

Abstract
Objective
One of the hallmarks of severe pneumonia and associated Acute Lung Injury (ALI) is neutrophil recruitment to the lung. Leptin is thought to be up-regulated in the lung following injury and to exert diverse effects on leukocytes, influencing both chemotaxis and survival. We hypothesized that pulmonary leptin contributes directly to the development of pulmonary neutrophilia during pneumonia and ALI.
Design
Controlled human and murine in vivo and ex vivo experimental studies.
Settings
Research laboratory of a university hospital.
Subjects
Healthy human volunteers and subjects hospitalized with bacterial and H1N1 pneumonia. C57Bl/6 and db/db mice were also used.
Interventions
Lung samples from patients and mice with either bacterial or H1N1 pneumonia and associated ALI were immunostained for leptin. Human bronchoalveolar-lavage (BAL) samples obtained after lipopolysaccharide (LPS)-induced lung injury were assayed for leptin. C57Bl/6 mice were examined after oropharyngeal aspiration of recombinant leptin alone or in combination with E.coli- or K.pneumonia-induced pneumonia. Leptin-resistant (db/db) mice were also examined using the E.coli model. BAL neutrophilia and cytokine levels were measured. Leptin-induced chemotaxis was examined in human blood- and murine marrow-derived neutrophils in vitro.
Measurements and Main Results
Injured human and murine lung tissue showed leptin induction compared to normal lung, as did human BAL following LPS instillation. BAL neutrophilia in uninjured and infected mice was increased and lung bacterial-load decreased by airway leptin administration, whereas BAL neutrophilia in infected leptin-resistant mice was decreased. In sterile lung injury by LPS, leptin also appeared to decrease airspace neutrophil apoptosis. Both human and murine neutrophils migrated towards leptin in vitro, and this required intact signaling through the JAK2/PI3K pathway.
Conclusion
We demonstrate that pulmonary leptin is induced in injured human and murine lungs and that this cytokine is effective in driving alveolar airspace neutrophilia. This action appears to be caused by direct effects of leptin on neutrophils.
Keywords: Neutrophil, Immunity, Innate, Pneumonia, Chemotaxis, Cytokines
Introduction
Acute Lung Injury (ALI) affects approximately 200,000 patients a year in the United States alone, with an associated mortality rate of between 37% and 60% [1]. Severe infection, particularly pneumonia, is the most frequent risk factor for ALI [2], and this syndrome is seen in response to a variety of bacterial and viral pulmonary pathogens. Neutrophils are key participants in the alveolar epithelial and endothelial injury that characterizes ALI, and are the predominant cells in alveolar edema fluid obtained from affected patients [3]. The normal response to pulmonary infection or injury induces the controlled recruitment of neutrophils to the lung, which are then rapidly eliminated through the induction of apoptosis and subsequent clearance by alveolar macrophages during the resolution phase of the inflammatory response [4]. In ALI, however, neutrophil recruitment is both exaggerated and persistent, and neutrophil apoptosis is delayed as a consequence of alveolar cytokines and growth factors, including IL-8 and G-CSF [5, 6]. This prolonged neutrophil life-span contributes significantly to the protracted lung injury seen in ALI patients [7]. Thus, the key neutrophil processes in ALI are: 1) cytokine-mediated neutrophil recruitment to the lung and 2) prolonged neutrophil survival in the lung leading to sustained inflammation and increased tissue damage [8–10].
Although traditionally considered a mediator of metabolic homeostasis [11], the adipokine and IL-6 family member leptin also appears to be released into the blood in the setting of systemic inflammation [12]. However, leptin’s overall role in inflammation remains unclear. Monocyte and macrophage activation, phagocytosis, and release of pro-inflammatory cytokines appear to be promoted by leptin during the innate immune response [13–15], and leptin may be required for these cells to clear bacterial pathogens [16]. In the case of neutrophils, surface expression of leptin receptor has been reported and it has been suggested that leptin may augment the functional responses of these cells, including chemotaxis [17], and may delay spontaneous neutrophil apoptosis [18].
Recent studies have shown up-regulated leptin expression in bronchial epithelial cells and alveolar macrophages in both human and murine lungs following chronic smoking-induced lung injury [19, 20], and that leptin is increased in BAL from ARDS patients [21]. Furthermore, it has been suggested that leptin receptor activation is important in the innate immune response to pneumonia [17, 18], as well as in the development of hyperoxia-induced ALI in mice [22].
Given the proposed effects of leptin on neutrophil function, we hypothesized that pulmonary leptin is an important factor during both pulmonary inflammatory response and ALI pathogenesis. In this study, we show for the first time that leptin expression is up-regulated in both human and murine lung tissues in response to bacterial and viral-induced lung injury, and further, present evidence that leptin is an important mediator of pulmonary neutrophilia in ALI that functions via leptin-augmented neutrophil chemotaxis to and possibly delayed clearance from the alveolar space.
Materials and Methods
Animals
Eight to twelve week old female C57Bl/6 mice (Harlan, Indianapolis, IN), and homozygous female leptin receptor-deficient mice (BKS db/db) and heterozygous littermates (Jackson Labs, Bar Harbor, ME), were housed in the animal facilities at the University of Vermont. All experimental animal procedures were approved by the University of Vermont Institutional Animal Care and Use Committee.
Human immunohistochemistry
Banked human lung specimens were analyzed from patients who died of bacterial (n=3) or influenza A (H1N1) (n=3) pneumonias with ALI/ARDS and compared to histologically normal lung tissue (n=3). A previously validated protocol for leptin-immunostaining using a rabbit-anti-human leptin antibody (Santa Cruz, Dallas, TX) was used to identify leptin-positive cells in both human and murine tissue [20].
Human pulmonary endotoxin injury
To determine human alveolar production of leptin in response to injury, alveolar lavage samples banked from a previous study of normal healthy volunteers (n=14) exposed to bronchoscopically-instilled endotoxin (LPS) versus saline were analyzed [23]. Briefly, subjects were exposed to endotoxin via bronchoscopic subsegmental-instillation (4ng/kg subject body weight in 10mL saline; E. coli strain O:113, NIH, Bethesda, MD) randomized to either the lingula or right middle lobe of the lung, which was immediately followed by instillation of 10mL sterile saline into a contralateral lung-subsegment. A second bronchoscopy was performed 16h later, and both the LPS- and saline-instilled subsegments were lavaged with 150mL saline. Previously reported lavage cell counts, and IgM and protein concentrations [23] were compared to lavage leptin levels, as determined by ELISA (R&D Systems), of the stored samples.
Murine exposures
Mice treated with pegylated recombinant murine leptin [19] were instilled (50μg in 100μl sterile PBS or vehicle-control) by oropharyngeal (o.p.) aspiration under isoflurane anesthesia. Murine influenza A (A/California/7/2009 H1N1, 3x103EIU), E. coli (O6:K2:H1 ATCC, 1x107CFU), and Klebsiella pneumonia (43816 serotype 2, ATCC, 2x103CFU) infections were performed by either o.p. aspiration (E.coli and K.pneumoniae) or nasal-instillation (H1N1), as described [24–26]. LPS lung injury was induced by nebulized LPS, as described [27].
Murine lung analysis and immunohistochemistry
Airspace lavage cell counts and cytokine levels, as well as whole-lung leptin mRNA levels and bacterial CFU where appropriate were determined at 24h (E.coli, K.pneumoniae) or 4d (H1N1) after exposure as described in the online supplement. Leptin immunostaining was performed on unlavaged, paraffin-embedded lungs from mice exposed to either E.coli (n=3) or H1N1 pneumonia (n=3), as well as matched saline-instilled control mice (n=3 for each condition), as described for human lung samples.
Neutrophil isolation
Human blood neutrophils were isolated by dextran sedimentation and discontinuous density-gradient centrifugation [28], and re-suspended in Krebs-Ringer-phosphate-dextrose buffer. Morphologically-mature murine bone marrow neutrophils were isolated by discontinuous density-gradient [29, 30].
Neutrophil chemotaxis
Neutrophil chemoattractant response to IL-8, KC (R&D), or recombinant human (R&D) or murine [19] leptin was examined using a modified Boyden chamber (Neuroprobe) [31] with or without pre-incubation of the neutrophils for 30min at 37°C with PI3K- or JAK2-inhibitor (Calbiochem).
Neutrophil apoptosis
Murine airspace neutrophil apoptosis was examined using TUNEL (R&D) and flow cytometry of lavaged mice, 6h after the induction of sterile lung injury by inhaled LPS [31].
Statistical analysis
Data were represented as mean ± SEM, and analysis of differences between experimental groups was performed by Student t test. Wilcoxon signed-rank test was used to analyze differences between lavage levels of leptin in subject-matched LPS- and saline-exposed lung subsegments. Spearman rank correlation was used to determine correlations between subjects’ lavage leptin, neutrophil, IgM, and protein levels using ‘delta’ values (LPS-treated lung subsegment minus saline-treated lung subsegment levels) for each subject [23]. One-way ANOVA was used for analysis of differences between conditions in chemotaxis assays of human and murine neutrophils. All analyses were performed using Prism5 software (GraphPad). Results with p<0.05 were considered statistically significant.
Results
Immunohistologically-detected leptin is increased in human lung tissues following infection and injury
To determine whether alveolar leptin content is increased in the human lung following infection and injury, lung tissue from patients with pneumonia and ALI due to either bacterial or viral (H1N1) infection, was examined by immunohistochemistry for leptin and compared to similarly-stained uninjured human lung samples. Limited leptin staining (blue) was present in the uninjured lung (Fig. 1A), whereas diffuse leptin staining was observed in lungs with bacterial and viral pneumonias (Figs. 1B and C). In particular, intense staining of airspace macrophages was detected following acute lung injury, a finding which has previously only been shown in response to chronic lung injury [19, 20].
Figure 1.
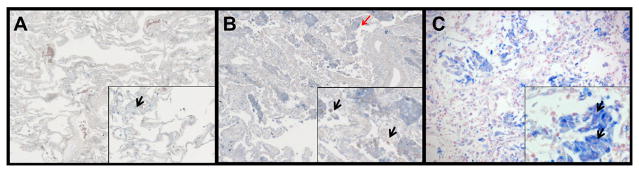
Leptin expression is augmented in the lung during ALI. Immunohistochemical examination was performed for leptin in normal human lung tissue (A), human lung tissue from an ALI patient (B), human lung tissue from an H1N1-infected patient (C). Leptin is indicated by blue staining and nuclei are counterstained in red. Limited leptin staining was observed in normal human lung tissue and was localized in alveolar macrophages. Increased leptin expression in airway macrophages (black arrows) and alveolar epithelium (red arrows) in lung tissue of both ALI and H1N1 infected lung tissue was observed. Original magnification x 200 (inserts x 400).
Airspace leptin levels increase following endotoxin-induced lung injury in humans
To further examine human alveolar leptin induction, we measured leptin protein levels in BAL samples from a previous study of bronchoscopic subsegmental LPS instillation in healthy volunteers (n=14) [23]. BAL samples obtained 16h after instillation of LPS showed significantly increased leptin levels in the LPS-exposed lung segments compared to saline-exposed contralateral lung segments of the same subject (Fig. 2A). Furthermore, a significant correlation was observed between airspace neutrophil accumulation and leptin levels (Fig. 2B). Total BAL protein concentrations, a measurement of both lung injury and capillary leak, also correlated with leptin release (Fig. 2C). Yet, BAL IgM levels, a more specific marker for capillary leak, did not correlate with leptin levels (Fig. 2D), suggesting that increased airspace leptin levels correlate with alveolar injury and not leakage of serum leptin into the airspace.
Figure 2.

Airspace leptin levels are increased by LPS exposure in humans. Human leptin was quantified in brochoalveolar lavage fluid from lung segments exposed to either saline (10mL) or LPS (4ng/kg in 10mL saline) 16 h after instillation (A). The increase in leptin expression was compared to the levels of brochoalveolar lavage neutrophils (B), total brochoalveolar lavage protein concentration (C), and IgM levels (D). n=14 in all groups. Data are presented as mean ± SEM, * p≤0.05 compared to control. Wilcoxon signed rank test was used to analyze differences between paired LPS and saline instilled subsegments from each subject (A), and Spearman rank correlation was used to analyze the relationship between LPS-treated subsegment lavage leptin, neutrophil, protein, and IgM levels, which were adjusted for the matched control subsegment in each subject (LPS-instilled subgement minus saline instilled subsegment).
Pulmonary leptin expression increases in murine models of both E. coli and influenza A pneumonia
To further examine the induction of leptin within the lung during infection, we determined leptin immunostaining in lung tissue as well as airspace leptin protein levels and whole lung leptin mRNA expression in mice. Limited leptin staining (blue) was present in the uninjured lung (Fig. 3A), whereas diffuse leptin staining was observed in lungs with bacterial and viral pneumonias (Figs. 3B and C) with intense staining of alveolar macrophages as well as epithelium, similar to the findings in human lung tissue. BAL leptin protein levels were significantly increased at both 24h after E.coli infection and 4d after H1N1 infection, compared to saline treated control mice (Fig. 3D). A similar effect was seen on relative leptin mRNA expression in lung tissue of these mice (Fig. 3E). Analysis of BAL leptin and BAL albumin levels (an indicator of the degree of serum leakage) showed no correlation in these experiments (data not shown), suggesting that airspace leptin is of pulmonary origin and does not reflect serum leptin leakage to a significant degree.
Figure 3.

Leptin levels are increased in murine lungs after bacterial or viral infection. Immunohistochemical examination was performed for leptin in normal murine lung tissue (n=5) (A), murine lung tissue infected with E. coli (n=5) (B), and murine H1N1 infected lung tissue (n=5) (C). Leptin is indicated by blue staining and nuclei are counterstained in red. Leptin staining was localized in alveolar macrophages (black arrows) and alveolar epithelium (red arrows). Original magnification x 200 (inserts x 400). Leptin levels were determined in either brochoalveolar lavage by ELISA (D) or lung tissue by qPCR (E) from mice exposed to either saline control (n=4), E. coli for 24h (n=8) or H1N1 for 4d (n=4). Data are presented as mean ± SEM, * p≤0.05, ** p≤0.01 and ***p≤0.001 compared to control.
Recombinant leptin augments airspace neutrophilia in injured and uninjured mice
We next investigated the effect of airspace leptin augmentation in models of E.coli- and K.pneumoniae-induced pneumonia. Pulmonary neutrophilia was significantly increased at 24h after E.coli infection with instilled leptin compared to E. coli infection without (Fig. 4A), and whole lung E.coli CFU were lower in the leptin-treated mice compared to controls (Fig. 4B). However, the latter difference did not reach statistical significance, likely related to the limited severity of the E.coli model. Using the more aggressive pulmonary pathogen K.pneumoniae [32, 33], leptin instillation significantly augmented bacterial clearance (Fig. 4D), while BAL neutrophil levels (Fig. 4C) were similar, possibly due to compensatory recruitment signals in the (sicker) control mice. To investigate whether the leptin-associated increase in BAL neutrophilia requires the induction of lung injury, we examined the effect of leptin airway-instillation on pulmonary neutrophil recruitment in uninjured mice. Leptin treatment led to a significant increase in total BAL neutrophil counts at both 6h and 24h compared to control (Fig. 4E), as well as increased albumin levels, indicative of injury and capillary leak (Fig. S1A). A similar trend was seen in E. coli and K. pneumoniae treated mice (Figs. S1B and C), despite decreased bacterial burden in the leptin-treated lungs (Figs. 4B and D).
Figure 4.

Leptin increases pulmonary neutrophilia in E. coli pneumonia injured as well as in uninjured mice. Total neutrophil counts in brochoalveolar lavage fluid were determined at 24h after o.p. aspiration of control (PBS) or recombinant pegylated leptin (50 μg) in E. coli (A) and K. pneumoniae (C) infected mice, and at 6 and 24h after o.p. aspiration of vehicle (PBS) or recombinant pegylated leptin (50μg) in uninjured mice (E). In addition, whole lung CFU were determined in the E.coli (B) and K.pneumoniae (D) infected mice. n=5 in all groups of uninjured mice; n=7 in the control group and n=8 in the pegylated-leptin treated group of E. coli pneumonia infected mice; n=10 in all groups of K. pneumonia infected mice. Data are presented as mean ± SEM, * p≤0.05 and ** p≤0.01 compared to vehicle-exposed mice.
Leptin does not induce alveolar release of inflammatory cytokines
To distinguish whether the effects of o.p.-instilled leptin on pulmonary neutrophilia were direct (i.e. acting on neutrophils themselves) or indirect (mediated through the induction of inflammatory cytokines), levels of inflammatory cytokines in BAL fluid from leptin- and control-treated uninjured and E.coli-infected mice were measured. No differences were observed in airspace cytokine levels between PBS- and leptin-treated animals in either uninjured (Figs. S2A and B) or E.coli-infected mice (Fig. S2C). These results suggest that leptin-associated augmentation of airspace neutrophilia may represent a direct effect of leptin on neutrophils themselves.
Defective leptin signaling is associated with decreased pulmonary neutrophilia following murine bacterial pneumonia
To determine whether leptin was not only sufficient but necessary for the development of pulmonary neutrophilia during infection and injury, we examined the effects of defective leptin signaling in E.coli- or K.pneumoniae-induced pneumonia in mice lacking leptin receptor (db/db). In contrast to our findings in wild type mice (Fig. 4E), leptin instillation alone in db/db mice did not result in significant airspace neutrophilia (Fig. S3). Pulmonary neutrophilia was significantly decreased at 24h after E. coli infection in db/db mice compared to their heterozygous littermates (Fig. 5A), and although whole lung E. coli CFU were higher in the db/db mice compared to controls (Fig. 5B), this difference did not reach statistical significance. In addition, BAL neutrophilic cytokine levels (IL-6, KC, G-CSF, TNF-α) were the same in db/db mice compared to littermate controls (Fig. 5C), suggesting that despite lower levels of neutrophils recruited to the airspace of db/db mice, the animals were still capable of handling this non-lethal pneumonia model. In the Klebsiella model, leptin receptor-deficiency led to significant impairment of bacterial clearance compared to littermate controls (Fig. 5E). Although the difference between BAL neutrophil levels in db/db compared littermate control mice was not significant, we found markedly increased levels of BAL cytokines in the db/db mice compared to control (Fig. 5F), suggesting that in the Klebsiella model, unlike the self-limited E. coli model, as infection worsens in the absence of leptin signaling it leads to the release of additional neutrophil recruitment signals.
Figure 5.

Defective leptin signaling decreases pulmonary neutrophilia in murine E. coli pneumonia and increases lung CFU levels in murine K. pneumoniae infection. Total neutrophil counts in brochoalveolar lavage fluid and whole lung CFU were determined at 24h after E. coli (A, B) or K. pneumonia (D, E) infection in leptin receptor-deficient (db/db) mice and heterozygote littermate controls. In addition, BAL cytokine levels were determined in db/db and control mice after E.coli (C) or K. pneumoniae (F) infections. n=11 in all groups of E.coli infected mice; n=7 in the control mice and n=6 in the db/db mice infected with K. pneumonia. Data are presented as mean ± SEM, * p≤0.05, ** p≤0.01, *** p≤0.001 and **** p≤0.0001 compared to control mice.
Leptin effects human and murine neutrophil chemotaxis in a dose-dependent manner
To establish whether a direct chemoattractant effect of leptin on neutrophils might contribute to its in vivo effects, we examined both murine and human neutrophil chemotaxis in vitro. A dose-dependent chemotaxis response to leptin was observed in both human and murine neutrophils (Figs. 6A and B). This effect appears to be greater in human neutrophils than mouse, although this may reflect the source and relative maturity of these cells (blood vs. bone marrow).
Figure 6.
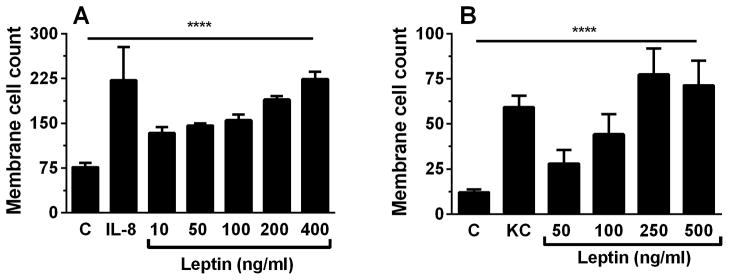
Leptin has a dose-dependent effect on both human and murine neutrophil migration. Human peripheral blood derived neutrophils (A) and murine isolated bone marrow-derived neutrophils (B) were exposed to control (C), IL-8 (100ng/mL) (human), KC (50ng/mL) (murine), and different concentrations of human and murine leptin in a modified Boyden chamber (n=4 for each condition). Data are presented as mean ± SEM. **** p≤0.0001 as performed with One-way ANOVA.
Signaling molecules Janus kinase 2 (JAK2) and phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) are critical for leptin-mediated neutrophil chemotaxis
To further dissect the leptin-induced chemotaxis response, we examined downstream targets of leptin signaling in isolated murine neutrophils. Inhibition of JAK2 and PI3K effectively abolished neutrophil chemotaxis towards leptin (Fig. 7), suggesting that neutrophil chemotaxis to leptin requires JAK2/PI3K signaling. Neutrophil response to the CXC chemokine KC did not require JAK2 signaling but was dependent on PI3K activation (Fig. 7), as has previously been demonstrated [34.].
Figure 7.

JAK2 and PI3K inhibition abolish leptin-mediated neutrophil chemotaxis. Murine bone-marrow derived neutrophils were pre-incubated with control, JAK2 (100μM) or PI3K (50μM) inhibitor (n=4 for each condition) and were exposed to buffer control, KC (50ng/mL) or different concentrations of murine leptin in a modified Boyden Chamber. Data are presented as mean ± SEM. # p≤0.05, ## p≤0.01, ### p≤0.001 compared to buffer control; ** p≤0.01, *** p≤0.001 compared to no inhibitor control.
Leptin delays murine airspace neutrophil apoptosis in vivo
As previous work has suggested that leptin may have an anti-apoptotic effect on neutrophils in vitro [18, 35], we examined whether it might also serve to increase airspace neutrophilia by delaying airspace neutrophil apoptosis in vivo. To address this possibility, we examined a less complex, sterile lung injury model using nebulized lipopolysaccharide to induce rapid recruitment of neutrophils to the lung. Detectable apoptotic neutrophils, which are typically scarce in the airspace due to their rapid clearance [36], were found at low levels in both groups but were significantly reduced in lung-injured mice treated with o.p. leptin compared to those treated with vehicle alone (Fig. 8).
Figure 8.
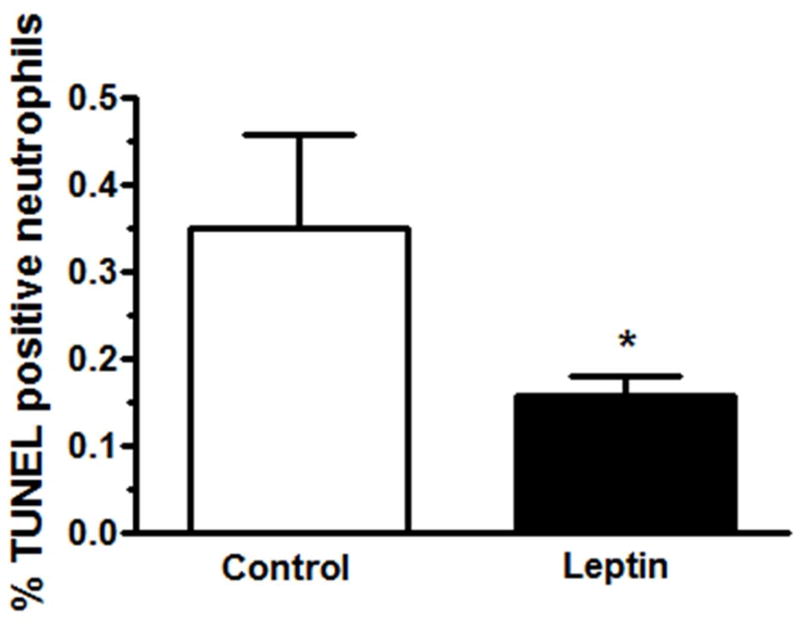
Leptin decreases murine neutrophil apoptosis in vivo. Apoptosis was determined in BAL neutrophils at 6h after LPS exposure and o.p. pegylated-leptin (50μg) or control (PBS) instillation by TUNEL staining (n=3 for control and n=9 for leptin treatment). Data are presented as mean ± SEM, * p≤0.05 compared to control treated mice.
Discussion
In this study we demonstrate the induction of leptin expression and release in both human and murine lung during pneumonia and acute lung injury. Furthermore, we describe leptin-mediated effects on neutrophil recruitment and potentially survival, suggesting a novel role for pulmonary leptin in the development and persistence of airspace neutrophilia in severe pneumonia and ALI.
Among its many effects, the adipokine leptin has recently been suggested to act as a neutrophil chemoattractant and an anti-apoptotic molecule in humans [17, 18]. Yet, the role of leptin in neutrophil recruitment in pneumonia and ALI has not previously been examined. Circulating leptin levels are increased in critical illness, and evidence suggests leptin expression increases is human lung tissue during chronic injury and inflammation [20, 37, 38], while leptin levels have been found to be elevated in BAL from hyperoxia-exposed mice and patients with acute lung injury [21, 22, 39]. Using immunohistochemistry, we observed intense leptin staining of both alveolar epithelium and macrophages in human subjects with either viral or bacterial-induced lung injury. We found similar leptin staining patterns in mouse models of both H1N1 and E. coli pneumonia, and levels of both leptin transcription and airspace release were increased. Furthermore, we found that endotoxin-induced lung injury in healthy human subjects leads to increased leptin content in BAL fluid, and that these increased leptin concentrations appear to correlate with both the degree of lung injury and levels of airspace neutrophilia. Thus, we show for the first time that pulmonary leptin up-regulation and airspace release is a response to infection and acute lung injury in both murine models and human disease.
The effects of leptin on pulmonary inflammatory responses remain poorly understood. Most relevant studies have examined mouse models of leptin- or leptin receptor-deficiency. Mancuso et al. demonstrated an impaired host defense in leptin deficient mice, and restoration of circulating leptin levels in these mice led to improved pulmonary bacterial clearance and survival [15, 16]. Others have shown that leptin infusion or instillation into wild type mice augments lung inflammation in models of allergic sensitization [40] and hyperoxia [22]. Furthermore, in recent studies of human ALI, high BAL leptin levels were reported to be associated with greater morbidity and mortality in patients with a normal BMI [21], suggesting a pathophysiological role for leptin in this disease.
Leptin has previously been suggested to act as a neutrophil chemoattractant [17, 41, 42] and anti-apoptotic [18, 35] in vitro, yet little has been reported on leptin’s effects on neutrophil behavior in vivo. In the present study, we show leptin to be an effective neutrophil chemoattractant not only in vitro but also in vivo. We also find in vivo evidence that leptin may further augment airspace neutrophilia in the context of injury through anti-apoptotic effects, similar to G-CSF [6, 7]. In this setting, we note that pneumonia-associated airspace neutrophilia is diminished in mice with impaired leptin signaling, while clearance of bacteria is significantly impaired. Mancuso et al. previously reported impaired bacterial clearance after K. pneumoniae infection in leptin-deficient (ob/ob) mice, suggesting an important role for leptin in the pulmonary antibacterial host defense [15]. Our data are in agreement with these findings; however, we show that not only the presence of leptin, but also intact leptin signaling is critical in the pulmonary host defense.
The signaling pathways underlying leptin-induced neutrophil chemotaxis have been poorly defined. In our work, signaling through the ‘canonical’ ObR/JAK2/STAT pathway was not detected [43]. We find instead that the ‘alternate’ JAK2/IRS-1 and the MAPK pathways [34] are required for leptin-induced neutrophil chemotaxis. Furthermore, this process requires PI3K, suggesting that JAK2 instead activates IRS1/2, which has been shown to activate PI3K in this context [44]. Further investigation will be required to fully delineate this process.
It has previously been suggested that increased airspace leptin levels following lung injury may be caused by increased capillary leak and consequent serum leptin extravasation into the airspace [40, 45]. In our examination of human airspace leptin response we used BAL IgM as a marker of capillary leak (Fig. 2) in order to avoid the confounding effects of using total protein, which reflects not only vascular leak, but is also an indicator of tissue injury. However, IgM is a larger protein compared to leptin, potentially allowing leptin to cross the alveolar/capillary membrane more readily than IgM following injury. Thus, although we present additional data that a pulmonary source of leptin exists (including immunohistology and whole lung quantitative PCR), our findings must be interpreted with caution. It should also be noted that, given unclear dilution factors related to airspace lavage and the critical role of microanatomical cytokine compartmentalization in the lung, the relevant airspace dosing of leptin is unknown.
Our findings not only suggest that leptin participates in the host response to pneumonia and the pathogenesis of ARDS, but may also shed light on the apparent effects of obesity and the accompanying leptin-resistance on both of these diseases. Obesity has been shown in both clinical studies and animal models to be associated with greater susceptibility to and poorer outcomes from pneumonia [46, 47]. Conversely, in the case of ALI/ARDS, we and others have shown that rising BMI reduces clinical mortality from this syndrome [48, 49], and have implicated a blunted inflammatory response during ALI in the obese [50]. Furthermore, we have recently reported that diet-induced obese mice have attenuated airspace neutrophilia in response to LPS-induced lung injury [27], which appears to be multifactorial in origin. In light of our current findings, we propose that the altered pulmonary innate immune response seen in obesity could in part reflect impairment of neutrophil leptin-response.
Conclusion
We demonstrate up-regulation of pulmonary leptin levels in both humans and mice following bacterial- and viral-induced pneumonia and ALI. Leptin enhances airspace neutrophilia, an effect that appears to be independent of secondary cytokine induction. This effect is in part mediated through leptin’s action as a neutrophil chemoattractant, but may also include leptin-driven effects on neutrophil survival. Taken together, the present study suggests an important role for leptin in the development of pulmonary neutrophilia following a wide range of insults. Further studies are needed to better understand the effects of leptin and its signaling cascades on both neutrophil development and neutrophil function in pulmonary inflammation.
Supplementary Material
Acknowledgments
This work was supported by Grants R01 HL084200, R01 HL089177, R01 HL090991 and NCRR P20RR15557 from the National Institutes of Health
Footnotes
Copyright Form Disclosures: Dr. Suratt’s institution received grant support from the National Institutes of Health (R01 HL084200). Dr. Suratt received support for article research from NIH. Dr. Burg’s institution received grant support from NIH (HL 084200). Dr. Berg received support for article research from NIH. Dr. Abraham’s institution received grant support from NIH-NHLBI. Dr. Nick’s institution received grant support from NIH. Dr. Dienz and his institution received grant support from NIH. Dr. Dienz is employed by University of Vermont. Dr. Mizgerd’s institution received grant support from NIH (research on pneumonia). Dr. Tavernier’s institution received grant support from U-Gent. Dr. Tavernier is employed by U-Gent. Dr. Poynter’s institution received grant support from NIH and FAMRI. The remaining authors have disclosed that they do not have any potential conflicts of interest.
References
- 1.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353(16):1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 2.Sheu CC, Gong MN, Zhai R, Bajwa EK, Chen F, Thompson BT, Christiani DC. The influence of infection sites on development and mortality of ARDS. Intensive Care Med. 2010;36(6):963–970. doi: 10.1007/s00134-010-1851-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steinberg KP, Milberg JA, Martin TR, Maunder RJ, Cockrill BA, Hudson LD. Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am J Respir Crit Care Med. 1994;150(1):113–122. doi: 10.1164/ajrccm.150.1.8025736. [DOI] [PubMed] [Google Scholar]
- 4.Vandivier RW, Henson PM, Douglas IS. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest. 2006;129(6):1673–1682. doi: 10.1378/chest.129.6.1673. [DOI] [PubMed] [Google Scholar]
- 5.Droemann D, Aries SP, Hansen F, Moellers M, Braun J, Katus HA, Dalhoff K. Decreased apoptosis and increased activation of alveolar neutrophils in bacterial pneumonia. Chest. 2000;117(6):1679–1684. doi: 10.1378/chest.117.6.1679. [DOI] [PubMed] [Google Scholar]
- 6.Matute-Bello G, Liles WC, Radella F, 2nd, Steinberg KP, Ruzinski JT, Hudson LD, Martin TR. Modulation of neutrophil apoptosis by granulocyte colony-stimulating factor and granulocyte/macrophage colony-stimulating factor during the course of acute respiratory distress syndrome. Crit Care Med. 2000;28(1):1–7. doi: 10.1097/00003246-200001000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Matute-Bello G, Liles WC, Radella F, 2nd, Steinberg KP, Ruzinski JT, Jonas M, Chi EY, Hudson LD, Martin TR. Neutrophil apoptosis in the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1997;156(6):1969–1977. doi: 10.1164/ajrccm.156.6.96-12081. [DOI] [PubMed] [Google Scholar]
- 8.Matute-Bello G, Martin TR. Science review: apoptosis in acute lung injury. Crit Care. 2003;7(5):355–358. doi: 10.1186/cc1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee WL, Downey GP. Neutrophil activation and acute lung injury. Curr Opin Crit Care. 2001;7(1):1–7. doi: 10.1097/00075198-200102000-00001. [DOI] [PubMed] [Google Scholar]
- 10.Suratt BT, Parsons PE. Mechanisms of acute lung injury/acute respiratory distress syndrome. Clin Chest Med. 2006;27(4):579–589. doi: 10.1016/j.ccm.2006.06.005. abstract viii. [DOI] [PubMed] [Google Scholar]
- 11.Muoio DM, Lynis Dohm G. Peripheral metabolic actions of leptin. Best Pract Res Clin Endocrinol Metab. 2002;16(4):653–666. doi: 10.1053/beem.2002.0223. [DOI] [PubMed] [Google Scholar]
- 12.La Cava A, Matarese G. The weight of leptin in immunity. Nat Rev Immunol. 2004;4(5):371–379. doi: 10.1038/nri1350. [DOI] [PubMed] [Google Scholar]
- 13.Zarkesh-Esfahani H, Pockley G, Metcalfe RA, Bidlingmaier M, Wu Z, Ajami A, Weetman AP, Strasburger CJ, Ross RJ. High-dose leptin activates human leukocytes via receptor expression on monocytes. J Immunol. 2001;167(8):4593–4599. doi: 10.4049/jimmunol.167.8.4593. [DOI] [PubMed] [Google Scholar]
- 14.Dixit VD, Mielenz M, Taub DD, Parvizi N. Leptin induces growth hormone secretion from peripheral blood mononuclear cells via a protein kinase C- and nitric oxide-dependent mechanism. Endocrinology. 2003;144(12):5595–5603. doi: 10.1210/en.2003-0600. [DOI] [PubMed] [Google Scholar]
- 15.Mancuso P, Gottschalk A, Phare SM, Peters-Golden M, Lukacs NW, Huffnagle GB. Leptin-deficient mice exhibit impaired host defense in Gram-negative pneumonia. J Immunol. 2002;168(8):4018–4024. doi: 10.4049/jimmunol.168.8.4018. [DOI] [PubMed] [Google Scholar]
- 16.Hsu A, Aronoff DM, Phipps J, Goel D, Mancuso P. Leptin improves pulmonary bacterial clearance and survival in ob/ob mice during pneumococcal pneumonia. Clin Exp Immunol. 2007;150(2):332–339. doi: 10.1111/j.1365-2249.2007.03491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caldefie-Chezet F, Poulin A, Vasson MP. Leptin regulates functional capacities of polymorphonuclear neutrophils. Free Radic Res. 2003;37(8):809–814. doi: 10.1080/1071576031000097526. [DOI] [PubMed] [Google Scholar]
- 18.Bruno A, Conus S, Schmid I, Simon HU. Apoptotic pathways are inhibited by leptin receptor activation in neutrophils. J Immunol. 2005;174(12):8090–8096. doi: 10.4049/jimmunol.174.12.8090. [DOI] [PubMed] [Google Scholar]
- 19.Vernooy JH, Bracke KR, Drummen NE, Pauwels NS, Zabeau L, van Suylen RJ, Tavernier J, Joos GF, Wouters EF, Brusselle GG. Leptin modulates innate and adaptive immune cell recruitment after cigarette smoke exposure in mice. J Immunol. 2010;184(12):7169–7177. doi: 10.4049/jimmunol.0900963. [DOI] [PubMed] [Google Scholar]
- 20.Vernooy JH, Drummen NE, van Suylen RJ, Cloots RH, Moller GM, Bracke KR, Zuyderduyn S, Dentener MA, Brusselle GG, Hiemstra PS, et al. Enhanced pulmonary leptin expression in patients with severe COPD and asymptomatic smokers. Thorax. 2009;64(1):26–32. doi: 10.1136/thx.2007.085423. [DOI] [PubMed] [Google Scholar]
- 21.Jain M, Budinger GR, Lo A, Urich D, Rivera SE, Ghosh AK, Gonzalez A, Chiarella SE, Marks K, Donnelly HK, et al. Leptin promotes fibroproliferative acute respiratory distress syndrome by inhibiting peroxisome proliferator-activated receptor-gamma. Am J Respir Crit Care Med. 2011;183(11):1490–1498. doi: 10.1164/rccm.201009-1409OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bellmeyer A, Martino JM, Chandel NS, Scott Budinger GR, Dean DA, Mutlu GM. Leptin resistance protects mice from hyperoxia-induced acute lung injury. Am J Respir Crit Care Med. 2007;175(6):587–594. doi: 10.1164/rccm.200603-312OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nick JA, Coldren CD, Geraci MW, Poch KR, Fouty BW, O’Brien J, Gruber M, Zarini S, Murphy RC, Kuhn K, et al. Recombinant human activated protein C reduces human endotoxin-induced pulmonary inflammation via inhibition of neutrophil chemotaxis. Blood. 2004;104(13):3878–3885. doi: 10.1182/blood-2004-06-2140. [DOI] [PubMed] [Google Scholar]
- 24.Dienz O, Rud JG, Eaton SM, Lanthier PA, Burg E, Drew A, Bunn J, Suratt BT, Haynes L, Rincon M. Essential role of IL-6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol. 2012;5(3):258–266. doi: 10.1038/mi.2012.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizgerd JP, Lupa MM, Kogan MS, Warren HB, Kobzik L, Topulos GP. Nuclear factor-kappaB p50 limits inflammation and prevents lung injury during Escherichia coli pneumonia. Am J Respir Crit Care Med. 2003;168(7):810–817. doi: 10.1164/rccm.200303-412OC. [DOI] [PubMed] [Google Scholar]
- 26.Wargo MJ, Gross MJ, Rajamani S, Allard JL, Lundblad LK, Allen GB, Vasil ML, Leclair LW, Hogan DA. Hemolytic phospholipase C inhibition protects lung function during Pseudomonas aeruginosa infection. Am J Respir Crit Care Med. 2011;184(3):345–354. doi: 10.1164/rccm.201103-0374OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kordonowy LL, Burg E, Lenox CC, Gauthier LM, Petty JM, Antkowiak M, Palvinskaya T, Ubags N, Rincon M, Dixon AE, et al. Obesity is associated with neutrophil dysfunction and attenuation of murine acute lung injury. Am J Respir Cell Mol Biol. 2012 doi: 10.1165/rcmb.2011-0334OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haslett C, Guthrie LA, Kopaniak MM, Johnston RB, Jr, Henson PM. Modulation of multiple neutrophil functions by preparative methods or trace concentrations of bacterial lipopolysaccharide. Am J Pathol. 1985;119(1):101–110. [PMC free article] [PubMed] [Google Scholar]
- 29.Suratt BT, Petty JM, Young SK, Malcolm KC, Lieber JG, Nick JA, Gonzalo JA, Henson PM, Worthen GS. Role of the CXCR4/SDF-1 chemokine axis in circulating neutrophil homeostasis. Blood. 2004;104(2):565–571. doi: 10.1182/blood-2003-10-3638. [DOI] [PubMed] [Google Scholar]
- 30.Suratt BT, Young SK, Lieber J, Nick JA, Henson PM, Worthen GS. Neutrophil maturation and activation determine anatomic site of clearance from circulation. Am J Physiol Lung Cell Mol Physiol. 2001;281(4):L913–921. doi: 10.1152/ajplung.2001.281.4.L913. [DOI] [PubMed] [Google Scholar]
- 31.Petty JM, Sueblinvong V, Lenox CC, Jones CC, Cosgrove GP, Cool CD, Rai PR, Brown KK, Weiss DJ, Poynter ME, et al. Pulmonary stromal-derived factor-1 expression and effect on neutrophil recruitment during acute lung injury. J Immunol. 2007;178(12):8148–8157. doi: 10.4049/jimmunol.178.12.8148. [DOI] [PubMed] [Google Scholar]
- 32.Standiford TJ, Kunkel SL, Greenberger MJ, Laichalk LL, Strieter RM. Expression and regulation of chemokines in bacterial pneumonia. J Leukoc Biol. 1996;59(1):24–28. doi: 10.1002/jlb.59.1.24. [DOI] [PubMed] [Google Scholar]
- 33.Sordi R, Menezes-de-Lima O, Della-Justina AM, Rezende E, Assreuy J. Pneumonia-induced sepsis in mice: temporal study of inflammatory and cardiovascular parameters. Int J Exp Pathol. 2013 doi: 10.1111/iep.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bjorbaek C, Uotani S, da Silva B, Flier JS. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem. 1997;272(51):32686–32695. doi: 10.1074/jbc.272.51.32686. [DOI] [PubMed] [Google Scholar]
- 35.Claycombe K, King LE, Fraker PJ. A role for leptin in sustaining lymphopoiesis and myelopoiesis. Proc Natl Acad Sci U S A. 2008;105(6):2017–2021. doi: 10.1073/pnas.0712053105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aldawood A, Arabi Y, Dabbagh O. Association of obesity with increased mortality in the critically ill patient. Anaesth Intensive Care. 2006;34(5):629–633. doi: 10.1177/0310057X0603400501. [DOI] [PubMed] [Google Scholar]
- 37.Koc E, Ustundag G, Aliefendioglu D, Ergenekon E, Bideci A, Atalay Y. Serum leptin levels and their relationship to tumor necrosis factor-alpha and interleukin-6 in neonatal sepsis. J Pediatr Endocrinol Metab. 2003;16(9):1283–1287. doi: 10.1515/jpem.2003.16.9.1283. [DOI] [PubMed] [Google Scholar]
- 38.Bruno A, Pace E, Chanez P, Gras D, Vachier I, Chiappara G, La Guardia M, Gerbino S, Profita M, Gjomarkaj M. Leptin and leptin receptor expression in asthma. J Allergy Clin Immunol. 2009;124(2):230–237. 237 e231–234. doi: 10.1016/j.jaci.2009.04.032. [DOI] [PubMed] [Google Scholar]
- 39.Barazzone-Argiroffo C, Muzzin P, Donati YR, Kan CD, Aubert ML, Piguet PF. Hyperoxia increases leptin production: a mechanism mediated through endogenous elevation of corticosterone. Am J Physiol Lung Cell Mol Physiol. 2001;281(5):L1150–1156. doi: 10.1152/ajplung.2001.281.5.L1150. [DOI] [PubMed] [Google Scholar]
- 40.Shore SA, Schwartzman IN, Mellema MS, Flynt L, Imrich A, Johnston RA. Effect of leptin on allergic airway responses in mice. J Allergy Clin Immunol. 2005;115(1):103–109. doi: 10.1016/j.jaci.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 41.Montecucco F, Bianchi G, Gnerre P, Bertolotto M, Dallegri F, Ottonello L. Induction of neutrophil chemotaxis by leptin: crucial role for p38 and Src kinases. Ann N Y Acad Sci. 2006;1069:463–471. doi: 10.1196/annals.1351.045. [DOI] [PubMed] [Google Scholar]
- 42.Ottonello L, Gnerre P, Bertolotto M, Mancini M, Dapino P, Russo R, Garibotto G, Barreca T, Dallegri F. Leptin as a uremic toxin interferes with neutrophil chemotaxis. J Am Soc Nephrol. 2004;15(9):2366–2372. doi: 10.1097/01.ASN.0000139321.98029.40. [DOI] [PubMed] [Google Scholar]
- 43.Zarkesh-Esfahani H, Pockley AG, Wu Z, Hellewell PG, Weetman AP, Ross RJ. Leptin indirectly activates human neutrophils via induction of TNF-alpha. J Immunol. 2004;172(3):1809–1814. doi: 10.4049/jimmunol.172.3.1809. [DOI] [PubMed] [Google Scholar]
- 44.Lakatos HF, Burgess HA, Thatcher TH, Redonnet MR, Hernady E, Williams JP, Sime PJ. Oropharyngeal aspiration of a silica suspension produces a superior model of silicosis in the mouse when compared to intratracheal instillation. Exp Lung Res. 2006;32 (5):181–199. doi: 10.1080/01902140600817465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mancuso P. Obesity and lung inflammation. J Appl Physiol. 2010;108(3):722–728. doi: 10.1152/japplphysiol.00781.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baik I, Curhan GC, Rimm EB, Bendich A, Willett WC, Fawzi WW. A prospective study of age and lifestyle factors in relation to community-acquired pneumonia in US men and women. Arch Intern Med. 2000;160(20):3082–3088. doi: 10.1001/archinte.160.20.3082. [DOI] [PubMed] [Google Scholar]
- 47.Mancuso P. Obesity and respiratory infections: Does excess adiposity weigh down host defense? Pulm Pharmacol Ther. 2012 doi: 10.1016/j.pupt.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Brien JM, Jr, Phillips GS, Ali NA, Lucarelli M, Marsh CB, Lemeshow S. Body mass index is independently associated with hospital mortality in mechanically ventilated adults with acute lung injury. Crit Care Med. 2006;34(3):738–744. doi: 10.1097/01.CCM.0000202207.87891.FC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martino JL, Stapleton RD, Wang M, Day AG, Cahill NE, Dixon AE, Suratt BT, Heyland DK. Extreme obesity and outcomes in critically ill patients. Chest. 2011;140(5):1198–1206. doi: 10.1378/chest.10-3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stapleton RD, Dixon AE, Parsons PE, Ware LB, Suratt BT. The association between BMI and plasma cytokine levels in patients with acute lung injury. Chest. 2010;138(3):568–577. doi: 10.1378/chest.10-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.