagenesis of the HRE. Mutation of the HRE core sequence from ACGTG to AATCG (Fig. 3D) was associated with a significant attenuation of the stretch-inducibility of the ADORA2B promoter (Fig 3E). Taken together, these studies implicate a functional HRE within the ADORA2B that regulates promoter activity during cyclic mechanical stretch.
Figure 3. Functional HIF-1 binding to the ADORA2B in cyclic mechanical stretch.

A) Map of cloned ADORA2B promoter showing relative positions of the HRE (1) (−642 to −647), along with SP1 (2) (−528 to 534bp), E2F (3) (−402 to −406bp) and AP2 (4) (−223 to −225bp). B) Map of cloned ADORA2B HRE within the promoter showing relative positions of the HIF binding site (HBS) and the HIF ancillary site (HAS). C) HMEC-1 monolayers were transfected with plasmids containing either a vector expressing the full-length ADORA2B promoter (1095bp) or one containing a truncated promoter region lacking the HRE (477bp). An empty vector (pGL3) was used as control. Cells were exposed to cyclic mechanical stretch for 24 h and subsequently assessed for luciferase activity (P<0.001, n = 4). All luciferase expression was normalized to Renilla. D) Map of the cloned ADORA2B HRE within the promoter showing the native HRE vs. the mutational change introduced within the HBS. E) HMEC-1 monolayers were transfected with plasmids expressing full-length ADORA2B or HIF-1 site-mutant (1095bpΔHIF), as well as the empty vector (pGL3), and exposed to stretch for 24h. Luciferase activity was assessed and normalized to Renilla expression (P<0.001, n = 4). Data are mean ± SEM.
HIF1A is responsible for the transcriptional induction of the ADORA2B during conditions of cyclic mechanical stretch
Having identified a functional HRE within the ADORA2B promoter, we next went on to examine the role of HIF transcription factors in this response. Based on previous studies showing expression and functional regulation of HIF1A in the lung (35, 39), we next examined previously characterized pulmonary epithelial cell lines (A549/Calu-3) with stable repression of HIF-1α utilizing a short hairpin RNAi approach (30, 35). Stretch-inducibility of ADORA2B transcript (Fig. 4A) or protein level (Fig. 4B) remain robust in control-transfected cells (HIF-1α Scr) exposed to different time periods of cyclic mechanical stretch. Strikingly, stretch-inducibility of the ADORA2B is almost completely abolished in Calu-3 cells with siRNA-mediated HIF-1α repression (HIF-1α KO). Taken together, these studies provide the first evidence for a selective role of HIF-1 in the stretch-inducibility of the ADORA2B.
Figure 4. Role of HIF-1α in stretch-induced ADORA2B expression in vitro.

Use of epithelial cell lines (A549/Calu-3), one stably expressing HIF-1α siRNA (HIF-1 KO) and one expressing scrambled (scr) siRNA. Cells were exposed to stretch (30%) for the indicated time course. Shown here are transcript levels of ADORA2B (A) and ADORA2B protein expression (Western blot) (B) both normalized to β-actin. C) ChIP assay analysis was performed with an antibody against HIF-1α to define and confirm the consensus binding sequence within the ADORA2B promoter, in control and stretched HMEC-1 cells. Shown here is the PCR amplification product at 374bp and D) the fold change in PCR expression. Data are mean ± SEM from (n = 3).
As a next step, we performed a transcription factor binding assay of the ADORA2B promoter during cyclic mechanical stretch conditions to test for direct HIF-1α binding to the ADORA2B promoter. For this purpose we performed ChIP utilizing a HIF-1α antibody for immunoprecipitation, and a primer set amplifying the area within the ADORA2B promoter containing the HRE. These studies demonstrated the presence of a band at 374bp only in cells that had been exposed to cyclic mechanical stretch and not in control conditions, indicating that HIF-1α is binding directly to the HRE within the ADORA2B promoter.
As a next step, we utilized a pharmacological inhibitor for HIF-1 activity during cyclic mechanical stretch in vitro. In these studies we utilized Echinomycin (35, 40) to study HIF-1 and ADORA2B responses. In fact, we observed dose-dependent inhibition of HIF-1 activity with increasing Echinomycin concentrations (Fig. 5A). Consistent with our previous studies indicating HIF-1α in the regulation of the ADORA2B during cyclic mechanical stretch, these findings were paralleled by abolished induction of the ADORA2B transcript (Fig 5A) and protein levels (Fig. 5B) with pharmacological HIF inhibition. Taken together, these studies indicate that HIF-1 regulates ADORA2B induction during conditions of cyclic mechanical stretch in vitro.
Figure 5. Consequences of pharmacological HIF inhibition with echinomycin on ADORA2B expression during cyclic mechanical stretch in vitro.

Different concentrations of the HIF-1α inhibitor Echinomycin were used to assess its effect on A) ADORA2B transcript and protein (B) (both normalized to β-actin); after treatment with increasing concentrations of Echinomycin (0, 3, 30 and 300 nM) and exposure to 24h stretch at 30%. Data are mean ± SEM (n = 3).
HIF-1 is responsible for transcriptional induction of the ADORA2B during VILI in vivo
Having demonstrated a functional role of HIF-1α in the stretch-inducibility of the ADORA2B, we next examined the functional role of HIF-1 during transcriptional regulation of the ADORA2B during ALI in vivo. For this purpose, we utilized a previously described model for inducing ALI in mice by means of high-pressure mechanical ventilation (36). Previous studies in this model had shown that gene-targeted mice for the ADORA2B suffer from more severe pulmonary edema and lung inflammation (27). As a first step, we utilized Echinomycin as a pharmacologic inhibitor of HIF activity in vivo. As shown in Fig. 6A, Echinomycin treatment was associated with abolished HIF-1α activity in mice exposed to 3h of high-pressure mechanical ventilation. As such, induction of the ADORA2B transcript (Fig. 6A) and ADORA2B protein (Fig. 6B) were abolished following pharmacologic inhibition of HIF-1. To recapitulate these findings in a genetic model, we examined pulmonary expression of the ADORA2B in mice with conditional deletion of Hif1α in lung epithelium during ALI. In fact, Hif1af/fSurfactantCre+ mice showed attenuated HIF-1α stabilization during ALI in vivo (Fig. 7B), in conjunction with abolished induction of ADORA2B transcript (Fig. 7A) and protein (Fig. 7B) upon exposure to ALI. Together, these findings implicate HIF-1 in the transcriptional induction of the ADORA2B during ALI induced by mechanical ventilation in vivo.
Figure 6. Consequences of pharmacological HIF-1 inhibition on A2B expression during acute lung injury.
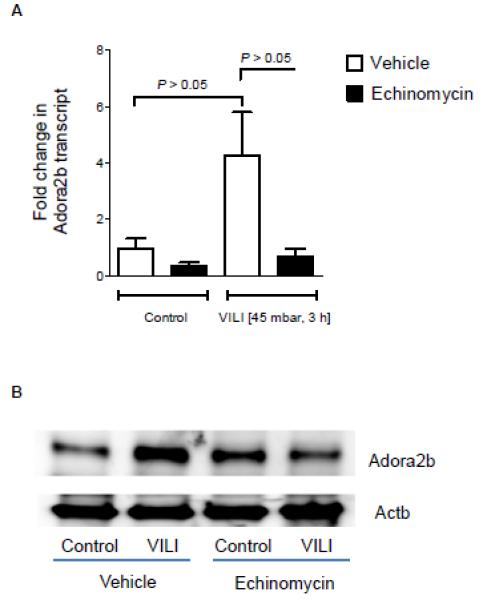
To study the effect of pharmacologic HIF-1α inhibition on ADORA2B protein and transcript levels, C57Bl6/J mice were treated with 30μg Echinomycin i.p. 1h before being exposed to high pressure ventilation; BL6 mice were ventilated in a pressure controlled setting over 0 or 3 h at 45mbar inspiratory pressure (control mice ventilated at 15 mbar). Lungs were excised, flash frozen and lysed; proteins were resolved by SDS-PAGE. Membranes were probed with an anti-ADORA2B (A) antibody and the same membrane was re-probed with β-actin antibody as a control for protein loading. ADORA2B transcript was calculated relative to β-actin (B; p<0.05). Results are presented as mean ± SEM, derived from n = 3 in each condition.
Figure 7. ADORA2B expression in “whole-body” induced Hif1aflox;CreER+mice during acute lung injury.

Hif1af/f SPC-rtA Tet-O-Cre+ mice and littermate controls without the floxed HIF allele were matched in age, gender and weight (as control). In these transgenic mice Cre recombinase is under the control of a doxycyclin-inducible cre-mediated recombination system. One week following cre-induction by doxycycline treatment, mice were ventilated (pressure controlled ventilation) for 0 or 3 h at an inspiratory pressure of 45 mbar (control mice were ventilated at 15mbar). Lungs were excised, flash frozen and lysed. A) ADORA2B transcript at 0 and 3 h ventilation and representative Western blot of B) HIF-1α and ADORA2B protein expression, membranes being re-probed for β-actin to control for protein loading and ADORA2B transcript calculated relative to β-actin (B; P<0.001). Results are presented as mean ± SEM, derived from n = 3 in each condition.
Discussion
Previous studies have implicated adenosine signaling events in endogenous lung protection during ALI (5, 17, 20, 26, 27, 41). These findings are supported by increased extracellular adenosine production and signaling events during ventilator-induced lung injury (8, 27), endotoxin-induced lung injury (17, 20), or lung injury induced by chemical irritants such as bleomycin (15). At present, there is only very little known about transcriptional mechanisms that govern pulmonary AR expression levels during ALI. In the present studies, we pursued the hypothesis that transcriptional mechanisms may regulate AR expression during cyclic mechanical stretch in vitro, or during ALI in vivo. Initial screening studies took advantage of a micro-array analysis of pulmonary epithelia exposed to cyclic mechanical stretch. These studies revealed a selective and robust induction of the ADORA2B. In fact, studies in different pulmonary epithelial cell lines confirmed time- and stretch-dose-dependent ADORA2B induction. Studies utilizing promoter constructs, site-directed mutagenesis, loss- and gain-of-function or transcription factor binding assays, identified HIF-1 as the key regulator of ADORA2B-induction during stretch conditions. Moreover, in vivo studies of ALI provided multiple levels of evidence for HIF-1 in the transcriptional induction of the ADORA2B during ventilator-induced ALI in vivo. Taken together, these findings implicate previously unknown signaling pathways in the regulation of pulmonary AR signaling during ALI.
The present studies implicate a functional role for HIF1A in the regulation of adenosine-elicited lung protection during ALI, thereby indirectly implicating HIF1A in lung protection. The hypothesis that HIF1A functions to dampen ALI is supported by a recent study, where the authors hypothesized that stretch conditions—such as those that occur during mechanical ventilation—result in transcriptional adaptation of alveolar epithelial cells—the innermost lining of the lungs. Indeed, they identified an unexpected involvement of the transcription factor hypoxia-inducible factor HIF1A in lung protection. A genome-wide screen revealed a transcriptional response similar to hypoxia signaling. Surprisingly, they found that stabilization of hypoxia-inducible factor 1A (HIF1A) during stretch conditions in vitro or during ventilator-induced ALI in vivo occurs under normoxic conditions. Extension of these findings identified a functional role for stretch-induced inhibition of succinate dehydrogenase (SDH) in mediating normoxic HIF1A stabilization, concomitant increases in glycolytic capacity, and improved tricarboxylic acid (TCA) cycle function. Pharmacologic studies with HIF activator or inhibitor treatment implicated HIF1A-stabilization in attenuating pulmonary edema and lung inflammation during ALI in vivo. Systematic deletion of HIF1A in the lungs, endothelia, myeloid cells, or pulmonary epithelia linked these findings to alveolar-epithelial HIF1A. In vivo analysis of 13C-glucose metabolites utilizing liquid-chromatography tandem mass-spectrometry demonstrated that increases in glycolytic capacity, improvement of mitochondrial respiration, and concomitant attenuation of lung inflammation during ALI were specific for alveolar-epithelial expressed HIF1A. These preclinical findings highlight the potential for pharmacological HIF1A stabilization in the treatment of acute lung injury (35). These findings are also consistent with other studies indicating that elevations of succinate levels can function as HIF activators via normoxic inhibition of PHDs (42–44).
Due to their large surface areas, mucosal organs such as the intestine or the lungs are particularly prone to hypoxia-elicited inflammation (13). Here, several studies have implicated HIF-1 in the transcriptional induction of molecular pathways that dampen hypoxia-elicited inflammation. For example, a recent study found that the neuronal guidance molecule netrin-1 is expressed by mucosal epithelia, and HIF-dependent induction of netrin-1 dampens hypoxia-induced mucosal inflammation in the intestine and the lungs (13). Such findings are consistent with previous studies that have demonstrated a transcriptional role for hypoxia in directly regulating the adenosine pathway. During mucosal hypoxia, HIF-elicited changes in gene expression result in increased production of adenosine by the 5'-ecto-nucleotidase (CD73) enhanced adenosine signaling events due to hypoxia-induction of the ADORA2B, in conjunction with attenuated adenosine uptake (34) and metabolism (12). This pathway has been implicated in experimental studies of acute lung injury induced by mechanical ventilation (8, 27) or polymicrobial lung infection (4, 5, 45). In fact, the latter study demonstrated that inhibition of hypoxia-induced enhancement of adenosine production and signaling by utilizing high inspired oxygen concentrations is associated with higher mortality in the model used (5). The authors concluded that oxygenation with high inspired oxygen concentrations may inhibit the physiological tissue-protective mechanisms elicited by hypoxia-signaling and may thereby exacerbate lung injury (5). Consistent with the role of HIF-1 in attenuating acute mucosal inflammation, several studies have implicated the ADORA2B in dampening tissue inflammation in acute models of injury, including ventilator induced lung injury, polymicrobial sepsis, or myocardial ischemia, or acute kidney injury (27, 41, 46, 47).
Presently, there are only a few studies that show mechanisms of how ADORA2B protein expression is regulated in response to environmental stimuli. As such, previous studies had identified transcriptionally regulated alterations of ADORA2B expression during hypoxia-elicited inflammation. These studies demonstrated selective induction of the ADORA2B following exposure of human vascular endothelia to ambient hypoxia. In contrast, transcript levels of other ARs were either repressed or unaltered (10). Subsequent studies identified a previously unrecognized binding site for HIF-1 within the promoter region of the ADORA2B (33). Additional studies investigating the promoter activity, functional chromatin binding and HIF-1 loss-of-function studies demonstrated a critical role of HIF-1 in mediating hypoxia-associated induction of the ADORA2B (33). Other studies demonstrated HIF-1-dependent induction of the ADORA2B during myocardial ischemia (41). Similarly, a recent study identified a transcriptionally regulated pathway elicited by hypoxia involving HIF-2-dependent induction of the A2AAR (48). While these studies demonstrate transcriptionally regulated alterations of AR gene expression, a recent study failed to demonstrate alterations of ADORA2B promoter activity elicited by inflammatory mediators (17). In contrast, this study indicated that increases in ADORA2B following exposure to inflammatory stimuli involve alterations in mRNA stability.
In contrast to the beneficial effects of increased adenosine production and signaling during ALI, there is some evidence suggesting a potentially detrimental role of chronically elevated adenosine levels (49–51). For example, levels of adenosine are increased in the lungs of asthmatics, and correlate with the degree of inflammatory insult (49). At present, it is not entirely clear whether such elevations of adenosine are part of a protective pathway to dampen lung inflammation, or play a provocative role of adenosine in asthma or chronic obstructive pulmonary disease (46). For example, mice incapable of extracellular adenosine generation (cd73−/− mice) exhibit a more severe phenotype in bleomycin-induced lung injury, indicating a protective role of extracellular adenosine signaling in this chronic model of lung disease (15). In contrast, adenosine-deaminase (ADA)-deficient mice develop signs of chronic lung inflammation in association with dramatically elevated pulmonary adenosine levels. In fact, ADA-deficient mice die within weeks after birth from severe respiratory distress, and pharmacological studies suggest that attenuation of adenosine signaling through the ADORA2B may reverse the severe pulmonary phenotypes in ADA-deficient mice (49). To address these findings on a genetic level, a very elegant study examined the contribution of ADORA2B signaling in this model via a genetic approach by generating ADA/ADORA2B double-knockout mice (16). The authors' initial hypothesis was that genetic removal of the ADORA2B from ADA-deficient mice would lead to diminished pulmonary inflammation and damage. Unexpectedly, ADA/ADORA2B double-knockout mice exhibited enhanced pulmonary inflammation and airway destruction. Marked loss of pulmonary barrier function and excessive airway neutrophilia are thought to contribute to the enhanced tissue damage observed. These findings support an important protective role for ADORA2B signaling during acute stages of lung disease (16). Importantly, however, a recent study brings these findings together by pointing out the existence of distinct roles for the ADORA2B dependent on the time-course of the disease (acute versus chronic stages of bleomycin-induced lung injury) .
Taken together, the present studies of ALI induced by cyclic mechanical stretch in pulmonary epithelia in vitro or by mechanical ventilation of mice in vivo, indicate a critical role for the transcription factor HIF in the regulation of pulmonary adenosine signaling events.
Acknowledgements
The authors acknowledge Carol M. Aherne, Susie Reithel, and Eric T. Clambey for technical assistance and manuscript discussion.
Grant support: The present manuscript is supported by United States National Institutes of Health grants R01 DK097075, R01-HL0921, R01-DK083385, R01- HL098294, POIHL114457-01 and a grant by the Crohn's and Colitis Foundation of America (CCFA) to HKE, K08 HL102267 to TE, R01-DK50189, R01-DK095491 and R01-HL60569 to SPC
Abbreviations
- (ALI)
Acute lung injury
- (ADORA2B)
A2B adenosine receptor
- (HRE)
Hypoxia responsive element
- (HIF-1)
Hypoxia-inducible factor-1
- (HMEC-1)
Human microvascular endothelial cells-1
- (ChIP)
Chromatin immunoprecipitation
- (VILI)
Ventilator-induced lung injury
- (HPAEpiC)
human primary alveolar epithelial cells
- (HAS)
HIF ancillary site
- (KO)
knockout
- (Scr)
scrambled
Footnotes
Disclosure The authors have no financial conflicts of interest.
References
- 1.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364:656–665. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ware LB, Matthay MA. The Acute Respiratory Distress Syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 3.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and Outcomes of Acute Lung Injury. N Engl J Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 4.Sitkovsky MV, Lukashev D, Apasov S, Kojima H, Koshiba M, Caldwell C, Ohta A, Thiel M. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annual Review of Immunology. 2004;22:657–682. doi: 10.1146/annurev.immunol.22.012703.104731. [DOI] [PubMed] [Google Scholar]
- 5.Thiel M, Chouker A, Ohta A, Jackson E, Caldwell C, Smith P, Lukashev D, Bittmann I, Sitkovsky MV. Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol. 2005;3:e174. doi: 10.1371/journal.pbio.0030174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taylor CT. Interdependent roles for hypoxia inducible factor and nuclear factor-kappaB in hypoxic inflammation. J Physiol. 2008;586:4055–4059. doi: 10.1113/jphysiol.2008.157669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor CT, Colgan SP. Hypoxia and gastrointestinal disease. J Mol Med. 2007;85:1295–1300. doi: 10.1007/s00109-007-0277-z. [DOI] [PubMed] [Google Scholar]
- 8.Eckle T, Fullbier L, Wehrmann M, Khoury J, Mittelbronn M, Ibla J, Rosenberger P, Eltzschig HK. Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J Immunol. 2007;178:8127–8137. doi: 10.4049/jimmunol.178.12.8127. [DOI] [PubMed] [Google Scholar]
- 9.Eltzschig HK, Abdulla P, Hoffman E, Hamilton KE, Daniels D, Schonfeld C, Loffler M, Reyes G, Duszenko M, Karhausen J, Robinson A, Westerman KA, Coe IR, Colgan SP. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J. Exp. Med. 2005;202:1493–1505. doi: 10.1084/jem.20050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, Robson SC, Colgan SP. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eckle T, Faigle M, Grenz A, Laucher S, Thompson LF, Eltzschig HK. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood. 2008;111:2024–2035. doi: 10.1182/blood-2007-10-117044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morote-Garcia JC, Rosenberger P, Kuhlicke J, Eltzschig HK. HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood. 2008;111:5571–5580. doi: 10.1182/blood-2007-11-126763. [DOI] [PubMed] [Google Scholar]
- 13.Rosenberger P, Schwab JM, Mirakaj V, Masekowsky E, Mager A, Morote-Garcia JC, Unertl K, Eltzschig HK. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat Immunol. 2009;10:195–202. doi: 10.1038/ni.1683. [DOI] [PubMed] [Google Scholar]
- 14.Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, Colgan SP. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J. Exp. Med. 2004;200:1395–1405. doi: 10.1084/jem.20040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Volmer JB, Thompson LF, Blackburn MR. Ecto-5′-nucleotidase (CD73)-mediated adenosine production is tissue protective in a model of bleomycin-induced lung injury. J Immunol. 2006;176:4449–4458. doi: 10.4049/jimmunol.176.7.4449. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Y, Mohsenin A, Morschl E, Young HW, Molina JG, Ma W, Sun CX, Martinez-Valdez H, Blackburn MR. Enhanced airway inflammation and remodeling in adenosine deaminase-deficient mice lacking the A2B adenosine receptor. J Immunol. 2009;182:8037–8046. doi: 10.4049/jimmunol.0900515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schingnitz U, Hartmann K, Macmanus CF, Eckle T, Zug S, Colgan SP, Eltzschig HK. Signaling through the A2B adenosine receptor dampens endotoxin-induced acute lung injury. J Immunol. 2010;184:5271–5279. doi: 10.4049/jimmunol.0903035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reutershan J, Cagnina RE, Chang D, Linden J, Ley K. Therapeutic anti-inflammatory effects of myeloid cell adenosine receptor A2a stimulation in lipopolysaccharide-induced lung injury. J Immunol. 2007;179:1254–1263. doi: 10.4049/jimmunol.179.2.1254. [DOI] [PubMed] [Google Scholar]
- 19.Eckle T, Koeppen M, Eltzschig HK. Role of extracellular adenosine in acute lung injury. Physiology (Bethesda) 2009;24:298–306. doi: 10.1152/physiol.00022.2009. [DOI] [PubMed] [Google Scholar]
- 20.Reutershan J, Vollmer I, Stark S, Wagner R, Ngamsri KC, Eltzschig HK. Adenosine and inflammation: CD39 and CD73 are critical mediators in LPS-induced PMN trafficking into the lungs. FASEB J. 2009;23:473–482. doi: 10.1096/fj.08-119701. [DOI] [PubMed] [Google Scholar]
- 21.Eltzschig HK. Adenosine: an old drug newly discovered. Anesthesiology. 2009;111:904–915. doi: 10.1097/ALN.0b013e3181b060f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grenz A, Homann D, Eltzschig HK. Antioxid Redox Signal. 2011. Extracellular Adenosine - a “Safety Signal” that Dampens Hypoxia-Induced Inflammation during Ischemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park SW, Chen SW, Kim M, Brown KM, D′Agati VD, Lee HT. Protection against acute kidney injury via A1 adenosine receptor-mediated Akt activation reduces liver injury after liver ischemia and reperfusion in mice. J Pharmacol Exp Ther. 2010 doi: 10.1124/jpet.110.166884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang D, Zhang Y, Nguyen HG, Koupenova M, Chauhan AK, Makitalo M, Jones MR, St Hilaire C, Seldin DC, Toselli P, Lamperti E, Schreiber BM, Gavras H, Wagner DD, Ravid K. The A2B adenosine receptor protects against inflammation and excessive vascular adhesion. J Clin Invest. 2006;116:1913–1923. doi: 10.1172/JCI27933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cronstein BN, Daguma L, Nichols D, Hutchison AJ, Williams M. The adenosine/neutrophil paradox resolved: human neutrophils possess both A1 and A2 receptors that promote chemotaxis and inhibit O2 generation, respectively. J Clin Invest. 1990;85:1150–1157. doi: 10.1172/JCI114547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 27.Eckle T, Grenz A, Laucher S, Eltzschig HK. A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J Clin Invest. 2008;118:3301–3315. doi: 10.1172/JCI34203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma AK, Laubach VE, Ramos SI, Zhao Y, Stukenborg G, Linden J, Kron IL, Yang Z. Adenosine A2A receptor activation on CD4+ T lymphocytes and neutrophils attenuates lung ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 2009 doi: 10.1016/j.jtcvs.2009.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor CT, McElwain JC. Ancient atmospheres and the evolution of oxygen sensing via the hypoxia-inducible factor in metazoans. Physiology (Bethesda) 2010;25:272–279. doi: 10.1152/physiol.00029.2010. [DOI] [PubMed] [Google Scholar]
- 30.Haeberle HA, Durrstein C, Rosenberger P, Hosakote YM, Kuhlicke J, Kempf VA, Garofalo RP, Eltzschig HK. Oxygen-independent stabilization of hypoxia inducible factor (HIF)-1 during RSV infection. PLoS ONE. 2008;3:e3352. doi: 10.1371/journal.pone.0003352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eltzschig HK, Faigle M, Knapp S, Karhausen J, Ibla J, Rosenberger P, Odegard KC, Laussen PC, Thompson LF, Colgan SP. Endothelial catabolism of extracellular adenosine during hypoxia: the role of surface adenosine deaminase and CD26. Blood. 2006;108:1602–1610. doi: 10.1182/blood-2006-02-001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, Colgan SP. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 2004;104:3986–3992. doi: 10.1182/blood-2004-06-2066. [DOI] [PubMed] [Google Scholar]
- 33.Kong T, Westerman KA, Faigle M, Eltzschig HK, Colgan SP. HIF-dependent induction of adenosine A2B receptor in hypoxia. Faseb J. 2006;20:2242–2250. doi: 10.1096/fj.06-6419com. [DOI] [PubMed] [Google Scholar]
- 34.Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. 2009;136:607–618. doi: 10.1053/j.gastro.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 35.Eckle T, Brodsky K, Bonney M, Packard T, Han J, Borchers CH, Mariani TJ, Kominsky DJ, Mittelbronn M, Eltzschig HK. HIF1A Reduces Acute Lung Injury by Optimizing Carbohydrate Metabolism in the Alveolar Epithelium. PLoS Biol. 2013;11:e1001665. doi: 10.1371/journal.pbio.1001665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eckle T, Fullbier L, Grenz A, Eltzschig HK. Usefulness of pressure-controlled ventilation at high inspiratory pressures to induce acute lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2008;295:L718–724. doi: 10.1152/ajplung.90298.2008. [DOI] [PubMed] [Google Scholar]
- 37.Aherne CM, Kewley EM, Eltzschig HK. The resurgence of A2B adenosine receptor signaling. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbamem.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lukashev DE, Smith PT, Caldwell CC, Ohta A, Apasov SG, Sitkovsky MV. Analysis of A2a receptor-deficient mice reveals no significant compensatory increases in the expression of A2b, A1, and A3 adenosine receptors in lymphoid organs. Biochem Pharmacol. 2003;65:2081–2090. doi: 10.1016/s0006-2952(03)00158-8. [DOI] [PubMed] [Google Scholar]
- 39.Yu AY, Frid MG, Shimoda LA, Wiener CM, Stenmark K, Semenza GL. Temporal, spatial, and oxygen-regulated expression of hypoxia-inducible factor-1 in the lung. Am J Physiol. 1998;275:L818–826. doi: 10.1152/ajplung.1998.275.4.L818. [DOI] [PubMed] [Google Scholar]
- 40.Werth N, Beerlage C, Rosenberger C, Yazdi AS, Edelmann M, Amr A, Bernhardt W, von Eiff C, Becker K, Schafer A, Peschel A, Kempf VA. Activation of hypoxia inducible factor 1 is a general phenomenon in infections with human pathogens. PLoS One. 2010;5:e11576. doi: 10.1371/journal.pone.0011576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. 2012;367:2322–2333. doi: 10.1056/NEJMra1205750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O′Neill LA. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koivunen P, Hirsila M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem. 2007;282:4524–4532. doi: 10.1074/jbc.M610415200. [DOI] [PubMed] [Google Scholar]
- 44.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 45.Sitkovsky M, Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat Rev Immunol. 2005;5:712–721. doi: 10.1038/nri1685. [DOI] [PubMed] [Google Scholar]
- 46.Zhou Y, Schneider DJ, Morschl E, Song L, Pedroza M, Karmouty-Quintana H, Le T, Sun CX, Blackburn MR. Distinct roles for the A(2B) adenosine receptor in acute and chronic stages of bleomycin-induced lung injury. J Immunol. 2011;186:1097–1106. doi: 10.4049/jimmunol.1002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Csoka B, Nemeth ZH, Rosenberger P, Eltzschig HK, Spolarics Z, Pacher P, Selmeczy Z, Koscso B, Himer L, Vizi ES, Blackburn MR, Deitch EA, Hasko G. A2B adenosine receptors protect against sepsis-induced mortality by dampening excessive inflammation. J Immunol. 2010;185:542–550. doi: 10.4049/jimmunol.0901295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ahmad A, Ahmad S, Glover L, Miller SM, Shannon JM, Guo X, Franklin WA, Bridges JP, Schaack JB, Colgan SP, White CW. Adenosine A2A receptor is a unique angiogenic target of HIF-2alpha in pulmonary endothelial cells. Proc Natl Acad Sci U S A. 2009;106:10684–10689. doi: 10.1073/pnas.0901326106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blackburn MR, Vance CO, Morschl E, Wilson CN. Adenosine receptors and inflammation. Handb Exp Pharmacol. 2009:215–269. doi: 10.1007/978-3-540-89615-9_8. [DOI] [PubMed] [Google Scholar]
- 50.Karmouty-Quintana H, Zhong H, Acero L, Weng T, Melicoff E, West JD, Hemnes A, Grenz A, Eltzschig HK, Blackwell TS, Xia Y, Johnston RA, Zeng D, Belardinelli L, Blackburn MR. The A2B adenosine receptor modulates pulmonary hypertension associated with interstitial lung disease. FASEB J. 2012 doi: 10.1096/fj.11-200907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, Dai Y, Wen J, Zhang W, Grenz A, Sun H, Tao L, Lu G, Alexander DC, Milburn MV, Carter-Dawson L, Lewis DE, Eltzschig HK, Kellems RE, Blackburn MR, Juneja HS, Xia Y. Detrimental effects of adenosine signaling in sickle cell disease. Nat Med. 2011;17:79–86. doi: 10.1038/nm.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]