

Abstract
DNA replication in all organisms requires polymerases to synthesize copies of the genome. DNA polymerases are unable to function on a bare template and require a primer. Primases are crucial RNA polymerases that perform the initial de novo synthesis, generating the first 8–10 nucleotides of the primer. Although structures of archaeal and bacterial primases have provided insights into general priming mechanisms, these proteins are not well conserved with heterodimeric (p48/p58) primases in eukaryotes. Here, we present X-ray crystal structures of the catalytic engine of a eukaryotic primase, which is contained in the p48 subunit. The structures of p48 reveal eukaryotic primases maintain the conserved catalytic prim fold domain, but with a unique sub-domain not found in the archaeal and bacterial primases. Calorimetry experiments reveal Mn2+ but not Mg2+ significantly enhances the binding of nucleotide to primase, which correlates with in vitro higher catalytic efficiency. The structure of p48 with bound UTP and Mn2+ provides insights into the mechanism of nucleotide synthesis by primase. Substitution of conserved residues involved in either metal or nucleotide binding altered nucleotide binding affinities, and yeast strains containing the corresponding Pri1p substitutions were not viable. Our results revealed two residues (S160 and H166) in direct contact with the nucleotide that were previously unrecognized as critical to the human primase active site. Comparing p48 structures to those of similar polymerases in different states of action suggests changes that would be required to attain a catalytically competent conformation capable of initiating dinucleotide synthesis.
Keywords: DNA replication, DNA primase, manganese, X-family DNA polymerase
INTRODUCTION
In all cells, de novo DNA replication begins with separation of the parental DNA strands by a hexameric replicative DNA helicase, a step that is tightly coordinated with protection of the template by a single-strand DNA-binding (SSB) protein 1,2,3. Initiation of DNA synthesis requires release of SSB to enable access to the DNA, synthesis of an intial [RNA] primer by a primase, and extension of the primer by a DNA polymerase. Primases play a special role in the initiation of DNA synthesis because they are the only enzymes capable of initiating synthesis de novo on ssDNA lacking a primer.
In bacteria, the DnaB-like helicase, DnaG-like primase, and SSB protein constitute a ‘primosome’, a dynamic complex coordinated primarily by multivalent interactions among the proteins 4. Although the DnaG-type primases have been structurally well characterized and the functional architecture of several bacterial primosomes has been investigated in detail 5,6,7,8,9,10,11,12, eukaryotic primosomes remain poorly understood. Importantly, eukaryotic replicative helicases translocate 3′ to 5′ on the leading strand template, rather than 5′ to 3′ on the lagging strand template as in bacteria, so fundamental differences in their primase and primosome architecture are expected.
Indeed, unlike single chain bacterial primases, eukaryotic primases are heterodimers of catalytic (p48) and regulatory (p58) subunits functioning within a heterotetrameric DNA polymerase α-primase complex 5,6,13. The initial primer synthesis occurs in three steps: slow formation of dinucleotide, rapid extension to 7–10 ribonucleotides, and direct transfer of primed template into the active site of DNA polymerase α (pol α). Extension of the RNA primer into RNA-DNA primers of ~30 nucleotides by pol α is required before handoff to a processive DNA polymerase 5,6,7.
Structurally, archaeal primases fall in the Archaeo-Eukaryotic Primase (AEP) superfamily, a diverse group of nucleotidyl transferases that also includes bacterial polymerases involved in nonhomologous end-joining (NHEJ) repair 14,15,16,17. Eukaryotic primases share significant functional homology with structurally characterized X-family DNA polymerases, which like the archaeal primases contribute to NHEJ 18. However, the only high-resolution structural information available for eukaryotic primases is for the 4Fe-4S cluster domains from the human p58 19,20 and budding yeast Pri2p regulatory subunits 21. Here we describe high-resolution crystal structures of human p48 along with biophysical and genetic data to characterize the distinct structural and functional properties of the catalytic subunit of eukaryotic primases. The data are discussed and interpreted in light of structural and functional differences and similarities with members of the AEP superfamily and X-family polymerases.
RESULTS AND DISCUSSION
Three-dimensional structures of human p48
To obtain structures of the human DNA primase p48 catalytic subunit at atomic resolution, we engineered a stable construct lacking 12 residues at the C-terminus (p48ΔC). This construct was crystallized and the structure determined by molecular replacement using a partially refined structure of the catalytic subunit of budding yeast primase (Pri1p) (PDB ID 4LIM) as the search model. In the 3.2 Å p48ΔC structure, no density was observed for a region spanning residues 360-380, indicating that these residues were not well ordered in the crystal (Fig. S1). A p48ΔC variant lacking residues 360-379 (p48ΔL) was then prepared. Crystals of this construct diffracted to 1.7 Å. The p48ΔL structure was determined by molecular replacement using the p48ΔC structure as the search model and refined to an Rfree/Rwork of 17.5%/19.8% with excellent geometry (Table 1).
Table 1.
Crystallographic data collection and refinement statistics of the catalytic subunit of eukaryotic primases.a
| p48ΔCb | p48ΔL | p48ΔL•UTP•Mn2+ | |
|---|---|---|---|
| Data collection | |||
| Space group | P43212 | C2 | P43212 |
| Cell dimensions (a, b, c [Å], α, β, γ [°]) | 79.9, 79.9, 147.3, 90, 90, 90 | 101.8, 71.9, 84.7, 90, 122, 90 | 79.3, 79.3, 148.2, 90, 90, 90 |
| Wavelength (Å) | 0.9787 | 0.9787 | 0.9787 |
| Resolution (Å) | 50–3.2 | 50–1.7 | 50–2.6 |
| Reflections | |||
| Total | 384,027 | 878,214 | 1,018,091 |
| Unique | 8,409 | 56,665 | 15,266 |
| Rsym (%) | 11.3 (51.7) | 6.6 (41.6) | 7.6 (45.4) |
| <I/σI> | 25.6 (6.1) | 23.4 (3.3) | 37.6 (6.1) |
| Completeness (%) | 99.9 (100) | 98.7 (89.7) | 100 (100) |
| Redundancy | 13.4 | 5.9 (4.7) | 14.2 (14.6) |
| Refinement | |||
| Rwork/Rfree (%) | 28.12/31.64 | 17.49/19.80 | 21.36/24.84 |
| No. residues | |||
| Protein | 367 | 371 | 370 |
| Nucleotide | 0 | 0 | 16 |
| Solvent | 1 | 325 | 34 |
| Zinc | 1 | 1 | 1 |
| Manganese | 0 | 0 | 2 |
| Average B factor (Å2) | |||
| Protein | 127.8 | 35.3 | 42.9 |
| Solvent | - | 41.2 | 40.8 |
| Nucleotide | - | - | 61.4 |
| Rmsd bonds (Å) | 0.009 | 0.009 | 0.011 |
| Rmsd angles (°) | 1.706 | 1.235 | 1.327 |
| Ramachandran (%) | |||
| Most favored | 96 | 94 | 95 |
| Additionally allowed | 4 | 6 | 5 |
Values in parenthesis refer to the highest resolution shell.
Used for p48ΔL structure determination only; no structural conclusion were drawn from this structure.
The p48ΔL structure reveals two domains, a conserved catalytic domain and a smaller helical domain (Fig. 1). The catalytic domain, encompassing residues 1-190 and 303-408, is composed of 13 α-helices, 8 β-strands, and a zinc-binding motif, and occupies the common primase (Prim) fold (Fig. 2). This similarity includes the conserved positioning of the key catalytic residues D109, D111, and D306 (p48) and of the 3′ nucleotide-binding motif SGXRG, which is located on a highly conserved loop connecting strands β5 and β6 (Fig. S2) 22,23,24.
Fig. 1.

Crystal structure of p48ΔL. Structure of p48ΔL with catalytic domain, helical domain, C-terminal helix, and zinc-binding motif colored pink, blue, yellow, and green, respectively.
Fig. 2.
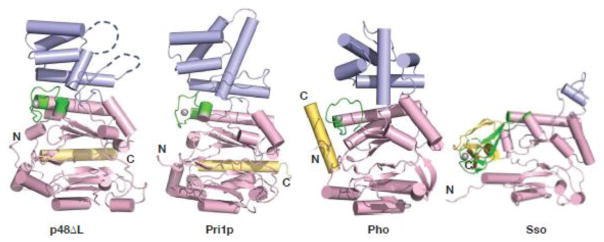
Comparison of the p48 fold with lower eukaryotic and archaeal folds. Structures of primases (p48ΔL; Pri1p (PDB ID: 4LIM); Pho PriS (PDB ID: 1V34); Sso (PDB ID: 1ZT2)) are oriented by alignment of the respective to the zinc-binding motif and colored as in Figure 1. The gray spheres are zinc atoms.
Interestingly, electron density for a bound citrate ion derived from the crystallization buffer was observed in the active site. Carboxylate groups of the citrate ion are bound to conserved basic residues (R162, K318, and H324) in the active site, suggesting that they mimic the β- and γ-phosphate groups of a nucleotide at this site. This is reminiscent of the phosphate and sulfate counterions occupying the nucleotide-binding sites in the structures of Pho PriS and Sso primase (or PriS), respectively 23,24.
The eukaryotic primases are distinguished by structural elements outside of the core region. The catalytic domains of both eukaryotic and archaeal primases contain a zinc ion, but their zinc-binding motifs are not well conserved in sequence or structure. Even the relative location of this motif varies, as is most evident in the Sso primase structure (Fig. 2). In bacterial primases, the zinc-binding motif constitutes an independent structural domain distant from the catalytic domain that has a critical role in recognizing template DNA 25. In contrast, the zinc-binding motif of eukaryotic primases is integrated into the catalytic domain and therefore may not play the same functional role as in bacterial primases.
Eukaryotic and archaeal catalytic domains also differ in the position of the C-terminal helix, which in both p48 and Pri1p is located at the apex of the flange in the catalytic domain. In contrast, the C-terminus of Pho PriS projects away from the fold adjacent to the zinc-binding motif, and in Sso PriS, the C-terminus contains a combination of α/β secondary structure elements. In an attempt to evaluate the importance of the C-terminal helix in p48, we generated a truncated construct lacking 40 C-terminal residues, but the protein was completely insoluble. This observation is consistent with the hydrophobic contacts between the C-terminal helix and β5, particularly F395 and F398 with L156 (Fig. S3), and suggests that the C-terminal helix may have a role in maintaining the structural integrity of the catalytic domain.
The most notable structural difference between the eukaryotic and archaeal catalytic subunits lies in the small helical domain (Fig. 2). In p48, this domain (residues 191-303) includes a five-helix bundle and a small and a long disordered region spanning residues 205-207 and 277-290, respectively. Although poor density was observed for residues 236-276, we were able to model helices α11–α13 into the density. This region has a high average B factor (94.4), suggesting that this domain has a higher degree of flexibility relative to the catalytic domain. The presence of the disordered region between helices 13 and 14 in p48 contrasts with the well-formed long helix that leads from the small helical domain regions of Pri1p and Pho PriS structures, and the short helix in the structure of Sso PriS. Consistent with these structural differences, the sequence of the small helical domain is not well conserved between the archaeal and the eukaryotic primases (Fig. S2). This dissimilarity argues against a direct role in catalysis for this domain and instead points to a possible role in modulating primase activity in the primosome or replisome.
Mn2+ but not Mg2+ stimulates binding of nucleotides to primase
To begin to use the p48 structure to obtain insights into the mechanism of primer synthesis, we turned to examining binding of nucleotides and catalytic metals using isothermal titration calorimetry (ITC). In experiments to measure p48 affinity for UTP and catalytic metals Mg2+ or Mn2+, binding was weak and only lower bounds of ~200 μM could be estimated for the dissociation constants (Kd). Since the full activity of primase requires both nucleotides and catalytic metals, experiments were designed in which the protein was pre-loaded with metal. An experiment performed in this manner provides an apparent Kd because with three components in the system there are three binary equilibria: protein-metal, protein-nucleotide, and nucleotide-metal. Differences in the apparent Kd of the protein for nucleotide can be estimated when the protein is pre-loaded with metal, as long as all other parameters are held constant. In this case, the affinities for UTP are known to be very similar (70 μM and 77 μM, respectively)26.
UTP binding experiments were performed for p48 with a 3-fold molar excess of Mg2+ or Mn2+. Markedly different effects were observed for the two metals: the addition of Mg2+ had no apparent effect on the very weak binding of UTP in the absence of metal, whereas the binding of UTP was significantly enhanced by Mn2+ with an apparent Kd of 11 μM (Fig. 3A; Table 2). The inability of Mg2+ to stimulate nucleotide binding to p48 contrasts directly with the metal-dependent nucleotide binding to bacterial primases 27. Mn2+ has been shown to stimulate the activity of human primase, even in the presence of Mg2+, primarily by reducing the KM for nucleotide binding28. To confirm that the stimulation of UTP binding by Mn2+ was not unique to the isolated catalytic subunit, the experiment was repeated for the intact p48/p58 primase dimer and a similar level of stimulation was observed (Fig. S4; Table 2). Moreover, Mn2+ also stimulated binding of ATP, CTP, and GTP to p48 (Fig. S5; Table 2), consistent with the known lack of nucleotide specificity of primase 28. We note a similar stimulatory effect of Mn2+ on catalytic activity and fidelity has been observed in X and Y-family DNA polymerases including pol ι, pol β, pol μ, and pol λ 29,30,31,32.
Fig. 3.

Binding of UTP and Mn2+ by p48. (A) ITC isotherm showing the raw (upper) and integrated (lower) heat changes for UTP binding to p48 (30 μM) in the presence of Mn2+ (100 μM). (B) Crystal structure of p48ΔL•UTP•Mn2+ colored by subdomain as in Figure 1, and including metals, UTP and key conserved residues. The insert shows an expansion of the active site region. The triphosphate moiety of UTP is shown in ball and stick representation. The manually positioned sugar and base moieties of UTP are shown as black lines. (C) Simulated annealing (mFo-DFc) omit electron density map contoured at 3σ (green) and 5σ (purple) showing both metals and the triphosphate of UTP.
Table 2.
Apparent dissociation constants for the binding of NTPs to wild-type and mutant p48.1
| Protein | Kdapp (μM) |
|---|---|
|
| |
| p48-UTP | |
| WT | 11 |
| p48ΔL | 12.5 ± 0.72 |
| D109N | 74 |
| D111N | 54 |
| S160A | NB |
| R163A | NB |
| H166A | NB |
| D306N | 48 |
| H315A | 22 |
| K318A | 49 |
| p48-ATP WT | 13 |
| p48-CTP WT | 15 |
| p48-GTP WT | 13 |
| p48/p58-UTP | 20 |
Abbreviations: WT, wild-type; NB, no measurable binding; ±, standard deviation.
Experiments performed in a buffer containing 20 mM HEPES (pH 7.2) and 100 mM NaCl with protein concentrations of 20–30 μM and 75–100 μM MnCl2.
Structure of the Mn2+•UTP complex
To obtain further insight into the mechanism of priming, crystallization trials were set up for p48ΔL in complex with metal and nucleotide. The metal-dependence of nucleotide binding characterized by ITC suggested pursuing the complex with UTP and Mn2+ for structural analysis and indeed, the p48ΔL•UTP•Mn2+ complex was crystallized. The structure was determined by molecular replacement using the 1.7 Å p48ΔL structure as a search model. The p48ΔL•UTP•Mn2+ model was refined against data to 2.6 Å resolution to an Rfree/Rwork of 21.4%/24.8% (Table 1). The presence of a single region of contiguous difference density at the active site enabled placement of one UTP molecule and two Mn2+ ions (I and II) into the structure (Fig. 3B, C). Alignment of the p48ΔL•UTP•Mn2+ and p48ΔL structures revealed no significant differences (RMSD 0.57 Å), indicating that the binding of UTP and Mn2+ does not alter the overall structure of the protein.
The ligands of Mn(I) include the α-, β-, and γ-phosphates of the UTP, and the side chain of catalytic residue D109 (Fig. 3B). Mn(II) ligands include one oxygen atom from the α-phosphate and two water molecules, with partial occupancy of a fourth coordination site by D109 (Fig. 3B, C). Clear electron density was observed for the UTP triphosphate (Fig. 3C). However, only limited density was observed for the ribose ring and none for the uracil base, suggesting that these portions of the nucleotide remain flexible in the absence of template DNA. After refinement with only the triphosphate of UTP we manually placed the UTP ribose ring and uracil base into the structure, so that an overall sense of where the UTP binds is provided. In the structure, the triphosphate moiety is stabilized by interactions with side chains of conserved residues S160, R162, R163, H166, and K318. Although the positioning of the ribose ring of UTP is not established, we note that H315 and K318 are in the general vicinity and may have a role in further stabilizing the binding of the nucleotide. The most important new finding in the structure of the p48ΔL•UTP•Mn2+ ternary complex is that two residues (S160 and H166) in direct contact with UTP were previously unrecognized as critical to the recruitment and positioning of nucleotide in the human primase active site.
To test our interpretation of the structure of the ternary complex, we performed a mutational analysis of primase residues contacting the nucleotide and metal ions, including D109N, D111N, S160A, R163A, H166A, D306N, and K318A (Fig. 3B). We also tested the conserved H315A based on its position in the general vicinity of the UTP. In a first step, we determined whether these residues were essential for cell viability (Fig. 4A). A yeast plasmid shuffle system was employed because p48 and Pri1p have highly conserved sequences and great similarity in the conformation of their active sites (Figs. 2 and S2). These yeast viability assays showed that the key catalytic/metal binding residues (D111, D113, D314; p48 D109, D111, D306), as well as the conserved arginines of the SGXRG motif (R164, R165; p48 R162, R163) and K326 (p48 K318) are all essential for viability. Substitutions in the two new residues we found that contact UTP in the structure were also lethal in yeast (S162A, H168A; p48 S160A, H166A), as well as His323 (p48 H315), confirming their importance for primase activity. To rule out lack of expression or structural problems as the cause for mutants lacking the ability to support growth, the expression and solubility of the mutant proteins was characterized. Figure 4B shows that each of the mutant Pri1 proteins is expressed and at least somewhat soluble. We note that S162A is present at somewhat lower level than the other mutants, but the available data do not allow us to distinguish if this arises due to lower levels of expression or poorer solubility. However, the corresponding mutant in human p48, S160A, expressed normally in bacteria and behaved normally during purification and ITC experiments, which lends support to the explanation that the lesser amount of S162A is due to a lower level of expression. In summary, these yeast viability data confirm and extend biochemical analysis of mouse p48 variants18,33, indicating that the conserved interactions among nucleotide, metals and the protein identified in the p48ΔL•UTP•Mn2+ structure are essential for replisome function in cells.
Fig. 4.

Metal and NTP binding Pri1p residues are essential for cell viability. (A) Serial dilutions of mutant yeast strains grown at RT on synthetic complete media (SC), or on SC containing 5-fluoroorotic acid (FOA). FOA is toxic in cells expressing URA3/PRI1, which selects for loss of the plasmid and reveals whether the mutant Pri1 allele on the TRP1 plasmid is sufficient for yeast growth. (B) Solubility of the mutant Pri1p proteins. Proteins are visualized by α-HA western blot, with α-tubulin as a loading control.
To obtain deeper mechanistic insights, ITC was used to directly assay the effect of these substitutions on nucleotide binding to Mn2+-bound p48 (Table 2). Among these mutants, S160A, R163A, and H166A showed no measurable nucleotide binding affinity, whereas D109N, D111N, D306N, H315A, and K318A exhibited a 2 to 6-fold reduction relative to the wild type protein. These results in combination with insights from the structure suggest that S160, R163, and H166 directly bind the 3′-NTP, whereas H315, K318 and the metal ligands (D109, D111, D306) aid in stabilization of the nucleotide.
Our ITC study shows that Mn2+, but not Mg2+, stimulates nucleotide binding, which provides one physical rationale for the higher in vitro catalytic efficiency of primase in the presence of Mn2+ relative to Mg2+28. The structure of the p48ΔL•UTP•Mn2+ complex shows that the Mn2+ ions and the UTP are stabilized by multiple interactions with each other and the protein (Fig. 3). Apparently, the differences in the properties of the two metals result in fewer stabilizing interactions for the UTP molecule. Higher resolution structures, especially with bound DNA template, would provide valuable insights into the origin of the differences in nucleotide binding for the two metals and set the stage for additional biochemical analyses to investigate how this factor contributes to the higher catalytic efficiency of primase with Mn2+. Nevertheless, despite the somewhat provocative evidence from in vitro studies, the importance of Mn2+ for the in vivo function of primase (and a number of polymerases) remains uncertain since the level of free Mg2+ is substantially higher than free Mn2+ 34,35.
Structural similarities of p48 and DNA polymerases
The prevailing view of eukaryotic priming is based on kinetic data and mutagenesis-based biochemistry of calf, murine, and human primases 18,36,37. Although full priming activity requires the p48•p58 complex, the availability of the p48ΔL•UTP•Mn2+ structure provides an important new starting point for refining current models. The initial steps of priming involve binding of the template ssDNA followed by the addition of the 3′-NTP and two metal ions. Previous studies have shown that D109, D111 and D306 play a role in catalysis and that R162 and R163 are important for binding of 3′-NTP 33. Our studies reveal that two additional residues, S160 and H166, are critical for binding the 3′-NTP (Fig. 3).
Primer synthesis occurs only after binding of the 5′-NTP, which has 10-fold lower affinity than the 3′-NTP 36,37. In a mutational study of human primase based on the sequence conservation with X-family polymerases, R304 was found to play a key role in binding of the 5′-NTP and in catalysis 18. Surprisingly, R304 is completely sequestered from the surface in both the p48ΔL and p48ΔL•UTP•Mn2+ structures. Moreover, R304 and the key metal ligand D111 both contact the zinc-binding motif (Fig. S5). This observation suggests that the zinc motif will be reoriented during priming so that D111 and R304 can directly engage the active site.
The comparison of the p48ΔL•UTP•Mn2+ structure with those of AEP and X-family polymerases in different pre-catalytic states can be used to generate models for the conformational changes required to generate a catalytically competent p48 active site. A DALI search with the p48ΔL•UTP•Mn2+ structure identified significant structural similarity to the polymerase domain of AEP polymerase Ligase D (LigD(pol)) with a Z score of 14.9 and a Cα RMSD of 3.7 Å for the LigD(pol)•DNA•UTP•Mn2+ complex 38. The similarity of the catalytic centers is evident in the overlay shown in Figure S7. Because the LigD(pol) structure contains template DNA, the similarity of these two structures argues that the 3′-nucleotide is in a catalytically active conformation. However, the LigD(pol) structure represents an pre-catalytic intermediate because it lacks primer on the template.
There are no structures of AEP family polymerases with a p48-like fold that contain all components including primed template. So to address the potential effect of primed template, we generated a homology model using a structure of the X-family DNA polymerase λ that has a Mg2+ ion, a Mn2+ ion, 3′-NTP (dUpnpp), and template ssDNA 39. Pol λ was chosen because conservation of sequence in the active sites suggests that the pol λ catalytic domain has functional homology to p48 18. Indeed, superposition of the catalytic centers of p48 and pol λ reveals a close alignment of the metal ions and the two internal β strands that contain the conserved catalytic residues D109 and D111, despite a significant difference in the structures at the outer edges of the active site (Fig. 5A, B). Notably, the UTP bound in the p48 active site occupies the same position as the 3′-dUpnpp in pol λ (Fig. 5C). Furthermore, the first nucleotide of the primer in the pol λ structure is positioned close to Mn(II) and the catalytic residue D306 in the p48 structure. The conserved structural positions of the metals and of the 3′-NTP support our proposal that the 3′-NTP occupies a catalytically active conformation.
Fig. 5.

Structural alignment of p48 and pol λ catalytic domains and a model for structural changes to activate p48. (A, B) Core catalytic regions from the structures of p48ΔL•UTP•Mn2+ and the pol λ complex, respectively, including conserved catalytic residues, dUpnpp, UTP and primed DNA (PDB ID: 2PFO). Mn2+ ions in (A) are magenta spheres. Mn2+ and Mg2+ ions in (B) are light and dark green spheres, respectively. (C) Overlay of the active sites from (A) and (B). Metals and key acidic residues are labeled. (D) Proposed conformational changes in p48 required to activate primer synthesis, highlighting the zinc-binding motif (green), template ssDNA (gray), 5′-NTP (yellow), and Mn2+ ions (magenta). Pink dashed arrows show the movement of the side chains of catalytically important residues. The green dashed arrow indicates the shift of the zinc-binding motif.
The structural similarity of the active sites in the structures of p48ΔL•UTP•Mn2+ and the pol λ complex provides a basis for modeling the structural changes in p48 as it shifts into a catalytically competent conformation. The key changes needed are the repositioning of the catalytically important residues D111, R304, and D306 and the generation of interactions to stabilize the 5′-NTP (Fig. 5D). In the p48ΔL•UTP•Mn2+ structure, both D111 and R304 have direct contacts with residues in the zinc motif and are completely sequestered from the surface. The comparison to the pol λ structure suggests that binding of template DNA will trigger a significant structural rearrangement of the zinc motif, which would enable changes in the conformations of these two key residues (Fig. 5D). These conformational changes would result in repositioning of R304 to contact the 5′-NTP and reorientation of the side chains of D111 and D306 to chelate metals, which in turn would position metal (II) to interact with the 3′-OH of 5′-NTP. Based on this model, we suggest that although the mechanism of eukaryotic primer synthesis has long remained poorly understood due to lack of high-resolution structures, the similarities we find between the active site of human p48 to LigD(pol) and pol λ argue that eukaryotic primases utilize a similar mechanism for dinucleotide synthesis.
CONCLUDING REMARKS
In the course of preparing the revised version of this manuscript, crystal structures of p48 in complex the N-terminal domain of p58 (p58N) were published40. One was a 2.7 Å structure of the free protein, which was similar overall to our 1.7 Å structure of free p48ΔL with an RMSD over all backbone atoms common to the two structures of 0.37 Å (Fig. S8). A 3.0 Å structure of p48-p58N soaked with UTP•Mg2+ was also very similar overall to our 2.7 Å structure of p48ΔL•UTP•Mg2+with an RMSD of 0.36 Å over all backbone atoms common to the two structures. The relatively low RMSDs in both the absence and presence of catalytic metals and nucleotide indicates that the interaction with the regulatory p58 subunit has only a very modest effect on the conformation of the catalytic p48 subunit. The interpretations of the two structural analyses of p48 lead to very similar conclusions, extending to the design of mutants for functional analysis. Although different approaches were used, our cell viability data are in full agreement with the biochemical results in that report and together, these constitute a highly complementary and thorough analysis of the key meal and nucleotide binding residues in the primase active site.
MATERIALS AND METHODS
Protein expression and production
Expression and purification of recombinant human primase heterodimer was described previously 41,42. Full length p48 and two p48 constructs p48ΔC (residues 1–408) and p48ΔL (p48ΔC lacking residues 360–379) were sub-cloned into pBG100 (Vanderbilt Center for Structural Biology) containing an N-terminal hexahistidine tag and H3C protease cleavage site and expressed in Escherichia coli BL21 (DE3) cells (Novagen). Cells were grown at 37 °C to an of 0.6–0.7, induced with 0.5 M isopropyl 1-thio-β-D-galactopyranoside, and grown at 18 OD600 °C overnight. Cells were resuspended in buffer containing 20 mM Tris (pH 8.0), 500 mM NaCl, 20 mM imidazole, and 0.1% NP40, and lysed using an Avestin EmulsiFlex C3 homogenizer. Protein was purified by nickel affinity chromatography (GE Healthcare). After removal of the hexahistidine tag by H3C protease, the protein was further purified by heparin sepharose and S200 gel filtration chromatography (GE Healthcare). Mutants of p48 were generated using site-directed mutagenesis and proteins were purified as described above.
Crystallization and structure determination
The p48 proteins were concentrated to 10 mg/ml in sample buffer containing 20 mM HEPES (pH 7.2), 150 mM NaCl, and 2 mM DTT. p48ΔL•UTP•Mn2+ complexes were assembled by mixing p48ΔL and UTP at a 1:3 molar ratio in sample buffer containing 2 mM MnCl2 and incubated at 4°C for 30 min prior to crystallization. Note that no nucleotide was present in crystals grown in the presence of UTP alone or UTP•Mg2+. All crystals were grown by sitting drop vapor diffusion at 21°C. p48ΔC and p48ΔL were crystallized by mixing 2 μl of the protein solution with 2 μl of reservoir solution containing 200 mM sodium citrate (pH 8.0) and 20% PEG 3350, and flash frozen in mother liquor containing 20% glycerol prior to data collection. Crystals of p48ΔL•UTP•Mn2+ complex were grown from drops composed of equal volumes of protein and mother liquor containing 200 mM potassium/sodium tartrate and 20% PEG 3350. X-ray diffraction data were collected at the 21-ID-F beam line at the Advanced Photon Source (Argonne, IL) and processed by HKL2000 43. Crystallographic statistics and space group information are shown in Table S1. Crystals of p48ΔC, p48ΔL and p48ΔL•UTP•Mn2+ each contained one molecule in the asymmetric unit. Phases of p48ΔC were determined by molecular replacement using the Pri1p structure (4LIM) as a search model in the program PHASER 44 and refined using Phenix/Coot. The low resolution of the p48ΔC structure limited our ability to draw conclusions from this structure, but the quality of the model was sufficient to generate a molecular replacement solution for p48ΔL. For the ternary complex, UTP and Mn2+ were located by the presence of 3σ Fo-Fc difference density and verified by Fo-Fc annealed and 2Fo-Fc composite omit maps.
Isothermal Titration Calorimetry
p48/58 heterodimer and p48 were exchanged into 20 mM HEPES (pH 7.2) and 100 mM NaCl prior to the experiment. Binding experiments were performed using a MicroCal VP-isothermal titration calorimeter by first injecting 2 μl of 400 μM nucleotide into 20–30 μM of protein contained in the sample cell, followed by 28 additional 10 μl injections of nucleotide. A three-fold molar excess of MnCl2 relative to the protein concentration was added to the protein solution prior to titrations. The binding data were analyzed using Origin 7.0. Thermodynamic parameters and binding constants (Kd) were calculated by fitting the data to a single-site binding model using a nonlinear least-squares fitting algorithm.
Yeast Strain Construction
The CYC1 terminator from pRS414-ADH45 was cloned into pRS41646 (pRS416-CycT). The PRI1 promoter and coding sequence beginning 274 basepairs upstream and ending at the stop codon was amplified from S288C47 (Mat α SUC2 gal2 mal mel flo1 flo8-1 hap1) and cloned into pRS416-CycT (pDA197). To create a knockout cassette for homologous recombination, approximately 300 bp of PRI1 flanking sequences were cloned into pAG3248 (pDA196). To generate S. cerevisiae knockout strain YDA205 (W303-1a + pri1Δ::HPHMx4 + pDA197), the PRI1 coding sequence was replaced in yeast strain W303-1a49 with the HPHMx4 cassette48 from NotI-linearized pDA196 while covered by plasmid pDA197. Integrants were selected by growth on YPD with 300 μg/ml hygromycin B. Correct integration was verified by phenotype, PCR, and southern blot.
pDA342 contains the native PRI1 promoter and terminator sequences from strain S288C in pRS41446 and allows the addition of an N-terminal HA2 tag. Specifically, the PRI1 promoter sequence from 274 bases upstream was cloned into pRS414 as a XbaI/BamHI fragment. The PRI1 terminator sequence to 451 nucleotides after the stop codon was cloned as a PstI/XhoI. To create the HA2 tag, oligos were annealed and cloned into the BamHI site. The PRI1 coding sequence was amplified from S288C as a BamHI/PstI fragment and cloned into pDA342. Mutations in the PRI1 coding sequence were generated by overlap extension PCR (modified from reference 50) and cloned into pDA342, generating a series of TRP1 marked plasmids.
Plasmid Shuffle
YDA205 was transformed with a series of TRP1 marked plasmids bearing a WT or mutant pri1 allele, selected by growth on synthetic complete media minus uracil and tryptophan (SC-UW), and passaged to SC-W. Liquid cultures in SC-W were grown to stationary phase at room temperature (RT) (~24 °C) with vigorous shaking. Cells were counted by hemocytometer and 1:3 serial dilutions (starting from 1×107 cells/ml), spotted onto SC and SC+FOA (5-fluoroorotic acid) plates, and grown at RT. FOA is toxic in cells expressing URA3/PRI1, selecting for loss of the plasmid and revealing whether the mutant Pri1 allele on the TRP1 plasmid is sufficient for yeast growth. Cells transformed with empty plasmid (pRS414)46 served as a negative control.
Yeast Protein Extracts
Cultures of S. cerevisiae YDA205 containing plasmids encoding a WT PRI1 and an HA2-tagged pri1 mutant allele were grown to mid-log phase in SC-UW, and lysed in 50 mM Tris-Cl pH 7.9; 200 mM NaCl; 0.5 mM EDTA; 10% glycerol; 1 mM DTT; 0.2 mM PMSF; 1x Roche complete protease inhibitor using glass beads. A 15 μg sample of each extract was separated by SDS-PAGE, transferred to NitroPure (GE), and HA2-Pri1p was detected by western blotting using rabbit polyclonal anti-HA (Abcam ab9110). Equal loading of samples was verified by blotting with rat monoclonal anti-tubulin (Abcam YOL1/34).
Supplementary Material
Highlights.
High resolution structures of the catalytic subunit of eukaryotic primases
The catalytic subunit of human primase contains the conserved primase fold
Structural insight into why Mn2+ but not Mg2+ stimulates nucleotide binding
Identified three previously unrecognized critical active site residues
New insights obtained into the mechanism of eukaryotic priming
Acknowledgments
This manuscript is dedicated to the memory of our dear colleague Ellen Fanning, whose passion for research inspired us all. We thank Dr. James M. Berger for early access to the Pri1p structure, LS-CAT staff for help with collection of diffraction data, Dr. Suraj Adhikary for help with structure determination and refinement, and Dr. Nicholas P. George for the p48 H315A construct. This work was supported by NIH grants GM65484 and CA92584 to WJC, GM52948 to EF, GM080570 to BFE, ES00267 to the Vanderbilt Center in Molecular Toxicology, and CA6868485 to the Vanderbilt-Ingram Cancer Center.
Footnotes
ACCESSION NUMBERS
PDB ID: 4LIM (Pri1p), 4LIK (p48ΔL) and 4LIL (p48ΔL•UTP•Mn2+).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Masai H, Matsumoto S, You Z, Yoshizawa-Sugata N, Oda M. Eukaryotic chromosome DNA replication: where, when, and how? Annu Rev Biochem. 2010;79:89–130. doi: 10.1146/annurev.biochem.052308.103205. [DOI] [PubMed] [Google Scholar]
- 2.Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu Rev Biochem. 2002;71:333–374. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- 3.Benkovic SJ, Valentine AM, Salinas F. Replisome-mediated DNA replication. Annu Rev Biochem. 2001;70:181–208. doi: 10.1146/annurev.biochem.70.1.181. [DOI] [PubMed] [Google Scholar]
- 4.Langston LD, Indiani C, O’Donnell M. Whither the replisome: emerging perspectives on the dynamic nature of the DNA replication machinery. Cell Cycle. 2009;8:2686–2691. doi: 10.4161/cc.8.17.9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frick DN, Richardson CC. DNA primases. Annu Rev Biochem. 2001;70:39–80. doi: 10.1146/annurev.biochem.70.1.39. [DOI] [PubMed] [Google Scholar]
- 6.Kuchta RD, Stengel G. Mechanism and evolution of DNA primases. Biochim Biophys Acta. 2010;1804:1180–1189. doi: 10.1016/j.bbapap.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keck JL, Berger JM. Primus inter pares (first among equals) Nat Struct Biol. 2001;8:2–4. doi: 10.1038/82996. [DOI] [PubMed] [Google Scholar]
- 8.Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 9.Corn JE, Berger JM. Regulation of bacterial priming and daughter strand synthesis through helicase-primase interactions. Nucleic Acids Res. 2006;34:4082–4088. doi: 10.1093/nar/gkl363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corn JE, Pelton JG, Berger JM. Identification of a DNA primase template tracking site redefines the geometry of primer synthesis. Nat Struct Mol Biol. 2008;15:163–169. doi: 10.1038/nsmb.1373. [DOI] [PubMed] [Google Scholar]
- 11.Patel SS, Pandey M, Nandakumar D. Dynamic coupling between the motors of DNA replication: hexameric helicase, DNA polymerase, and primase. Curr Opin Chem Biol. 2011;15:595–605. doi: 10.1016/j.cbpa.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keck JL, Roche DD, Lynch AS, Berger JM. Structure of the RNA polymerase domain of E. coli primase. Science. 2000;287:2482–2486. doi: 10.1126/science.287.5462.2482. [DOI] [PubMed] [Google Scholar]
- 13.Johansson E, Macneill SA. The eukaryotic replicative DNA polymerases take shape. Trends Biochem Sci. 2010;35:339–347. doi: 10.1016/j.tibs.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 14.Aravind L, Koonin EV. Prokaryotic homologs of the eukaryotic DNA-end-binding protein Ku, novel domains in the Ku protein and prediction of a prokaryotic double-strand break repair system. Genome Res. 2001;11:1365–1374. doi: 10.1101/gr.181001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weller GR, Doherty AJ. A family of DNA repair ligases in bacteria? FEBS Lett. 2001;505:340–342. doi: 10.1016/s0014-5793(01)02831-9. [DOI] [PubMed] [Google Scholar]
- 16.Iyer LM, Koonin EV, Leipe DD, Aravind L. Origin and evolution of the archaeo-eukaryotic primase superfamily and related palm-domain proteins: structural insights and new members. Nucleic Acids Res. 2005;33:3875–3896. doi: 10.1093/nar/gki702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramsden DA. Polymerases in nonhomologous end joining: building a bridge over broken chromosomes. Antioxid Redox Signal. 2011;14:2509–2519. doi: 10.1089/ars.2010.3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirk BW, Kuchta RD. Arg304 of human DNA primase is a key contributor to catalysis and NTP binding: primase and the family X polymerases share significant sequence homology. Biochemistry. 1999;38:7727–7736. doi: 10.1021/bi990247c. [DOI] [PubMed] [Google Scholar]
- 19.Vaithiyalingam S, Warren EM, Eichman BF, Chazin WJ. Insights into eukaryotic DNA priming from the structure and functional interactions of the 4Fe-4S cluster domain of human DNA primase. Proc Natl Acad Sci USA. 2010;107:13684–13689. doi: 10.1073/pnas.1002009107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agarkar VB, Babayeva ND, Pavlov YI, Tahirov TH. Crystal structure of the C-terminal domain of human DNA primase large subunit: implications for the mechanism of the primase-polymerase alpha switch. Cell Cycle. 2011;10:926–931. doi: 10.4161/cc.10.6.15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sauguet L, Klinge S, Perera RL, Maman JD, Pellegrini L. Shared active site architecture between the large subunit of eukaryotic primase and DNA photolyase. PLoS One. 2010;5:e10083. doi: 10.1371/journal.pone.0010083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Augustin MA, Huber R, Kaiser JT. Crystal structure of a DNA-dependent RNA polymerase (DNA primase) Nat Struct Biol. 2001;8:57–61. doi: 10.1038/83060. [DOI] [PubMed] [Google Scholar]
- 23.Lao-Sirieix SH, Nookala RK, Roversi P, Bell SD, Pellegrini L. Structure of the heterodimeric core primase. Nat Struct Mol Biol. 2005;12:1137–1144. doi: 10.1038/nsmb1013. [DOI] [PubMed] [Google Scholar]
- 24.Ito N, Nureki O, Shirouzu M, Yokoyama S, Hanaoka F. Crystal structure of the Pyrococcus horikoshii DNA primase-UTP complex: implications for the mechanism of primer synthesis. Genes Cells. 2003;8:913–923. doi: 10.1111/j.1365-2443.2003.00693.x. [DOI] [PubMed] [Google Scholar]
- 25.Lee SJ, Zhu B, Akabayov B, Richardson CC. Zinc-binding domain of the bacteriophage T7 DNA primase modulates binding to the DNA template. J Biol Chem. 2012;287:39030–39040. doi: 10.1074/jbc.M112.414151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zea CJ, Camci-Unal G, Pohl NL. Thermodynamics of binding of divalent magnesium and manganese to uridine phosphates: implications for diabetes-related hypomagnesaemia and carbohydrate biocatalysis. Chem Cent J. 2008;2:15. doi: 10.1186/1752-153X-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rymer RU, Solorio FA, Tehranchi AK, Chu C, Corn JE, Keck JL, Wang JD, Berger JM. Binding mechanism of metalNTP substrates and stringent-response alarmones to bacterial DnaG-type primases. Structure. 2012;20:1478–1489. doi: 10.1016/j.str.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirk BW, Kuchta RD. Human DNA primase: anion inhibition, manganese stimulation, and their effects on in vitro start-site selection. Biochemistry. 1999;38:10126–10134. doi: 10.1021/bi990351u. [DOI] [PubMed] [Google Scholar]
- 29.Blanca G, Shevelev I, Ramadan K, Villani G, Spadari S, Hubscher U, Maga G. Human DNA polymerase lambda diverged in evolution from DNA polymerase beta toward specific Mn(++) dependence: a kinetic and thermodynamic study. Biochemistry. 2003;42:7467–7476. doi: 10.1021/bi034198m. [DOI] [PubMed] [Google Scholar]
- 30.Wang TS, Eichler DC, Korn D. Effect of Mn2+ on the in vitro activity of human deoxyribonucleic acid polymerase beta. Biochemistry. 1977;16:4927–4934. doi: 10.1021/bi00641a029. [DOI] [PubMed] [Google Scholar]
- 31.Frank EG, Woodgate R. Increased catalytic activity and altered fidelity of human DNA polymerase iota in the presence of manganese. J Biol Chem. 2007;282:24689–24696. doi: 10.1074/jbc.M702159200. [DOI] [PubMed] [Google Scholar]
- 32.Martin MJ, Garcia-Ortiz MV, Esteban V, Blanco L. Ribonucleotides and manganese ions improve non-homologous end joining by human Polmu. Nucleic Acids Res. 2013;41:2428–2436. doi: 10.1093/nar/gks1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Copeland WC, Tan X. Active site mapping of the catalytic mouse primase subunit by alanine scanning mutagenesis. J Biol Chem. 1995;270:3905–3913. doi: 10.1074/jbc.270.8.3905. [DOI] [PubMed] [Google Scholar]
- 34.Csernoch L, Bernengo JC, Szentesi P, Jacquemond V. Measurements of intracellular Mg2+ concentration in mouse skeletal muscle fibers with the fluorescent indicator mag-indo-1. Biophys J. 1998;75:957–967. doi: 10.1016/S0006-3495(98)77584-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brandt M, Schramm VL. In: Manganese in Metabolism and Enzyme Function. Schramm VL, Wedler FC, editors. Academic Press; New York: 1986. pp. 3–16. [Google Scholar]
- 36.Copeland WC, Wang TS. Enzymatic characterization of the individual mammalian primase subunits reveals a biphasic mechanism for initiation of DNA replication. J Biol Chem. 1993;268:26179–26189. [PubMed] [Google Scholar]
- 37.Sheaff RJ, Kuchta RD. Mechanism of calf thymus DNA primase: slow initiation, rapid polymerization, and intelligent termination. Biochemistry. 1993;32:3027–3037. doi: 10.1021/bi00063a014. [DOI] [PubMed] [Google Scholar]
- 38.Brissett NC, Martin MJ, Pitcher RS, Bianchi J, Juarez R, Green AJ, Fox GC, Blanco L, Doherty AJ. Structure of a preternary complex involving a prokaryotic NHEJ DNA polymerase. Mol Cell. 2011;41:221–231. doi: 10.1016/j.molcel.2010.12.026. [DOI] [PubMed] [Google Scholar]
- 39.Garcia-Diaz M, Bebenek K, Krahn JM, Pedersen LC, Kunkel TA. Role of the catalytic metal during polymerization by DNA polymerase lambda. DNA Repair (Amst) 2007;6:1333–1340. doi: 10.1016/j.dnarep.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kilkenny ML, Longo MA, Perera RL, Pellegrini L. Structures of human primase reveal design of nucleotide elongation site and mode of Pol alpha tethering. Proc Natl Acad Sci U S A. 2013;110:15961–15966. doi: 10.1073/pnas.1311185110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weiner BE, Huang H, Dattilo BM, Nilges MJ, Fanning E, Chazin WJ. An iron-sulfur cluster in the C-terminal domain of the p58 subunit of human DNA primase. J Biol Chem. 2007;282:33444–33451. doi: 10.1074/jbc.M705826200. [DOI] [PubMed] [Google Scholar]
- 42.Copeland WC. Expression, purification, and characterization of the two human primase subunits and truncated complexes from Escherichia coli. Protein Expr Purif. 1997;9:1–9. doi: 10.1006/prep.1996.0665. [DOI] [PubMed] [Google Scholar]
- 43.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 44.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mumberg D, Muller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 1995;156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- 46.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mortimer RK, Johnston JR. Genealogy of principal strains of the yeast genetic stock center. Genetics. 1986;113:35–43. doi: 10.1093/genetics/113.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goldstein AL, Pan X, McCusker JH. Heterologous URA3MX cassettes for gene replacement in Saccharomyces cerevisiae. Yeast. 1999;15:507–511. doi: 10.1002/(SICI)1097-0061(199904)15:6<507::AID-YEA369>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 49.Rothstein RJ. One-step gene disruption in yeast. Methods Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- 50.Lee J, Shin MK, Ryu DK, Kim S, Ryu WS. Insertion and deletion mutagenesis by overlap extension PCR. Methods Mol Biol. 2010;634:137–146. doi: 10.1007/978-1-60761-652-8_10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.