

Abstract
Antimicrobial peptides (AMPs) are host-defense agents capable of both bacterial membrane disruption and immunomodulation. However, the development of natural AMPs as potential therapeutics is hampered by their moderate activity and susceptibility to protease degradation. Herein we report lipidated cyclic γ-AApeptides that have potent antibacterial activity against clinically relevant Gram-positive and Gram-negative bacteria, many of which are resistant to conventional antibiotics. We show that lipidated cyclic γ-AApeptides mimic the bactericidal mechanism of AMPs by disrupting bacterial membranes. Interestingly, they also harness the immune response and inhibit lipopolysaccharide (LPS) activated Toll-Like Receptor 4 (TLR4) signaling, suggesting that lipidated cyclic γ-AApeptides have dual roles as novel antimicrobial and anti-inflammatory agents.
Keywords: γ-AApeptides, peptidomimetics, antimicrobial activity, anti-inflammatory activity
INTRODUCTION
Rapidly emerging resistance of antibiotics is a major public health concern.1 As such, natural antibiotics including antimicrobial peptides have captured researchers’ attention. Antimicrobial peptides (AMPs) are short cationic amphiphilic peptides present in almost every living organism.2 Due to their broad-spectrum antimicrobial activity, AMPs are a promising new class of drug candidates.2 AMPs are effective due to their ability to form a globally amphipathic structure with segregated hydrophobic and cationic regions, capable of disrupting bacterial membranes.2,3 Such disruption is rooted in the physical properties of bacterial membranes, making the development of resistance difficult.4 Additionally, AMPs are known as host defense peptides (HDPs) for their role in modulating host innate immunity and diminishing septic responses after bacterial infection.2,5 The immunomodulatory activities of AMPs are speculated to be a crucial contributor to host defense, since the direct antimicrobial activity of AMPs is often weak under physiological conditions.2,6,7 One of the most important immune responses against bacterial infection is lipopolysaccharide (LPS)-activated Toll-like receptor 4 (TLR4) signaling. LPS is a characteristic component of Gram-negative bacterial cell walls that activates the immune response through TLR4. As such, AMPs may have dual roles as both antibiotic and anti-inflammation agents in the treatment of pathogen invasions. As host inflammatory responses to bacterial infection can lead to deadly septic shock, the current treatment for severe infections involves the administration of both anti-bacterial and anti-inflammatory drugs simultaneously. Therefore, the dual functional roles of AMPs have potential as new generation of anti-infective therapeutics.
Nonetheless, AMPs are susceptible to proteolytic cleavage,8 hampering their development into novel therapeutics. Peptidomimetics as an alternative approach may circumvent these problems. This is because peptidomimetics, with modified peptide backbones, are more stable against protease degradation. Examples of successful antimicrobial peptidomimetics include β-peptides,9–11 peptoids,12,13 and others.4,8,14–24 Similar to AMPs, these peptidomimetics are designed to form globally amphipathic structures upon interaction with lipid bilayers, disrupting bacterial membranes. Although many peptidomimetics have broad-spectrum antibacterial activity, the reports of immunomodulatory responses are very rare.8,24 Tew et. al. identified that AMP mimics antagonize lipoteichoic acid (LTA)-activated Toll-like receptor 2 (TLR2) signaling.24 Here we present a new class of immunomodulatory antimicrobial peptidomimetics, which inhibit TLR signaling without ligand interaction.
We recently developed a new class of peptidomimetics termed “γ-AApeptides” based on a chiral peptide nucleic (PNA) backbone.25,26 Due to the versatility of γ-AApeptides, we were able to synthesize potent antimicrobials with broad-spectrum activities against both Gram-positive and Gram-negative bacteria.19,22,23,26 The design of antimicrobial γ-AApeptides is straightforward, which is achieved through joining of amphipathic γ-AApeptide building blocks. Herein, we report the design and synthesis of a new generation: lipidated cyclic γ-AApeptides. We show that this class of compounds has potent and broad-spectrum antimicrobial activity against both Gram-positive and Gram-negative bacteria, including community-acquired multidrug-resistant pathogens. Interestingly, several lipidated cyclic γ-AApeptides inhibit Toll-Like Receptor 4 (TLR4) signaling and block production of the proinflammatory cytokine tumor necrosis factor-α (TNF-α), implying the potential for a new generation of antibiotic agents with dual functionality.
RESULTS AND DISCUSSION
We have shown previously that antimicrobial AApeptides with potent and broad-spectrum activity can be designed and prepared easily by joining amphipathic AApeptide building blocks together.19,22,23 This is because AApeptides formed by this approach are capable of adopting globally amphipathic structures, which are crucial for the disruption of bacterial membranes.19,22,23 Moreover, cyclization22 or lipidation23 can increase their potency. Cyclization reduces structure motility and facilitates bacterial membrane disruption;22 while lipidation encourages antimicrobial agent interactions with membranes.23 We therefore speculated that γ-AApeptides that are both cyclized and lipidated would be more potent in killing bacterial pathogens. In fact, lipidated cyclic peptides polymyxin27 and daptomycin28 have been used as “last-resort” antibiotics to treat infections caused by Gram-negative and Gram-positive bacteria, respectively. However, the exploration of antimicrobial peptidomimetics with lipidated cyclic structural motifs is scarce. To test our hypothesis, a series of lipidated cyclic γ-AApeptides (Figure 1) containing different numbers of amphiphilic γ-AApeptide building blocks were designed and synthesized on the solid phase. Their antibacterial activity was then measured against a range of multi-drug resistant Gramnegative and Gram-positive bacteria (Table 1).
Figure 1.

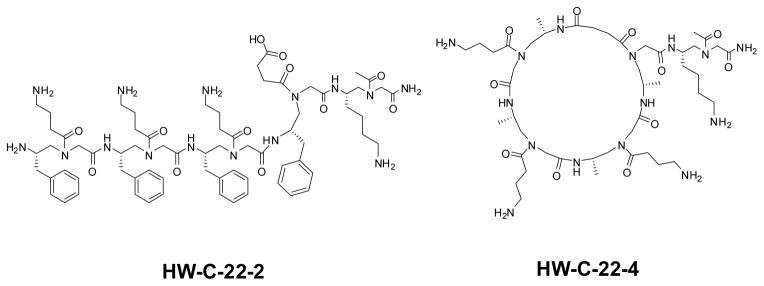
The structures of lipidated cyclic γ-AApeptides. Two γ-AApeptides, HW-C-22-2 and HW-C-22-4, which is either linear or cyclic and do not contain lipid tails, were used for comparison. Compounds are named according to their notebook codes.
Table 1.
The antimicrobial and hemolytic activities of lipidated cyclic γ-AApeptides. The microbial organisms used are E. coli (ATCC 25922), K. pneumoniae (ATCC 13383), P. aeruginosa (ATCC 27853), Methicillin-resistant S. epidermidis (RP62A), Vancomycin-resistant E. faecalis (ATCC 700802), and Methicillin-resistant S. aureus (ATCC 33591). The minimum inhibitory concentration (MIC) is the lowest concentration that completely inhibits growth after 24 h. HC50 is the concentration causing 50% hemolysis. Pexiganan8,29 and previously reported cyclic γ-AApeptide HW-B-1322 are included for comparison. YL-36, the compound with the most potent and broad-spectrum activity, is shaded in grey.
| Oligomers | MIC, μg/mL (μM) | Hemolysis μg/mL (μM) | |||||
|---|---|---|---|---|---|---|---|
| Gram-negative | Gram-positive | ||||||
| E. coli | K. pneumoniae | P. aeruginosa | S. epidermidis | E. faecalis | S. aureus | HC50 | |
| HW-B-73 | >25 (16) | 20 (12) | >25 (16) | 20 (12) | >25 (16) | 20 (12) | - |
| HW-B-77 | >25 (13) | 20 (10) | >25 (13) | >25 (13) | >25 (13) | >25 (13) | - |
| HW-B-78 | >25 (11) | 20 (9) | >25 (11) | >25 (11) | >25 (11) | >25 (11) | - |
| YL-1 | >25 (16) | 8 (5) | 20 (12) | 2 (1) | 5 (2) | 5 (2) | 250 (160) |
| YL-4 | >25 (13) | >25 (13) | >25 (13) | >25 (13) | >25 (13) | >25 (13) | 250 (130) |
| YL-12 | 5 (3) | 5 (3) | 10 (6) | 5 (3) | 5 (3) | 10 (6) | 180 (114) |
| YL-29 | >25 (14) | 10 (5) | 25 (14) | 5 (3) | 5 (3) | 5 (3) | 150 (82) |
| YL-34 | 10 (6) | 3 (2) | 10 (6) | 1 (0.6) | 2 (1) | 2 (1) | 200 (129) |
| YL-36 | 2 (1) | 3 (2) | 5 (3) | 1 (0.6) | 3 (2) | 2 (1) | 100 (65) |
| HW-B-13 | 20 (10) | 8 (4) | 8 (4) | 2 (1) | 5 (3) | 1 (0.5) | 100 (50) |
| Pexiganan | 8 (3) | 8 (3) | 16 (6) | 8 (3) | 32 (12) | 16 (6) | 120 (45) |
We initially designed and synthesized lipidated cyclic γ-AApeptides (HW-B-73, -77 and -78, Figure 1) in which the lipid tail C16 is directly connected to the ring structure. Unfortunately, these sequences only possess weak activity against bacteria. We hypothesize that this is because the lipid tail on the ring structure has limited orientation, therefore cannot position itself for membrane insertion even after the amphipathic ring contacts the bacterial membranes. Indeed, a trend was apparent in other membrane-active lipidated cyclic peptide antibiotics, including daptomycin and polymyxin B, which contain at least two amino acid residues connecting both lipid tails and ring structures.27,28 To test our hypothesis, we moved the lipid tail outside the cyclic ring, leading to the sequence YL-1. As expected, YL-1 showed strong activity against Gram-positive bacteria, although it only contains three amphiphilic building blocks. To determine how ring size affects antibacterial activity, we then included more amphiphilic building blocks into the ring structure. However, an additional amphiphilic building block (YL-4) did not boost activity. A similar phenomenon has been observed in cyclic peptoids12 and other cyclic peptide antimicrobial agents,30,31 which normally contain 6–8 residues in their ring structures. In fact, YL-4 has weaker antimicrobial activity than YL-1, which may due to the increased flexibility of the larger ring. This trend is also seen in HW-B-73, HW-B-77, and HW-B-78.
We have previously observed that the exact amphiphilic structures can affect the antimicrobial activity of a sequence.20 The reversal of cationic and hydrophobic groups results in antimicrobial agents with different potency.20 It appears that while the overall amphipathicity dominates antimicrobial activity, the precise distribution of functional groups in molecules will affect the strength of peptidomimetic interaction with bacterial membranes. As such, YL-12, containing one reversed amphiphilic building block in relation to YL-1, was prepared. This sequence has improved antimicrobial activity against both Gram-negative and Gram-positive bacteria. The reversal of additional building blocks leads to the most potent lipidated cyclic γ-AApeptides YL-34 and YL-36. Particularly, YL-36, with a small amphipathic ring and a C16 lipid tail, shows very potent activity against all tested drug-resist Gram-positive and Gram-negative strains. It not only has much improved antimicrobial activity over the AMP Pexiganan, but is also superior when compared with the previously reported cyclic γ-AApeptide HW-B-13 of much larger ring size, especially against Gram-negative pathogens.
We hypothesize that lipidated cyclic γ-AApeptides are able to kill bacteria through membrane disruption analogous to AMPs, as they possess globally cationic amphipathic structures and broad-spectrum activity against both Gram-positive and Gram-negative bacteria. As such, a fluorescence microscopy experiment was carried out to study the ability of the most potent lead YL-36 to affect the membranes of S. aureus (Gram-positive) and E. coli (Gram-negative). Briefly, both bacteria were stained with the membrane permeable dye 4′,6-diamidino-2-phenylindole (DAPI) and the non-permeable dye propidium iodide (PI) in the absence or presence of YL-36 (Figure 2). YL-36 treatment resulted in PI becoming visible using fluorescence microscopy, suggesting bacterial membranes of both S. aureus and E. coli were damaged. Aggregation of S. aureus after treatment with YL-36 is observed, which is generally believed to arise from the loss of membrane potential after the disruption of membranes.19–23,26
Figure 2.

Fluorescence micrographs of S. aureus (A) and E. coli (B) that are treated/(or no treatment) with 5 μg/mL lipidated cyclic γ-AApeptide YL-36 for 2 h. a1, control, no treatment, DAPI stained; a2, control, no treatment, PI stained; b1, YL-36 treatment, DAPI stained; b2, YL-36 treatment, PI stained.
In addition to bacterial membrane disruption, some AMPs are speculated to modulate the immune system through TLR signaling.32 Since lipidated cyclic γ-AApeptides were designed to mimic AMPs, we anticipated that they may be also capable of harnessing immune responses. As such, YL-1, YL-12, YL-29, YL-34, and YL-36 were also assessed for the ability to modulate the LPS-induced TLR4 signaling response by measuring nitric oxide production (Figure 3a).33 Nitric oxide is produced as a downstream inflammatory factor in all TLR signaling, and is part of the global inflammatory response. Figure 3a shows that lipidated cyclic γ-AApeptides are a potent class of anti-TLR4 signaling agents, capable of reducing nitric oxide production. To further investigate the anti-inflammatory capabilities of lipidated cyclic γ-AApeptides, we chose the most potent antimicrobial agent YL-36 for further investigation. The IC50 of YL-36 was obtained by treating RAW 264.7 cells with varying concentrations of YL-36 in concert with 20 ng/mL LPS. The resulting inflammatory response was monitored through measurement of nitric oxide production.
Figure 3.

a. Nitric oxide production in the presence of the lipidated cyclic γ-AApeptides YL-1, YL-12, YL-29, and YL-36. Two γ-AApeptides lacking alkyl tails, either linear (HW-C-22-2) or cyclic (HW-C-22-4), were included to demonstrate the importance of γ-AApeptide lipidation on their TLR activities. RAW 264.7 cells/well were treated with 20 ng/mL LPS and varying concentrations of lipidated cyclic γ-AApeptides in a 96-well plate. Data is normalized to 20 ng/mL LPS as 100% activation, and untreated cells as 0% activation. b. Dose-dependent inhibition of nitric oxide production by YL-36. This experiment was performed as described in 3a, and a dose response curve was obtained for calculation of an IC50 value.
YL-36 shows an inhibitory effect of the nitric oxide signaling with an IC50 value of 1.98 μM (Figure 3b). HW-C-22-2 and HW-C-22-4 demonstrate that lipidation increases the effectiveness of γ-AApeptides as TLR signaling antagonists, as neither construct is active under the tested concentrations (Figure 3a and 3b). Nitric oxide is produced by TLR activation, and correlates to the level of inflammatory response. Additionally, YL-36 shows little cytotoxicity up to 100 μM (Figure S3), suggesting the inhibition of nitric oxide is not due to the toxicity of YL-36. As such, we further explored the ability of YL-36 to inhibit downstream NF-κB activation and the production of pro-inflammatory cytokines. Inhibition of the NF-κB activation (Figure 4a) was determined by a previously developed secreted embryonic alkaline phosphatase (SEAP) assay.34 Furthermore, the pro-inflammatory cytokine tumor necrosis factor-α (TNF-α) is also strongly inhibited (IC50 = 2.05 μM) (Figure 4b).35 Both NF-κB and TNF-α are produced due to TLR activation. Their inhibition by YL-36 shows that TLR signaling is being inhibited in a dose dependent fashion. Neither HW-C-22-2 nor HW-C-22-4 shows inhibitory activities, further demonstrating anti-inflammatory activity depends on the lipidation of γ-AApeptides.
Figure 4.

a. NF-κB activation in HEK 293 cells in the presence of various γ-AApeptides. HEK 293 cells were stably transfected with TLR4 and its accessory proteins, as well as a secreted embryonic alkaline phosphatase (SEAP) reporter gene. Cells were plated in 96-well plate with 40,000 cells/well and treated with 20 ng/mL LPS and YL-36. Data is normalized to 20 ng/mL LPS as 100% activation, and untreated cells as 0% activation. Increasing concentrations of YL-36 decrease transcription of SEAP in a dose-dependent fashion. HW-C-22-2 and HW-C-22-4 were included for comparison. b. YL-36 reduces production of TNF-α. TNF-α production was measured with a monoclonal mouse ELISA. Data is normalized to 20 ng/mL LPS as 100% activation, and untreated cells as 0% activation. Increasing concentrations of YL-36 result in decreased production of TNF-α. HW-C-22-2 and HW-C-22-4 were included for comparison.
Tew et. al. has previously elegantly identified antimicrobial agents that reduce TLR-induced inflammatory response.8,24 Interestingly, our lipidated cyclic γ-AApeptides appear to have a different mechanism of inhibition, not interacting with the TLR ligands. If γ-AApeptides interacted with LPS, pre-treatment with YL-36 prior to LPS addition would abolish its anti-TLR4 activities. Our data though demonstrates that both pre-treatment and co-treatment with γ-AApeptides and LPS results in complete antagonism of nitric oxide production (Figure 5), indicating that γ-AApeptides do not bind to LPS directly.
Figure 5.

YL-36 inhibits TLR4 activation independent of LPS binding. Raw 264.7 cells were incubated for 30 minutes with 10 μM YL-36. After incubation, pre-treatment cells were washed with medium and activated with 20 ng/mL LPS. Co-treatment cells were not washed, and were treated with 20 ng/mL LPS while YL-36 remained in the medium. Antagonism for both cell treatments is 100%, suggesting that YL-36 does not bind to the ligand as a mode of inhibition. Data is normalized to 20 ng/mL LPS as 100% activation, and untreated cells as 0% activation.
CONCLUSION
As a new class of antimicrobial peptidomimetics that mimic AMPs, lipidated cyclic γ-AApeptides exhibit potent and broad-spectrum activity against a range of multi-drug resistant Gram-negative and Gram-positive bacteria. Maybe even more importantly, lipidated cyclic γ-AApeptides also imitate AMPs in their immunomodulatory capabilities. These γ-AApeptides have been shown to antagonize the LPS activated NF-κB signaling response, and to potently suppress release of harmful pro-inflammatory cytokines including tumor necrosis factor-α (TNF-α). Additionally, lipidated cyclic γ-AApeptides do not bind to TLR ligands, suggesting a novel mechanism for immunomodulation. These peptidomimetics provide an exciting new approach to treat bacterial infections by exerting dual-functional roles: they are a new generation of antibiotic agents that kill both multi-drug resistant Gram-positive and Gram-negative bacteria directly, as well as anti-inflammatory agents through harnessing immune responses. Furthermore, as deregulation of TNF-α is also related to diseases such as cancer and lupus erythematosus,36,37 lipidated cyclic γ-AApeptides may also provide other therapeutic applications in the future.
EXPERIMENTAL SECTION
General experimental methods
Rink amide MBHA resins (200–400 mesh, 0.7 mmol/g) were purchased from Chem-Impex Int’l Inc. Other chemicals were ordered from either Sigma-Aldrich or Fisher Scientific, and used without further purification. 1H NMR spectra of the building blocks were obtained on an Agilent DD800 instrument. The solid phase syntheses of the lipidated cyclic γ-AApeptides were carried out in a peptide reaction vessel on a Burrell Wrist-Action shaker, were analyzed and purified using an analytical and preparative Waters HPLC system, respectively. The final products were dried in a Labcono lyophilizer. Molar masses were identified using a Bruker AutoFlex MALDI-TOF mass spectrometer.
Solid phase synthesis of lipidated cyclic γ-AA peptides
The syntheses of lipidated cyclic γ-AApeptides were carried out on the solid phase as reported previously.19,22 For each coupling cycle, 20% Piperidine in DMF was used to remove the Fmoc protecting group, followed by the coupling of 1.5 equiv of building blocks with 4 equiv of HOBT (1-hydroxybenzotriazole monohydrate)/DIC (diisopropylcarbodiimide) in DMF for 6 h. The allyl group was removed by 0.2 equiv of Pd(PPh3)4 in the presence of 10 equiv of PhSiH3/CH2Cl2 (2 h for each, repeated twice). The exposed carboxyl group reacted with the N-terminus of the sequence to complete the cyclization using PyBOP/HOBT/DIPEA/DMF. The lipidated γ-peptides was cleaved from solid support in 50:48:2 TFA/CH2Cl2/TIS (triisopropylsilane) for 2 h. The solvent was evaporated and the sequences were analyzed and purified using a Waters HPLC system monitored at 215 nm. The desired fractions were collected and lyophilized, and their molecular weights were confirmed by the Bruker AutoFlex MALDITOF mass spectrometer.
Antimicrobial assays.19,22
The lipidated cyclic γ-AApeptides were tested for their antimicrobial activity against various microbial organisms including E. coli (ATCC 25922), K. pneumonia (ATCC 13383), multi-drug resistant P. aeruginosa (ATCC 27853), Methicillin-resistant S. epidermidis (MRSE, RP62A), Vancomycin-resistant E. faecalis (ATCC 700802), Methicillin-resistant S. aureus (ATCC 33592). The highest concentration of the tested AA-peptides was 25 μg/mL. The bacteria in 5 mL of medium were grown at 37 °C overnight and then diluted to make a suspension of approximate 1 × 106 CFU/mL. Aliquots of 50 QL of bacterial suspension were mixed with 50 QL of medium containing different concentrations of lipidated cyclic γ-AA-peptides. The plate was incubated at 37 °C overnight with cell growth monitored by a Biotek Synergy HT microtiter plate reader under the 600 nm wavelength. MIC was determined when the lowest concentration of the compounds inhibit the cell growth completely in 24 h. The results were repeated at least three times with duplicates for each time.
Hemolysis assay.19,22
Freshly drawn, K2 EDTA treated human red blood cells (hRBCs) were washed with PBS buffer twice and centrifuged at 1000g for 10 min. After the clear supernatant was removed, the cell pellets were mixed with serial diluted lipo-cyclic γ-AApeptides in a 96-well plate. The plate was incubated at 37 °C for 1 h and centrifuged at 3500 rpm for 10 min. The supernatant was separated and diluted in PBS, and the absorbance was detected at 360 nm using a Biotek Synergy TH plate reader. % hemolysis = (Abssample − AbsPBS)/(AbsTriton − AbsPBS) × 100%. 0% hemolysis (negative control) was determined by mixing blood with PBS and 100% hemolysis (positive control) was determined by mixing blood with Triton X-100 (final concentration 0.1%). The results were repeated at least three times with duplicates for each time.
Fluorescence microscopy.19,22
DAPI (4′, 6-Diamidino-2-phenylindole dihydrochloride, Sigma, >98%) and PI (Propidium iodide, Sigma) were used to stain the bacteria cells of E. coli or S. aureus. DAPI is a DNA binding dye staining all bacterial cells regardless of their viabilities, and PI is an ethidium derivative which only can pass through damaged bacterial membranes and intercalates with their nucleic acids. Briefly, bacteria in mid-logarithmic phase were incubated with lipidated cyclic γ-AA peptides (2 × MIC) for 2 h, and then were centrifuged at 3000g for 15 min. The bacteria cell pellets were separated then incubated with PI, followed by washing and incubation with DAPI (each dye incubation was performed at 0 °C for 15 minutes in dark). Controls were bacteria culture without peptides following the same procedure described above. The stained bacteria cells were observed under Zeiss Axio Imager Zloptical microscope using the 100X oil-immersion objective.
Fluorescent Detection of Nitric Oxide.34,35
Raw 264.7 (Mouse leukaemic monocyte macrophage cell line) cells were grown in RPMI 1640 medium containing 1% L-glutamine, 1% Penicillin/streptomycin and 10% fetal bovine serum (FBS). Cells were plated in a 96-well plate at 75,000 cells/well in complete RPMI 1640 medium, and allowed to grow overnight at 37°C and 5% CO2 in a humidified incubator. The media was removed, and cells were placed in unsupplemented RPMI 1640 medium. 20 ng/mL LPS and the appropriate concentration of lipidated cyclic γ-AApeptides (20 mM stock solutions in PBS) were added to a final volume of 200 μL. PBS controls were included in each experiment. Plates were then incubated for 24 h, and then 100 μL of media was transferred to a flat black 96-well microfluor plate (Thermo Scientific, MA, USA). Following that, 10 μL of 0.05 mg/mL 2,3-diaminonamthalene in 0.62 M HCl was added to the media and incubated for 20 minutes in the dark. The reaction was quenched with 5 μL of 3.0 M NaOH, and the plate was read on a Beckman Coulter DTX880 plate reader (Beckman Coulter, CA, USA). Data was collected with excitation at 360 nm and emission at 430 nm. Data was normalized with the ligand only control as 100% activation, and the untreated cells as 0% activation. Fold inhibition = [(Sample 430 nm − Untreated cells 430 nm)/(Ligand Control 430 nm − Untreated cells 430 nm)]. The EC50 values were calculated graphically using OriginPro v8.6 software.
Secreted Embryonic Alkaline Phosphatase (SEAP) Reporter of NF-κB Transcription.34,35
HEK293 (Human Embryonic Kidney 293) cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) in the presence of 1% Penicillin/streptomycin, 10% fetal bovine serum (FBS), and 1% L-glutamine. HEK293 cells are stably transfected with human TLR4, as well as the required accessory proteins MD-2 and CD14. Moreover, the cells also possess an optimized alkaline phosphatase reporter gene under the control of a NF-κB inducible promoter. 38 Cells were first plated in a 96-well plate at 40,000 cells/well in complete DMEM medium and allow to grow overnight at 37°C and 5% CO2 in a humidified incubator. Then the media was removed, and cells were placed in Optimem + 0.5% FBS medium. 20 ng/mL LPS and the appropriate concentration of lipidated cyclic γ-AApeptides were added to a final volume of 200 μL. PBS buffer was included as control. Plates were then incubated for 24 h, and then medium was assayed per the instructions of the Phospha-Light™ SEAP Reporter Gene Assay System (Applied Biosystems, NY, USA). The plate was read on a Beckman Coulter DTX880 plate reader (Beckman Coulter, CA, USA). Data was collected with luminescence at 430 nm. Data was normalized with the ligand only control as 100% activation, and the untreated cells as 0% activation. Fold inhibition = [(Sample 430 nm − Untreated cells 430 nm)/(Ligand Control 430 nm − Untreated cells 430 nm)]. The EC50 values were calculated graphically using OriginPro v8.6 software.
Enzyme-Linked Immunosorbent Assay (ELISA) Detection of TNF-α.34,35
Raw 264.7 (Mouse leukaemic monocyte macrophage cell line) cells were grown in RPMI 1640 medium with 10% fetal bovine serum (FBS), 1% Penicillin/streptomycin, and 1% L-glutamine. Cells were plated in a 96-well plate at 75,000 cells/well in complete RPMI 1640 medium and allowed to grow overnight at 37°C and 5% CO2 in a humidified incubator. Then media was removed, and cells were placed in unsupplemented RPMI 1640 medium. 20 ng/mL LPS and the appropriate concentration of lipidated cyclic γ-AApeptides (20 mM stock solutions) were added to a final volume of 200 μL. PBS was included as the control. Plates were then incubated for 24 hours, and then samples were assayed for TNF-α per the method outlined in the BD Biosciences Mouse TNF (Mono/Mono) ELISA Set (BD Biosciences, CA, USA). The plate was read on a Beckman Coulter DTX880 plate reader (Beckman Coulter, CA, USA). Data was collected with absorbance at 450 nm. Data was normalized with the ligand only control as 100% activation, and the untreated cells as 0% activation. Fold inhibition = [(Sample 450 nm − Untreated cells 450 nm)/(Ligand Control 450 nm − Untreated cells 450 nm)]. The EC50 values were calculated graphically using OriginPro v8.6 software.
Crystal Violet Toxicity Assay.34,35
Cells which were treated with compound for nitric oxide experimentation were also tested for compound toxicity using crystal violet stain. Cells were fixed for 20 minutes in 4% paraformaldehyde after the media was removed. After fixing, formaldehyde was removed and cells were incubated for 1h with 0.05% crystal violet stain. After incubation, cells were rinsed with deionized water and reconstituted in 100% methanol for 10 minutes. The plate was read on a Beckman Coulter DTX880 plate reader (Beckman Coulter, CA, USA). Data was collected with absorbance at 535 nm. Data was normalized with the untreated cells control as 100% survival, and the blank wells as 0% survival. Fold inhibition = [(Sample 535 nm − Blank 535 nm)/(Untreated cells 535 nm − Blank 535 nm)].
Supplementary Material
Acknowledgments
This work was supported by the USF start-up fund (J. Cai) and NIH (GM101279 and GM103843) (H. Y).
Footnotes
Supporting Information includes Figures S1–S3, Tables S1, experimental methods, HPLC traces and NMR data. This material is available free of charge via the Internet at http://pubs.acs.org
Contributor Information
Hang Yin, Email: hubert.yin@colorado.edu.
Jianfeng Cai, Email: jianfengcai@usf.edu.
References
- 1.Levy SB, Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med. 2004;10:S122–129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 2.Hancock RE, Sahl HG. Antimicrobial and host-defense peptides as new antiinfective therapeutic strategies. Nat Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 3.Marr AK, Gooderham WJ, Hancock RE. Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr Opin Pharmacol. 2006;6:468–472. doi: 10.1016/j.coph.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 4.Choi S, Isaacs A, Clements D, Liu D, Kim H, Scott RW, Winkler JD, DeGrado WF. De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc Natl Acad Sci U S A. 2009;106:6968–6973. doi: 10.1073/pnas.0811818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scott MG, Gold MR, Hancock REW. Interaction of cationic peptides with lipoteichoic acid and gram-positive bacteria. Infect Immun. 1999;67:6445–6453. doi: 10.1128/iai.67.12.6445-6453.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hancock REW, Brown KL, Mookherjee N. Host defence peptides from invertebrates - emerging antimicrobial strategies. Immunobiology. 2006;211:315–322. doi: 10.1016/j.imbio.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 7.Bowdish DME, Davidson DJ, Hancock REW. A re-evaluation of the role of host defence peptides in mammalian immunity. Curr Protein Pep Sci. 2005;6:35–51. doi: 10.2174/1389203053027494. [DOI] [PubMed] [Google Scholar]
- 8.Thaker HD, Som A, Ayaz F, Lui DH, Pan WX, Scott RW, Anguita J, Tew GN. Synthetic Mimics of Antimicrobial Peptides with Immunomodulatory Responses. J Am Chem Soc. 2012;134:11088–11091. doi: 10.1021/ja303304j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karlsson AJ, Pomerantz WC, Weisblum B, Gellman SH, Palecek SP. Antifungal activity from 14-helical beta-peptides. J Am Chem Soc. 2006;128:12630–12631. doi: 10.1021/ja064630y. [DOI] [PubMed] [Google Scholar]
- 10.Karlsson AJ, Pomerantz WC, Neilsen KJ, Gellman SH, Palecek SP. Effect of sequence and structural properties on 14-helical beta-peptide activity against Candida albicans planktonic cells and biofilms. ACS Chem Biol. 2009;4:567–579. doi: 10.1021/cb900093r. [DOI] [PubMed] [Google Scholar]
- 11.Karlsson AJ, Flessner RM, Gellman SH, Lynn DM, Palecek SP. Polyelectrolyte multilayers fabricated from antifungal beta-peptides: design of surfaces that exhibit antifungal activity against Candida albicans. Biomacromolecules. 2010;11:2321–2328. doi: 10.1021/bm100424s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang ML, Shin SB, Benson MA, Torres VJ, Kirshenbaum K. A comparison of linear and cyclic peptoid oligomers as potent antimicrobial agents. ChemMedChem. 2012;7:114–122. doi: 10.1002/cmdc.201100358. [DOI] [PubMed] [Google Scholar]
- 13.Chongsiriwatana NP, Patch JA, Czyzewski AM, Dohm MT, Ivankin A, Gidalevitz D, Zuckermann RN, Barron AE. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc Natl Acad Sci. 2008;105:2794–2799. doi: 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Claudon P, Violette A, Lamour K, Decossas M, Fournel S, Heurtault B, Godet J, Mely Y, Jamart-Gregoire B, Averlant-Petit MC, Briand JP, Duportail G, Monteil H, Guichard G. Consequences of isostructural main-chain modifications for the design of antimicrobial foldamers: helical mimics of host-defense peptides based on a heterogeneous amide/urea backbone. Angew Chem Int Ed. 2010;49:333–336. doi: 10.1002/anie.200905591. [DOI] [PubMed] [Google Scholar]
- 15.Hua J, Scott RW, Diamond G. Activity of antimicrobial peptide mimetics in the oral cavity: II. Activity against periopathogenic biofilms and anti-inflammatory activity. Mol Oral Microbiol. 2010;25:426–432. doi: 10.1111/j.2041-1014.2010.00591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hua J, Yamarthy R, Felsenstein S, Scott RW, Markowitz K, Diamond G. Activity of antimicrobial peptide mimetics in the oral cavity: I. Activity against biofilms of Candida albicans. Mol Oral Microbiol. 2010;25:418–425. doi: 10.1111/j.2041-1014.2010.00590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srinivas N, Jetter P, Ueberbacher BJ, Werneburg M, Zerbe K, Steinmann J, Van der Meijden B, Bernardini F, Lederer A, Dias RL, Misson PE, Henze H, Zumbrunn J, Gombert FO, Obrecht D, Hunziker P, Schauer S, Ziegler U, Kach A, Eberl L, Riedel K, DeMarco SJ, Robinson JA. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science (New York, N Y. 2010;327:1010–1013. doi: 10.1126/science.1182749. [DOI] [PubMed] [Google Scholar]
- 18.Obrecht D, Robinson JA, Bernardini F, Bisang C, DeMarco SJ, Moehle K, Gombert FO. Recent progress in the discovery of macrocyclic compounds as potential antiinfective therapeutics. Curr Med Chem. 2009;16:42–65. doi: 10.2174/092986709787002844. [DOI] [PubMed] [Google Scholar]
- 19.Niu Y, Padhee S, Wu H, Bai G, Harrington L, Burda WN, Shaw LN, Cao C, Cai J. Identification of gamma-AApeptides with potent and broad-spectrum antimicrobial activity. Chem Commun. 2011;47:12197–12199. doi: 10.1039/c1cc14476f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Padhee S, Hu Y, Niu Y, Bai G, Wu H, Costanza F, West L, Harrington L, Shaw LN, Cao C, Cai J. Non-hemolytic alpha-AApeptides as antimicrobial peptidomimetics. Chem Commun. 2011;47:9729–9731. doi: 10.1039/c1cc13684d. [DOI] [PubMed] [Google Scholar]
- 21.Hu Y, Amin MN, Padhee S, Wang R, Qiao Q, Ge B, Li Y, Mathew A, Cao C, Cai J. Lipidated Peptidomimetics with Improved Antimicrobial Activity. ACS Med Chem Lett. 2012;3:683–686. doi: 10.1021/ml3001215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu H, Niu Y, Padhee S, Wang RE, Li Y, Qiao Q, Ge B, Cao C, Cai J. Design and synthesis of unprecedented cyclic gamma-AApeptides for antimicrobial development. Chem Sci. 2012;3:2570–2575. [Google Scholar]
- 23.Niu Y, Padhee S, Wu H, Bai G, Qiao Q, Hu Y, Harrington L, Burda WN, Shaw LN, Cao C, Cai J. Lipo-gamma-AApeptides as a New Class of Potent and Broad-Spectrum Antimicrobial Agents. J Med Chem. 2012;55:4003–4009. doi: 10.1021/jm300274p. [DOI] [PubMed] [Google Scholar]
- 24.Som A, Navasa N, Percher A, Scott RW, Tew GN, Anguita J. Identification of Synthetic Host Defense Peptide Mimics That Exert Dual Antimicrobial and Anti-Inflammatory Activities. Clin Vacc Immunol. 2012;19:1784–1791. doi: 10.1128/CVI.00291-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niu Y, Wu H, Li Y, Hu Y, Padhee S, Li Q, Cao C, Cai J. AApeptides as a new class of antimicrobial agents. Org Biomol Chem. 2013;11:4283–4290. doi: 10.1039/c3ob40444g. [DOI] [PubMed] [Google Scholar]
- 26.Niu Y, Wang RE, Wu H, Cai J. Recent development of small antimicrobial peptidomimetics. Future Med Chem. 2012;4:1853–1862. doi: 10.4155/fmc.12.111. [DOI] [PubMed] [Google Scholar]
- 27.Zavascki AP, Goldani LZ, Li J, Nation RL. Polymyxin B for the treatment of multidrug-resistant pathogens: a critical review. J Antimicrob Chemother. 2007;60:1206–1215. doi: 10.1093/jac/dkm357. [DOI] [PubMed] [Google Scholar]
- 28.Weis F, Beiras-Fernandez A, Schelling G. Daptomycin, a lipopeptide antibiotic in clinical practice. Curr Opin Investig Drugs. 2008;9:879–884. [PubMed] [Google Scholar]
- 29.Ge Y, MacDonald DL, Holroyd KJ, Thornsberry C, Wexler H, Zasloff M. In vitro antibacterial properties of pexiganan, an analog of magainin. Antimicrob Agents Chemother. 1999;43:782–788. doi: 10.1128/aac.43.4.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu G, Abraham T, Rapp J, Vastey F, Saad N, Balmir E. Daptomycin: evaluation of a high-dose treatment strategy. Int J Antimicrob Agents. 2011;38:192–196. doi: 10.1016/j.ijantimicag.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 31.Vaara M. Novel derivatives of polymyxins. J Antimicrob Chemother. 2013;68:1213–1219. doi: 10.1093/jac/dkt039. [DOI] [PubMed] [Google Scholar]
- 32.Mookherjee N, Brown KL, Bowdish DME, Doria S, Falsafi R, Hokamp K, Roche FM, Mu RX, Doho GH, Pistolic J, Powers JP, Bryan J, Brinkman FSL, Hancock REW. Modulation of the TLR-mediated inflammatory response by the endogenous human host defense peptide LL-37. J Immunol. 2006;176:2455–2464. doi: 10.4049/jimmunol.176.4.2455. [DOI] [PubMed] [Google Scholar]
- 33.Cheng K, Wang XH, Yin H. Small-Molecule Inhibitors of the TLR3/dsRNA Complex. J Am Chem Soc. 2011;133:3764–3767. doi: 10.1021/ja111312h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slivka PF, Shridhar M, Lee GI, Sammond DW, Hutchinson MR, Martinko AJ, Buchanan MM, Sholar PW, Kearney JJ, Harrison JA, Watkins LR, Yin H. A Peptide Antagonist of the TLR4-MD2 Interaction. Chembiochem. 2009;10:645–649. doi: 10.1002/cbic.200800769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang ST, Cheng K, Wang XH, Yin H. Selection, synthesis, and anti-inflammatory evaluation of the arylidene malonate derivatives as TLR4 signaling inhibitors. Bioorg Med Chem. 2012;20:6073–6079. doi: 10.1016/j.bmc.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 37.Sabry A, Sheashaa H, El-husseini A, Mahmoud K, Eldahshan KF, George SK, Abdel-Khalek E, El-Shafey EM, Abo-Zenah H. Proinflammatory cytokines (TNF-alpha and IL-6) in Egyptian patients with SLE: Its correlation with disease activity. Cytokine. 2006;35:148–153. doi: 10.1016/j.cyto.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 38.Hutchinson MR, Zhang YN, Brown K, Coats BD, Shridhar M, Sholar PW, Patel SJ, Crysdale NY, Harrison JA, Maier SF, Rice KC, Watkins LR. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4) Eur J Neurosci. 2008;28:20–29. doi: 10.1111/j.1460-9568.2008.06321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.