

Abstract
Neurofibromatosis Type 1 (NF1) is an autosomal disorder that affects neural crest-derived tissues, leading to a wide spectrum of clinical presentations. Patients commonly present with plexiform neurofibromas, benign but debilitating growths that can transform into malignant peripheral nerve sheath tumors (MPNSTs), a main cause of mortality. Currently, surgery is the primary course of treatment for MPNST, but with the limitation that these tumors are highly invasive. Radiation therapy is another treatment option, but is undesirable because it can induce additional mutations. MPNST patients may also receive doxorubicin as therapy, but this DNA-intercalating agent has relatively low tumor specificity and limited efficacy. In this study, we exploited a robust genetically-engineered mouse model of MPNST that recapitulates human NF1 associated MPNST to identify a novel small chemical compound that inhibits tumor cell growth. Compound 21 (Cpd21) inhibits growth of all available in vitro models of MPNST and human MPNST cell lines, while remaining non-toxic to normally-dividing Schwann cells or mouse embryonic fibroblasts. We show that this compound delays the cell cycle and leads to cellular apoptosis. Moreover, Cpd21 can reduce MPNST burden in a mouse allograft model, underscoring the compound’s potential as a novel chemotherapeutic agent.
Keywords: Neurofibromatosis Type1, NF1, Malignant Peripheral Nerve Sheath Tumor, MPNST, small molecule, apoptosis
Introduction
Neurofibromatosis type 1 (NF1) is an autosomal dominant genetic disorder that affects 1 in 3500 people (1). Patients typically present with a constellation of signs, including café-au-lait pigmentation spots, benign neurofibromas, axillary or inguinal freckling, optic gliomas, Lisch nodules in the eye, bone abnormalities, and multiple additional less penetrant manifestations (2). Furthermore, NF1 patients are predisposed to malignant peripheral nerve sheath tumors (3), which are a significant source of mortality.
Neurofibromas are benign tumors that grow on the skin (dermal) or inside the body (plexiform) (4). Neurofibromas have complex cellularity including Schwann lineage cells, fibroblasts, perineurial cells, mast cells, collagen deposits, and physical proximity to peripheral nerves. Plexiform neurofibromas appear in 30% of NF1 patients (5), and although they retain the capacity to enlarge throughout life, many lines of evidence point to an embryonic origin (4). Dermal neurofibromas can arise from a type of adult stem cell called skin-derived precursors (SKPs) (4), while plexiform neurofibromas arise from embryonic neural crest lineage Schwann cell progenitors (6). Whether the two cell types are related or the same remains unclear (7).
NF1 patients can also develop malignant tumors, including leukemia, rhabdomyosarcoma and neuroblastoma in children (2). The most common malignancy, however, is MPNST, which arises from and appears in, approximately 9–21% of patients with plexiform neurofibromas (8). MPNSTs are initially difficult to detect, metastasize widely, and have a poor prognosis (9). Mutation of Nf1, which encodes a RAS-GTPase activating enzyme (10), is known to activate RAS downstream effectors including the ERK, PI3K, and mTOR pathways (11–15). The two most common cancer-associated mutations present in MPNSTs in addition to Nf1 mutation are the tumor suppressors p53 (16–18) and CDKN2 (16, 19–23). Mice harboring mutated Nf1 and p53 or Nf1 and Cdkn2 spontaneously develop MPNST (24, 25). In addition to Nf1 and its downstream effector Ras, the conditional deletion of Pten also leads to MPNST in mice (26).
In this study, we exploit a robust, model of MPNST that histologically and molecularly recapitulates human MPNST. We used primary cells from these MPNSTs to screen for compounds that specifically arrest tumor cell growth. Such compounds would not only have potential as a platform for novel therapeutic development but also for probing the biology of MPNST. Elucidating the mechanism of action of such compounds could identify MPNST specific pathways and point to therapeutic opportunities. We report a novel small molecule, compound 21 (Cpd21 or identification number SW106065), which induces apoptosis in all models of MPNST tested but spares normal cells and tissues. Moreover, we demonstrate that this compound can decrease the tumor burden of MPNST in a mouse allograft model.
Methods
Cell and Tissue Samples
S462 and SNF96.2 cells were a gift from Karen Cichowski. Schwann cells (ScienCell) were cultured in Schwann Cell Medium (ScienCell), and were immortalized with Myc-retrovirus (27).
High-throughput screening
sMPNST and Schwann cells (400 cells/well) were seeded in 384-well plates, and grown overnight. Compounds (final concentration of 2.5 µM) were added and incubated for 96h. CellTiter-Glo™ (Promega) was added, and luminescence was quantified using Envision 2102 Multilabel Reader (Perkin Elmer).
Soft Agar Assay
A cellular layer of 0.6% Bacto-agar in MPNST media was plated. Cells were trypsinized, pelleted, resuspended, mixed in 1:1 ratio with 0.6% agar to a final density of 1 × 104 cells/mL, and plated on top. After incubation for 10 days, colonies were stained with 0.5 mL 0.005% crystal violet.
Cell Cycle Analysis
BrdU (10 µM) was added to cells for 30 min. Cells were collected, resuspended in 100 uL PBS, fixed in 5 mL ice cold ethanol, and then stored overnight at 4°C. 2N HCl/Triton X-100 was added to denature DNA. After neutralization, samples were incubated with 50 uL 0.5% Tween 20/1%BSA/PBS, 20 uL anti-BrdU-FITC (BD Biosciences), and 5 uL of 10 mg/mL RNAse overnight. Samples were pelleted, resuspended in 1 mL 5 ug/mL propidium iodide (Sigma), and analyzed by flow cytometry.
Western Blotting
Cells were collected, trypsinized, and lysed with 600 uL RIPA buffer. Samples were run on 5–20% SDS-PAGE gel (BioRad). The following primary antibodies were used: Cyclin D1, H3 (Millipore), Caspase 3, pERK, ERK, pAKT, AKT (Cell Signaling), PARP (Novus Biologicals and Millipore), αTubulin (Sigma), and GAPDH (Santa Cruz).
Annexin V Staining
Floating and adherent cells were resuspended in Binding Buffer (0.01 M Hepes/NaOH (pH 7.4), 0.14 M NaCl, 2.5 mM CaCl2), and processed according to manufacturer (MACS Miltenyi Biotec). 300 uL of suspension was added to 5 uL Annexin V antibody (MACS Miltenyi Biotec) and 2.5 uL propidium iodide, and analyzed by flow cytometry.
Metabolic Stability Studies
Cpd21 levels for metabolic stability and pharmacokinetic studies were monitored by LC-MS/MS with AB/Sciex (Applied Biosystems) 3200 Qtrap mass spectrometer coupled to Shimadzu Prominence LC. Cpd21 was detected with the mass spectrometer in positive MRM (multiple reaction monitoring) mode by following the precursor to fragment ion transition 205.0 to 111.1. An Agilent C18 XDB 5 micron packing column (50 × 4.6 mm) was used for chromatography. For S9 studies, Cpd21 (2 µM) was incubated in a 1 mL incubation volume with 1 mg of murine CD-1 S9 (combined cytosol and microsome) fractions (Celsis/In Vitro Technologies) and Phase I (the NADPH regenerating system) cofactors (Sigma) for 0–240 min. Reactions were quenched by mixing the incubation mixture with 1 mL of methanol/0.2 ng/µL n-benzylbenzamide/0.2% formic acid. The quenched mixture was vortexed for 15 s, incubated at RT for 10 min and spun for 5 min at 986×g. Supernatants were spun in a refrigerated microcentrifuge for 5 min at 16,100×g. The second supernatant was transferred to an HPLC vial and analyzed by LC-MS/MS.
Pharmacokinetic Studies
6–7 week old NCR-nu/nu female mice were implanted with 2.5×105 MPNST cells in the left flank, subcutaneously. When tumors reached 800 mm3, animals were injected with Cpd21 at 20 mg/kg in 0.2 ml 10% ethanol, 10% PEG400, 80% of a solution of 5% dextrose, pH 7.4. Blood was drawn at 0, 10, 30, 60, 180, 360, 1440 min using the anticoagulant, ACD (acidified citrate dextrose) and plasma isolated by centrifugation. 100 µL plasma was mixed with 200 µL acetonitrile containing 0.15% formic acid and 37.5 ng/mL n-benzylbenzamide IS. Samples were vortexed 15 s, incubated at RT for 10 min and spun twice at 16,100×g at 4°C. Amount of Cpd21 present in plasma was quantified by LC-MS/MS to determine the rate of clearance from mouse blood. A Cpd21 standard curve was generated using blank plasma spiked with known Cpd21 concentrations and processed as described above. Concentrations of Cpd21in each time-point sample were quantified using Analyst 1.4.2. The limit of detection (LOD) was defined as 3-fold above the signal obtained from blank plasma. The limit of quantitation (LOQ) was defined as the lowest concentration at which back calculation yielded a concentration within 20% of theoretical. The LOQ for plasma was 5 ng/mL and 1 ng/mL for tumor. Back-calculation of points on the standard curve yielded values within 15% of theoretical over 4 orders of magnitude (5000 to 5 or 1 ng/mL). Pharmacokinetic parameters were calculated using the noncompartmental analysis tool of WinNonLin (Pharsight).
Bioluminescent Imaging
sMPNST-luc+ cells (2×505 cells/200 uL) were injected in both flanks of nude mice. Mice were injected with 40 mg/kg Cpd21 or vehicle (10% ethanol, 10% PEG400, and 80% of 5% dextrose in water) IP twice per day for 4 weeks. Mice were injected with 160 uL 20 mg/mL D-Luciferin Potassium Salt (Perkin Elmer) in 0.9% sterile NaCl, and total flux was quantified using IVIS Lumina II (Caliper Life Sciences) weekly.
Immunohistochemistry
Tissues were fixed in 10% formalin and paraffin embedded. After sectioning, immunohistochemical studies were performed as previously described (29). The following primary antibodies were used: S100 (28), GAP43 (Abcam), Ki67 (Thermo Scientific), Caspase 3 (Cell Signaling), and PARP (Novus Biologicals).
Results
Nf1 and p53 deficient Skin-Derived Precursors (SKPs) form MPNST
NF1-associated dermal and plexiform neurofibromas are histologically compatible despite having significantly different natural history and tumor progression properties. Recently, we reported that SKPs are the cell-of-origin of dermal neurofibromas (4). Given the potential in mice for neural crest progenitors to give rise to MPNST after additional loss of the p53 tumor suppressor, we tested the consequences of dual Nf1 and p53 loss of function (NP) in SKPs. SKPs from mice with Nf1f/f;p53f/f genotype were cultured and infected with adenovirus containing Cre-GFP (Ad-Cre) to induce tumor suppressor recombination (30) (Figure 1Aa). The majority of infected cells displayed green fluorescence, indicating effective viral entry and expression (Figure 1Ab). We confirmed effective Cre-mediated recombination of all floxed alleles by PCR analysis (Figure 1Ab).
Figure 1. MPNSTs can be Generated from SKPs that are Deficient in Nf1 and p53.

(A) a. SKPs are isolated from the skin of cis-Nf1f/f;p53f/f mice, cultured, and infected with Ad-Cre-GFP. b. Infected SKPs were screened for expression of GFP, and genotyped for the deletion of Nf1. Lane 1 represents Nf1f/f control. Lane 2 represents wildtype Nf1+/+ control. Lane 3 represents Nf1−/− control. Lane 4 represents Nf1−/− SKPs that were infected with Ad-Cre-GFP. c. These Nf1−/−;p53−/− SKPs are then autologously transplanted back into the original mouse. d. Resultant tumors are transplanted into nude mice via xenotransplantation. e. RT-qPCR results. Lanes 1–4 represent MPNSTs derived from cis-Nf1+/−;p53+/− mice. Lanes 5–7 represent MPNSTs derived from PLP-CreERT-Nf1f/f;p53f/f mice that have been injected with tamoxifen to induce recombination. Lanes 8–11 represent sMPNSTs. (B) a. Hematoxylin and eosin staining of sMPNST. b–e. Immunohistochemical staining for S100B in sMPNST (b) and skin (c), and for GAP43 in sMPNST (d) and skin (e). Scale bars represent 100 µm.
The Nf1−/−;p53−/− cells (NP SKPs) were reimplanted into the same mouse from which they were derived (autograft) and highly aggressive, malignant tumors developed in all cases (Figure 1Ac). These tumors could be successfully cultured as either spheres or adherent cells (data not shown), or allografted into nude mice (Figure 1Ad). These tumors (termed sMPNST, for SKP derived MPNST) were assayed for human MPNST-associated genes including embryonic Schwann cell markers, S100 (24, 25), Krox20 (6), PLP (31), and GAP43 (32, 33); developing glia marker, Dhh (34); and neural-crest-associated marker, snail (35). The RT-qPCR data demonstrated expression of all markers in all sMPNSTs analyzed (Figure 1Ae). Thus, sMPNSTs exhibit molecular characteristics of human NF1 associated MPNST.
Hematoxylin and eosin (H&E) staining of sMPNSTs showed presence of spindle-shaped cells that interweave in a poorly-differentiated, wavy appearance, reminiscent of human MPNST histology (Figure 1Ba). Additionally, the commonly used MPNST markers, S100 and GAP43, were present (Figure 1Bb–1Be). Therefore, our results indicate that SKPs, cells that give rise to neurofibromas upon loss of Nf1, can also give rise to MPNST with additional loss of p53. The facility of generating and explanting sMPNSTs made this a favorable system for performing chemical compound screens.
Novel MPNST inhibitory Small Molecule
We hold the concern that over time in culture, tumor cells will drift from their original intrinsic cellular and molecular programs in unpredictable ways. These uncontrollable and unmeasurable changes may inadvertently alter fundamental properties of the original tumor cells. To mitigate these concerns, the relative facility of generating primary cultures from sMPNSTs afforded the advantage of propagating sufficient numbers of cells for a high-throughput screening while keeping the cell passage number low. We performed a limited high-throughput, small-molecule screen using sMPNST cells produced above in an extension of a related ongoing large scale screen in our research group. We carried out a 200,000 compound screen on primary mouse glioblastoma multiforme (GBM) cells that are deficient for Nf1, p53, and Pten (KL and LFP, unpublished data). This original GBM screen yielded 4480 compounds, identified as inhibitory to GBM cell growth. We reasoned that since sMPNST cells are deficient in two of these same tumor suppressors, some compounds might have conserved sMPNST cell growth inhibitory properties.
A well-established luminescence assay for measuring ATP levels (an indication of cellular metabolic activity) was adopted (CellTiter-Glo™, Promega), and performed 96 hours after compound exposure. 1515 compounds exhibited a greater than 20% decrease in ATP levels at 2.5 µM (Figure 2A and 2B). To exclude compounds that exert general cell toxicity, or perturb the generic cell cycle or mitotic machinery, we counter-screened the 1515 compounds against Myc-immortalized Schwann cells (27, 36) and eliminated compounds that showed greater than 20% lower ATP levels. The resultant 119 compounds were rescreened on sMPNSTs with a cutoff at 70% inhibition. 28 compounds passed this test and were further tested in dose response studies ranging from 125 nM to 20 µM. The twenty-first compound (Figure 2C) (Cpd21 or SW106065) inhibited ATP consumption of sMPNST and all other models of MPNST tested (Figure 2D) with an EC50 of 1 µM. We extended the functional analysis of Cpd21 to additional cell lines. Human MPNST cell lines, S462 (Figure 2E) and SNF96.2 (Figure 2F), were assayed for dose-dependent growth, and EC50 concentrations of 439.0 and 753.6 nM, respectively, were determined. In contrast, NIH3T3 cells and wildtype Schwann cells showed a considerably higher dose resistance (Figure 2G). We also tested sMPNST response to Cpd21 in anchorage-independent soft agar growth assays and found that Cpd21 inhibited colony formation (Figure 2H and 2I). This effect was also observed in MPNST cells derived from cis-Nf1+/−;p53+/− mice (Figure 2J and 2K). Thus Cpd21 has general toxic activity on Nf1 and p53 deficient MPNST cells but not on diverse non-tumorigenic, mitotically active fibroblasts or Schwann cells.
Figure 2. Identification of Cpd21 by high-throughput, small-molecule screening and Validation.

(A) Summary of screening procedure. (B) High-throughput screening data for sMPNST cells (upper panels) and Schwann cells (lower panels). Intensity of blue or red signal correlates with degree of inhibition or induction of ATP levels by small molecules, respectively. (C) Structure of Cpd21. (D) Dose-response curves of Cpd21 (0.125, 0.25, 0.5, 1, 2.5, 5, 10, and 20 µM) in sMPNST cells, MEF, Schwann cells, and MPNST cells from multiple murine models. (E, F) Dose-response curves of Cpd21 treatment on human MPNST cell lines, S462 and SNF96.2, respectively, compared with mouse NIH3T3 cells and sMPNST cells. (G) Dose-response curves of Cpd21 treatment on wildtype Schwann cells, compared with sMPNST and MPNST from cis-Nf1f/f;p53f/f mice. (H) Soft agar assay of Cpd21-treated (0.25, 0.5, 1, 2.5, or 5 µM) sMPNST cells compared with DMSO. Arrows indicate colonies, which may be out of plane in agar. Scale bars represent 100 µm. (I) Quantification of H. (J) Soft agar assay of MPNST cells from cis-Nf1+/−;p53+/− mice, and that were treated with DMSO or Cpd21 at 0.25, 0.5, 1, 2.5, or 5 µM. Arrows indicate colonies, which may be out of plane in agar. Scale bars represent 100 µm. (K) Quantification of J. All values represent the mean±standard deviation. Student’s t-test used for significance testing (*p<0.05, **p<0.01, ***p<0.001).
Cpd21 Delays the Cell Cycle in sMPNST
To address the mechanism of action of Cpd21, we treated sMPNST cells at 0.25, 0.5, 1, 2.5, and 5 µM for 24 h, and pulsed with BrdU for 30 min. Subsequent flow cytometry analysis revealed a decreased percentage of cells in S phase, and a corresponding increased percentage in G1/G0 and G2/M, compared to treatment with DMSO alone (Figure 3A).
Figure 3. Effect of Cpd21 on Cell Cycle Machinery.

(A) Effect of Cpd21 on cell cycle distribution. sMPNST cells were treated with DMSO, D, or Cpd21 (0.25, 0.5, 1, 2.5 or 5 µM) for 24 h, pulsed with BrdU for 30 min, and analyzed by flow cytometry. Percentage of cells in each phase is shown. (B, C, D) sMPNST cells were treated with DMSO, D, or with Cpd21 (0.25, 0.5, 1, 2.5, or 5 µM) for 24 h and RNA harvested for qRT-PCR for cyclins (B), cdks (C), or CDKIs (D). (E) Western blot for cyclin D1 in sMPNST cells that were treated with DMSO, D, or Cpd21 (0.25, 0.5, 1, and 2.5 µM). All values represent the mean±standard deviation. Student’s t-test used for significance testing (*p<0.05, **p<0.01, ***p<0.001).
Several cell cycle regulatory genes exhibited decreased mRNA in the presence of Cpd21 including: cyclin A2, cyclin B1, cyclin D1, cyclin E, cdk4, and cdk6 (Figure 3B and 3C). Increased levels of cdkn1a and cdkn2a mRNA were observed in a dose-dependent manner (Figure 3D). Also, levels of Cyclin D1 protein, which is known to interact with cdk4/6 in regulating G1/S progression, were decreased (37, 38) (Figure 3E). These results indicate that following Cpd21 exposure, cells in S phase are underrepresented, consistent with cell cycle delay and an arrest of the mitotic machinery.
We examined Cpd21-treated sMPNST cells for apoptosis by annexin V-FITC-conjugated flow cytometry. Tumor cells treated with Cpd21 concentrations at or above 0.5 µM showed a statistically-significant increase in the percentage of apoptotic cells (Figure 4A). At the EC50 concentration of 1 µM, 11.23±1.56% of the cells were annexin V positive, compared to 5.02% ± 0.83% in DMSO-treated control cells.
Figure 4. Cpd21 Induces Apoptosis in vitro.

(A) Annexin V staining as detected by flow cytometry. sMPNST cells were treated with DMSO, D, or Cpd21 (0.25, 0.5, 1, and 2.5 µM). sMPNST cells were treated with staurosporine, St, as positive control. (B–F) Western blotting for caspase 3 cleavage in sMPNST cells (B), PARP cleavage in sMPNST cells (C), caspase 3 cleavage in MPNSTs from cis-Nf1+/−;p53+/− mice (D), caspase 3 cleavage in human MPNST cell line, S462 (E), caspase 3 cleavage in Myc-SC (F), and caspase 3 cleavage in MEF cells (G). Each cell type was treated with staurosporine, St, as a positive control. Total caspase 3 (T. Casp 3) and cleaved caspase 3 (Cl. Casp 3) were assayed in each case. All values represent the mean±standard deviation. Student’s t-test used for significance testing (*p<0.05, **p<0.01, ***p<0.001).
We also performed Western blotting of Cpd21-treated sMPNST cells and observed Caspase 3 (Figure 4B) and PARP (Figure 4C) cleavage, consistent with induced apoptosis. We further validated this mechanism in MPNST cells cultured from spontaneous MPNSTs from our cis-Nf1+/−;p53+/− mouse model (Figure 4D), and in the human MPNST cell line, S462 (Figure 4E). Other cell types, including Myc-immortalized Schwann cells (Figure 4F) and MEF cells (Figure 4G) did not undergo caspase 3 cleavage. Together, these data indicate that, in vitro, Cpd21 induced mitotic arrest of MPNST cells that ultimately results in cellular apoptosis, and that this phenotype was not observed in untransformed dividing cells.
Cpd21 Decreases Tumor Burden and Induces Apoptosis in vivo
To determine whether Cpd21 could be directly tested in vivo, we sought to define the metabolic properties in vitro according to previously published methods (39, 40). To examine stability, Cpd21 was incubated with purified, hepatic, enzymatic S9 fraction over a period of 240 min, and measured over time (Figure 5A). The half-life in the S9 fraction is 22.65 min. We also incubated Cpd21 with cultured hepatocytes and found that the half-life is 121.6 min (Figure 5B). These assays demonstrate that Cpd21 would have reasonable stability in vivo.
Figure 5. Effect of Cpd21 in vivo.

(A and B) Cpd21 was incubated with murine, hepatic S9 fractions (A) or murine hepatocyte (B) for indicated times. Amount of Cpd21 remaining was quantified by mass spectrometry. (C) Cpd21 was injected at 20 mg/kg IP and quantified in plasma or tumor over time. PK parameters were quantified using the noncompartmental analysis tool in WinNonlin (Pharsight). (D) sMPNST-luc+ cells were injected subcutaneously in nude mice. Cpd21 (40 mg/kg) or vehicle was injected IP twice per day for 4 weeks. Mice were imaged every 7 days. Images shown are from week 4. (E) Quantification of tumor images in D by normalizing total flux at week 4 with baseline flux (n=10 for vehicle, n=8 for Cpd21). (F) BrdU staining of Cpd21- or vehicle-treated tumors (G) Quantification of F. (H) Immunohistochemistry of Ki67. (I) Quantification of H. (J) Immunohistochemistry of cleaved caspase 3. (K) Quantification of J. (L) Immunohistochemistry of cleaved PARP. (M) Quantification of L. (N) Western blotting for caspase 3 and PARP cleavage in tumor lysates. All microscopic images taken at 20×. Scale bars represent 100 µm. All values represent the mean±standard deviation. Student’s t-test used for significance testing (*p<0.05, **p<0.01, ***p<0.001).
We next sought to define the pharmacokinetic (PK) properties of Cpd21 according to previously published methods (40). sMPNST (2.5 × 105) cells were injected subcutaneously into nude mice, and when tumors reached 800 mm3, a single dose of Cpd21 was injected IP at 20 mg/kg. Plasma and tumor samples were collected after 10, 30, 90, 180, 360, and 960 min and assayed by mass spectrometry (Figure 5C). We observed that Cpd21 rapidly distributes from the plasma into sMPNST tissues, and that the half-life in sMPNST tissues is 6–8 hours once the compound is completely distributed. Importantly, Cpd21 concentration remains above the EC50 concentration of 1 µM (204 ng/mL) in the sMPNSTs for over 6 hours. These results indicate that Cpd21 is successfully delivered to the tumor tissue in adequate concentrations and with an adequate half-life for in vivo studies.
We next determined a dose tolerable for chronic in vivo administration of Cpd21. Cpd21 dissolved in a mixture of ethanol, PEG400, and dextrose was delivered intraperitoneally (IP) at 20 or 40 mg/kg twice per day, into nude mice over the course of two weeks for comparison with vehicle administration. These concentrations were selected because 40 mg/kg is the maximum solubility in the aforementioned formulation. Over this period, the mice were weighed daily and observed for signs of toxicity. Neither concentration of Cpd21 produced measurable weight loss or distress in the mice (data not shown). Therefore, we selected 40 mg/kg twice per day IP as the dosing regimen for efficacy studies.
We then tested the effects of Cpd21 administration on tumor burden in vivo. Nude mice were injected with sMPNST cells carrying a luciferase gene (MPNST-luc+) to permit non-invasive in vivo tumor growth assessment by luminescence. One week after allograft, recipient mice were IP injected with either 40 mg/kg of Cpd21 or vehicle twice daily over 4 weeks, and mice were imaged weekly. We observed that treatment with Cpd21 reduced the tumor burden as measured by total flux normalized to baseline flux levels (Figure 5D and 5E).
BrdU was injected into mice prior to necropsy. Both BrdU and Ki67 evaluation indicated a significant decrease in tumor cell proliferation following Cpd21 treatment (Figure 5F–5I). These results are consistent with our observations that Cpd21 inhibits the cell cycle in vitro, resulting in a decrease in the proliferative capacity of sMPNSTs.
Consistent with in vitro data, Cpd21-treated sMPNSTs exhibited elevated activated caspase 3 and PARP (Figure 5J–5M). These results were confirmed by Western blotting to detect cleaved caspase 3 and PARP in tumors from our in vivo model (Figure 5N). Taken together, our results demonstrate that Cpd21 can be delivered to mice in concentrations to sufficiently penetrate sMPNST tissue, and inhibit tumor development.
To extend the previous observation that chronic Cpd21 exposure did not appear to cause general toxicity, we examined various tissues following necropsy for histological evidence of Cpd21 induced toxicity. In addition to tumor tissue (Figure 6A), we examined liver (Figure 6B), kidney (Figure 6C), brain (Figure 6D), skin (Figure 6E), and sciatic nerve (Figure 6F), and found no evidence of abnormal histology. Activated caspase 3 could only be detected in tumor samples (Figure 6G) and not in other tissues examined (Figure 6H–L). In contrast to other chemotherapeutic agents such as doxorubicin that induce toxicity in a variety of tissues (41), Cpd21 appears to act most notably on malignant tumor tissue.
Figure 6. Apoptotic effect of Cpd21 is specific to tumor tissue.
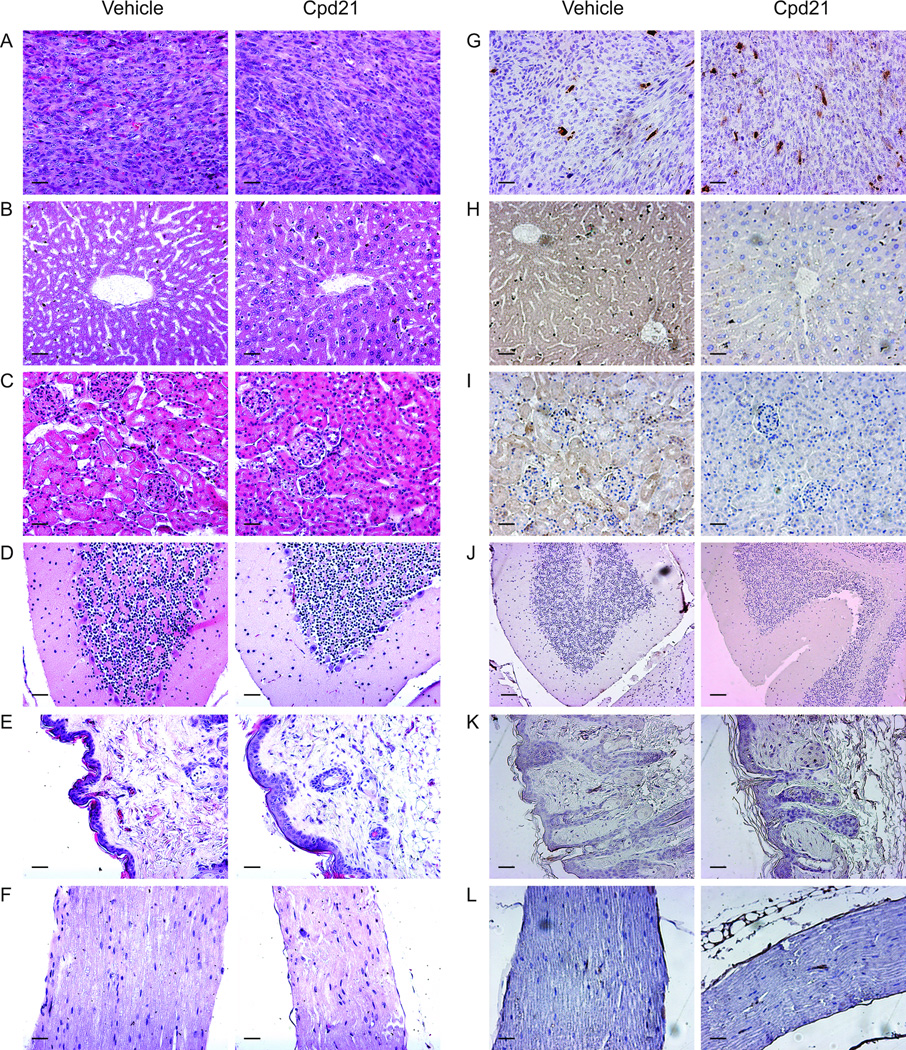
(A–F) H&E staining of tumors (A), liver (B), kidney (C), brain (D), skin (E), and sciatic nerve (F) from mice treated with either Cpd21 or vehicle. (G–L) Immunohistochemitry for cleaved caspase 3 is only detected in tumor tissue (G), but not in liver (H), kidney (I), brain (J), skin (K), or sciatic nerve (L). All images taken at 20×. Scale bars represent 100 µm.
Interaction with Other Anticancer Agents
In addition to doxorubicin, MPNSTs have been shown to be sensitive to pathway-specific inhibitors of MAPK/ERK (42), PI3K/AKT (43), mTOR (43–45), and CXCR4 (7). We therefore examined whether Cpd21 in conjunction with doxorubicin or specific inhibitors, including U0126, a MEK inhibitor (42); LY294002, an AKT inhibitor (43); rapamycin, an mTOR inhibitor (43–45); and AMD3100, a CXCR4 inhibitor (7), might better inhibit sMPNST cell growth as assessed by ATP consumption (46). We found the inhibition of the PI3K pathway combined with Cpd21 treatment resulted in a decrease in cellular growth and survival that is greater than either inhibitor alone (Figure 7A and 7B). Phospho-AKT Western blots of sMPNST cells treated with LY294002 at 20 µM for 30 or 60 minutes verified effective pAKT inhibition in sMPNST cells (Figure 7C). Inhibition of the MEK/ERK pathway combined with Cpd21 treatment did not produce detectable additive or synergistic effects (Figure 7D). Phospho-ERK inhibition by U0126 was verified by Western blotting (Figure 7E). Inhibition of the mTOR pathway combined with Cpd21 treatment did not produce detectable additive or synergistic effects (Figure 7F). In fact, sMPNST cellular growth seemed relatively resistant to rapamycin, despite exhibiting decreased pS6 levels after rapamycin treatment (Figure 7G). None of the other tested reagents produced measurable additive or synergistic effects (data not shown). We extended the above studies of dual LY294002 and Cpd21 treatment to MPNST cells derived from cis-Nf1+/−;p53+/− mice, and again, the combined effect of both inhibitors is greater than that of each individual inhibitor (Figure 7H). We tested the effects of both inhibitors on phosphorylation of AKT, and found that pAKT was inhibited only when LY294002 is present (Figure 7C and 7I). Thus, in contrast to published reports of successful application of MEK inhibitors to suppress NF1 based solid tumors and leukemias, in the context of Cp21, co-inhibition of the PI3K pathway was most effective in impeding tumor cell growth and survival.
Figure 7. Cpd21 Interacts with PI3K Inhibitor.

(A) ATP assay following combined treatment of Cpd21 and LY294002 in sMPNST cells. Synergism determined by Loewe method. (B) Phase contrast images of sMPNST cells treated with DMSO, Cpd21 alone, LY294002 (LY) alone, or a combination of both inhibitors. Images taken at 20×. Scale bars represent 100 µm. (C) Western blotting for pAKT in sMPNST cells treated with 20 µM LY294002 for 30 or 60 minutes. (D) ATP assay following combined treatment of Cpd21 and U0126 in sMPNST cells. (E) Western blotting for pERK in sMPNST cells treated with 10 µM U0126 for 30 minutes. (F) ATP assay following combined treatment of Cpd21 and LY294002 in sMPNST cells. (G) Western blotting for pS6 in sMPNST cells treated with 0.05 µM (50 nM) rapamycin for 30 or 60 min. (H) ATP assay following combined treatment of MPNST cells from cis-Nf1+/−;p53+/− mice with Cpd21 and LY294002. Synergism determined by Loewe method. (I) Western blotting for pAKT and AKT in sMPNST cells treated with Cpd21 alone, LY294002 alone, or with both inhibitors. All graphs plotted in micromolar concentrations.
Discussion
Malignant peripheral nerve tumors are the major source of mortality in NF1. Like most malignant sarcomas, these tumors are resistant to all current chemotherapeutic and radiotherapeutic strategies. It has long been recognized that MPNSTs harbor tumor suppressor mutations in addition to Nf1 loss. Notably, loss-of-function mutations of the p53 tumor suppressor have been reported (16–18). This has led to the successful generation of spontaneous mouse models of MPNST by mutation of both Nf1 and p53 tumor suppressor genes (24, 25). A further refinement of MPNST modeling came about by identifying SKPs as a source for dermal neurofibromas in the skin and by the capacity of SKPs to engender plexiform neurofibromas when placed in the sciatic nerve (4). Additive mutation of p53 together with Nf1 turned SKPs into progenitors of MPNST, thus facilitating high throughput screens for chemical compounds that might inhibit early passage cell growth.
We identified Cpd21 as a small molecule with inhibitory properties on cultured mouse and human MPNST cells. It was a surprise that this compound originating from a 200,000 chemical compound library, had, without chemical modification a repertoire of desirable in vivo properties including low micromolar EC50, adequate stability, manageable toxicity, and adequate accumulation in solid tumor tissue. These serendipitous favorable properties allowed allograft analysis of antitumor efficacy and demonstrated a significant reduction in tumor burden by this compound. Future efforts to assess the effectiveness of Cp21 on human MPNST xenografts will provide additional insight into the potential for this compound as a therapeutic agent.
At present we cannot identify the specific mechanism whereby Cpd21 exerts its inhibitory action on MPNST cells. Compound exposure to tumor cells results in a cell cycle delay manifested by depletion of certain cell cycle related molecules and eventually leading to cellular apoptosis. A similar course of events is seen when tumor-bearing mice are treated with compound. However, the precise molecular target and mechanistic mode of compound action remains a topic of investigation. Empirical chemical compound screens to identify cancer cell inhibitory compounds have been applied for several decades. A significant proportion of such empirically derived cancer cell inhibitory small molecules that have reached the clinic, have turned out to mechanistically impinge on various aspects of cell division including mitotic spindle machinery, cell cycle machinery and DNA replication and repair machinery. These cellular properties are not unique to cancer cells and are shared by many normally dividing cells in the body that are essential for organ health. Chemotherapeutic toxicity to normal cells represents a major impediment to effective cancer treatment. In this regard, our screen departs from traditional high throughput screens in that we limited our cells assayed to early passage cells rather than to established cell lines. It is also noteworthy that the cellular specificity of Cpd21 renders it relatively innocuous to the normally dividing cells we tested (MEFs, Myc-Schwann Cells, and wildtype SKPs). This feature is consistent with the idea that the compound target(s) relates to the “cancer state” of the MPNST cells rather than to more general properties of mitotically active cells. Although we currently do not know whether Cpd21 is also effective against the benign plexiform neurofibromas, it is entirely possible that these slowly-dividing tumor cells may also be susceptible to Cpd21 activity if they share common pathways with MPNSTs that are potential target(s) of Cpd21.
Recent advances in MPNST research have pointed to additional potential therapeutic targets including the CXCR4 cytokine axis (7), the mTOR pathways (44), and the RAS/MEK/ERK pathways (13, 47–50). In these studies, AMD3100 and rapamycin have been shown to inhibit MPNSTs from the cis-Nf1+/−;p53+/− mouse model, and ERK inhibitors, such as U0126, have been verified in a variety of human and mouse derived MPNSTs. Interestingly, when such compounds were tested in conjunction with Cp21, we found no additive value. Instead, blockade of the PI3K pathway did cooperate with Cpd21 action to inhibit tumor growth. This intriguing result points to the potential plasticity of cancer cells and to the complexity of tumor pathway interactions. For example, it may be that Cpd21 activity somehow unveils a synthetic dependence on PI3Kinase signaling that is not present in the absence of compound. Continued studies of Cpd21, its specific mode of action and the mechanistic basis for its activity on MPNST cells but not on non-tumorigenic cells may shed light on novel anticancer pathways.
Acknowledgements
We thank Michael White for helpful discussions throughout this project, and Karen Cichowski for sharing the human MPNST cell lines. We also thank all members in Luis Parada’s laboratory and in Lu Le’s laboratory for helpful discussions. This work was partially supported by funding from Texas Neurofibromatosis Foundation, the National Cancer Institute (Grant# R01 CA166593 to L.Q.L.), the National Institute of Health (Grant# P50NS052606 to L.F.P.), CPRIT (RP 100782 to L.F.P), and the Simmons Cancer Center Support Grant# 5P30 CA 142543-03. L.Q.L. is a Disease-Oriented Clinical Scholar and holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund. L.F.P. is an American Cancer Society Research Professor.
References
- 1.Pulst SM. Prenatal diagnosis of the neurofibromatoses. Clin Perinatol. 1990;17:829–844. [PubMed] [Google Scholar]
- 2.Brems H, Beert E, de Ravel T, Legius E. Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis type 1. Lancet Oncol. 2009;10:508–515. doi: 10.1016/S1470-2045(09)70033-6. [DOI] [PubMed] [Google Scholar]
- 3.Riccardi VM. Neurofibromatosis: past, present, and future. N Engl J Med. 1991;324:1283–1285. doi: 10.1056/NEJM199105023241812. [DOI] [PubMed] [Google Scholar]
- 4.Le LQ, Shipman T, Burns DK, Parada LF. Cell of origin and microenvironment contribution for NF1-associated dermal neurofibromas. Cell Stem Cell. 2009;4:453–463. doi: 10.1016/j.stem.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huson SM, Harper PS, Compston DA. Von Recklinghausen neurofibromatosis. A clinical and population study in south-east Wales. Brain. 1988;111(Pt 6):1355–1381. doi: 10.1093/brain/111.6.1355. [DOI] [PubMed] [Google Scholar]
- 6.Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296:920–922. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mo W, Chen J, Patel A, et al. CXCR4/CXCL12 Mediate Autocrine Cell- Cycle Progression in NF1-Associated Malignant Peripheral Nerve Sheath Tumors. Cell. 2013;152:1077–1090. doi: 10.1016/j.cell.2013.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002;62:1573–1577. [PubMed] [Google Scholar]
- 9.Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57:2006–2021. doi: 10.1002/1097-0142(19860515)57:10<2006::aid-cncr2820571022>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 10.Ballester R, Marchuk D, Boguski M, et al. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell. 1990;63:851–859. doi: 10.1016/0092-8674(90)90151-4. [DOI] [PubMed] [Google Scholar]
- 11.Le LQ, Parada LF. Tumor microenvironment and neurofibromatosis type I: connecting the GAPs. Oncogene. 2007;26:4609–4616. doi: 10.1038/sj.onc.1210261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeClue JE, Papageorge AG, Fletcher JA, et al. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell. 1992;69:265–273. doi: 10.1016/0092-8674(92)90407-4. [DOI] [PubMed] [Google Scholar]
- 13.Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992;356:713–715. doi: 10.1038/356713a0. [DOI] [PubMed] [Google Scholar]
- 14.Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res. 2005;65:2755–2760. doi: 10.1158/0008-5472.CAN-04-4058. [DOI] [PubMed] [Google Scholar]
- 15.Weiss B, Bollag G, Shannon K. Hyperactive Ras as a therapeutic target in neurofibromatosis type 1. Am J Med Genet. 1999;89:14–22. [PubMed] [Google Scholar]
- 16.Birindelli S, Perrone F, Oggionni M, et al. Rb and TP53 pathway alterations in sporadic and NF1-related malignant peripheral nerve sheath tumors. Lab Invest. 2001;81:833–844. doi: 10.1038/labinvest.3780293. [DOI] [PubMed] [Google Scholar]
- 17.Menon AG, Anderson KM, Riccardi VM, et al. Chromosome 17p deletions and p53 gene mutations associated with the formation of malignant neurofibrosarcomas in von Recklinghausen neurofibromatosis. Proc Natl Acad Sci U S A. 1990;87:5435–5439. doi: 10.1073/pnas.87.14.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jhanwar SC, Chen Q, Li FP, Brennan MF, Woodruff JM. Cytogenetic analysis of soft tissue sarcomas. Recurrent chromosome abnormalities in malignant peripheral nerve sheath tumors (MPNST) Cancer Genet Cytogenet. 1994;78:138–144. doi: 10.1016/0165-4608(94)90081-7. [DOI] [PubMed] [Google Scholar]
- 19.Nielsen GP, Stemmer-Rachamimov AO, Ino Y, Møller MB, Rosenberg AE, Louis DN. Malignant Transformation of Neurofibromas in Neurofibromatosis 1 Is Associated with CDKN2A/p16 Inactivation. The American Journal of Pathology. 1999;155:1879–1884. doi: 10.1016/S0002-9440(10)65507-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berner JM, Sorlie T, Mertens F, et al. Chromosome band 9p21 is frequently altered in malignant peripheral nerve sheath tumors: studies of CDKN2A and other genes of the pRB pathway. Genes Chromosomes Cancer. 1999;26:151–160. [PubMed] [Google Scholar]
- 21.Kourea HP, Orlow I, Scheithauer BW, Cordon-Cardo C, Woodruff JM. Deletions of the INK4A gene occur in malignant peripheral nerve sheath tumors but not in neurofibromas. Am J Pathol. 1999;155:1855–1860. doi: 10.1016/S0002-9440(10)65504-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buchstaller J, McKeever Paul E, Morrison Sean J. Tumorigenic Cells Are Common in Mouse MPNSTs but Their Frequency Depends upon Tumor Genotype and Assay Conditions. Cancer Cell. 2012;21:240–252. doi: 10.1016/j.ccr.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perrone F, Tabano S, Colombo F, et al. p15INK4b, p14ARF, and p16INK4a inactivation in sporadic and neurofibromatosis type 1-related malignant peripheral nerve sheath tumors. Clin Cancer Res. 2003;9:4132–4138. [PubMed] [Google Scholar]
- 24.Vogel KS, Klesse LJ, Velasco-Miguel S, Meyers K, Rushing EJ, Parada LF. Mouse tumor model for neurofibromatosis type 1. Science. 1999;286:2176–2179. doi: 10.1126/science.286.5447.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cichowski K, Shih TS, Schmitt E, et al. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999;286:2172–2176. doi: 10.1126/science.286.5447.2172. [DOI] [PubMed] [Google Scholar]
- 26.Gregorian C, Nakashima J, Dry SM, et al. PTEN dosage is essential for neurofibroma development and malignant transformation. Proceedings of the National Academy of Sciences. 2009;106:19479–19484. doi: 10.1073/pnas.0910398106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 28.Rallis E, Ragiadakou D. Giant plexiform neurofibroma in a patient with neurofibromatosis type I. Dermatol Online J. 2009;15:7. [PubMed] [Google Scholar]
- 29.Alcantara Llaguno S, Chen J, Kwon CH, et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. 2009;15:45–56. doi: 10.1016/j.ccr.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biernaskie JA, McKenzie IA, Toma JG, Miller FD. Isolation of skin-derived precursors (SKPs) and differentiation and enrichment of their Schwann cell progeny. Nat Protoc. 2006;1:2803–2812. doi: 10.1038/nprot.2006.422. [DOI] [PubMed] [Google Scholar]
- 31.Mayes DA, Rizvi TA, Cancelas JA, et al. Perinatal or Adult Nf1 Inactivation Using Tamoxifen-Inducible PlpCre Each Cause Neurofibroma Formation. Cancer Res. 2011;71:4675–4685. doi: 10.1158/0008-5472.CAN-10-4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stemple DL, Anderson DJ. Isolation of a stem cell for neurons and glia from the mammalian neural crest. Cell. 1992;71:973–985. doi: 10.1016/0092-8674(92)90393-q. [DOI] [PubMed] [Google Scholar]
- 33.Kioussi C, Gruss P. Making of a Schwann. Trends in Genetics. 1996;12:84–86. doi: 10.1016/0168-9525(96)81411-9. [DOI] [PubMed] [Google Scholar]
- 34.Wu J, Williams JP, Rizvi TA, et al. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell. 2008;13:105–116. doi: 10.1016/j.ccr.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandes KJ, McKenzie IA, Mill P, et al. A dermal niche for multipotent adult skin-derived precursor cells. Nat Cell Biol. 2004;6:1082–1093. doi: 10.1038/ncb1181. [DOI] [PubMed] [Google Scholar]
- 36.Ridley AJ, Paterson HF, Noble M, Land H. Ras-mediated cell cycle arrest is altered by nuclear oncogenes to induce Schwann cell transformation. EMBO J. 1988;7:1635–1645. doi: 10.1002/j.1460-2075.1988.tb02990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bates S, Bonetta L, MacAllan D, et al. CDK6 (PLSTIRE) and CDK4 (PSK-J3) are a distinct subset of the cyclin-dependent kinases that associate with cyclin D1. Oncogene. 1994;9:71–79. [PubMed] [Google Scholar]
- 38.Tam SW, Theodoras AM, Shay JW, Draetta GF, Pagano M. Differential expression and regulation of Cyclin D1 protein in normal and tumor human cells: association with Cdk4 is required for Cyclin D1 function in G1 progression. Oncogene. 1994;9:2663–2674. [PubMed] [Google Scholar]
- 39.Lu J, Ma Z, Hsieh J-C, et al. Structure–activity relationship studies of small-molecule inhibitors of Wnt response. Bioorganic & Medicinal Chemistry Letters. 2009;19:3825–3827. doi: 10.1016/j.bmcl.2009.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tso SC, Qi X, Gui WJ, et al. Structure-based design and mechanisms of allosteric inhibitors for mitochondrial branched-chain alpha-ketoacid dehydrogenase kinase. Proc Natl Acad Sci U S A. 2013;110:9728–9733. doi: 10.1073/pnas.1303220110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brenner DE, Wiernik PH, Wesley M, Bachur NR. Acute doxorubicin toxicity relationship to pretreatment liver function, response, and pharmacokinetics in patients with acute nonlymphocytic leukemia. Cancer. 1984;53:1042–1048. doi: 10.1002/1097-0142(19840301)53:5<1042::aid-cncr2820530505>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 42.DeClue JE, Heffelfinger S, Benvenuto G, et al. Epidermal growth factor receptor expression in neurofibromatosis type 1-related tumors and NF1 animal models. J Clin Invest. 2000;105:1233–1241. doi: 10.1172/JCI7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zou CY, Smith KD, Zhu Q-S, et al. Dual targeting of AKT and mammalian target of rapamycin: A potential therapeutic approach for malignant peripheral nerve sheath tumor. Molecular Cancer Therapeutics. 2009;8:1157–1168. doi: 10.1158/1535-7163.MCT-08-1008. [DOI] [PubMed] [Google Scholar]
- 44.De Raedt T, Walton Z, Yecies Jessica L, et al. Exploiting Cancer Cell Vulnerabilities to Develop a Combination Therapy for Ras-Driven Tumors. Cancer Cell. 2011;20:400–413. doi: 10.1016/j.ccr.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johansson G, Mahller YY, Collins MH, et al. Effective in vivo targeting of the mammalian target of rapamycin pathway in malignant peripheral nerve sheath tumors. Molecular Cancer Therapeutics. 2008;7:1237–1245. doi: 10.1158/1535-7163.MCT-07-2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Straetemans R, O'Brien T, Wouters L, et al. Design and analysis of drug combination experiments. Biom J. 2005;47:299–308. doi: 10.1002/bimj.200410124. [DOI] [PubMed] [Google Scholar]
- 47.Katz D, Lazar A, Lev D. Malignant peripheral nerve sheath tumour (MPNST): the clinical implications of cellular signalling pathways. Expert Rev Mol Med. 2009;11:e30. doi: 10.1017/S1462399409001227. [DOI] [PubMed] [Google Scholar]
- 48.Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. 2001;104:593–604. doi: 10.1016/s0092-8674(01)00245-8. [DOI] [PubMed] [Google Scholar]
- 49.Rahrmann EP, Watson AL, Keng VW, et al. Forward genetic screen for malignant peripheral nerve sheath tumor formation identifies new genes and pathways driving tumorigenesis. Nat Genet. 2013 doi: 10.1038/ng.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jessen WJ, Miller SJ, Jousma E, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest. 2013;123:340–347. doi: 10.1172/JCI60578. [DOI] [PMC free article] [PubMed] [Google Scholar]