

Abstract
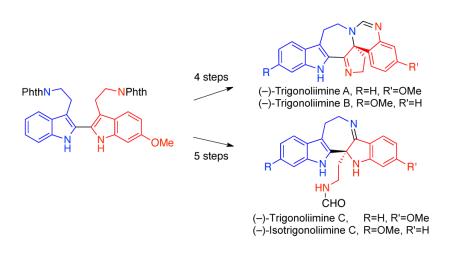
A full account of our concise and enantioselective total syntheses of all known (−)-trigonoliimine alkaloids is described. Our retrobiosynthetic analysis of these natural products enabled identification of a single bistryptamine precursor as a precursor to all known trigonoliimines through a sequence of transformations involving asymmetric oxidation and reorganization. Our enantioselective syntheses of these alkaloids enabled the revision of the absolute stereochemistry of (−)-trigonoliimines A, B, and C. We report that trigonoliimines A, B, C, and structurally related compounds showed weak anticancer activities against HeLa and U-937 cells.
Introduction
Bisindole alkaloids containing 2,2′-connectivity constitute a large number of natural products which includes fascaplycins and indolocarbazole glycopeptides. 1 , 2 The potent antitumor activity of indolocarbazole glycopeptides such as rebeccamycin3 and stauroporine4 have stimulated the development of synthetic analogs of this family of natural products for the development of anticancer agents.5 Additionally, recent studies have shown possible application of these natural products as selective and sensitive chemical sensors based on their unique chemiluminescence properties.6
In 2010, Hao and coworkers reported the isolation of 2,2′-linked bistryptamine alkaloids (+)-trigonoliimines A (1) and B (2) along with (−)-trigonoliimine C (3) from the leaves of Trigonostemon lii that Y. T. Chang collected in Yunnan province of China.7 From their biological studies of these alkaloids, 1 showed modest activity in an anti-HIV assay (EC50 = 0.95 μg/mL, TI = 7.9). Fascinated by their unique molecular architecture, we initiated a synthetic program aimed to provide a general solution to trigonoliimine alkaloids in enantiomerically enriched form. In 2011, we completed the first total synthesis of all known (−)-trigonoliimines A (1), B (2) and C (3) using a synthetic strategy based on asymmetric oxidation and reorganization of a single heterodimeric bistryptamine and revised the absolute stereochemistry of all (−)-trigonoliimines. 8 In a contemporaneous study, Tambar and coworkers reported the total synthesis of (±)-trigonoliimine C,9 and shortly after, Hao and coworkers published the synthesis of the pentacyclic skeleton of (±)-trigonoliimine C using a similar oxidation and 1,2-alkyl shift strategy.10 Interest toward these fascinating natural products has continued and Shi and coworkers reported the synthesis of the desmethoxy hexacyclic skeleton of (±)-trigonoliimines A and B using carbanion mediated seven-membered ring formation as a key step.11 In 2012, Zhu and coworkers reported the total synthesis of (±)-trigonoliimine B using a late stage Bischler–Napieralski reaction.12 This strategy was also applied in Hao and coworkers’ synthesis of the hexacyclic skeleton of (±)-trigonoliimines A and B.13 Most recently, Hao and coworkers reported the total synthesis of (±)-trigonoliimine A using a Strecker reaction for the formation of the C20 tetrasubstituted carbon center and a Houben–Hoesch14 type reaction for the seven-membered ring forming cyclization.15 Herein, we detail the full account of our synthetic approach to the trigonoliimine alkaloids, which currently remains the only solution to access all known trigonoliimines in enantiomerically enriched form. We also report anticancer activities of trigonoliimines A–C, and structurally related alkaloids against HeLa (human cervical cancer) and U-937 (human lymphoma) cell lines.
Results and Discussion
Design plan for the total synthesis
Oxidation and rearrangement of 2,3-disubstituted indoles has served as an efficient strategy to access the indoxyl moiety and has been applied in the total synthesis of various alkaloids.16 In 2008, our laboratory proposed a synthetic strategy to access calycanthaceous alkaloids through oxidation and rearrangement of a 2,2′-bistryptamine derivative.17,18 Our continued interest in this area yielded an efficient and stereocontrolled synthetic strategy involving oxidation and rearrangement of 2,3-disubstituted indoles, especially those with an aryl substituent at the 2-position, for efficient access to oxindole products.19 Despite outstanding advancements in the area of asymmetric oxidation,20 enantioselective oxidation of 2,3-disubstituted indoles remains a challenging problem.21,22 Recently, in collaboration with the Miller group, we reported an asymmetric oxidation of 2,3-disubstituted indole for the formation of enantiomerically enriched hydroxyindolenines using an aspartyl based peptide catalyst.23,24
With these considerations in mind, the molecular structure of trigonoliimine alkaloids (Figure 1) drew our immediate attention upon their isolation due to their possible structural relevance to intermediates in our primary investigations toward chimonanthine (S6, Scheme S1).17 Our unified strategy for the enantioselective total synthesis of all known trigonoliimines was based on the hypothesis that bistryptamine heterodimer 15 may serve as a common biosynthetic precursor to these alkaloids (Scheme 1). We envisioned that mono-oxidation of bistryptamine 15 would generate hydroxyindolenines 9 and 10, intermediates that could serve as a branching point from which to access the two distinct molecular architectures present in the trigonoliimine alkaloid family (Scheme 1).
Figure 1.
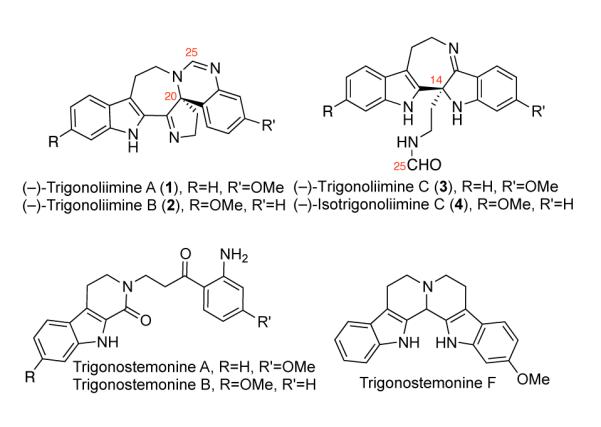
Structure of (−)-trigonoliimines A–C (1–3) with revised absolute stereochemistry (reference 8) and other representative trigonostemon alkaloids.
Scheme 1.

Retrosynthetic analysis of (−)-trigonoliimines A–C (1–3) and isotrigonoliimine C (4).
To assemble the amidine moiety present in trigonoliimines A (1) and B (2), we designed a late stage N-formylation and intramolecular condensation of azepanes 5 and 6. We planned to access azepanes 5 and 6 from aminals 7 and 8 via ionization and stereoretentive intramolecular trapping of carbinol C20 by N12, and subsequent rearrangement. The requisite cis-fused aminals 7 and 8 could result from intramolecular cyclization of hydroxyindolenines 9 and 10, respectively. Retrosynthetic analysis of trigonoliimine C (3) and isotrigonoliimine C (4) commenced with formylations of N24 in imines 11 and 12. The seven-membered imine moiety would be derived from an intramolecular condensation of N12 to C15 of indoxyls 13 and 14, respectively. Indoxyls 13 and 14 would be accessible via a stereospecific Wagner–Meerwein type 1,2-alkyl shift25 of hydroxyindolenines 9 and 10, the common intermediates that serve as gateways to access all trigonoliimines.
Our biosynthetically inspired retrosynthesis predicted the S-configuration on the stereogenic centers of trigonoliimines A–C (1–3) resulting from common hydroxyindolenines 9 and 10 with S-configuration at their stereogenic centers (Scheme 1). Interestingly, the isolation report predicted the S-configuration at the stereogenic centers of (+)-trigonoliimines A and B (Class A) and the R-configuration at the stereogenic center of (−)-trigonoliimine C (Class B),7 suggesting that these two classes of trigonoliimines stem from enantiomeric hydroxyindolenine precursors. This provided further impetus to develop an enantioselective synthesis of these alkaloids as we could begin to address these questions by accessing synthetic samples of enantiomerically enriched trigonoliimines A–C.
Synthesis of (±)-19-desmethoxy-trigonoliimine C
In the initial stages of our studies, in order to quickly evaluate the plausibility of our retrosynthetic analysis of trigonoliimines, we focused on a desmethoxy trigonoliimine model system starting from tryptamine 16 (Scheme 2).26 Recently, Hao and Liu reported their synthetic studies of a similar desmethoxy model system.10 When phthalimide protected tryptamine 16 was treated with trifluoroacetic acid (TFA), the desired dimer 17 was formed in quantitative yield.27 Heterodimer 17 could be oxidized to homodimeric 2,2′-bistryptamine 18 in the presence of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in tetrahydrofuran in 58% yield. Subsequent treatment of bistryptamine 18 with Davis’ oxaziridine 19 afforded hydroxyindolenine (±)-20 in quantitative yield. After further attempts to optimize this synthetic sequence, we found that treatment of heterodimer 17 with Davis’ oxaziridine 19 (2.2 equiv) afforded the desired hydroxyindolenine (±)-20 in 89% yield. Notably, treatment of 17 with excess Davis’ oxaziridine 19 (4 equiv) did not result in the formation of over-oxidized products consistent with our original hypothesis.17
Scheme 2.
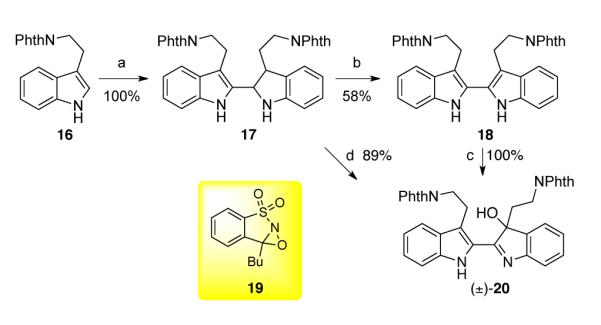
Synthesis of hydroxyindolenine (±)-20. Conditions: (a) TFA, 23 °C; (b) DDQ, THF, 23 °C; (c) 19 (1.1 equiv), CH2Cl2, 23 °C; (d) 19 (2.2 equiv), CH2Cl2, 23 °C.
With ready access to hydroxyindolenine (±)-20, we investigated the Wagner–Meerwein type 1,2-alkyl shift25 to form indoxyl (±)-21, which possesses the structural backbone of trigonoliimine C (3, Scheme 1). When hydroxyindolenine (±)-20 was treated with scandium trifluoromethanesulfonate [Sc(OTf)3] in toluene at 85 °C,18 the desired indoxyl 21 was obtained in 36% yield along with oxindole 22 (32% yield, Scheme 3) as a byproduct. Interestingly, treatment of hydroxyindolenine 20 with 20 equivalent of hydrazine in refluxing methanol afforded the cyclized hydroxyaminal 23 as a major isomer in 99% yield. While the 1H NMR analysis of aminal 23 in deuterated chloroform was consistent with the structure shown in Scheme 3, 1H NMR analysis of the same sample in deuterated methanol revealed the presence of multiple species consistent with structural flexibility of this intermediate (vide infra).
Scheme 3.
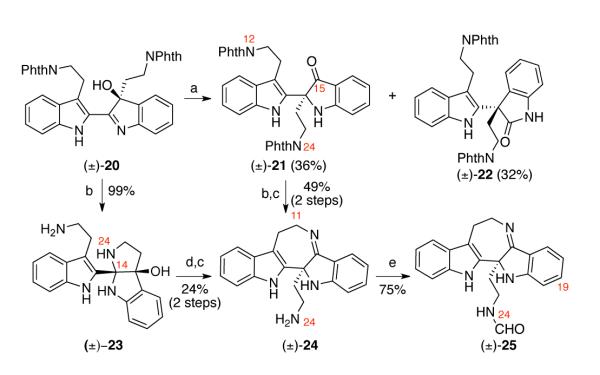
Synthesis of (±)-19-desmethoxy-trigonoliimine C (25). Conditions: (a) Sc(OTf)3, PhMe , 85 °C; (b) NH2NH2•H2O, MeOH, 80 °C; (c) Ti(OEt)4, THF, 40 °C; (d) Sc(OTf) , MeCN, PhMe (2:1), 81 °C;28 3 (e) N-Formylimidazole, THF, 23 °C.
With synthetic access to indoxyl (±)-21, we were set to answer the question of whether the intramolecular condensation would occur between N12–C15 or N24–C15. Hydrazinolysis of indoxyl (±)-21 and subsequent treatment of the resulting crude mixture with titanium tetraethoxide yielded seven-membered imine (±)-24 in 49% yield over two steps (vide infra). Notably, we did not observe the formation of the 5-membered imine resulting from the condensation of N24 with the ketone moiety (vide infra), but noticed a broadening of 1H NMR peaks at C11 possibly due to a rapid equilibration between imine and aminal structures. Interestingly, imine (±)-24 could also be accessed from hydroxyaminal (±)-23 upon heating with Sc(OTf)328 followed by treatment of the resulting indoxyl intermediate with titanium ethoxide. When pentacycle 24 was treated with N-formylimidazole, N24 was formylated to give (±)-19-desmethoxy-trigonoliimine C (25) in 75% yield. 1H NMR peaks of the formylated product (±)-25 were sharper and better resolved compared to intermediate 24, consistent with the decreased nucleophilicity of N24.
Synthesis of the heterodimeric 2,2′-bistryptamine
Promising results described above in the initial phase of this program prompted investigation of the synthesis of heterodimeric 2,2′-bistryptamine 15 (Scheme 1), which can be derived from the union of tryptamine 16 and 6-methoxy tryptamine 26.27b,29 Initial heterodimerization attempts with tryptamine 16 and 6-methoxytryptamine 26 under ionic conditions yielded dimeric tryptamine 17 as a single diastereomer in 22% yield (Scheme 4). However, no desired heterodimer could be obtained in any synthetically useful amount. Hence, for the selective union of two tryptamine subunits, we envisioned the use of a metal-catalyzed and directed cross-coupling reaction.
Scheme 4.
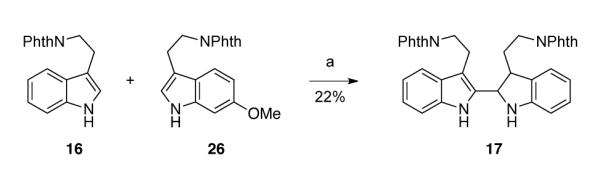
Attempted heterodimerization of tryptamines 16 and 26 under ionic condition. Conditions: (a) TFA, 23 °C.
The union of two indole subunits at their 2,2′-junction by virtue of a transition metal-catalyzed cross-coupling reaction is well-precedented.30,31 However, to the best of our knowledge, there had been no reports that cross-couple two N1-unprotected indole subunits. Hence, we focused our attention on direct assembly of two N1-unprotected tryptamine subunits by application of the Suzuki–Miyaura cross-coupling reaction.32
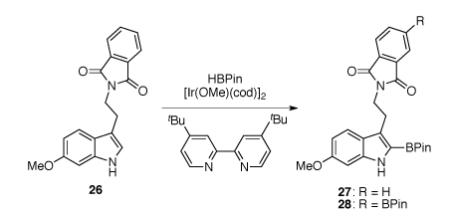
For the synthesis of the boronate coupling partner, we planned to achieve a direct C2 borylation of 6-methoxytryptamine 26 by using an iridium based catalyst system developed by Ishiyama, Miyaura, and Hartwig (Table 1).33 Initial attempts with pinacolborane (1 equiv) in the presence of 5 mol% of iridium catalyst and 10 mol% of bispyridine ligand in tetrahydrofuran at 70 °C did not afford any desired product (Table 1, entry 1). When 6-methoxytryptamine 26 was treated with pinacolborane (2.6 equiv) at 80 °C, only diborylated product 28 was obtained in 93% yield (Table 1, entry 2). After optimization (Table 1, entries 2–5), we found that 6-methoxytryptamine 26 could be converted selectively to monoborylated tryptamine 27 in 67% yield by exposure to pinacolborane (2.2 equiv), 10 mol% of iridium catalyst, and 10 mol% of bispyridine ligand in dichloromethane at 23 °C (Table 1, entry 6).
Table 1.
Optimization of the iridium-catalyzed borylation at C2 of 6-methoxytryptamine 26.
| Entry | HBPin equiv |
Ir catalyst (mol%) |
bipyridyl ligand (mol%) |
temp (°C) |
solvent | resulta |
|---|---|---|---|---|---|---|
| 1 | 1 | 5 | 10 | 70 | THF | No reaction |
| 2 | 2.6 | 3 | 6 | 80 | THF | 28: 93% |
| 3 | 2 | 5 | 10 | 45 | THF | 27:28 = 2.2:1b, 48% |
| 4 | 2 | 5 | 10 | 45 | CH2Cl2 | 27: 55% |
| 5 | 2.5 | 5 | 10 | 45 | CH2Cl2 | 27:28 = 4.5:1b, 66% |
| 6 | 2.2 | 10 | 10 | 23 | CH2Cl2 | 27: 67% |
Isolated yield.
Ratio of 27 to 28 was determined by 1H NMR.
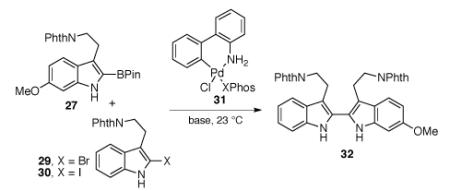
We next sought conditions that enabled the efficient union of two tryptamine subunits to form 2,2′-bistryptamine 32. Initial cross-coupling attempts using various palladium sources in conjunction with XPhos34 and potassium phosphate in toluene–water mixture at 110 °C provided the desired product 32, albeit in low and variable yields (7–44%). Hence, we sought to perform this carbon–carbon bond formation under milder conditions. When bromide 29 and boronic ester 27 were treated with the Buchwald biphenyl precatalyst 31 and potassium phosphate in toluene–water mixture at 23 °C,35 the desired bistryptamine heterodimer 32 was formed in 40% yield (Table 2, entry 1). We discovered that the addition of a halophilic silver salt to the reaction mixture induced a notable increase in product yield (Table 2, entries 6 and 7).36 Notably, the use of Ag2CO3 (2.0 equiv) as the sole base resulted in a lower yield (33%, Table 2, entry 8) of the desired product 32. When we used Ag3PO4 (2.0 equiv) as the sole base additive, we were able to obtain the desired product in 63% yield (Table 2, entry 9), possibly due to the two-fold role of this base as both a halophile and ideal base additive.
Table 2.
Optimization of the Suzuki–Miyaura cross-coupling reaction for the formation of 2,2′-bistryptamine 32.
| entry | halide | base | solvent | yield (%)b |
|---|---|---|---|---|
| 1 | 29 | K3PO4 | PhMe, H2O (5:1) | 40 |
| 2 | 29 | KOH | PhMe, H2O (5:1) | 30 |
| 3 | 29 | Tl2CO3 | THF, H2O (5:1) | 8 |
| 4 | 30 | K3PO4 | PhMe, H2O (5:1) | 31 |
| 5 | 30 | K2CO3 | PhMe, H2O (5:1) | 19 |
| 6c | 30 | K3PO4 + Ag(OTf) | PhMe, H2O (5:1) | 46 |
| 7d | 30 | K3PO4 + Ag2CO3 | PhMe, H2O (5:1) | 60 |
| 8 | 30 | Ag2CO3 | PhMe, H2O (5:1) | 33 |
| 9 | 30 | Ag3PO4 | PhMe, H2O (5:1) | 63 |
| 10 | 29 | Ag3PO4 | PhMe, H2O (5:1) | 64 |
| 11e | 30 | Ag3PO4 | PhMe, H2O (5:1) | 56 |
Uniform conditions unless otherwise noted: Precatalyst 31 (20 mol%), base (2.0 equiv), halide (1 equiv), boronate (1.2 equiv), 23 °C.
Isolated yield (%).
K3PO4 (2.0 equiv), Ag(OTf) (6.0 equiv).
K3PO4 (2.0 equiv), Ag2CO3 (1.0 equiv).
Precatalyst 31 (10 mol%)
Oxidation of bistryptamine 32
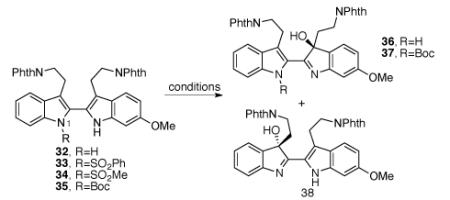
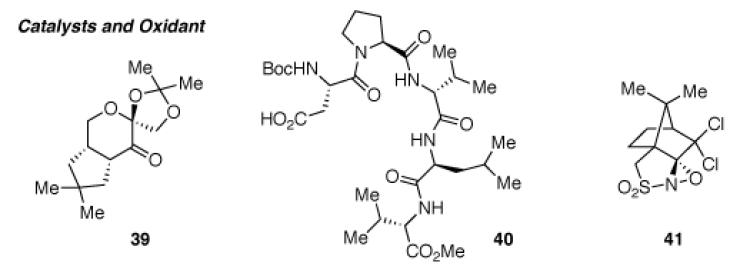
Unlike the oxidation of homodimeric bistryptamine 18 used in our model system (Scheme 2), 32 consists of two indole rings with different electronic properties, raising an interesting question regarding the regioselectivity of the following oxidation event. When bistryptamine 32 was treated with Davis’ oxaziridine 19 at 0 °C, we obtained hydroxyindolenines (±)-36 and (±)-38 (36:38 = 1:1) in 91% combined yield (Table 3, entry 1). Interestingly, exposure of neat bistryptamine 32 to air for 12 d resulted in the formation of hydroxyindolenines (±)-36 and (±)-38 (36:38 = 1.5:1) in 27% combined yield along with 65% of unreacted starting material 32 (Table 3, entry 2). Notably, the presence of regioisomeric pairs is commonly observed in the trigonostemon alkaloids family (Figure 1).37 Hence, we reasoned that the formation of both regioisomers during the oxidation of bistryptamine precursor might explain the biodiversity observed in the trigonostemon alkaloids family. Notably, modification of bistryptamine 32 through an N1-protection with various electron withdrawing groups greatly altered their chemical properties toward oxidation. Phenylsulfonyl, methylsulfonyl, and t-butoxycarbonyl protection of N1 of 32 completely suppressed their oxidation under the conditions previously described using Davis’ oxaziridine 19 (Table 3, entries 3,4, and 6). When substrate 35 was treated with DMDO, the desired hydroxyindolenine could be formed as a single regioisomer, albeit in only 32% yield (Table 3, entry 7). However, substrate 35 was found to be inert under representative asymmetric oxidation conditions developed by Shi38 and Sharpless39 (Table 3, entries 8 and 9).
Table 3.
Oxidation of 2,2′-bistryptamine derivatives.
| Entry | substrate | condition | yield (product) | % ee (product) |
|---|---|---|---|---|
| 1 | 32 | 19, CH2Cl2, 0 °C | 91% (36:38 = 1:1)a | - |
| 2 | 32 | air, neat, 23 °C, 12 d | 27% (36:38 = 1.5:1)a,b | - |
| 3 | 33 | 19, CH2Cl2, 23→50 °C | No reaction | - |
| 4 | 34 | 19, CH2Cl2, 23 °C | No reaction | - |
| 5 | 34 | mCPBA, CH2Cl2, 23 °C, 3 h | decomposition | - |
| 6 | 35 | 19, CH2Cl2, 23→65 °C | No reaction | - |
| 7 | 35 | DMDO, acetone, 23 °C, 11 h | 32% (37) | - |
| 8 | 35 | Shi epoxidationc | No reaction | - |
| 9 | 35 | Sharpless asymmetric dihydroxylationd |
No reaction | - |
| 10 | 32 | Sharpless asymmetric dihydroxylationd |
39% (36:38 = 1.7:1)a | <3 (36), <3 (38) |
| 11 | 32 |
40 (10 mol%),DMAP, H2O2, DIC, CHCl3, 0 °C, 24.5 h |
49% (36:38 = 1.3:1)a | 20 (36), <5 (38) |
| 12 | 32 | 41, CH2Cl2, −30→23 °C | 95% (36:38 = 2.2:1)a | 96 (36), 96 (38) |
|
| ||||
| ||||
The ratio of 36 to 38 was determined by 1H NMR.
65% of unreacted starting material 33 was isolated.
Shi catalyst (39), Na2B2O7•H2O, n-Bu4NHSO4, oxone, K2CO3, EDTA, H2O, DMM, MeCN.
AD-mix- α , methanesulfonamide, t-BuOH, H2O.
When unprotected bistryptamine 32 was oxidized under Sharpless39 or Miller23 asymmetric oxidation conditions, hydroxyindolenines (+)-36 and (+)-38 could be obtained albeit in low enantiomeric excess (Table 3, entries 10 and 11). Gratifyingly, we discovered that (+)-((8,8-dichlorocamphoryl)sulfonyl) oxaziridine 41, originally reported by the Davis research group,40 afforded hydroxyindolenines (+)-36 and (+)-38 (36:38 = 2.2:1) in 95% yield and with an outstanding level of enantioselection for both regioisomers (96% ee, Table 3, entry 12).8 We envisioned that hydroxyindolenine (+)-36 would serve as a synthetic precursor for trigonoliimines A (1) and C (3) and hydroxyindolenine (+)-38 as a precursor for trigonoliimine B (2) and isotrigonoliimine C (4).
Total synthesis of (−)-trigonoliimines A and B
We next aimed to synthesize (−)-trigonoliimines A (1) and B(2) from hydroxyindolenines (+)-36 and (+)-38. Consistent with our model studies (Scheme 3), hydrazinolysis of hydroxyindolenines (+)-36 and (+)-38 afforded the cis-fused aminals (+)-7 and (+)-8 (7:8 = 2.2:1) in 99% combined yield through intramolecular cyclizations (Scheme 5). Aminals (+)-7 and (+)-8 were separable at this stage to enabled their chemical examination independently. However, during scale up they were easily processed together and separated at a more practical stage in the synthesis (vide infra, Scheme 7).
Scheme 5.
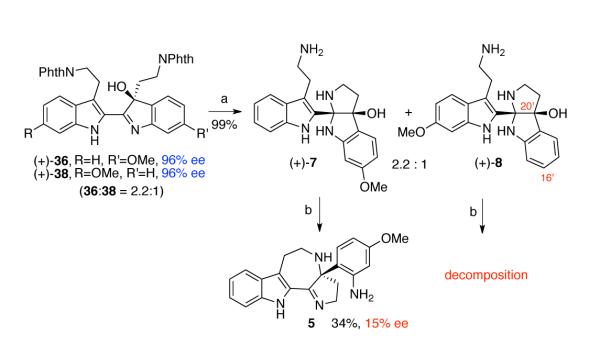
Formation and attempted stereoselective rearrangement of hydroxyaminals (+)-7 and (+)-8. Conditions: (a) NH2NH2•H2O, MeOH, 80 °C, 99%; (b) TFE, 105 °C.
Scheme 7.

Stereoselective formation of azepanes (−)-5 and (−)-6.
When aminal (+)-7 (96% ee) was heated at 105 °C in trifluoroethanol (TFE), rearranged azepane 5 was obtained in 34% yield, albeit with significantly eroded enantiomeric excess (15% ee). On the other hand, when aminal (+)-8 (96% ee) was subjected to identical reaction conditions, only decomposition of the starting material was observed. In the latter case, we reasoned that the absence of the electron donating methoxy group at C16′ results in less efficient ionization of hydroxyl group at C20′. While the NMR analysis of aminals (+)-7 and (+)-8 in deuterated chloroform was consistent with cis-fused pentacyclic structures, analysis of the same synthetic sample in deuterated methanol showed the presence of multiple species, consistent with formation of aminal and imine isomers (Scheme 6). We rationalized that the erosion of ee was due to a low level of diastereoselection during the N12–C24 bond formation followed by reversible ionization of C20 in the form of intermediate 44. Alternatively, the C20 dehydroxylation of intermediate 42, or the corresponding solvent adduct, would result in overall N12–C20 bond formation with low level of stereochemical control (Scheme 6).
Scheme 6.
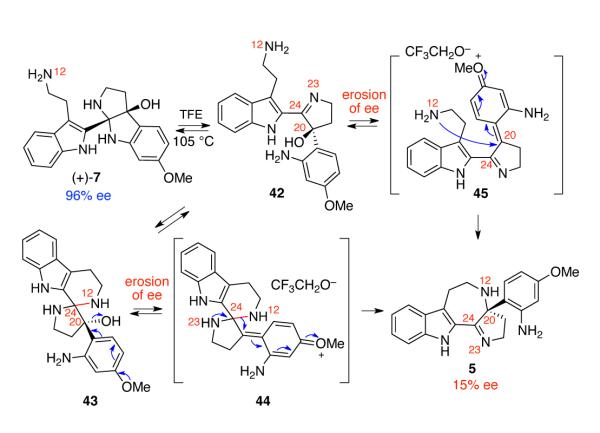
Plausible mechanism for the erosion of enantiomeric excess during the formation of 5.
Based on these observations we reasoned that the activation of the hydroxyl groups of aminals (+)-7 and (+)-8 under mild conditions in an aprotic solvent would be ideal in order for this transformation to proceed with minimal erosion of enantiomeric excess. Hence, we treated a solution of aminals (+)-7 and (+)-8 (7:8 = 2.2:1) in dichloromethane with Martin sulfurane41 at −78 °C, which provided the desired azepanes (−)-5 and (−)-6 in 47% combined yield (28% and 19% yield, respectively, Scheme 7). Azepanes (−)-5 and (−)-6 could be readily separated using flash column chromatography at this stage. Importantly, azepanes (−)-5 and (−)-6 were obtained with minimal erosion of enantiomeric excess (94% ee and 95% ee, respectively). A plausible mechanism for this transformation involves an activation of hydroxyl moiety of the cis-fused aminals (+)-7 and (+)-8 upon treatment with Martin sulfurane to give iminium ions 48 and 49. The fused bicyclic structure of intermediates 48 and 49 allows N12 to attack only from the α-face, resulting in a stereoretentive cyclization. The expulsion of the aniline nitrogen from aminal intermediates 50 and 51 would result in the formation of azepanes (−)-5 and (−)-6. X-ray crystal structure analysis of pentacycle (−)-6 (Scheme 7), the direct precursor of (−)-trigonoliimine B (2), unambiguously confirmed the molecular structure and assigned the S-configuration at C20, corroborating the stereoretentive mode of N12–C20 cyclization.
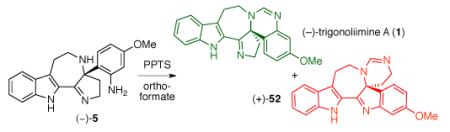
For the completion of the total synthesis of (−)-trigonoliimine A (1), we initially treated the azepane (−)-5 with a catalytic amount of pyridinium p-toluenesulfonate (PPTS) in trimethylorthoformate at 50 °C (Table 4, entry 1).42 Under these conditions, we were able to obtain the first synthetic sample of (−)-trigonoliimine A (1), albeit in low yield (16%). A possible pathway for the formation of (−)-trigonoliimine A (1) (Path A) and byproduct (+)-52 (Path B) from azepane (−)-5 is shown in Scheme 8. Conversion of (−)-5 to (−)-1 involves imidate formation at N13 followed by cyclization with N12.43 Interestingly, the major product formed under these conditions was hexacyclic imine 52 (61%, Scheme 8). Initial reaction of N23 of azepane (−)-5 with the oxocarbenium ion generated from trimethylorthoformate may form the iminium ion 56 (Path B), and the resulting electrophilic C24 would be trapped with N13 to form intermediate 57. The N23 released from the aminal moiety of 57 will give the pentacyclic intermediate 58. Intramolecular cyclization resulting from the N12 attack to the imidate moiety of 58 followed by methanol elimination would yield the isomeric hexacyclic byproduct (+)-52.
Table 4.
Completion of the total synthesis of (−)-trigonoliimine A (1).
| Entry | PPTS equiv |
orthoformate + solvent | temp (°C) |
1 (%) | 52 (%) |
|---|---|---|---|---|---|
| 1 | 0.1 | CH(OMe)3 (solvent) | 50 | 16 | 61 |
| 2 | 2 | CH(Oi-Pr)3, CH2Cl2 (1:1) | 45 | 49 | 37 |
| 3 | 3 | CH(Oi-Pr)3 (10 equiv), CH2Cl2 | 23 | 82 | 15 |
Scheme 8.

Plausible mechanism for the formation of (−)-trigonoliimine A (1) and byproduct (+)-52.
In order to suppress the undesired reactivity at N23 of (−)-5, we examined its pretreatment with PPTS (2 equiv) followed by exposure to triisopropylorthoformate, a more sterically crowded reagent. As expected, the desired (−)-trigonoliimine A (1) was obtained as a major product (49%, Table 4, entry 2). After further optimization, we found that pre-treatment of azepane (−)-5 with PPTS (3 equiv) in dichloromethane at 23 °C, followed by addition of triisopropylorthoformate (10 equiv) afforded the desired (−)-trigonoliimine A (1) in 82% yield {[α]D24= −294 (c 0.24, CHCl3)} (Table 4, entry 3). Under identical reaction conditions, azepane (−)-6 was converted to (−)-trigonoliimine B (2) in 94% yield {[α]D24= −352 (c 0.32, CHCl3), Scheme 9}. All 1H and 13C NMR data for our synthetic (−)-trigonoliimines A (1) and B (2) matched those provided in the isolation report,7 confirming the molecular structure of these alkaloids.
Scheme 9.
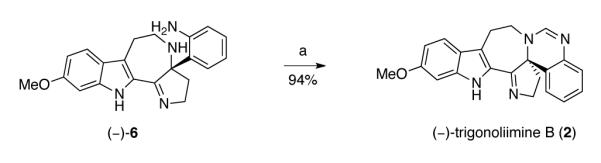
Total synthesis of (−)-trigonoliimine B (2). Conditions: (a) CH(OiPr)3, PPTS, CH2Cl2, 23 °C.
Total synthesis of (−)-trigonoliimine C and (−)-isotrigonoliimine C
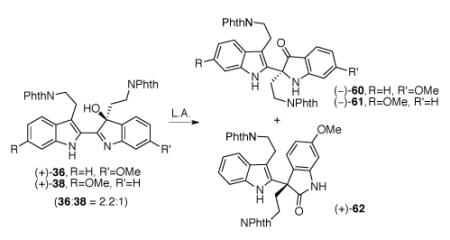
We also wishes to gain access to (−)-trigonoliimine C (3) and (−)-isotrigonoliimine C (4) from the same versatile hydroxyindolenines (+)-36 and (+)-38 described above via a divergent synthetic path employing a Wagner–Meerwein type 1,2-alkyl rearrangement.25 Literature precedent and prior synthetic studies from our laboratory suggested that hydroxyindolenines derived from the oxidation of 2-aryl-3-alkyl-disubstituted indoles can rearrange to the corresponding indoxyls, oxindoles, or mixtures of both depending on the reaction conditions.19,44 Hence, we turned our attention to finding conditions that would favor the selective formation of indoxyls (−)-60 and (−)-61 over the oxindole byproducts (Table 5). Initially, we examined various lanthanide trifluoromethanesulfonates as additives for the rearrangement of hydroxyindolenines (+)-36 and (+)-38 (36:38 = 2.2:1, Table 5). In general, we were able to obtain indoxyls (−)-60 and (−)-61 in moderate yield (Table 5, entries 3–9, entries 11–16, entries 18–20). The use of methanol as solvent did not provide the desired indoxyl products in the presence of europium trifluoromethanesulfonate at 65 °C (Table 5, entry 10). We observed that the choice of solvent had a significant effect in products ratio. When acetonitrile, acetonitrile–toluene mixture, or tetrahydrofuran was used as solvent indoxyl (−)-60 was obtained as a major product. On the other hand, when toluene or 1,2-dichloroethane was used as solvent indoxyl (−)-61 was formed in slight excess (Table 5, entries 11, 13–16). The major byproduct observed in these reactions was oxindole (+)-62 (34% for Table 5, entry 16). Oxindole byproduct (+)-62 did not show any reactivity upon treatment with three equivalents of lanthanum trifluoromethanesulfonate in 1,2-dichloroethane at 65 °C. Similarly, when the indoxyl (−)-60 was treated with three equivalents of lanthanum trifluoromethanesulfonate in 1,2-dichloroethane at 65 °C, no reactivity was observed.
Table 5.
Rearrangement of hydroxyindolenines (+)-36 and (+)-38 in the presence of various Lewis acids.
| Entry | L.A. (equiv.) | solvent | temp (°C) |
time (h) |
% yield of indoxyls (60:61)a |
|---|---|---|---|---|---|
| 1 | Sc(OTf)3 (3) | MeCN | 65 | 45 | 12 (60 only) |
| 2 | Dy(OTf)3 (3) | MeCN | 65 | 45 | 35 (1.7:1) |
| 3 | La(OTf)3 (3) | MeCN | 65 | 39 | 50 (1.6:1) |
| 4 | Pr(OTf)3 (3) | MeCN | 65 | 14 | 42 (2:1) |
| 5 | Ho(OTf)3 (3) | MeCN | 65 | 39 | 44 (2:1) |
| 6 | Eu(OTf)3 (3) | MeCN | 65 | 14 | 50 (1.3:1) |
| 7 | Nd(OTf)3 (3) | MeCN | 65 | 14 | 41 (1.4:1) |
| 8 | Gd(OTf)3 (3) | MeCN | 65 | 14 | 45 (1.4:1) |
| 9 | Eu(OTf)3 (3) | PhMe, MeCN (1:1) | 65 | 24 | 46 (1.3:1) |
| 10 | Eu(OTF)3 (3) | MeOH | 65 | 45 | no reaction |
| 11 | La(OTf)3 (3) | PhMe | 90 | 14 | 40 (1:1.2) |
| 12 | La(OTf)3 (3) | THF | 65 | 55 | 49 (1.3:1) |
| 13 | La(OTf)3 (3) | ClCH2CH2Cl | 65 | 19 | 49 (1:1.2) |
| 14 | La(OTf)3 (1) | ClCH2CH2Cl | 65 | 19 | 57 (1:1.1) |
| 15 | La(OTf)3 (0.5) | ClCH2CH2Cl | 65 | 15 | 52 (1:1.1) |
| 16 | La(OTf)3 (0.5) | PhMe | 80 | 19 | 56 (1:1.2) |
| 17 | La(OTf)3 (0.5) | ClCH2CH2Cl | 23 | 72 | no reaction |
| 18 | Eu(OTf)3 (0.5) | MeCN | 65 | 18 | 42 (1.5:1) |
| 19 | La(OTf)3 (0.5) | MeCN | 65 | 42 | 52 (1.3:1) |
| 20 | Eu(OTf)3 (0.5) | MeCN | 72 | 38 | 48 (1.2:1) |
The ratio of 60 to 61 was determined by 1H NMR.
Due to the non-trivial separation of regioisomeric hydroxyindolenines (+)-36 and (+)-38 by flash column chromatography, prep-HPLC enabled us to access pure samples of both hydroxyindolenines (+)-36 and (+)-38 for their separate characterization and evaluation of chemical properties. When hydroxyindolenine (+)-36 was treated with three equivalents of europium trifluoromethanesulfonate in acetonitrile at 72 °C, indoxyls (−)-60 and (−)-61 (48%, 60:61 = 4.5:1) and oxindole (+)-62 (50%) were obtained (Scheme 10).45 On the other hand, upon treatment with three equivalents of europium trifluoromethanesulfonate in acetonitrile at 72 °C, hydroxyindolenine (+)-38 afforded indoxyl (−)-61 as the sole product in 62% yield (Scheme 10).
Scheme 10.
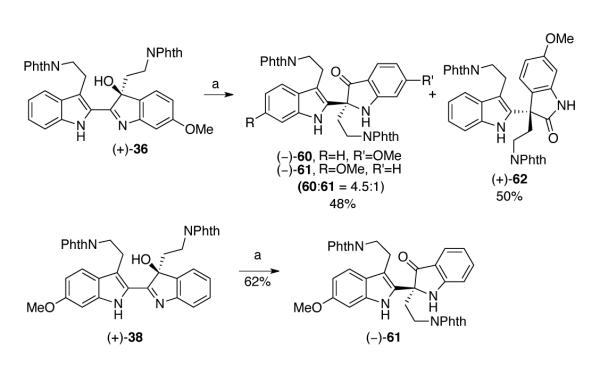
Eu(OTf)3 mediated rearrangement of (+)-hydroxyindolenines (+)-36 and (+)-38. Conditions: (a) Eu(OTf)3, MeCN, 72 °C.
While most of the reaction conditions that utilized lanthanide Lewis acids produced oxindoles as byproducts during the rearrangement of hydroxyindolenines (+)-36 and (+)-38, promising results were obtained during our study directed toward the total synthesis of (−)-trigonoliimine A (1). Inspired by the transformation of aminal (+)-7 to azepane 5 upon heating in TFE (Scheme 5), we attempted to directly access trigonoliimine A (1) by exposing formylated hydroxyindolenine 63 under the same reaction conditions. Interestingly, when the formylated hydroxyindolenine 63 was heated in TFE at 105 °C, indoxyl 64 was selectively formed in 59% yield (Scheme 11).
Scheme 11.
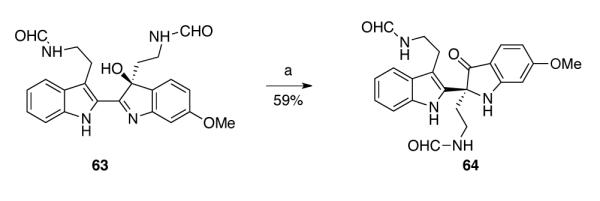
Selective rearrangement of hydroxyindolenine 63 to indoxyl 64. Conditions: (a) TFE, 105 °C.
Based on these observations, we heated hydroxyindolenines (+)-36 and (+)-38 (36:38 = 2.2:1) in TFE at 102 °C and obtained indoxyls (−)-60 and (−)-61 (60:61 = 2.2:1) selectively in 93% yield without any formation of the undesired oxindole (Scheme 12). X-ray crystal structure analysis of the enantiomerically enriched (96% ee) C18-bromide derivative of (−)-60 enabled us to unequivocally assign the S-configuration at C14.8
Scheme 12.
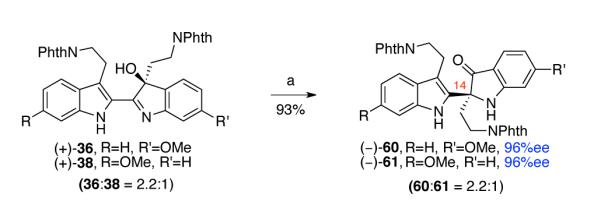
Selective 1,2-alkyl shift of hydroxyindolenines (+)-36 and (+)-38. Conditions: (a) TFE, 102 °C.
The synthetic utility of asymmetric oxidation followed by stereospecific rearrangement of 2-aryl-3-alkyl-indole derivatives holds great promise for other applications as illustrated in Scheme 13. The 1,2-alkyl shift of hydroxyindolenine (+)-65, obtained through Miller’s asymmetric oxidation of the corresponding 2-aryl tryptamine,23 gave indoxyl (+)-66, which constitutes the core structure of hinckdentine A (67).46 Similarly, when tryptophol derivative 68 was subjected to Miller’s asymmetric oxidation23 followed by base mediated 1,2-alkyl shift sequence, indoxyl (−)-70 was obtained in 79% ee, providing a formal synthesis of (−)-hinckdentine (67, Scheme 13).42 Notably, the indoxyl 70 in racemic form has served as a key intermediate in Kawasaki’s total synthesis of (±)-hinckdentine (67).42
Scheme 13.
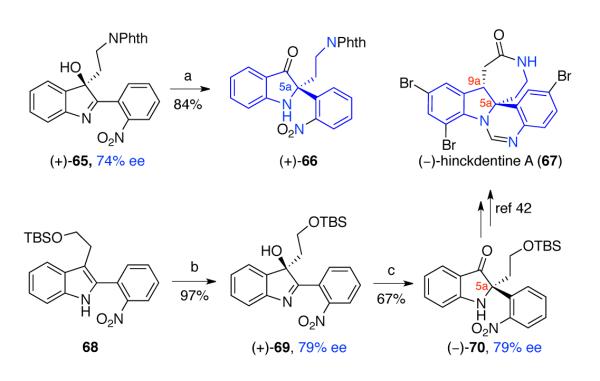
Further synthetic application of asymmetric oxidation and 1,2-alkyl shift of 2,3-disubstituted indole derivatives. Conditions: (a) HFIP, HCOOH (10:1), 90 °C; (b) 40, DMAP, H2O2, DIC, CHCl3, 0 °C; (c) t-BuOK, t-BuOH, 23 °C.
With robust access to indoxyls (−)-60 and (−)-61 secured, we next investigated their hydrazinolysis. When indoxyl (−)-60 was treated with ten equivalents of hydrazine hydrate in methanol at 80 °C, indoxyl 71 was obtained in 81% yield (Scheme 14). However, when indoxyl (−)-61 was exposed to the same reaction conditions, lower and variable yields (8–45%) of the indoxyl 72 were obtained (Scheme 14). We reasoned that the absence of the methoxy group at C19 of indoxyl 72 rendered the carbonyl group more electrophilic, which resulted in the formation of polar hydrazone byproducts.
Scheme 14.
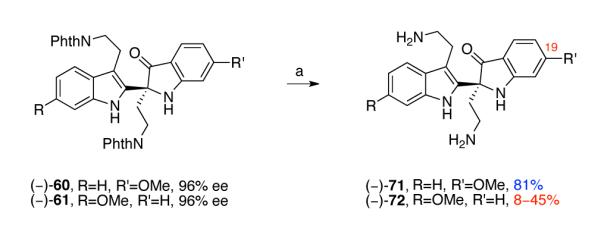
Hydrazinolysis of indoxyls (−)-60 and (−)-61. Conditions: (a) NH2NH2•H2O, MeOH, 90 °C.
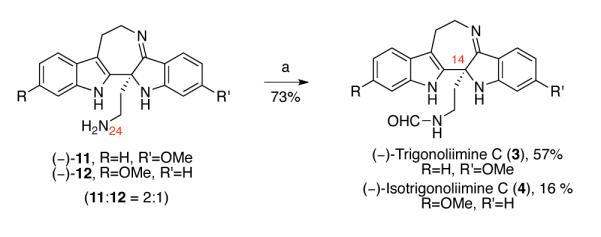
Hence, in order to achieve higher efficiency and practicality, we envisioned performing the hydrazynolysis of indoxyls (−)-60 and (−)-61 and the condensation of the resulting intermediates in a one-pot, two-step procedure. We predicted that indoxyls (−)-71, (−)-72, and hydrazone byproducts formed during the hydrazinolysis of indoxyls (−)-60 and (−)-61 would be viable substrates for the subsequent titanium ethoxide-mediated condensation reaction. Indeed, treatment of indoxyls (−)-60 and (−)-61 (60:61 = 2.2:1) with ten equivalents of hydrazine hydrate in methanol at 80 °C, followed by concentration of the reaction mixture, and addition of the tetrahydrofuran solution of titanium ethoxide and heating at 42 °C afforded the desired pentacyclic imines (−)-11 and (−)-12 (11:12 = 2:1) in 61% yield over two steps (Scheme 15). Consistent with our earlier observations in the model system (Scheme 3) and the molecular structure of the natural product trigonoliimine C (3), we did not observe the formation of the 5-membered imine resulting from the condensative cyclization of N24 onto the ketone moiety (Figure 2). Notably, DFT calculation (B3LYP, 6.31G*) of the equilibrium geometries and energy of 7-membered imine 11 and 5-membered imine 73 predicted that imine 11 is 9.4 kcal/mol lower in energy (Figure 2).47
Scheme 15.
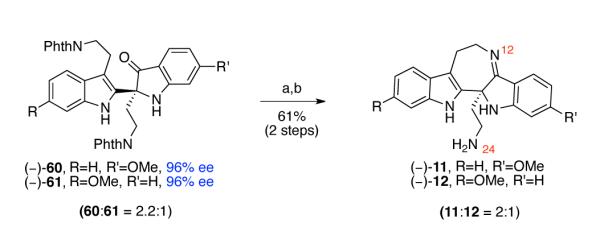
One-pot, two-step synthesis of imines (−)-11 and (−)-12. Conditions: (a) NH2NH2•H2O, MeOH, 80 °C; (b) Ti(OEt)4, THF, 42 °C.
Figure 2.
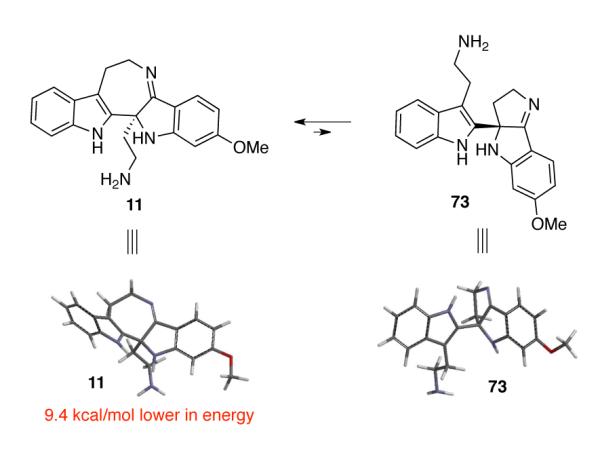
Calculated equilibrium geometries of 7-membered imine 11 and 5-membered imine 73.
Consistent with the model studies (Scheme 3), treatment of imines (−)-11 and (−)-12 (11:12 = 2:1) with freshly prepared N-formyl imidazole afforded (−)-trigonoliimine C (3) {[α]D24= −147 (c 0.12, CHCl3)} and (−)-isotrigonoliimine C (4) ([α]D24 = −220 (c 0.10, CHCl3)} in 57% and 16% yield, respectively. All 1H and 13C NMR data of our synthetic (−)-trigonoliimines C (3) matched those provided in the isolation report.1 While regioisomeric intermediates mentioned above could be separated earlier in the synthetic sequence for independent examination,8 the ease of separation of (−)-trigonoliimine C (3) and (−)-isotrigonoliimine C (4) by silica gel column chromatography allowed a practical processing of the earlier intermediates as mixtures until the final stage of the synthesis.48
The sign and magnitude of specific rotation of our synthetic trigonoliimines in conjunction with our X-ray crystal structure data provided valuable information regarding the stereochemistry of these alkaloids. Firstly, our synthetic trigonoliimines A–C (1–3) derived from hydroxyindolenines (+)-36 and (+)-38 exhibited negative signs in their specific rotation (Table 6). However, naturally occurring trigonoliimines A (1) and B (2) were reported to have a positive sign in their specific rotations whereas trigonoliimine C (3) was reported to have a negative sign in its specific rotation. These observations, in conjunction with our X-ray crystal structures, which unambiguously corroborated the absolute stereochemistry of our synthetic (−)-trigonoliimines A–C (1–3), enabled the revision of the absolute stereochemistry of all trigonoliimines (Figure 1). Additionally, all of our synthetic (−)-trigonoliimines A–C (1–3) showed a significantly larger magnitude of specific rotations compared to those reported for the naturally isolated samples (Table 6). Importantly, the enantiomeric excess of our samples has been quantified by HPLC analysis of enantiomerically enriched samples of several intermediates against readily available racemic samples from our exploratory studies in this program. A potential explanation for the smaller magnitude of the specific rotations of the naturally occurring trigonoliimines is their possible natural formation via partial resolution after a non-selective oxidation of a precursor such as bistryptamine 15 (Scheme 1). Our observations regarding the propensity of bistryptamine 32 to undergo air oxidation (Table 3, entry 2) lends further credence to this hypothesis.8,49
Table 6.
Specific rotation values of natural and synthetic trigonoliimines A–C (1–3).
| Entry | Alkaloids | Natural ([α]10D) | Synthetic ([α]24D)a |
|---|---|---|---|
| 1 | Trigonoliimine A | +13.3 (c 0.3, CHCl3) | −294 (c 0.24, CHCl3, 94% ee) |
| 2 | Trigonoliimine B | +5.0 (c 0.5, CHCl3) | −352 (c 0.32, CHCl3, 95% ee) |
| 3 | Trigonoliimine C | −4.8 (c 0.45, CHCl3) | −147 (c 0.12, CHCl3, 96% ee) |
% ee of the key precursor for the corresponding synthetic trigonoliimine.
Testing of all known Trigonoliimines and Structurally Related Alkaloids for their Anticancer Activity
Despite the numerous synthetic efforts targeting the trigonoliimine class of natural products, only one study has investigated their biological activity.7 This report identified modest anti-HIV activity for trigonoliimine A (0.95 μg/mL, 2.7 μM). Inspired by the potent anticancer activities of indolocarbazole glycopeptides with 2,2′-connectivity,2 we set out to test the anticancer activities of trigonoliimines A–C and various synthetic intermediates generated from this synthetic program.
Our synthetic work has enabled a study of the anti-cancer activities of all known trigonoliimines as well as numerous advanced intermediates, many of which share similar cores to the natural products. Thirteen compounds were identified as preliminary hits for their ability to induce cell death and were carried on for further testing in two cancer cell lines (U-937, human lymphoma, and HeLa, human cervical cancer). The 72-hour IC50 values in both U-937 and HeLa cells for the lead compounds is shown in Table 7. In general, most of the compounds, including trigonoliimines A–C, showed weak activity (10–50 μM) in both cell lines.
Table 7.
Anticancer activity of all known trigonoliimines and structurally related alkaloids.a
| Entry | Compound | HeLa (μM) |
U-937 (μM) |
|---|---|---|---|
| 1 | (−)-1 | >50 | 44 ± 9 |
| 2 | (±)-1 | >50 | 21.3 ± 4.5 |
| 3 | (−)-2 | >50 | 34.6 ± 7.1 |
| 4 | (−)-3 | >50 | 20.3 ± 3.1 |
| 5 | (±)-3 | >100 | 29.6 ± 9.5 |
| 6 | (−)-4 | >50 | >35 |
| 7 | (−)-11•CD3CO2D | 29.7 ± 3.1 | 10.9 ± 0.2 |
| 8 | (−)-12•CD3CO2D | >100 | 38.6 ± 9.5 |
| 9 | (±)-23 | >50 | 19.2 ± 4.4 |
| 10 | (±)-24 | >30 | 10.3 ± 3.0 |
| 11 | (±)-24•CF3CO2D | 8.6 ± 1.8 | 0.73 ± 0.05 |
| 12 | (+)-52 | >100 | 35.6 ± 9.2 |
| 13 | 68 | 3.6 ± 1.1 | 7.9 ± 0.5 |
| 14 | (±)-69 | 13.6 ± 6.3 | 22.8 ± 2.2 |
| 15 | (+)-69b | 35.8 ± 8.6 | >50 |
72-hour IC50 values in HeLa (human cervical cancer) or U-937 (human lymphoma) cell lines as monitored by SRB (HeLa) or MTS (U-937). Error is standard deviation of the mean, n=3. SRB: sulforhodamine B; MTS: 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium.
43% ee.
TFA-d1 salt of amine (±)-24, a precursor to trigonoliimine C (3), showed excellent activity in U-937 cells (730 nM). However, when comparing it to free amine of (±)-24 and acetic acid-d4 salt of (−)-11 and (−)-12, which are all structurally related compounds, the TFA-d1 salt of (±)-24 was much more active than would be predicted from their subtle structural differences. As TFA is not able to induce cancer cell death on its own, this suggests that the activity of the TFA salt of amine (±)-24 arises from a specific interaction resulting from the TFA salt. This is consistent with previous reports that TFA and TFA salts affect cell proliferation and protein synthesis in cells.50 Tryptophol derivative 68 was reasonably active against both U-937 and HeLa cells. However, 68 contains a nitroaromatic group which is known to behave promiscuously in cells.51 Hence, further biological studies would be necessary to unequivocally address anti-cancer activities of these compounds.
Conclusion
We have described the development of our synthetic strategy for the enantioselective total synthesis of all known (−)-trigonoliimines A (1), B (2), and C (3). Rapid access to the key hydroxyindolenines (+)-36 and (+)-38 was enabled by a Suzuki–Miyaura cross-coupling reaction using Buchwald’s precatalyst 31 in conjunction with silver phosphate at ambient temperature, followed by a highly enantioselective oxidation at the enantiodetermining branching point of our syntheses. Stereoretentive and condensative cyclization of cis-fused aminals (+)-7 and (+)-8, obtained from the phthalimide deprotection of (+)-36 and (+)-38, gave azepanes (−)-5 and (−)-6 as the direct precursors of (−)-trigonoliimines A (1) and B (2). Hence, the total synthesis of (−)-trigonoliimines A (1) and B (2) were achieved in seven steps from commercially available material. Our succinct enantioselective syntheses of (−)-trigonoliimines C (3) and (−)-isotrigonoliimine C (4) are each eight steps from commercially available material and draw on a selective Wagner–Meerwein type 1,2-alkyl shift of hydroxyindolenines (+)-36 and (+)-38, which gave indoxyls (−)-60 and (−)-61.
Our enantioselective approach to these natural products allowed the revision of the absolute stereochemistry of alkaloids (−)-1–3 and raised interesting questions related to the apparent low enantiomeric excess of the naturally occurring trigonoliimines. Furthermore, our synthetic efforts have enabled the testing of these alkaloids and several advanced intermediates in two cancer cell lines, revealing mild activity for certain members of this class. The concise total synthesis of (−)-trigonoliimines A–C (1–3) highlights the power of retrobiosynthetic analysis52 as a source of inspiration in antithetic simplification of natural product targets.
Experimental Section
Experimental procedures and full characterization data for previously reported key compounds
For complete experimental procedures and full characterization data of all (−)-trigonoliimine alkaloids A–C (1–3, respectively), please see the supporting information of our previous report of the total synthesis of all (−)-trigonoliimines.8 For complete experimental procedures and full characterization data of the key compounds (−)-4, (−)-5, (−)-6, (+)-7, (+)-8, (−)-11, (−)-12, 26, 27, 30, 32, (+)-36, (+)-38, (−)-60 and (−)-61 discussed in our fully optimized route, please see the supporting information of our previous report of the total synthesis of all (−)-trigonoliimines.8
(±)-2,2′-((3′-Hydroxy-1H,3′H-[2,2′-biindole]-3,3′-diyl)bis(ethane-2,1-diyl))bis(isoindoline-1,3-dione) (20)
A solution of Davis’ oxaziridine 19 (145 mg, 0.606 mmol, 2.2 equiv) in anhydrous dichloromethane (2.5 mL) at 0 °C was transferred by cannula to a solution of indoline derivative 17 (164 mg, 0.276 mmol, 1 equiv) in anhydrous dichloromethane (9 mL) at 0 °C under an atmosphere of argon. The reaction mixture was allowed to gently warm to 23 °C. After 11 h, the reaction mixture was concentrated under reduced pressure and the residue was purified by flash column chromatography (silica gel: diam. 3 cm, ht. 13 cm; eluent: 4% acetone in dichloromethane) to afford hydroxyindolenine (±)-20 (121 mg, 73.6%) as a yellow solid. 1H NMR (500.4 MHz, CDCl3, 21 °C): δ 9.35 (s, 1H), 7.71 (dd, J = 5.5, 3.0 Hz, 2H), 7.61 (dd, J = 5.5, 3.0 Hz, 2H), 7.56 (dd, J = 5.6, 2.9 Hz, 2H), 7.51 (dd, J = 5.4, 3.2 Hz, 2H), 7.46 (d, J = 2.9 Hz, 1H), 7.44 (d, J = 4.0 Hz, 1H), 7.28 (d, J = 7.5 Hz, 1H), 7.11 (dt, J = 7.5, 1.2 Hz, 1H), 7.09 (d, J = 6.7 Hz, 1H), 7.02 (app-t, J = 7.5 Hz, 2H), 6.85 (ddd, J = 7.9, 7.0, 0.9 Hz, 1H), 4.08–4.02 (m, 2H), 3.64–3.58 (m, 2H), 3.54–3.48 (m, 1H), 3.46–3.39 (m, 2H), 2.68 (app-quintet, J = 7.1 Hz, 1H), 2.30 (app-quintet, J = 6.8 Hz, 1H). 1H NMR (500.4 MHz, DMSO-d6, 21 °C): δ 10.93 (s, 1H), 7.75–7.67 (m, 6H), 7.62 (dd, J = 5.5, 3.0 Hz, 2H), 7.60 (d, J = 8.3 Hz, 1H), 7.50 (d, J = 7.6 Hz, 2H), 7.44 (d, J = 7.6 Hz, 1H), 7.32 (dt, J = 7.6, 1.0 Hz, 1H), 7.18 (dt, J = 7.4, 0.7 Hz, 1H), 7.14 (app-t, J = 8.0 Hz, 1H), 6.92 (app-t, J = 7.8 Hz, 1H), 6.51 (s, 1H), 4.05–3.92 (m, 2H), 3.47 (t, J = 6.8 Hz, 2H), 3.38–3.23 (m, 2H), 2.55–2.49 (m, 1H), 2.33–2.27 (m, 1H). 13C NMR (125.8 MHz, CDCl3, 21 °C): δ 173.2, 168.7, 168.0, 154.2, 138.3, 137.0, 133.8, 133.8, 132.4, 132.0, 130.4, 128.4, 127.8, 126.1, 124.9, 123.2, 123.0, 122.6, 121.4, 120.2, 119.8, 119.2, 112.0, 86.3, 38.6, 35.4, 33.4, 24.4. 13C NMR (100.6 MHz, DMSO-d6, 21 °C): δ 174.1, 167.8, 167.2, 153.9, 139.8, 136.9, 134.0, 134.0, 131.7, 131.4, 129.3, 127.6, 127.6, 125.5, 123.6, 122.8, 122.6, 122.6, 120.4, 119.3, 118.8, 117.0, 113.0, 85.2, 37.9, 35.4, 32.8, 23.8. FTIR (neat) cm−1: 3411 (br-s), 1771 (m), 1708 (s), 1548 (m), 1397 (s), 717 (s). HRMS (ESI, TOF) (m/z): calc’d for C36H26N4O5, [M+H]+: 595.1976, found: 595.1986. TLC (5% acetone in dichloromethane), Rf: 0.4 (CAM, UV).
(±)-2-(2-(2-(3-(2-(1,3-Dioxoisoindolin-2-yl)ethyl)-1H-indol-2-yl)-3-oxoindolin-2-yl)ethyl)isoindoline-1,3-dione (21)
Scandium (III) trifluoromethanesulfonate (11.6 mg, 0.0230 mmol, 1.50 equiv) was added to a solution of hydroxyindolenine (±)-20 (9.1 mg, 15 μmol, 1 equiv) in toluene (1.5 mL) at 23 °C and the resulting reaction mixture was warmed to 85 °C. After 18 h, the reaction mixture was allowed to cool to 23 °C. The reaction mixture was diluted with dichloromethane (8 mL) and saturated aqueous sodium bicarbonate solution (8 mL) was added to the reaction mixture and the layers were separated. The aqueous layer was extracted with dichloromethane (8 mL), and the combined organic layers were dried over anhydrous sodium sulfate, and were concentrated under reduced pressure. The sample of the crude residue was purified by flash column chromatography (silica gel: diam. 1.5 cm, ht. 10 cm; eluent: 1.3% dichoromethane in acetone to 10% dichloromethane in acetone) to afford indoxyl (±)-21 (3.3 mg, 36%) and oxindole (±)-22 (2.9 mg, 32%) as pale yellow solids. 1H NMR (500.4 MHz, CDCl3, 21 °C): δ 9.08 (s, 1H), 7.87 (dd, J = 5.5, 3.0 Hz, 2H), 7.73 (dd, J = 5.5, 3.0 Hz, 2H), 7.61 (dd, J = 5.5, 2.9 Hz, 2H), 7.54–7.47 (m, 5H), 7.22 (app-t, J = 8.9 Hz, 2H), 7.05 (ddd, J = 8.1, 7.0, 1.1 Hz, 1H), 6.97 (ddd, J = 7.9, 7.0, 1.0 Hz, 1H), 6.79 (s, 1H), 6.77 (ddd, J = 7.8, 7.1, 0.8 Hz, 1H), 3.89–3.74 (m, 4H), 3.13 (ddd, J = 10.3, 6.8, 3.1 Hz, 2H), 2.69 (app-quintet, J = 7.4 Hz, 1H), 2.43–2.38 (m, 1H). 13C NMR (125.8 MHz, CDCl3, 21 °C): δ 201.0, 168.8, 168.3, 161.1, 138.5, 135.5, 134.3, 134.0, 132.4, 131.7, 130.5, 128.6, 125.4, 123.6, 123.2, 122.6, 119.8, 119.3, 118.4, 118.2, 113.1, 111.2, 109.1, 67.8, 39.0, 37.0, 33.7, 24.3. FTIR (neat) cm−1: 3381 (br-s), 1770 (s), 1719 (s), 1616 (s), 1397 (s), 1103 (w), 751 (s). HRMS (ESI, TOF) (m/z): calc’d for C36H26N4O5, [M+H]+: 595.1976, found: 595.1987. TLC (5% acetone in dichloromethane), Rf: 0.66 (CAM, UV).
(±)-2-(7,12,12b,13-Tetrahydro-6H-azepino[3,2-b:4,5-b′]diindol-12b-yl)ethanamine (24)
Hydrazine monohydrate (33.0 μL, 0.669 mmol, 15.0 equiv) was added via syringe to a solution of indoxyls (±)-21 (26.5 mg, 0.0446 mmol, 1 equiv) in methanol (5 mL) under an atmosphere of argon at 23 °C, and the reaction flask was equipped with a reflux condenser, and the reaction set-up was sealed under an atmosphere of argon and heated to 80 °C. After 90 min, the pale yellow homogeneous reaction mixture was allowed to cool to 23 °C and the volatiles were removed under reduced pressure to afford a pale yellow solid. A solution of titanium ethoxide (41.5 μL, 0.178 mmol, 4.00 equiv) in anhydrous tetrahydrofuran (5 mL) was added via syringe to the yellow solid under an atmosphere of argon, and the resulting mixture was warmed to 40 °C. After 3 h, the reaction mixture was concentrated under reduced pressure. The crude residue adsorbed onto silica gel (2.5 g) was dry loaded and purified by flash column chromatography (silica gel: diam. 2 cm, ht. 10 cm; eluent: 23% chloroform, 5.1% methanol, 0.6% ammonium hydroxide in dichloromethane) to afford imine (±)-24 (6.9 mg, 49%, 2 steps) as a yellow solid. 1H NMR (500.4 MHz, CD3OD, 21 °C)53: δ 7.83 (ddd, J = 8.3, 1.2, 0.8 Hz, 1H), 7.71 (ddd, J = 8.6, 7.0, 1.2 Hz, 1H), 7.51 (td, J = 7.9, 1.0 Hz, 1H), 7.42 (td, J = 8.2, 0.9 Hz, 1H), 7.21 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H) 7.11 (td, J = 8.6, 0.8 Hz, 1H), 7.08 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H) 6.97 (ddd, J = 8.4, 6.9, 0.8 Hz, 1H) 4.68 (td, J = 7.9, 1.0 Hz, 1H), 4.13 (ddd, J = 14.5, 4.1, 3.1 Hz, 1H) 3.38–3.24 (m, 2H), 3.17–3.14 (m, 1H), 3.01–2.92 (m, 2H), 2.88–2.82 (m, 1H). 13C NMR (125.8 MHz, CD3OD, 21 °C)53: δ 183.2, 162.3, 143.7, 137.5, 129.4, 126.3, 124.6, 124.6, 122.4, 121.1, 119.2, 115.1, 114.5, 112.5, 111.8, 70.0, 44.9, 37.7, 36.4, 24.7. FTIR (neat) cm−1: 3272 (br-m), 1675 (s), 1636 (s), 1202 (s), 1134 (s), 723 (m). HRMS (ESI, TOF) (m/z): calc’d for C20H20N4, [M+H]+: 317.1761, found: 317.1768. TLC (18% methanol, 2% ammonium hydroxide in chloroform), Rf: 0.59 (CAM, UV).
(±)-19-Desmethoxy-trigonoliimine C (25)
Freshly prepared N-formyl imidazole54 solution (0.058 M solution in tetrahydrofuran, 0.40 mL, 23 μmol, 1.1 equiv) was added dropwise via syringe to a solution of amine (±)-24 (6.9 mg, 22 μmol, 1 equiv) in tetrahydrofuran (1 mL) at 23 °C and placed under an argon atmosphere. After 1.5 h, saturated aqueous sodium bicarbonate solution (4.5 mL) was added and the resulting mixture was diluted with dichloromethane (4.5 mL), and the layers were separated. The aqueous layer was extracted with dichloromethane (4.5 mL), and the combined organic layers were dried over anhydrous sodium sulfate, and were concentrated under reduced pressure. The sample of the crude residue was purified by flash column chromatography (silica gel: diam. 2 cm, ht. 10 cm; eluent: 4% methanol, 0.4 % ammonium hydroxide in chloroform) to afford (±)-19-desmethoxyl-trigonoliimine C (25, 5.6 mg, 75%) as a yellow solid. 1H NMR (500.4 MHz, CDCl3, 21 °C): δ 7.94 (s, 1H), 7.59 (ddd, J = 7.8, 1.3, 0.7 Hz, 1H), 7.43 (td, J = 7.9, 1.0 Hz, 1H), 7.33–7.30 (m, 2H), 7.10 (ddd, J = 8.2, 7.0, 1.1 Hz, 1H), 7.00 (ddd, J = 7.9, 7.0, 1.0 Hz, 1H) 6.84 (td, J = 8.1, 0.8 Hz, 1H), 6.77 (ddd, J = 7.9, 7.0, 0.8 Hz, 1H) 4.46 (ddd, J = 14.0, 11.9, 2.5 Hz, 1H) 4.09 (td, J = 12.1, 3.6 Hz, 1H), 3.29–3.23 (m, 2H) 3.16 (td, J = 16.9, 3.2 Hz, 1H) 2.75 (ddd, J = 15.3, 9.0, 5.1 Hz, 1H) 2.40 (ddd, J = 15.0, 9.1, 5.0 Hz, 1H). 13C NMR (125.8 MHz, CDCl3, 21 °C): δ 176.5, 164.0, 157.6, 137.1, 135.5, 132.2, 129.9, 124.7, 124.2, 123.1, 120.2, 120.1, 118.9, 112.5, 111.9, 110.6, 68.0, 48.5, 40.7, 35.3, 24.4. FTIR (neat) cm−1: 3285 (br-m), 2923 (m), 1642 (s), 1611 (m), 1469 (m), 1375 (m), 1317 (m), 1141 (w), 743 (m). HRMS (ESI, TOF) (m/z): calc’d for C21H21N4O [M+H]+: 345.1710, found: 345.1732. TLC (18% methanol, 2% ammonium hydroxide in chloroform), Rf: 0.69 (CAM, UV).
Hexacyclic imine (+)-52
Anhydrous dichloromethane (4 mL) was added via syringe to a flask charged with pentacycle (−)-5 (14.0 mg, 40.4 μmol, 1 equiv) and pyridinium p-toluenesulfonate (PPTS, 31.0 mg, 0.121 mmol, 3.00 equiv) at 23 °C under an atmosphere of argon to form a bright yellow solution. After 2 min, triisopropyl orthoformate (93.0 μL, 0.404 mmol, 10.0 equiv) was added to the reaction mixture. After 1h, saturated aqueous sodium bicarbonate solution (6 mL) was added and the resulting mixture was diluted with dichloromethane (2 mL), and the layers were separated. The aqueous layer was extracted with dichloromethane (2 × 6 mL), the combined organic layers were dried over anhydrous sodium sulfate, and were concentrated under reduced pressure. The crude residue was purified by flash column chromatography (silica gel: diam. 2 cm, ht. 12 cm; eluent: 1.8% methanol, 0.2 % ammonium hydroxide in chloroform to 4.5% methanol, 0.5 % ammonium hydroxide in chloroform) to afford (−)-trigonoliimine A (1, 12 mg, 82%) as a pale yellow solid. Hexacyclic imine (+)-52 (2.2 mg, 15%) was also obtained as a yellow solid byproduct. Structural assignment of (+)-52 utilized additional information from gCOSY, HSQC and HMBC. 1H NMR (500.4 MHz, CD3OD, 21 °C): δ 7.63 (s, 1H, C25′H), 7.58 (d, J = 8.1 Hz, 1H, C7′H), 7.46 (d, J = 8.3 Hz, 1H, C4′H), 7.29 (dt, J = 7.6, 0.8 Hz, 1H, C6′H), 7.24 (d, J = 8.2 Hz, 1H, C18′H), 7.15 (d, J = 2.3 Hz, 1H, C15′H), 7.11 (t, J = 7.5 Hz, 1H, C5′H), 6.86 (dd, J = 8.2, 2.3 Hz, 1H, C17′H), 3.95 (app-dd, J = 13.3, 4.0 Hz, 1H, C11′H), 3.85 (s, 3H, OMe), 3.64–3.53 (m, 2H, C22′H2), 3.21 (app-t, J = 5.3 Hz, 2H, C10′H2), 3.19–3.12 (m, 1H, C11′H2), 1.82 (dt, J = 12.4, 5.1 Hz, 1H, C21′H), 1.36 (dd, J = 12.4, 3.4 Hz, 1H, C21′H), 2.96 (ddd, J = 16.9, 12.1, 4.3 Hz, 1H, C10′H), 2.19–2.13 (m, 1H, C21′H), 2.06 (dd, J = 12.0, 5.8 Hz, 1H, C21′H). 13C NMR (125.8 MHz, CD3OD, 21 °C): δ 177.7 (C24′), 163.1 (C16′), 155.8 (C14′), 151.2 (C25′), 133.3 (C8′), 136.0 (C19′), 129.4 (C9′), 128.3 (C2′), 126.2 (C16′), 124.5 (C18′), 121.7 (C3′), 121.1 (C5′), 120.7 (C7′), 113.1 (C4′), 112.6 (C17′), 108.3 (C15′), 75.6 (C20′), 56.3 (C27′), 51.2 (C11′), 41.1 (C22′), 34.0 (C21′), 30.9 (C ). FTIR (neat) cm−1 10′ : 3057 (br-s), 2929 (br-s), 1632 (s), 1581 (s), 1483 (s), 1334 (s), 1147 (s), 1027 (m), 735 (s). HRMS (ESI, TOF) (m/z): calc’d for C22H21N4O, [M+H]+: 357.1710, found: 357.1717. [α]D24: +151 (c 0.43, MeOH). TLC (9% methanol, 1% ammonium hydroxide in chloroform), Rf: 0.15 (CAM, UV).
(R)-2-(2-(3-(3-(2-(1,3-Dioxoisoindolin-2-yl)ethyl)-1H-indol-2-yl)-6-methoxy-2-oxoindolin-3-yl)ethyl)isoindoline-1,3-dione (62)
Lanthanum (III) trifluoromethanesulfonate (22.7 mg, 38.7 μmol, 0.500 equiv) was added to a solution of hydroxyindolenines (+)-36 and (+)-38 (36:38, 2.2:1, 48.5 mg, 77.6 μmol, 1 equiv) in toluene (7.7 mL) at 23 °C and the resulting reaction mixture was warmed to 80 °C. After 19.5 h, the reaction mixture was allowed to cool to 23 °C, saturated aqueous sodium bicarbonate solution (15 mL) and ethyl acetate (15 mL) were added and the layers were separated. The aqueous layer was extracted with dichloromethane (15 mL). The combined organic layers were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The residue was purified by flash column chromatography (silica gel: diam. 2 cm, ht. 10 cm; eluent: 3.3% acetone in dichloromethane to 10% acetone in dichloromethane) to afford indoxyls (−)-60 and (−)-61 (60:61, 1:1.2, 27 mg, 56%) and oxindole (+)-62 (17 mg, 34%) as a mixture of yellow solids. 1H NMR (500.4 MHz, CDCl3, 21 °C): δ 9.68 (s, 1H), 8.93 (s, 1H), 7.78 (dd, J = 5.3, 3.1 Hz, 2H), 7.70–7.65 (m, 5H), 7.61 (dd, J = 5.5, 3.1 Hz, 2H), 7.45 (d, J = 8.1 Hz, 1H), 7.16–7.05 (m, 3H), 6.60 (d, J = 2.3 Hz, 1H), 6.44 (dd, J = 8.2, 2.3 Hz, 1H), 4.06–4.00 (m, 1H), 3.97–3.91 (m, 1H), 3.69 (s, 3H), 3.64–3.51 (m, 2H), 2.83–2.76 (m, 2H), 2.70–2.64 (m, 1H), 2.57–2.51 (m, 1H), 2.71–2.66 (m, 1H), 2.37–2.32 (m, 1H). 13C NMR (125.8 MHz, CDCl3, 21 °C): δ 179.6, 168.6, 168.3, 160.6, 142.1, 135.4, 134.2, 133.9, 132.3, 132.1, 131.6, 129.2, 125.3, 123.5, 123.5, 123.1, 122.3, 119.8, 118.7, 111.4, 109.8, 108.2, 98.3, 55.6, 51.6, 37.7, 34.7, 33.9, 23.9. FTIR (neat) cm−1: 3335 (br-s), 3020 (m) 2361 (w), 1771 (m), 1709 (s), 1633 (m), 1397 (s), 1032 (w), 718 (s). HRMS (DART, TOF) (m/z): calc’d for C37H29N4O6, [M+Na]+: calculated: 647.1901, found: 647.1914. [α]D24: +93.6 (c 0.24, CHCl3). TLC (14% acetone in dichloromethane), Rf: 0.34 (CAM, UV).
(R)-3-(2-((tert-Butyldimethylsilyl)oxy)ethyl)-2-(2-nitrophenyl)-3H-indol-3-ol (69)
To a glass vial charged with tryptophol derivative 68 (22.6 mg, 57.0 μmol, 1 equiv) and catalyst 40 (3.7 mg, 5.7 μmol, 0.10 equiv) was added DMAP (0.35 mg, 2.9 μmol, 0.050 equiv) in chloroform (200 μL) and the reaction mixture was cooled to 0 °C. After 3 min, hydrogen peroxide (30 % w/w in water, 7.0 μL, 68 μmol, 1.2 equiv) was added to the reaction mixture. After 9 min, additional chloroform (80 μL) was added to the reaction mixture. After 4 min, DIC (10.7 μL, 68.4 μmol, 1.20 equiv) was added to the reaction mixture and the resulting dark orange solution was placed in a cryogenic cooler adjusted to 0 °C. After 22 h, the reaction mixture was diluted with chloroform (0.5 mL) and directly purified by flash column chromatography (silica gel: diam. 2.0 cm, ht. 11 cm; eluent: 13% ethyl acetate in hexanes) to afford hydroxyindolenine (+)-69 (23 mg, 97%) as a colorless oil. Hydroxyindolenine (+)-69 was found to have 89.5:10.5 er by chiral HPLC analysis [Chiralpak IC 0.5 mL/min; 80% hexanes, 20% isopropanol; tR(major) = 12.4 min, tR(minor) = 10.1 min]. 1H NMR (500.4 MHz, CDCl3, 21 °C): δ 8.15 (dd, J = 7.7, 1.4 Hz, 1H), 7.93 (dd, J = 8.1, 1.1 Hz, 1H), 7.66 (dt, J = 7.6, 1.3 Hz, 1H), 7.58 (appt-dt, J = 7.7, 1.5 Hz, 1H), 7.51–7.49 (m, 1H), 7.46–7.45 (m, 1H), 7.35 (dt, J = 7.6, 1.4 Hz, 1H), 7.27 (dt, J = 7.4, 1.1 Hz, 1H), 5.11 (s, 1H), 3.98 (ddd, J = 10.6, 7.9, 4.1 Hz, 1H), 3.77 (ddd, J = 10.7, 6.1, 4.6 Hz, 1H), 2.21 (ddd, J = 12.6, 8.0, 4.6 Hz, 1H), 2.03 (ddd, J = 10.26, 6.1, 4.2 Hz, 1H), 0.88 (s, 9H), 0.04 (s, 3H), 0.04 (s, 3H). 13C NMR (125.8 MHz, CDCl3, 21 °C): δ 177.6, 152.9, 149.4, 140.5, 132.5, 130.9, 130.4, 129.8, 129.6, 127.1, 124.6, 123.0, 122.1, 89.1, 61.1, 37.4, 26.0, 18.2, −5.5, −5.6. FTIR (neat) cm−1: 3406 (s), 2955 (s), 2857 (m), 1649 (w), 1538 (s), 1471 (m), 1361 (m), 1258 (m), 1984 (m), 838 (s). HRMS (DART, TOF) (m/z): calc’d for C22H29N2O4Si, [M+H]+: 413.1891, found: 413.1890. [α]D24 : +136.1 (c 0.49, CHCl3). TLC (25% ethyl acetate in hexanes), Rf: 0.43 (CAM, UV).
(R)-2-(2-((tert-Butyldimethylsilyl)oxy)ethyl)-2-(2-nitrophenyl)indolin-3-one (70)
To a solution of hydroxyindolenine (+)-69 (26.2 mg, 63.5 μmol, 1 equiv) in freshly distilled tert-butanol (1.6 mL) under argon was added a solution of potassium tert-butoxide (3.8 mg, 32 μmol, 0.50 equiv) in tert-butanol (1.6 mL) via syringe at 23 °C. After 16.5 h, saturated aqueous ammonium chloride solution (3 mL) was added and the resulting mixture was diluted with water (4.5 mL) and ethyl acetate (6 mL) and the layers were separated. The aqueous layer was extracted with ethyl acetate (2 × 6 mL), and the combined organic layers were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The sample of the crude residue was purified by flash column chromatography (silica gel: diam. 2.0 cm, ht. 1 cm; eluent: 13% ethylacetate in hexanes) to afford indoxyl (−)-70 (18 mg, 67%) as a yellow oil. Indoxyl (−)-70 was found to have 89.5:10.5 er by chiral HPLC analysis [Chiralpak IC 0.5 mL/min; 80% hexanes, 20% isopropanol; tR(major) = 14.9 min, tR(minor) = 24.8 min]. 1H NMR (500.4 MHz, CDCl3, 21 °C): δ 7.79 (appt-d, J = 7.4 Hz, 1H), 7.61 (td, J = 7.7, 0.6 Hz, 1H), 7.53–7.50 (m, 1H), 7.48 (ddd, J = 8.3, 7.1, 1.3 Hz, 1H), 7.36–7.35 (m, 2H), 6.90 (appt-t, J = 7.8 Hz, 1H), 6.86 (td, J = 8.2, 0.7 Hz, 1H), 6.13 (s, 1H), 3.79–3.72 (m, 2H), 2.53 (ddd, J = 14.9, 6.8, 2.9 Hz, 1H), 2.08 (ddd, J = 14.8, 9.7, 4.9 Hz, 1H), 0.84 (s, 9H), −0.09 (s, 3H), −0.10 (s, 3H). 13C NMR (125.8 MHz, CDCl3, 21 °C): δ 201.5, 160.8, 150.9, 137.7, 131.0, 130.9, 129.7, 128.5, 125.3, 123.9, 120.6, 119.8, 113.7, 70.6, 60.1, 40.2, 26.0, 18.2, −5.7. FTIR (neat) cm−1: 3350 (m), 2954 (m), 2929 (m), 1705 (s), 1617 (s), 1533 (s), 1484 (m), 1369 (m), 1324 (m), 1258 (m), 1096 (s), 836 (s), 778 (s). HRMS (DART, TOF) (m/z): calc’d for C22H29N2O4Si, [M+H]+: 413.1891, found: 413.1903. [α]D24: −79.5 (c 0.15, CHCl3). TLC (17% ethyl acetate in hexanes), Rf: 0.32 (CAM, UV).
Cell Culture Information
U-937 and HeLa cells were grown in RPMI-1640 supplemented with fetal bovine serum (10%, FBS) and antibiotics (100 μg/mL penicillin and 100 U/mL streptomycin). Cells were incubated at 37 °C in a 5% CO2, 95% humidity atmosphere.
IC50 Value Determination for HeLa Cells using Sulforhodamine B (SRB)
HeLa cells were added into 96-well plates (1,500 cells/well) in 100 μL media and were allowed to adhere for 2–3 hours. Compounds were solubilized in DMSO as 100× stocks, added directly to the cells (100 μL final volume), and tested over a range of concentrations (0.25 μM to 100 μM) in triplicate (1% DMSO final) on a third-log scale. DMSO and cell-free wells served as the live and dead control, respectively. After 72 hours of continuous exposure, the plates were evaluated using the SRB colorimetric assay as described previously.55 Briefly, media was removed from the plate, and cells were fixed by the addition of 100 μL cold 10% trichloroacetic acid in water. After incubating at 4 °C for an hour, the plates were washed in water and allowed to dry. Sulforhordamine B was added as a 0.057% solution in 1% acetic acid (100 μL), and the plates were incubated at room temperature for 30 minutes, washed in 1% acetic acid, and allowed to dry. The dye was solubilized by adding 10 mM Tris base solution (pH 10.5, 200 μL) and incubating at room temperature for 30 minutes. Plates were read at λ = 510 nm.
IC50 Value Determination for U-937 Cells using MTS
In a 96-well plate, compounds were pre-added as DMSO stocks (1% final) in triplicate to achieve final concentrations of 0.25 μM to 100 μM on a third-log scale. DMSO and cell-free wells served as the live and dead control, respectively. U-937 (5,000 cells/well) cells were distributed in 100 μL media to the compound-containing plate. After 72 hours, cell viability was assessed by adding 20 μL of a PMS/MTS solution56 to each well, allowing the dye to develop at 37 °C until the live control had processed MTS, and reading the absorbance at λ = 490 nm.
Supplementary Material
Scheme 16.
Total synthesis of (−)-trigonoliimine C (3) and (−)-isotrigonoliimine C (4). Conditions: (a) N-Formylimidazole, THF, 23 °C.
Acknowledgment
We acknowledge financial support by NIH-NIGMS (GM074825) and the University of Illinois. S.H. acknowledges EMD Serono and Kenneth M Gordon summer graduate fellowships. K.C.M. is a National Science Foundation predoctoral fellow and a Robert C. and Carolyn J. Springborn graduate fellow.
Footnotes
Supporting Information Available. General procedures, supplementary discussion, and copy of 1H, 13C NMR spectra and HPLC data. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Footnotes
- 1.Bergman J, Koch E, Pelcman B. Tetrahedron. 1995;51:5631–5642. [Google Scholar]
- 2.For a review on indolocarbazole natural products, see: Sánchez C, Méndez C, Salas JA. Nat. Prod. Rep. 2006;23:1007–1045. doi: 10.1039/b601930g.
- 3.Gallant M, Link JT, Danishefsky SJ. J. Org. Chem. 1993;58:343–349. [Google Scholar]
- 4.Link JT, Raghavan S, Danishefsky SJ. J. Am. Chem. Soc. 1995;117:552–553. [Google Scholar]
- 5 (a).Davis PD, Hallam TJ, Harris W, Hill CH, Lawton G, Nixon JS, Smith JL, Vesey DR, Wilkinson SE. Bioorg. Med. Chem. Lett. 1994;4:1303–1308. [Google Scholar]; (b) Vice SF, Bishop WR, McCombie SW, Dao H, Frank E, Ganguly AK. Bioorg. Med. Chem. Lett. 1994;4:1333–1338. [Google Scholar]; (c) Animati F, Berettoni M, Bigioni M, Binaschi M, Cipollone A, Irrissuto C, Nardelli F, Olivieri L. Bioorg. Med. Chem. Lett. 2012;22:5013–5017. doi: 10.1016/j.bmcl.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 6.Nakazono M, Nanbu S, Uesaki A, Kuwano R, Kashiwabara M, Kaitsu K. Org Lett. 2007;9:3583–3586. doi: 10.1021/ol701431g. [DOI] [PubMed] [Google Scholar]
- 7.Tan C-J, Di Y-T, Wang Y-H, Zhang Y, Si Y-K, Zhang Q, Gao S, Hu X-J, Fang X, Li S-F, Hao X-J. Org. Lett. 2010;12:2370–2373. doi: 10.1021/ol100715x. [DOI] [PubMed] [Google Scholar]
- 8.Han S, Movassaghi M. J. Am. Chem. Soc. 2011;133:10768–10771. doi: 10.1021/ja204597k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qi X, Bao H, Tambar UK. J. Am. Chem. Soc. 2011;133:10050–10053. doi: 10.1021/ja203960b. [DOI] [PubMed] [Google Scholar]
- 10.Liu S, Hao X-J. Tetrahedron Lett. 2011;52:5640–5642. [Google Scholar]
- 11.Feng P, Fan Y, Xue F, Liu W, Li S, Shi Y. Org. Lett. 2011;13:5827–5829. doi: 10.1021/ol202475r. [DOI] [PubMed] [Google Scholar]
- 12.Buyck T, Wang Q, Zhu J. Org. Lett. 2012;14:1338–1341. doi: 10.1021/ol300249y. [DOI] [PubMed] [Google Scholar]
- 13.Qiu J, Zhang J-X, Liu S, Hao X-J. Tetrahedron Lett. 2013;54:300–302. [Google Scholar]
- 14.Gulati KC, Seth SR, Venkataraman K. Org. Synth. 1935;15:70–71. [Google Scholar]
- 15 (a).Zhao B, Hao X-Y, Zhang J-X, Liu S, Hao X-J. Org. Lett. 2013;15:528–530. doi: 10.1021/ol303344d. For another recent total synthesis of (±)-trigonoliimine C, see: Reddy BN, Ramana CV. Chem. Commun. 2013;49:9767–9769. doi: 10.1039/c3cc45512b.
- 16 (a).For elegant examples of the application of oxidation and rearrangement of 2,3-disubstituted indoles in complex synthesis, see: Finch N, Taylor WI. J. Am. Chem. Soc. 1962;84:3871–3877. Hutchison AJ, Kishi Y. J. Am. Chem. Soc. 1979;101:6786–6788. Williams RM, Glinka T, Kwast E, Coffman H, Stille JK. J. Am. Chem. Soc. 1990;112:808–821. Gueller R, Borschberg H. Helv. Chim. Acta. 1993;76:1847–1862. Stoermer D, Heathcock CH. J. Org. Chem. 1993;58:564–568. Cushing TD, Sanz-Cervera JF, Williams RM. J. Am. Chem. Soc. 1996;118:557–579. Ito M, Clark CW, Mortimore M, Goh JB, Martin SF. J. Am. Chem. Soc. 2001;123:8003–8010. doi: 10.1021/ja010935v. Baran PS, Corey EJ. J. Am. Chem. Soc. 2002;124:7904–7905. doi: 10.1021/ja026663t. Liu Y, McWhorter WW., Jr. J. Am. Chem. Soc. 2003;125:4240–4252. doi: 10.1021/ja021380m. Williams RM, Cox RJ. Acc. Chem. Res. 2003;36:127–139. doi: 10.1021/ar020229e. and references cited therein. Liu Y, McWhorter WW, Jr., Hadden CE. Org. Lett. 2003;5:333–335. doi: 10.1021/ol0202417. Baran PS, Richter JM. J. Am. Chem. Soc. 2005;127:15394–15396. doi: 10.1021/ja056171r. Poriel C, Lachia M, Wilson C, Davies JR, Moody CJ. J. Org. Chem. 2007;72:2978–2987. doi: 10.1021/jo062627r. Miller KA, Tsukamoto S, Williams RM. Nature Chem. 2009;1:63–68. doi: 10.1038/nchem.110.
- 17.Schmidt MA, Movassaghi M. Synlett. 2008:313–324. [Google Scholar]
- 18.See supporting information for details.
- 19.Movassaghi M, Schmidt MA, Ashenhurst JA. Org. Lett. 2008;10:4009–4012. doi: 10.1021/ol8015176. [DOI] [PubMed] [Google Scholar]
- 20 (a).Hentges SG, Sharpless KB. J. Am. Chem. Soc. 1980;102:4263–4264. [Google Scholar]; (b) Katsuki TK, Sharpless KB. J. Am. Chem. Soc. 1980;102:5974–5976. [Google Scholar]; (c) Zhang W, Loebach JL, Wilson SR, Jacobsen EN. J. Am Chem. Soc. 1990;112:2801–2803. [Google Scholar]; (d) Irie R, Noda K, Ito Y, Katsuki T. Tetrahedron Lett. 1991;32:1055–1058. [Google Scholar]; (e) Tu Y, Wang ZX, Shi Y. J. Am. Chem. Soc. 1996;118:9806–9807. [Google Scholar]; (f) Peris G, Jakobsche CE, Miller SJ. J. Am. Chem. Soc. 2007;129:8710–8711. doi: 10.1021/ja073055a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.For an asymmetric oxidation of 2,3-disubstituted indole using chiral auxiliary, see: Pettersson M, Knueppel D, Martin SF. Org. Lett. 2007;9:4623–4626. doi: 10.1021/ol702132v.
- 22.For biosynthetic studies of asymmetric oxidation of 2,3-disubstituted indole moiety of prenylated indole natural products, see: Li S, Finefield JM, Sunderhaus JD, McAfoos TJ, Williams RM, Sherman DH. J. Am. Chem. Soc. 2012;134:788–791. doi: 10.1021/ja2093212. and references therein.
- 23.Kolundzic F, Noshi MN, Tjandra M, Movassaghi M, Miller SJ. J. Am. Chem. Soc. 2011;133:9104–9111. doi: 10.1021/ja202706g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.For an application of aspartyl based asymmetric oxidation in total synthesis, see: Sun M, Hao X-Y, Liu S, Hao X-J. Tetrahedron Lett. 2013;54:692–694.
- 25 (a).Wagner G, Brickner W. Ber. 1899;32:2302–2325. [Google Scholar]; (b) Meerwein H, Van Emster K, Joussen J. Ber. 1922;55:2500–2528. [Google Scholar]
- 26.We finished the synthesis of the (±)-19-desmethoxy-trigonoliimine C (33) on June 2010, and focused on the enantioselective total synthesis of all trigonoliimines since then.
- 27 (a).Bergman J, Koch E, Pelcman B. Tetrahedron Lett. 1995;36:3945–3948. [Google Scholar]; (b) Gilbert EJ, Ziller JW, Van Vranken DL. Tetrahedron. 1997;53:16553–16564. [Google Scholar]
- 28.Treatment of hydroxyaminal 23 wih three equiv of Sc(OTf)3 in acetonitrile–toluene (2:1) at 81 °C provided a mixture of indoxyl and oxindole intermediates in good yield (indoxyl:oxindole = 4:1, >99%). However, nontrivial separation of these intermediates allowed only 31% isolated yield of a pure indoxyl intermediate.
- 29.For the synthesis of 6-methoxy tryptamine: see: Woodward RB, Bader FE, Bickel H, Frey AJ, Kierstead RW. Tetrahedron. 1958;2:1–57.
- 30 (a).For examples of C2–C2′ bond formation of two indoles using Stille cross-coupling reaction, see: Palmisano G, Santagostino M. Helv. Chim. Acta. 1993;76:2356–2366. Hudkins RL, Diebold JL, Marsh FD. J. Org. Chem. 1995;60:6218–6220. Danieli B, Lesma G, Martinelli M, Passarella D, Peretto I, Silvani A. Tetrahedron. 1998;54:14081–14088. Cai X, Snieckus V. Org. Lett. 2004;6:2293–2295. doi: 10.1021/ol049780x. Selvi S, Pu S-C, Cheng Y-M, Fang J-M, Chou P-T. J. Org. Chem. 2004;69:6674–6678. doi: 10.1021/jo049192x. La Regina G, Bai R, Maria Rensen W, Di Cesare E, Coluccia A, Piscitelli F, Famiglini V, Reggio A, Nalli M, Pelliccia S, Da Pozzo E, Costa B, Granata I, Porta A, Maresca B, Sorani A, Lisa Iannitto M, Santoni A, Li J, Miranda Cona M, Chen F, Ni Y, Brancale A, Dondio G, Vultaggio S, Varasi M, Mercurio C, Martini C, Hamel E, Lavia P, Novellino E, Silvestri R. J. Med. Chem. 2013;56:123–149. doi: 10.1021/jm3013097.
- 31 (a).For examples of C2–C2’ bond formation of two indoles using Suzuki–Miyaura cross-coupling reaction, see: Merlic CA, McInnes DM. Tetrahedron Lett. 1997;38:7661–7664. Merlic CA, You Y, McInnes DM, Zechman AL, Miller MM, Deng Q. Tetrahedron. 2001;57:5199–5212. Yamabuki A, Fujinawa H, Chosi T, Tohyama S, Matsumoto K, Ohmura K, Nobuhiro J, Hibino S. Tetrahedron Lett. 2006;47:5859–5861. Matsumoto K, Choshi T, Hourai M, Zamami Y, Sasaki K, Abe T, Ishikura M, Hatae N, Iwamura T, Tohyama S, Nobuhiro J. Bioorg. Med. Chem. Lett. 2012;22:4762–4764. doi: 10.1016/j.bmcl.2012.05.064.
- 32.Miyaura N, Suzuki A. J. Chem. Soc. Chem. Commun. 1979:866–867. [Google Scholar]
- 33 (a).Ishiyama T, Takagi J, Ishida K, Miyaura N, Anastasi NR, Hartwig JF. J. Am. Chem. Soc. 2002;124:390–391. doi: 10.1021/ja0173019. [DOI] [PubMed] [Google Scholar]; (b) Ishiyama T, Takagi J, Hartwig JF, Miyaura N. Angew. Chem. Int. Ed. 2002;41:3056–3058. doi: 10.1002/1521-3773(20020816)41:16<3056::AID-ANIE3056>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]; (c) Boller TM, Murphy JM, Hapke M, Ishiyama T, Miyaura N, Harwig JF. J. Am. Chem. Soc. 2005;127:14263–14278. doi: 10.1021/ja053433g. [DOI] [PubMed] [Google Scholar]
- 34.Huang X, Anderson KW, Zim D, Jiang L, Klapars A, Buchwald SL. J. Am. Chem. Soc. 2003;125:6653–6655. doi: 10.1021/ja035483w. [DOI] [PubMed] [Google Scholar]
- 35.Kinzel T, Zhang Y, Buchwald SL. J. Am. Chem. Soc. 2010;132:14073–14075. doi: 10.1021/ja1073799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karabelas Kostas, Hallberg A. J. Org. Chem. 1986;51:5286–5290. [Google Scholar]
- 37.Hu X-J, Di Y-T, Wang Y-H, Kong S-Y, Gao S, Li C-S, Liu H-Y, He H-P, Ding J, Xie H, Hao X-J. Planta Med. 2009;75:1157–1161. doi: 10.1055/s-0029-1185505. [DOI] [PubMed] [Google Scholar]
- 38.Zhu Y, Tu Y, Yu H, Shi Y. Tetrahedron Lett. 1998;39:7819–7822. [Google Scholar]
- 39.Sharpless KB, Amberg W, Bennani YL, Crispino GA, Hartung J, Jeong K-S, Kwong H-L, Morikawa K, Wang Z-M, Xu D, Zhang X-L. J. Org. Chem. 1992;57:2768–2771. [Google Scholar]
- 40 (a).Davis FA, Weismiller MC. J. Org. Chem. 1990;55:3715–3717. [Google Scholar]; (b) Davis FA. J. Org. Chem. 2006;71:8993–9003. doi: 10.1021/jo061027p. [DOI] [PubMed] [Google Scholar]
- 41.Martin JC, Arhart RJ. J. Am. Chem. Soc. 1971;93:4327–4329. [Google Scholar]
- 42.Higuchi K, Sato Y, Tsuchinochi M, Sugiura K, Hatori M, Kawasaki T. Org. Lett. 2009;11:197–199. doi: 10.1021/ol802394n. [DOI] [PubMed] [Google Scholar]
- 43.Alternative reaction mechanism for the formation of (−)-trigonoliimine A (1) involves the initial imidate formation at N12 of (−)-12 followed by condensation intramolecular condensation with N13.
- 44.Zhang X, Foote CS. J. Am. Chem. Soc. 1993;115:8867–8868. [Google Scholar]
- 45.For an excellent discussion of a similar scrambling of regioisomers during the 1,2-alkyl shift of a hydroxyindolenine, see: ref 9.
- 46.Blackman AJ, Hambley TW, Picker K, Taylor WC, Thirasasana N. Tetrahedron Lett. 1987;28:5561–5562. [Google Scholar]
- 47.Spartan ′06 was used for the calculations.
- 48.For an X-ray crystal structure of (−)-trigonoliimine C (3), see ref 8.
- 49.For a report of similar air oxidation of a 2,2′-bistryptamine derivative, see: ref 9.
- 50 (a).Ma TG, Ling YH, McClure GD, Tseng MT. J. Toxicol. Environ. Health. 1990;31:147–158. doi: 10.1080/15287399009531444. [DOI] [PubMed] [Google Scholar]; (b) Cornish J, Callon KE, Lin CQ-X, Xiao CL, Mulvey TB, Cooper GJS, Reid IR. Am. J. Physiol. Endocrinol. Metab. 1999;277:E779–E783. doi: 10.1152/ajpendo.1999.277.5.E779. [DOI] [PubMed] [Google Scholar]
- 51 (a).Rishton GM. Drug Discov. Today. 1997;2:382–384. doi: 10.1016/s1359644602025722. [DOI] [PubMed] [Google Scholar]; (b) Walters WP, Murcko MA. Adv. Drug Delivery Rev. 2002;54:255–271. doi: 10.1016/s0169-409x(02)00003-0. [DOI] [PubMed] [Google Scholar]; (c) Baell JB, Holloway GA. J. Med. Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 52.Movassaghi M, Siegel DS, Han S. Chem. Sci. 2010;1:561–566. doi: 10.1039/c0sc00351d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Acetic acid-d4 (1.1 equiv) was added, resulting in sharpening of peaks.
- 54.Staab HA, Polenski B. Liebigs Ann. Chem. 1962;655:95–102. [Google Scholar]
- 55.Vichai V, Kirtikara K. Nature Prot. 2006;1:1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 56.Cory AH, Owen TC, Barltrop JA, Cory JG. Cancer Commun. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.