

Abstract
The poor solubility of cisplatin (CDDP) often presents a major obstacle in the formulation of CDDP in nanoparticles (NPs) by traditional methods. We have developed a novel method for synthesizing CDDP NPs taking advantage of its poor solubility. By mixing two reverse microemulsions containing KCl and a highly soluble precursor of CDDP, cis-diaminedihydroplatinum (II), we have successfully formulated CDDP NPs with a controllable size (in the range of 12–75 nm) and high drug loading capacity (approximately 80 wt%). The formulation was done in two steps. The pure CDDP NPs were first stabilized for dispersion in an organic solvent by coating with 1, 2-dioleoyl-sn-glycero-3-phosphate (DOPA). Both x-ray photoelectron spectroscopy and 1H NMR data confirmed that the major ingredient of the DOPA-coated NPs was CDDP. After purification, additional lipids were added to stabilize the NPs for dispersion in an aqueous solution. The final NPs contain a lipid bilayer coating and are named Lipid-Pt-Cl (LPC) NPs, which showed significant antitumor activity both in vitro and in vivo. Thus, CDDP precipitate serves as the major material for assembling the novel NPs. This unique method of nanoparticle synthesis may be applicable in formulating other insoluble drugs.
Keywords: Cisplatin, Nanoparticle, Drug delivery, Nanoprecipitate, Microemulsion
1. Introduction
Cisplatin (CDDP) is a chemotherapy drug commonly used in the clinical setting. In clinics, maximum tolerated dose (MTD) of CDDP is significantly limited by nephrotoxicity [1, 2]. To improve patient care, carboplatin and oxaliplatin are administered, while altering the chloride leaving groups of CDDP with 1,2-diaminocyclohexane or an oxalate ligand compromises outcome [1].
In order to maintain the efficacy and reduce the nephrotoxicity of cisplatin, nanoparticulate CDDP formulations are very promising. Nanoparticulate CDDP formulations have been achieved through chelating CDDP with polymers and NPs [3–5], loading of a pro-drug in the poly(lactide-co-glycolide) (PLGA) nanoparticles (NPs) or encapsulating CDDP into liposomes [3, 6–13]. For example, CDDP was loaded into PLGA NPs by exploiting double emulsion technique, while the encapsulation and loading efficacy were low and burst release was often observed [14]. Dhar et al utilized a prodrug strategy, i.e., modified the hydrophobicity of CDDP, and therefore improved the encapsulation of CDDP into PLGA NPs [13, 15, 16]. Kataoka et al alternatively chelated CDDP positively charged platinum species to carboxylate-rich copolymers with a drug loading of 30 wt% and showed a strong relationship between the therapeutic efficacy and the size of carrier [3, 4]. Lipoplatin, a liposomal formulation, employed electrostatic interaction to load positively charged platinum into negatively charged 1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol sodium salt (DPPG)-lipid micelles [17, 18]. For Lipoplatin, reverse micelles were mixed with premade liposomes and homogenized by extrusion. Drug loading of Lipoplatin was reported to be 8.9 wt%. However, these formulations were for either prodrug or charged platinum, but not for native CDDP.
While the synthesis of CDDP (Scheme 1) is a well-documented reaction in the field of inorganic chemistry [19], the poor solubility of CDDP in both water and organic solvents significantly hinders the development of nanoparticulate formulations in a manner similar to the formulation of nanoparticles with hydrophobic drugs [20, 21]. In our lab, we have recently developed a Lipid coated Calcium Phosphate (LCP) platform to deliver diverse bioactive molecules, such as DNA, silencing RNA and gemcitabine triphosphate [22–24]. An outer layer of a cationic lipid 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt) (DOTAP) and high density of poly(ethylene glycol) (PEG) was coated on the calcium phosphate cores. The cationic lipid DOTAP allows the nanoparticles to be internalized by tumor cells more efficiently and to subsequently escape from the lysosomes. Additionally, a high density of PEGylation will help the nanoparticles avoid reticuloendothelial system (RES) system, improving drug pharmacokinetics and drug bioavailability. We found both components are critical for the successful delivery of drugs into tumors.
Scheme 1.
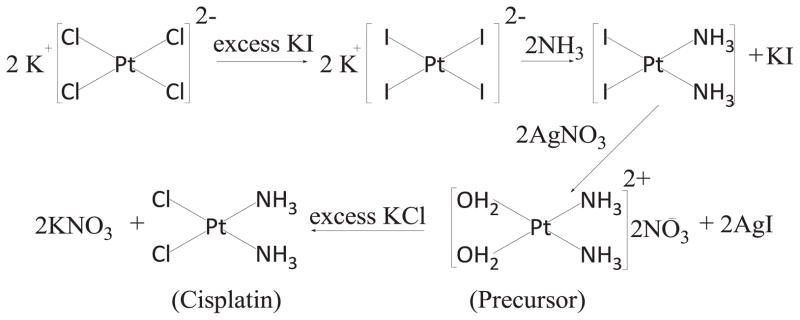
Classic synthesis route of CDDP.
Herein, we wish to replace calcium phosphate by CDDP as the core in order to make a CDDP nanoparticulate formulation. Our new formulation would be favorable due to its high drug loading capacity. Another aspect is that, our platform might be applicable to the manufacture of many other CDDP analog nanoparticulate formulations. Our platform is expected to improve the solubility of platinum based drug candidates with poor solubility, such as cis-diamminedibromoplatinum(II) and cis-diamminediiodoplatinum(II). Therefore, we hypothesize that: (1) CDDP can be encapsulated as a nanoprecipitate in a microemulsion and stabilized in an organic solvent with 1, 2-dioleoyl-sn-glycero-3-phosphate (DOPA); (2) DOPA-coated CDDP NPs can be further dispersed into aqueous solution by adding lipids to form the outer leaflet of the coating bilayer; (3) the lipid bilayer-coated CDDP NPs will show anti-cancer activity in vitro and in vivo.
2. Materials and methods
2.1. Materials
Lipids were purchased from Avanti Polar Lipids (Alabaster, AL). Dulbecco’s Modified Eagle Medium (DMEM), L-glutamine, penicillin Gsodium, streptomycin and fetal calf serum were purchased from Gibco®. 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[anisamide(polyethylene glycol)-2000] (DSPE-PEG-AA) was synthesized in our laboratory as previously reported [25]. 1-Hexanol was purchased from Alfa Aesar. Igepal® CO-520, triton™ X-100, cyclohexane, cisplatin and silver nitrate were obtained from Sigma-Aldrich (St Louis, MO) without further purification.
2.2. Cell lines
1205Lu cells were cultured in DMEM medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 20 mM of L-glutamine, 100 U/mL of penicillin G sodium, and 100 mg/mL of streptomycin at 37 °C in an atmosphere of 5% CO2 and 95% air.
2.3. Synthesis of cis-[Pt(NH3)2(H2O)2](NO3)2 precursor
To a suspension of CDDP (60 mg, 0.20 mmol) in 1.0 mL water, AgNO3 (66.2 mg, 0.39 mmol) was added. The mixture was heated at 60 °C for 3 h and then stirred overnight in a flask protected from light with aluminum foil. Afterwards, the mixture was centrifuged at 16,000 rpm for 15 min to remove the AgCl precipitate. The solution was then filtered using a 0.2 μm syringe filter. The concentration of cis-[Pt(NH3)2(H2O)2](NO3)2 was measured using inductively coupled plasma mass spectrometry (ICP-MS) and adjusted to 200 mM.
2.4. Preparation of LPC NPs
The synthesis route of LPC NPs is described in Scheme 2. First, 100 μL of 200 mM cis-[Pt(NH3)2(H2O)2](NO3)2 was dispersed in a solution composed of mixture of cyclohexane/Igepal CO-520 (71:29, V:V) and cyclohexane/triton-X100/hexanol (75:15:10, V:V:V) to form a well-dispersed, water-in-oil reverse micro-emulsion. Another emulsion containing KCl was prepared by adding 100 μL of 800 mM KCl in water into a separate 8.0 mL oil phase. One hundred μL of DOPA (20 mM) was added to the CDDP precursor phase and the mixture was stirred. Twenty minutes later, the two emulsions were mixed and the reaction proceeded for another 30 min. After that, 16.0 mL of ethanol was added to the micro-emulsion and the mixture was centrifuged at 12,000 g for at least 15 min to remove the cyclohexane and surfactants. After being extensively washed with ethanol 2–3 times, the pellets were re-dispersed in 3.0 mL of chloroform and stored in a glass vial for further modification.
Scheme 2.

Synthesis of lipid bilayer-coated CDDP NPs using a reverse microemulsion method.
To prepare the final NPs, 1.0 mL of LPC NPs core, 100 μL of 20 mM DOTAP/Cholesterol (molar ratio 1:1) and 50 μL of 10 mM DSPE–PEG-2000 or DSPE–PEG–AA were combined. After evaporating the chloroform, the residual lipids were dispersed in 1.0 mL of d-H2O.
2.5. Characterization of NPs
The zeta potential and particle size of LPC NPs were determined using a Malvern ZetaSizer Nano series (Westborough, MA). Transmission electron microscopy (TEM) images were acquired using a JEOL 100CX II TEM (JEOL, Japan). The LPC NPs were negatively stained with 2% uranyl acetate.
The drug-loading capacity and platinum content were measured using Varian 820-MS (Palo Alto, CA). Samples were digested in 70% HNO3 and diluted in water to a final acid content of 2%. Platinum concentration was determined using the 195Pt isotope.
The composition of DOPA-coated CDDP NPs was studied using X-ray photoelectron spectroscopy (XPS) (Kratos Axis Ultra DLD X-ray Photoelectron Spectrometer) and NMR (Varain Inova 400 NMR Spectrometer).
2.6. Cell toxicity assay
1205Lu cells were seeded in 96-well plates at a density of 2000 cells/well and incubated in 10% FBS of DMEM containing 100 U/mL penicillin, and 100 mg/mL streptomycin for 20 h. Then, the medium was removed and replaced by Opti-MEM containing CDDP or LPC NPs. Forty-eight hours later, a CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI) kit containing the tetrazolium compound MTS was used to assay cell viability according to the manufacturer’s protocols.
2.7. Cellular uptake
1205Lu cells (2 × 105) were seeded in 35 mm MatTek glass bottom dishes (MatTek Corporation, MA) 20 h before experiments. The cells were treated with NBD-PE labeled LPC NPs at a concentration of 100 μM platinum (Pt) at 37 °C for 4 h. The cells were subsequently washed twice with PBS. The nucleus was stained with Hoechest 33342 (Sigma, St Louis, MO), and lysosomes were stained by lysotracker red (Invitrogen, Carlsbad, CA). Then, the sample was observed by Olympus FV 1000-MPE microscope (Olympus, Japan).
To measure the amount of Pt in cells, cells were washed and lysed in order to determine their uptake of NPs. Samples were digested in 70% HNO3 and diluted in water to a final acid content of 2%.
2.8. In vivo anticancer efficacy evaluation
Animals were maintained in the Center for Experimental Animals (an AAALAC accredited experimental animal facility) at the University of North Carolina. All procedures involving experimental animals were performed in accordance with the protocols approved by the University of North Carolina Institutional Animal Care and Use committee and conformed to the Guide for the Care and Use of Laboratory Animals (NIH publication No. 86–23, revised 1985). Female athymic nude mice, 5–6 weeks old and weighing 18–22 g, were supplied by the University of North Carolina animal facility. 1205Lu xenograft tumors were developed through subcutaneous injection of approximately 5 million 1205Lu cells in female nude mice. 2.0 mg/kg of Pt was administered weekly by IV injection for CDDP and LPC NPs groups. Tumor growth and body weight were monitored. Tumor volume was calculated using the following formula: , with W being smaller than L. Finally, mice were sacrificed by CO2 inhalation. Tumors were collected after treatment and were formalin fixed and processed for TUNEL assay.
2.9. TUNEL assay
The tumors were fixed in 4.0% paraformaldehyde (PFA) and subsequently paraffin-embedded, to be sectioned at the UNC Lineberger Comprehensive Cancer Center Animal Histopathology Facility. To detect apoptotic cells in tumor tissues, a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, using a DeadEndTM Fluorometric TUNEL System (Promega, Madison, WI), was performed, following the manufacturer’s protocol. Cell nuclei, which were stained with green fluorescence, were defined as TUNEL-positive nuclei. TUNEL-positive nuclei were monitored using a fluorescence microscope (Nikon, Tokyo, Japan). The cell nuclei were stained with 4, 6-diaminidino-2-phenyl-indole (DAPI) (Vectashield, Vector Laboratories, Inc., Burlingame, CA). To quantify TUNEL-positive cells, green-fluorescence-positive cells were counted in three images taken at 40× magnification.
2.10. Statistical analysis
Quantitative data were expressed as mean ± SEM. The analysis of variance is completed using a one-way ANOVA. P < 0.05 was considered statistically significant.
3. Results and discussions
3.1. Synthesis and characterization of DOPA-coated CDDP Cores
CDDP NPs were synthesized in microemulsion during the reaction between KCl and its highly soluble cis-[Pt(NH3)2(H2O)2](NO3)2 precursor. To synthesize stable CDDP precipitates, DOPA, which is known to strongly interact with the platinum cation at the interface [26–28], was used. To maximize the yield of CDDP NPs, an excess of KCl was used to inhibit hydrolysis equilibrium. After CDDP was precipitated, CDDP cores were then coated with a hydrophobic layer of DOPA (Scheme 2). DOPA-coated CDDP NPs were purified in a manner similar to that of silica NPs, which are also synthesized in microemulsion. Ethanol was added to destroy the emulsion and precipitate CDDP NPs. DOPA-coated CDDP NPs precipitate was then collected by centrifugation and readily dispersed in chloroform, toluene or hexane. By adjusting the composition of the surfactant system, the size of the NPs can be altered between 12 to 75 nm in diameter (Fig. 1).
Fig. 1.

TEM images of DOPA-coated, CDDP NPs prepared using different surfactant systems. The surfactants used to create the microemulsion were a mixture of Igepal-520 system (Igepal-520: cyclohexane=30:70 (v/v)) and Triton X-100 system (Triton X-100: Hexanol: cyclohexane=15:10:75 (v/v/v); the volume ratio of Igepal-520 system to Triton X-100 system was 6:2 (A), 2:6 (B) and 0:8 (C) respectively.
X-ray photoelectron spectroscopy (XPS) was used to confirm the composition of DOPA-coated CDDP NPs (Fig. 2). The ratio of N:Pt:Cl was 2:1:1.8, and a small amount of phosphorous element was also present, as expected from the presence of DOPA (Fig. 2D). In addition, no potassium element was found. We also confirmed the composition of DOPA-coated CDDP NPs using 1H NMR spectra in DMF-d7 (shown in Fig. 3). The data illustrated that the major peaks of DOPA-coated NPs were consistent with those of CDDP; and therefore, we can conclude that NPs are core-shell structures with CDDP as the core and DOPA as a coating layer. The DOPA-coated CDDP NPs were expected to have high drug loading capacity. We used ICP-MS to measure the content of Pt and used NBD-labeled DOPA to measure the amount of DOPA on the surface of CDDP NPs. The results indicated that the yield of DOPA-coated CDDP NPs was approximately 45 ± 3 wt%. The NPs were also characterized with drug loading capacity as high as 93 ± 2 wt%.
Fig. 2.

XPS study of Pt 4f (A), N 1s (B), Cl 2p (C) and P 2p (D) on CDDP and DOPA-coated CDDP NPs.
Fig. 3.
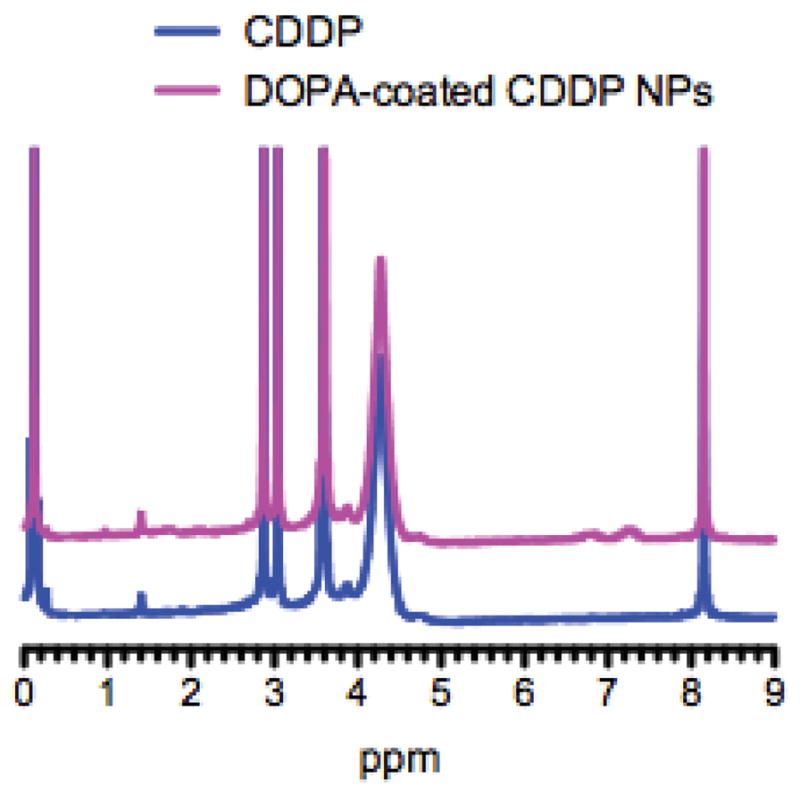
1H NMR spectra of CDDP and DOPA-coated CDDP NPs in DMF-d7.
3.2. Facile surface engineering of hydrophobic DOPA-coated CDDP NPs with outer leaflet lipids
To further disperse the hydrophobic DOPA-coated CDDP NPs into aqueous solution, additional lipids composed of DOTAP, cholesterol, DSPE-mPEG and DSPE-PEG-AA (molar ratio 4: 4: 1: 1) were used. These lipids self-assembled in water into the outer leaflet of the bilayer through a hydrophobic interaction with DOPA-coated CDDP NPs as a template [29, 30]. The DOPA layer served as inner leaflet of the asymmetrical bilayer coating CDDP core. Composition of outer leaflet lipids was carefully chosen to contain steric lipid (DSPE-mPEG) for prolonged circulation of NPs in the blood stream [31], DSPE-PEG-anisamide for vivid uptake of NPs by the tumor cells and DOTAP for rupturing endosomes [32, 33].
The final NPs are named Lipid-Pt-Cl (LPC) NPs. LPC NPs were then purified via centrifugation to remove free liposomes formed as the result of excess outer leaflet lipids. LPC NPs were negatively stained with uranyl acetate and examined using TEM (Fig. 4A). The clear core/membrane structure revealed the bilayer coating the surface of LPC NPs. As shown in Fig. 4A, size of the particles determined using TEM was approximately 15 nm, which was smaller than the results obtained from dynamic light scattering (DLS) (Fig. 4B). Zeta potential of the NPs was +15 ± 0.3 mV and DLS results showed that distribution of LPC NPs was narrow, with a PDI of 0.15 ± 0.02. The drug loading of LPC NPs was approximately 82 ± 5 wt%. Overall, these results indicate LPC NPs are well dispersed in aqueous solution and characterized by a high drug loading.
Fig. 4.
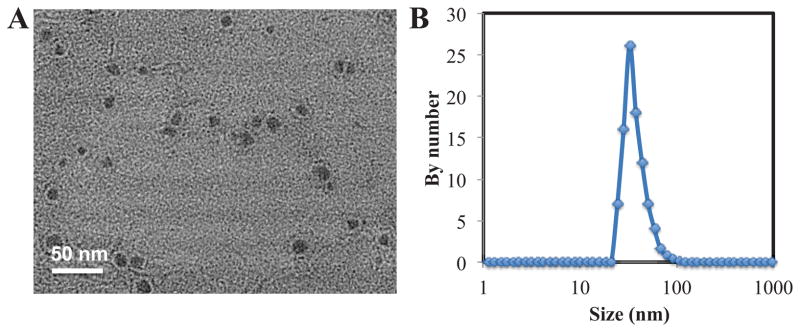
TEM image of CDDP NPs negatively stained using uranyl acetate (A) and size distribution by DLS (B) of LPC NPs.
Previously, a similar formulation, Lipoplatin, employs electrostatic interaction to load positively charged platinum into negatively charged DPPG-lipid micelles. Reverse micelles were mixed with premade liposomes and homogenized by extrusion [18]. Drug loading was reported to be inefficient, yielding a loading of 8.9 wt% cisplatin [17]. In contrast, LPC NPs employed a microemulsion system to synthesize a hydrophobic, pure cisplatin-based nanoparticle with tunable size for customized delivery, which further surface-functionalized with amphiphilic lipids to ensure successful evasion of reticuloendothelial system and efficient tumor targeting. LPC NPs achieved high loading capacity due to the solid, cisplatin-based core, producing nanoparticles containing up to 80 wt% drug. In addition, DOPA-coated CDDP NPs was prepared with the hydrophobic surface in organic solvent, allowing versatile coating and surface modification for a variety of purposes, in a manner similar to quantum dots and iron nanoparticles. We believe our platform may be of significant value to the research community as an alternative approach to develop nanoparticles made of the drug itself rather than loaded with drug as cargos.
3.3. Cell toxicity and cellular uptake of LPC NPs in 1205Lu cells
To test the anticancer efficacy of LPC NPs, we evaluated the performance of LPC NPs in 1205Lu melanoma cancer cells. DOPA-coated CDDP NPs with size of 12 nm were used to prepare LPC NPs for the evaluation of anti-cancer effect. As shown in Fig. 5A, the IC50 of CDDP and LPC NPs in 1205Lu cells was 12.4 ± 1.5 μM and 0.80 ± 0.2 μM respectively. Additionally, the empty liposome vesicles having a composition similar to that of the coating bilayer of LPC did not show any cytotoxicity (data not shown). The cellular uptake of LPC NPs was studied using confocal microscopy and ICP-MS. We labeled LPC NPs using fluorescent 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt) (NBD-PE) lipid and incubated them with cells. Some of the LPC NPs were co-localized with lysosomes as shown by the yellow areas (Fig. 6). However, while a large number of NPs were endocytosed, they showed little accumulation in the lysosomes; a phenomenon that is facilitated by the capability of DOTAP to enhance endosome escape. Quantitative data indicated that cellular uptake of LCP NPs was more efficient than free CDDP (Fig. 5B). LPC NPs delivering CDDP showed 11-fold increase in drug internalization over free CDDP, which explained the low IC50 of LPC NPs.
Fig. 5.
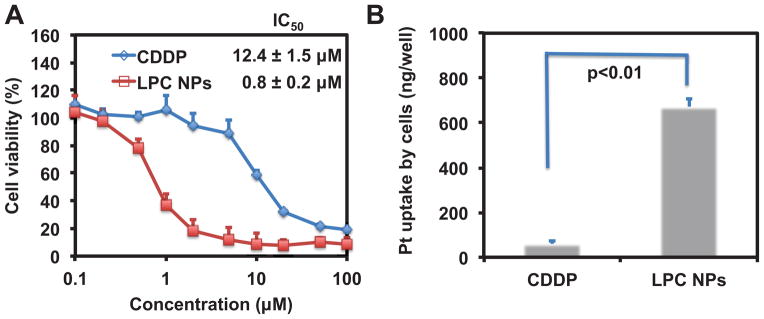
Cytotoxicity of free CDDP and LPC NPs in 1205Lu cells (A) and the amount of cell uptake of CDDP and LPC NPs in 1205Lu tumor cells quantified using ICP-MS (B). Data is expressed as the amount of the Pt drug associated with cells incubated with 100 μM CDDP or LPC NPs in 24 well plates. Each bar represents the mean ± SEM of three independent experiments. The analysis of variance was completed using a one-way ANOVA.
Fig. 6.
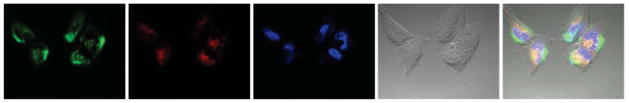
Cell uptake of LPC NPs in 1205Lu cell line imaged by confocal microscopy. The LPC NPs were labeled with NBD-PE lipid (green). Lysosome (red) and nucleus (blue) were stained by Lysotracker-Red and Hoechst 33342, separately.
3.4. In vivo anti-cancer efficacy of LPC NPs on 1205Lu xenograft tumor and TUNEL assay
Efficacy of LPC NPs in a xenograft tumor model was further evaluated. Fig. 7A illustrates that free CDDP only had a partial effect on tumor growth inhibition at the dose and dose schedule adopted in the experiment. In contrast, tumor growth was significantly suppressed when LPC NPs were administered intravenously. In addition, mice in the treatment groups did not exhibit significant weight loss (Fig. 7B). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay is a method for detecting DNA fragmentation occurring in apoptosis by labeling the terminal end of nucleic acids. TUNEL images (Fig. 7C) indicate that LPC NPs induced noticeably more apoptosis (32.1 ± 3.6%) than free CDDP (6.3 ± 0.8%), which is consistent with the observed efficacy of tumor inhibition. All in all, our data suggest that LPC NPs are both effective and safe in treating 1205Lu melanoma tumors. Biodistribution and lack of nephro- and hepato-toxicities of LPC NPs have been studied and published elsewhere [34].
Fig. 7.

Effects of free CDDP and LPC NPs on growth of 1205Lu tumors (A) and relative body weight (B). Free CDDP and LPC NPs were administered intravenously at a dose of 2.0 mg/kg Pt. After mice were sacrificed, tumor tissue was sectioned for TUNEL assay (C). The arrowheads indicate the time of injection. Tumor volume (TV) was calculated using the following formula: , with W < L. The results are shown as means ± SEM (error bars) of 5 animals per group and are representative of two independent experiments. The analysis of variance is completed using a one-way ANOVA. *, P < 0.05; **, P < 0.01.
4. Conclusions
Successful fabricated CDDP NPs are characterized with high drug loading capacity and optimal aqueous dispensability. Engineered via the microemulsion method and coated with DOPA and additional outer leaflet layers, LPC NPs exhibited significant antitumor effects both in vitro and in vivo. By adjusting the fabrication parameters, size of CDDP NPs can also be altered between 12–75 nm for optimal tumor accumulation. Note that CDDP is the major ingredient of LPC. As far as we know, this is a good example of using an unmodified drug as a material for the assembly of NPs. Synthesis of LPC NPs can be applied to formulate other insoluble drugs.
Acknowledgments
This work was supported by NIH grants CA129835, CA129421, CA151652, CA151455 and CA149363. We thank C. Michael Lin, Steven Glenn Plonk and Kelly Racette for their assistance in manuscript preparation and Chin-Ying Chung and Gavin Robertson for providing 1205Lu cell line.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yao X, Panichpisal K, Kurtzman N, Nugent K. Cisplatin nephrotoxicity: a review. Am J Med Sci. 2007;334:115–124. doi: 10.1097/MAJ.0b013e31812dfe1e. [DOI] [PubMed] [Google Scholar]
- 2.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int. 2008;73:994–1007. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- 3.Plummer R, Wilson RH, Calvert H, Boddy AV, Griffin M, Sludden J, Tilby MJ, Eatock M, Pearson DG, Ottley CJ, Matsumura Y, Kataoka K, Nishiya T. A Phase I clinical study of cisplatin-incorporated polymeric micelles (NC-6004) in patients with solid tumours. Br J Cancer. 2011;104:593–598. doi: 10.1038/bjc.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, Nishiyama N, Kataoka K. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815–823. doi: 10.1038/nnano.2011.166. [DOI] [PubMed] [Google Scholar]
- 5.Baba M, Matsumoto Y, Kashio A, Cabral H, Nishiyama N, Kataoka K, Yamasoba T. Micellization of cisplatin (NC-6004) reduces its ototoxicity in guinea pigs. J Control Release. 2012;157:112–117. doi: 10.1016/j.jconrel.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 6.Sengupta P, Basu S, Soni S, Pandey A, Roy B, Oh MS, Chin KT, Paraskar AS, Sarangi S, Connor Y, Sabbisetti VS, Kopparam J, Kulkarni A, Muto K, Amarasiriwardena C, Jayawardene I, Lupoli N, Dinulescu DM, Bonventre JV, Mashelkar RA, Sengupta S. Cholesterol-tethered platinum II-based supramolecular nanoparticle increases antitumor efficacy and reduces nephrotoxicity. Proc Natl Acad Sci USA. 2012;109:11294–11299. doi: 10.1073/pnas.1203129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harrington KJ, Rowlinson-Busza G, Syrigos KN, Vile RG, Uster PS, Peters AM, Stewart JS. Pegylated liposome-encapsulated doxorubicin and cisplatin enhance the effect of radiotherapy in a tumor xenograft model. Clin Cancer Res. 2000;6:4939–4949. [PubMed] [Google Scholar]
- 8.Zisman N, Dos Santos N, Johnstone S, Tsang A, Bermudes D, Mayer L, Tardi P. Optimizing Liposomal Cisplatin Efficacy through Membrane Composition Manipulations. Chemother Res Pract. 2011;2011 doi: 10.1155/2011/213848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zamboni W, Gervais A, Egorin M, Schellens JM, Zuhowski E, Pluim D, Joseph E, Hamburger D, Working P, Colbern G, Tonda M, Potter D, Eiseman J. Systemic and tumor disposition of platinum after administration of cisplatin or STEALTH liposomal-cisplatin formulations (SPI-077 and SPI-077 B103) in a preclinical tumor model of melanoma. Cancer Chemother Pharmacol. 2004;53:329–336. doi: 10.1007/s00280-003-0719-4. [DOI] [PubMed] [Google Scholar]
- 10.Vail DM, Kurzman ID, Glawe PC, O’Brien MG, Chun R, Garrett LD, Obradovich JE, Fred RM, 3rd, Khanna C, Colbern GT, Working PK. STEALTH liposome-encapsulated cisplatin (SPI-77) versus carboplatin as adjuvant therapy for spontaneously arising osteosarcoma (OSA) in the dog: a randomized multicenter clinical trial. Cancer Chemother Pharmacol. 2002;50:131–136. doi: 10.1007/s00280-002-0469-8. [DOI] [PubMed] [Google Scholar]
- 11.Seetharamu N, Kim E, Hochster H, Martin F, Muggia F. Phase II study of liposomal cisplatin (SPI-77) in platinum-sensitive recurrences of ovarian cancer. Anticancer Res. 2010;30:541–545. [PubMed] [Google Scholar]
- 12.Khiati S, Luvino D, Oumzil K, Chauffert B, Camplo M, Barthélémy P. Nucleoside–Lipid-Based Nanoparticles for Cisplatin Delivery. ACS Nano. 2011;5:8649–8655. doi: 10.1021/nn202291k. [DOI] [PubMed] [Google Scholar]
- 13.Dhar S, Kolishetti N, Lippard SJ, Farokhzad OC. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc Natl Acad Sci USA. 2011;108:1850–1855. doi: 10.1073/pnas.1011379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Avgoustakis K, Beletsi A, Panagi Z, Klepetsanis P, Karydas AG, Ithakissios DS. PLGA-mPEG nanoparticles of cisplatin: in vitro nanoparticle degradation, in vitro drug release and in vivo drug residence in blood properties. J Control Release. 2002;79:123–135. doi: 10.1016/s0168-3659(01)00530-2. [DOI] [PubMed] [Google Scholar]
- 15.Kolishetti N, Dhar S, Valencia PM, Lin LQ, Karnik R, Lippard SJ, Langer R, Farokhzad OC. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc Natl Acad Sci USA. 2010;107:17939–17944. doi: 10.1073/pnas.1011368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci USA. 2008;105:17356–17361. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boulikas T. Clinical overview on Lipoplatin: a successful liposomal formulation of cisplatin. Expert Opin Invest Drugs. 2009;18:1197–1218. doi: 10.1517/13543780903114168. [DOI] [PubMed] [Google Scholar]
- 18.Boulikas T. United States Patent 6,511,676. Therapy for human cancers using cisplatin and other drugs or genes encapsulated into liposomes. 2003
- 19.Alderden RA, Hall MD, Hambley TW. The discovery and development of cisplatin. J Chem Educ. 2006;83:728–734. [Google Scholar]
- 20.Aryal S, Hu CMJ, Zhang L. Polymer-Cisplatin Conjugate Nanoparticles for Acid-Responsive Drug Delivery. ACS Nano. 2009;4:251–258. doi: 10.1021/nn9014032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt (IV) prodrug-PLGA–PEG nanoparticles. Proc Natl Acad Sci USA. 2008;105:17356–17361. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Kim WY, Huang L. Systemic delivery of gemcitabine triphosphate via LCP nanoparticles for NSCLC and pancreatic cancer therapy. Biomaterials. 2013;34:3447–3458. doi: 10.1016/j.biomaterials.2013.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, Yang Y, Huang L. Calcium phosphate nanoparticles with an asymmetric lipid bilayer coating for siRNA delivery to the tumor. J Control Release. 2012;158:108–114. doi: 10.1016/j.jconrel.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu Y, Haynes MT, Wang Y, Liu F, Huang L. A highly efficient synthetic vector: nonhydrodynamic delivery of DNA to hepatocyte nuclei in vivo. ACS Nano. 2013;7:5376–5384. doi: 10.1021/nn4012384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banerjee R, Tyagi P, Li S, Huang L. Anisamide-targeted stealth liposomes: A potent carrier for targeting doxorubicin to human prostate cancer cells. Int J Cancer. 2004;112:693–700. doi: 10.1002/ijc.20452. [DOI] [PubMed] [Google Scholar]
- 26.Burger KN, Staffhorst RW, de Vijlder HC, Velinova MJ, Bomans PH, Frederik PM, de Kruijff B. Nanocapsules: lipid-coated aggregates of cisplatin with high cytotoxicity. Nat Med. 2002;8:81–84. doi: 10.1038/nm0102-81. [DOI] [PubMed] [Google Scholar]
- 27.Khiati S, Luvino D, Oumzil K, Chauffert B, Camplo M, Barthélémy P. Nucleoside–Lipid-Based Nanoparticles for Cisplatin Delivery. ACS Nano. 2011;5:8649–8655. doi: 10.1021/nn202291k. [DOI] [PubMed] [Google Scholar]
- 28.Velinova MJ, Staffhorst RW, Mulder WJ, Dries AS, Jansen BA, de Kruijff B, de Kroon AI. Preparation and stability of lipid-coated nanocapsules of cisplatin: anionic phospholipid specificity. Biochim Biophys Acta, Biomembr. 2004;1663:135–142. doi: 10.1016/j.bbamem.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 29.Lin CAJ, Sperling RA, Li JK, Yang TY, Li PY, Zanella M, Chang WH, Parak WJ. Design of an Amphiphilic Polymer for Nanoparticle Coating and Functionalization. Small. 2008;4:334–341. doi: 10.1002/smll.200700654. [DOI] [PubMed] [Google Scholar]
- 30.Chen T, Öçsoy I, Yuan Q, Wang R, You M, Zhao Z, Song E, Zhang X, Tan W. One-Step Facile Surface Engineering of Hydrophobic Nanocrystals with Designer Molecular Recognition. J Am Chem Soc. 2012;134:13164–13167. doi: 10.1021/ja304115q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klibanov AL, Maruyama K, Torchilin VP, Huang L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990;268:235–237. doi: 10.1016/0014-5793(90)81016-h. [DOI] [PubMed] [Google Scholar]
- 32.Nakagawa O, Ming X, Huang L, Juliano RL. Targeted Intracellular Delivery of Antisense Oligonucleotides via Conjugation with Small-Molecule Ligands. J Am Chem Soc. 2010;132:8848–8849. doi: 10.1021/ja102635c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu Y, Szoka FC. Mechanism of DNA Release from Cationic Liposome/DNA Complexes Used in Cell Transfection†,‡. Biochemistry. 1996;35:5616–5623. doi: 10.1021/bi9602019. [DOI] [PubMed] [Google Scholar]
- 34.Guo S, Wang Y, Miao L, Xu Z, Lin CM, Zhang Y, Huang L. Lipid-Coated Cisplatin Nanoparticles Induce Neighboring Effect and Exhibit Enhanced Anticancer Efficacy. ACS Nano. 2013 doi: 10.1021/nn403606m. [DOI] [PMC free article] [PubMed] [Google Scholar]