

Abstract
Previous studies in human patients and animal models have suggested that transglutaminase 2 (TG2) is upregulated in pulmonary hypertension (PH), a phenomenon that appears to be associated with the effects of serotonin (5-hydroxytryptamine; 5-HT) in this disease. Using chemical tools to interrogate and inhibit TG2 activity in vivo, we have shown that pulmonary TG2 undergoes marked post-translational activation in a mouse model of hypoxia-induced PH. We have also identified irreversible fluorinated TG2 inhibitors that may find use as non-invasive positron emission tomography probes for diagnosis and management of this debilitating, lifelong disorder. Pharmacological inhibition of TG2 attenuated the elevated right ventricular pressure but had no effect on hypertrophy of the right ventricle of the heart. A longitudinal study of pulmonary TG2 activity in PH patients is warranted.
INTRODUCTION
Many previous studies have associated the biogenic amine serotonin (5-hydroxy-tryptamine; 5-HT) with clinical and experimental pulmonary hypertension (PH). Herve et al first identified the presence of pulmonary arterial hypertension (PAH) with depleted platelets and elevated serum 5-HT in patients with platelet storage disease (1, 2). Elevated serum 5-HT has also been noted in other patients with PAH (3). The fawn hooded rat with elevated serum levels of 5-HT has been shown to develop spontaneous PH (4). Intracellular uptake of 5-HT, enabled by the serotonin transporter, is enhanced in patients with PAH (5). At a cellular level, 5-HT has also been shown to stimulate smooth muscle cell proliferation and contractility (6–8), thereby providing a rationale for its role in pulmonary vascular remodeling and tension generation in PAH. Importantly, genetic or pharmacological inactivation of Tph1, the lung-associated tryptophan hydroxylase isozyme that is rate-limiting in serotonin biosynthesis, reduces or eliminates hemodynamic changes produced in mice exposed to hypoxia (9–12). Because a distinct isozyme of Tph1 is involved in 5-HT metabolism in the CNS, and because the CNS is chemically insulated from the rest of the body by the blood brain barrier, tryptophan hydroxylase inhibitors are being evaluated as potential therapeutic agents for PAH.
Notwithstanding the compelling association of 5-HT metabolism and transport with PH, several mechanistic details remain poorly understood. For example, both genetically reduced (13) and augmented (14) serotonin transporter activities have been shown to correlate with experimental PH. More fundamentally, the localization of pathogenically relevant 5-HT pools and their precise mode of action are unknown. An intriguing development in this regard is the recent observation that transglutaminase 2 (TG2), a ubiquitous intracellular and extracellular enzyme, catalyzes site-specific post-translational attachment of serotonin to Gln residues on certain proteins (15–17) (Figure 1). These observations suggest a potential link between 5-HT and TG2 in the development of PH. Preliminary support for this hypothesis emerged from studies in one of our laboratories, showing that the well-known substrate of TG2, fibronectin, is serotonylated when the proliferation and migration of pulmonary artery smooth muscle cells is induced by 5-HT (18). More recently, we have also detected an increase in serotonylated fibronectin in the lungs as well as serum of experimental models of pulmonary hypertension, and also in serum of patients with PAH (19). These findings motivated us to definitively establish whether TG2 activity was indeed elevated in lungs at the onset of PH. This question is particularly germane because TG2 is predominantly maintained in a catalytically inactive state in most organs of mammalian body (including the lung); its activity is regulated by elaborate mechanisms that operate inside and outside the cell (for review, see Klock and Khosla, 2012). To address this question, we took advantage of small molecule tools and methods for modulating and visualizing TG2 activity in mammals, developed by our other laboratory (21, 22). Specifically, we had previously shown that single doses of the biotinylated amine (5-biotinylamido pentylamine; 5 BP) can be systemically administered to rodents. It is covalently attached to tissues only upon allosteric activation of TG2. Our earlier studies had also led to the discovery of ERW1041E (23), a small molecule inhibitor of TG2 that pharmacologically blocks activated TG2 in vivo in a dose-dependent manner (21, 22). The results of our collaborative efforts are reported here.
Figure 1. Transglutaminase activity and chemical tools for investigating its role in PAH.

Transglutaminase 2 (TG2) catalyzes the formation of an isopeptide bond between a protein or peptide substrate harboring a target Gln residue and an acyl acceptor substrate, which may be a Lys residue on another protein or peptide or even a small molecule amine (panel A). One such biogenic amine substrate is serotonin (5-hydroxytryptamine, 5-HT). 5-biotinamidopentylamine (5BP) is another (unnatural) substrate for TG2 that is used as a probe for TG2 activity in tissues (panel B). ERW1041E (Panel C) (1) is an irreversible, acyl-donor substrate-competitive inhibitor of TG2.
RESULTS
TG2 activity is induced in lungs in response to hypoxia
Consistent with earlier findings in the gut and liver (21, 22), we found that very little, if any, biotinylation was detected in the lungs isolated from control mice exposed to room air (Figure 2A), indicating that basal TG2 activity in lung is low. In contrast, exposure to 10.5% hypoxia induced TG2 activation in mouse lung tissue within as few as 5 days. Importantly, TG2 activity increased steadily over the duration of hypoxia treatment (e.g., compare activity at day 5 versus day 16, Figure 2A), when the onset of PH was observed. This elevated TG2 activity was not associated with an increase in TG2 expression (indicated via immunofluorescent staining of TG2), leading to the speculation that the observed increase in activity was due to the well-understood mechanism of post-translational TG2 activation via disulfide bond reduction (24–27). The ostensibly high TG2 activity in heart tissue in normoxia control animals precluded the measurement of dynamic TG2 activity in cardiac tissue (Supplemental Figure S1A).
Figure 2. Hypoxia induced TG2 activity in mice.

C57BL/6 mice were placed in normobaric hypoxic (FIO2 10.5%) chambers for 5–16 days or left in atmosphere as the normoxia-exposed animal controls. TG2 activity in the lung was measured using the nucleophilic substrate, 5BP, as described in the text. To demonstrate that the observed 5BP incorporation was due to TG2 activity, the TG2 inhibitor, ERW1041E, was contemporaneously administered in animals exposed to 16 days of hypoxia. Alexa Fluor 488 (green) was used to visualize TG2 protein, whereas Alexa Fluor 555 (red) corresponded to TG2 activity. (A) Hypoxia induced TG2 activation in lung tissue compared to normoxic treatments. White scale bars are 100[m in length. (B) Quantitation of specific TG2 activity using ImageJ. At least 30 images per condition were collected of which representative images are shown. Data are represented as average +/− standard error. All images were processed identically.
In addition to visual inspection, fluorescent images were quantified using the ImageJ software. Specifically, the average pixel intensity of TG2 (green) and 5BP (red) was calculated for each image. The relative TG2 activity was measured by dividing the average 5BP (red) intensity by the average TG2 (green) intensity and normalized to the normoxia control animals. In hypoxia-treated animals, a steady increase in TG2 activity was observed over a period of 16 days; TG2 activity was up to 4.5-fold higher in hypoxic versus control mice (Figure 2B). TG2 activity in heart was constitutively high in room-air exposed mice, and did not change with hypoxia exposure time (Supplemental Figure S1B).
We also extended our studies to include combination treatment with hypoxia and the anti-angiogenic agent SU-5416, as previously reported in experimental models of PH (12, 28). The basal TG2 activity was low in not only control mice but also in mice exposed only to SU-5416. Analogous to the above findings, combined exposure to hypoxia and SU-5416 resulted in more than 3-fold higher TG2 activity in mouse lung tissue (Figures 3A & B).
Figure 3. Duration of TG2 blockade in pulmonary hypertensive mice.

C57BL/6 mice were placed in normobaric hypoxic (FIO2 10.5%) chambers for 3 weeks or left in atmosphere as the normoxia-exposed animal controls. SU-5416 was given weekly to hypoxia exposed animals, as previously reported in experimental models of pulmonary hypertension (28). TG2 protein and activity in the lung was quantified, as described in the caption to Figure 2. (A) Hypoxia and SU-5416 induced TG2 activation in lung tissue compared to normoxia controls. (B) Quantitative analysis of specific TG2 activity in lungs using ImageJ. (C) The duration of TG2 blockade in response to a single IP injection of 50 mg/kg ERW1041E was estimated. ERW1041E or vehicle was administered 0.5, 6, 18, and 26 hours prior to sacrifice. At least 30 images per condition were collected of which representative images are shown. White scale bars are 100[m in length. (B and D) Data represented as average +/− standard error. All images were processed identically.
Inhibition of TG2 activity in mouse lung by ERW1041E
To verify that the observed 5BP incorporation in the above experiments was indeed due to TG2 activity, the irreversible active site directed TG2 inhibitor ERW1041E (22, 23) was co-administered. As shown in Figures 2 (A&B) and 3 (A&B), complete suppression of 5BP incorporation was observed in the mouse lung tissue. Because this compound has a greater than 10-fold specificity for TG2 over Factor XIIIa (discussed below), the TG2 isoform with appreciable activity in the lung (29), our data suggest that TG2 is induced in the mouse lung in response to hypoxia. This inference is also consistent with our previous discovery that serotonylated fibronectin is elevated in the lungs of mice and rats exposed to hypoxia (19).
In order to determine the duration for which ERW1041E was able to block TG2 activity, mice were exposed to hypoxia plus SU-5416 for 3 weeks, followed by treatment with a single intraperitoneal dose of 50 mg/kg ERW1041E. At various intervals from 0 to 26 h after ERW1041E administration, 100 mg/kg 5BP was administered intraperitoneally, followed by euthanasia 30 min later. Although TG2 activity remained mostly suppressed up to 6 h after ERW1041E administration, it gradually returned toward normal levels over the ensuing 26 hours of the study (Figures 3C & D). The t1/2 of TG2 inhibition was estimated as 12 ± 1 h (Figure 3D), suggesting that a twice-daily dose of ERW1041E could adequately suppress TG2 activity in a mouse model of hypoxia-induced chronic PH.
Effect of TG2 inhibition on measures of murine pulmonary hypertension
A 3-week study was performed in which mice were exposed to hypoxia plus SU-5416, along with a once- or twice-daily IP injection of 50 mg/kg ERW1041E. After 3 weeks, the animals were removed to normal air, and body weights (BW) as well as right ventricular systolic pressure (RVSP) were recorded. The animals were then euthanized, the heart dissected, and the right ventricle (RV) was separated from the left ventricle and septum (LV + S). The RV/BW and RV/LV+S ratios were calculated. Mice exposed to hypoxia plus SU-5416 for 3 weeks showed a strong increase in RVSP and RV weight normalized to BW or LV + S weight (Figure 4). Concurrent ERW1041E administration led to a significant reduction in RVSP (Figure 4A). Although the twice-daily dosing schedule showed mild reduction of RV/BW (Figure 4B), the difference was not statistically significant (p=0.087). Similarly, the difference in RV/LV + S values between ERW1041E treated and untreated animals was not statistically different (Figure 4C).
Figure 4. Hemodynamic measurements in pulmonary hypertensive mice treated with TG2 inhibitor.

C57BL/6 mice were placed in normobaric hypoxic (FIO2 10.5%) chambers for 3 weeks or left in atmosphere as the normoxia-exposed animal controls. SU-5416 was given weekly as described previously (28). Following three weeks of normoxia or hypoxia plus SU-5416, animals were removed to room air and anesthetized. (A) Right ventricular systolic pressure (RVSP) was measured as noted in Methods. Body weights (BW) were recorded. The animals were then euthanized. The heart was dissected and right ventricular (RV) and left ventricular plus septal (LV + S) weights were determined. (B) RV/BW and (C) RV/LV+S ratios were calculated. Values are expressed as means +/− SEM, n= 7 animals in each group. ERW1041E was given once per day (1X) or twice daily (2X) at 50 mg/kg. + p <0.05 vs normoxic; * p < 0.05 vs hypoxic/SU-5416.
Improved multigram-scale synthesis of ERW1041E
A large quantity of ERW1041E was required to initiate and verify reproducibility of the chronic rodent studies detailed above. It was therefore necessary to render the synthetic route to this enantiopure agent more scalable. In particular, in the previously published synthesis, the preparation of the nitrophenylcarbonate building block [(5a), step (e) in Scheme 1] which serves as the precursor to the carbamate moiety in (1) and its coupling to proline methyl ester [step (a)] proved unsatisfactory, both from the perspective of synthesis yields and the need for two chromatographic purification steps (23, 30).
Scheme 1.

(a) (5a), NMM (3 eq.), DMF ; (a*) (5b), NEt3 (1 eq.), DMAP (0.1 eq), DMF/DCM 3:2, overnight RT; (b) aq. LiOH, MeOH/THF; (c) EDCI, HOBt, NMM, (S)-DHI, DMF, (d) LiBH4, EtOH/THF, −78°C; (e) p-nitrophenyl chloroformate, NMM, DCM; (e*) 1,1′-carbonyldiimidazole, ACN, RT; (f) TFA
We therefore investigated the utility of 1,1′-carbonyldiimidazole (CDI) in place of p-nitrophenyl chloroformate for the introduction of the carbamate group. CDI appeared as the reagent of choice, given that it readily forms the desired imidazolyl carbamate (5b) with quinolyl-3-methanol at ambient conditions (31, 32). The desired carbamate precipitates in high purity and yield within a few minutes. When this imidazolyl carbamate was reacted with L-proline methyl ester hydrochloride in the presence of an equimolar amount of triethylamine and catalytic DMAP in dichloromethane/DMF overnight, extraction furnished the product in good yield and purity. The compound could be taken through the subsequent synthetic steps without additional purification, effectively eliminating two of three chromatographic purification steps.
Discovery of fluorinated ERW1041E analogs as potential positron emission tomography probes of pulmonary TG2 activity
When taken in conjunction with our recent finding of elevated serotonylated fibronectin in the serum of PAH patients (19), the above results in mouse models of experimental PH suggest that post-translationally activated TG2 in the lung may be a useful quantitative biomarker of disease progression in PAH and related PH disorders. Testing this hypothesis would require discovery of a fluorinated TG2 inhibitor binding exclusively to the catalytically active enzyme; such an agent could be used in conjunction with positron emission tomography (PET).
One strategy towards that end is the incorporation of bioorthogonal functional groups into a TG2 inhibitor that could be used to functionalize the compound with a PET-active nucleus. In fact, we have previously reported a set of irreversibly binding probes of the TG2 active site that included “clickable” alkyne or azide-functionality (e.g., 4) (30). Unfortunately, the clickable moiety could only be introduced at the expense of inhibitor potency. We therefore sought to investigate the feasibility of introducing appropriate modifications at other sites in ERW1041E.
Hydroxylation of the L-proline ring at the 3 and 4 positions was well tolerated in the trans stereochemistry (data not shown). In particular, the trans-4-hydroxy substitution had comparable potency to ERW1041E, as shown in Table 1. The 4-hydroxy group was therefore used as a functional handle to attach the desired propargyl group (3). We also prepared the 4-fluoro derivatives and, surprisingly, found that the 4-cis derivative (2a) was more potent than the corresponding trans diastereomer (2b). Given the strong conformational bias a 4-fluoro substituent imposes on the proline ring (33), this observation might be explained by opposing conformational and steric trends for the position of substituents on the proline ring.
Table 1. Specificity of ERW1041E (1) and analogs against TG2 and selected isoforms.
Inhibitors were profiled in the GDH coupled deamidation assay (40, 41), and the inhibition parameters kinh and Ki were obtained by fitting the progress curves to an irreversible inhibition model (42, 43).
| # | R |
Kinh/Ki [mM−1 min−1]
|
|||||
|---|---|---|---|---|---|---|---|
| TG2 | mTG2 | TG1 | TG3 | FXIIIa | |||
|
|
|
|
|
|
|
||
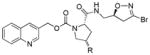
|
1 | -H | 18.5 | 6.1 | 16.9 | < 1 | 1.3 |
| 2a | -F (cis) | 20.9 | 4.6 | 19.4 | < 1 | 1.5 | |
| 2b | -F (trans) | 10.5 | 4.4 | 10.0 | < 1 | n/d | |
| 3 |
|
13.7 | 6.1 | 10.3 | < 1 | n/d | |
|
|
|
|
|
|
|
||
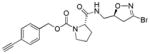
|
4 | 9.7 | 3.4 | 6.2 | < 1 | n/d | |
n/d = not determined
To assess their potential as TG2 specific probes, we profiled our inhibitors against major other human transglutaminase isoforms, specifically keratinocyte transglutaminase TG1, epidermal TG3, as well as the ubiquitous, blood-borne Factor XIIIa (34, 35). As summarized in Table 1, ERW1041E and its analogues possessed good specificity for TG2 over TG3 and FXIIIa, but were roughly equipotent towards TG1 in vitro, rendering these compounds as promising avenues for probe development.
DISCUSSION AND CONCLUSIONS
We have utilized a method developed by one of our laboratories (21, 22), in which 5BP is administered as a nucleophilic TG2 substrate, to assess in situ activity of TG2 in lungs of mice showing the onset of experimental pulmonary hypertension (PH). Although baseline activity of TG2 is very low or non-existent under room air conditions, it becomes markedly elevated by 16 days of exposure to 10.5 % O2 (Figure 2). This level of hypoxia leads to the development of pulmonary hypertension in these animals by 3 weeks of exposure. The marked elevation of TG2 activity detected at 16 days argues that TG2 activity is associated with this model PH, and is consistent with our previous studies in which we observed enhanced serotonylation of fibronectin in the lungs of hypoxic mice (19).
We also assessed whether the hypoxia induced TG2 activity could be pharmacologically blocked using the selective TG2 inhibitor, ERW1041E. To determine duration of inhibition, we first elevated TG2 activity by exposure of mice to hypoxia for 21 days and then gave a single injection of ERW1041E. Strong inhibition of TG2 activity was observed, when ERW1041E was administered at a dose of 50 mg/kg, although residual TG2 activity could be detected (Figure 3). A high degree of TG2 inhibition was obtained at 30 min after administration of ERW1041E. It remained reduced at 6 hours and gradually returned toward baseline at 18 and 26 hours after administration. From these experiments we concluded that TG2 activity could be considerably reduced but not eliminated for most of a 24-hour interval by twice daily injections. We estimated that the inhibitory half-life of ERW1041E was ca. 12 h, implying that a twice daily dose of ERW1041E would adequately reduce continual hypoxia mediated TG2 activity.
We next performed experiments to determine whether ERW1041E could modulate hemodynamic effects of exposures of mice to hypoxia in models of experimental pulmonary hypertension. Weekly injections of SU-5416 were introduced, as we (unpublished data) and others (28) have found a more profound effect of hypoxia on pulmonary hypertension in mice when this anti-angiogenic agent is used in combination with hypoxia. However, the combination did not significantly elevate TG2 activity compared to hypoxia treatment alone (Figure 3). Dosing of ERW1041E at both 24 and 12 h intervals produced significant reductions in RVSP but the effect on RV/BW was not statistically significant and the RV/LV + S remained unaltered. Any higher or more frequent dosing schedule would have been potentially problematic and costly to achieve. It remains to be determined if alternative methods, such as smooth muscle actin staining and vessel occlusion or nucleus defined area ratios, support the role of TG2 activity in PAH dependent vascular remodeling or hypertrophy, respectively. Future studies along these lines are being planned.. It is also of interest that relatively high TG2 activity was observed in the hearts of control mice. The mechanistic rationale for high cardiac TG2 activity is unknown.
Taken together, our findings suggest an association between elevated TG2 activity and experimental PH but not necessarily causation. While further studies are needed to discern whether or not small molecule inhibition of TG2 does or does not constitute a therapeutic strategy for the treatment of PAH, pulmonary TG2 activity is clearly identified as a promising biomarker for PAH disease states. Activity based probes, such as the ERW1041E analogs described here (Table 1), could therefore provide powerful tools for studying PAH. In particular, derivatives bearing bioorthogonal functionalities such as alkynes and fluorinated derivatives that can be adapted as PET probes, are desirable. Compounds 2a and 2b fall in the latter category, whereas compound 3 could be a platform compound for “click” reactions (for review, see Glaser and Robins, 2009).
Activity-based chemical tools have been used to demonstrate that the development of pulmonary hypertension in mice exposed to hypoxia elevates TG2 activity in the lungs of these animals. This is the first observation of this phenomenon, and makes a strong argument for further investigations into the relevance of this enzyme activity as a biomarker of pulmonary arterial hypertension. Derivatives of an irreversible TG2 inhibitor, ERW1041E, have also been engineered as potential probes of TG2 activity in vivo, and could find use in the context of disease imaging.
EXPERIMENTAL METHODS
Synthesis
(S)-quinolin-3-ylmethyl 2-((((S)-3-bromo-4,5-dihydroisoxazol-5-yl)methyl)carbamoyl)pyrrolidine-1-carboxylate (1, ERW1041E)
Commercial proline methyl ester hydrochloride (15.69 g, 95 mmol) was dissolved in 150 mL DMF and catalytic N,N-dimethylpyridin-4-amine (1.930 g, 15.79 mmol) was added, followed by triethylamine (11.01 ml, 79 mmol). The mixture was stirred and a white solid precipitated. Separately, quinolin-3-ylmethyl 1H-imidazole-1-carboxylate (5b) (20 g, 79 mmol) was dissolved in 100 mL methylene chloride and then added to the reaction mixture. The mixture was stirred overnight at room temperature, before the volatiles were evaporated to about 70 mL. A white solid was filtered off. The filtrate was diluted with 300 mL water and some hydrochloric acid (1 M) was added to reach pH 5–6. The cloudy mixture was diluted with 500 mL ethyl acetate, partitioned, and the organic layer washed with brine (4 × 150 mL). The organic layer was dried over sodium sulfate and evaporated under reduced pressure, yielding (S)-2-methyl 1-(quinolin-3-ylmethyl) pyrrolidine-1,2-dicarboxylate (21.5 g, 68.4 mmol, 87 % yield) that was used in the next step without prior purification.
The saponification of the intermediate ester and the subsequent amide coupling were performed similar to the previously reported protocol (23). The ester (21.5 g, 68.4 mmol) was thus dissolved in 200 mL THF/MeOH 1:1 and LiOH (1 M in water) was added in small aliquots until the starting material had disappeared by TLC (9:1 ethyl acetate / hexanes). An equivalent amount of hydrochloric acid (1 M) was added and the volatiles were removed under reduced pressure. The crude oil was diluted with lightly acidic water and extracted with ethyl acetate (4 × 150 mL). The combined organic extracts were dried over sodium sulfate and evaporated under reduced pressure, furnishing (S)-1-((quinolin-3-ylmethoxy)carbonyl) pyrrolidine-2-carboxylic acid (18.8 g, 62.6 mmol, 92 % yield) as a yellow oil.
An aliquot of the free acid (17.8 g, 59.3 mmol) that was used without purification from the previous step, EDCI·HCl (13.07 g, 68.2 mmol), and 1H-benzo[d][1,2,3]triazol-1-ol (8.01 g, 59.3 mmol) were dissolved in DMF (120 mL) and 4-methylmorpholine (NMM) (6.52 ml, 59.3 mmol) was added. The mixture was stirred for 10 min, and enantiomerically enriched (S)-(3-bromo-4,5-dihydroisoxazol-5-yl)methanamine (S-DHI)(10.61 g, 59.3 mmol) was added dropwise. The mixture was stirred at room temperature for 2 h before it was diluted with 250 mL water. This mixture was diluted with 600 mL ethyl acetate and extracted. The organic layer was washed with brine (4 × 200 mL), dried over sodium sulfate and evaporated under reduced pressure, furnishing the desired product in good yield and purity. The crude material was purified on a short silica gel column (10 cm diameter, 15 cm length) with a stepwise gradient of 600 mL 80% EtOAc/pentane, 600 mL 90% EtOAc/pentane, 1000 mL EtOAc, 600 mL 5% MeOH in EtOAc, 1000 mL 10% MeOH in EtOAc. After pooling all fractions containing product and evaporation of the volatiles, (S)-quinolin-3-ylmethyl 2-((((S)-3-bromo-4,5-dihydroisoxazol-5-yl)methyl)carbamoyl)pyrrolidine-1-carboxylate (22.55 g, 48.9 mmol, 82 % yield) was obtained as a viscous oil that foamed under high vacuum. A fine amorphous solid was obtained by crushing the solidified foam. 1H NMR (400 MHz, DMSO-d6, mixture of rotational isomers) δ 8.93 & 8.84 (2 d, J=1.9 Hz, 1H), 8.20–8.37 (m, 2H), 7.90–8.06 (m, 2H), 7.69–7.83 (m, 1H), 7.62 (t, J=7.6 Hz, 1H), 5.16–5.37 (m, 2H), 4.73 & 4.59 (2 m, 1H), 4.27 & 4.19 (dd, J=2.8, 8.2 Hz & dd, J=3.5, 8.4 Hz, 1H), 3.12–3.56 (m, 5H), 2.92–3.10 (m, 1H), 1.99–2.24 (m, 1H), 1.71–1.91 (m, 3H).
(2S,4S)-quinolin-3-ylmethyl 2-((((S)-3-bromo-4,5-dihydroisoxazol-5-yl)methyl)carbamoyl)-4-fluoropyrrolidine-1-carboxylate (2a)
Commercial (2S,4S)-1-(tert-butoxycarbonyl)-4-fluoropyrrolidine-2-carboxylic acid (0.1003 g, 0.430 mmol, 1 eq.), enantiomerically enriched S-DHI (0.0779 g, 0.436 mmol, 1 eq.), EDCI·HCl (0.1102 g, 0.575 mmol, 1.3 eq.), and 1-hydroxybenzotriazole hydrate (0.0784 g, 0.512 mmol, 1.2 eq.) were dissolved in DMF (2 mL) and stirred as NMM (0.06 mL, 0.055 mmol, 1.25 eq.) was added dropwise. The solution was stirred for 3 hours, then was diluted with ethyl acetate (150 mL) and washed with water (50 mL) and sodium bicarbonate solution (2 × 40mL). The organic layer was dried over sodium sulfate and evaporated under reduced pressure to yield crude tert-butyl (2S,4S)-2-((((S)-3-bromo-4,5-dihydroisoxazol-5-yl)methyl)carbamoyl)-4-fluoropyrrolidine-1-carboxylate) as a solution in residual DMF which was used without further purification.
The intermediate from the previous step was dissolved in DCM (4 mL) and stirred as TFA (1.0 mL) was added. Stirring was continued for 45 minutes when the reaction mixture was diluted with ethyl acetate (100 mL). This organic mixture was partitioned with sodium carbonate solution at a pH of 9. The organic layer was dried over sodium sulfate and evaporated by reduced pressure to yield crude (2S,4S)-N-(((S)-3-bromo-4,5-dihydroisoxazol-5-yl)methyl)-4-fluoropyrrolidine-2-carboxamide (0.128 g, 0.435 mmol, 101.2 % yield over 2 steps) which was used without further purification.
(2S,4S)-N-(((S)-3-bromo-4,5-dihydroisoxazol-5-yl)methyl)-4-fluoropyrrolidine-2-carboxamide (0.128 g, 0.435 mmol, 1 eq.) and building block (5a) (0.1215 g, 0.375 mmol, 0.85 eq.) were dissolved in DMF (2 mL) and stirred as NMM (0.07 mL, 0.640 mmol, 1.5 eq.) was added dropwise. The solution was stirred for 23 hours, then diluted with 125 mL of ethyl acetate and washed with sodium bicarbonate solution (2× 40 mL), sodium carbonate solution (30 mL), and brine (40 mL). The organic layer was dried over sodium sulfate, evaporated under reduced pressure, and then purified by column chromatography with a gradient ranging from 50% EtOAc:50% Pentane – 5% MeOH: 95% EtOAc to yield the title compound (0.0846 g, 0.177 mmol, 40.7% yield) as a viscous oil. 1H NMR (400 MHz, CDCl3, mixture of rotational isomers) δ 8.92 (br. s., 1H), 8.17 (br. s., 1H), 8.10 (d, J=8.4 Hz, 1H), 7.84 (d, J=7.5 Hz, 1H), 7.66–7.79 (m, 1H), 7.56 (t, J=7.4 Hz, 1H), 6.78 (br. s., 1H), 5.22–5.50 (m, 2H), 5.28 & 5.15 (2 t, J=3.3 Hz, 1H), 4.60–4.83 (m, 1H), 4.51 (d, J=10.1 Hz, 1H), 3.77–3.96 (m, 1H), 3.31–3.74 (m, 3H), 3.12–3.30 (m, 1H), 2.96–3.12 (m, 1H), 2.62 (br t, J=15.9 Hz, 1H), 2.21–2.43 (m, 1H). HRMS (ESI-QTOF) m/z: calculated for C20H21BrFN4O4 [M+H]+ 479.0725; found 479.07198.
(2S,4R)-quinolin-3-ylmethyl 2-((((S)-3-bromo-4,5-dihydroisoxazol-5-yl)methyl)carbamoyl)-4-fluoropyrrolidine-1-carboxylate (2b)
Compound (2b) was prepared by the method used to prepare (2a), furnishing the product as a viscous oil (0.0608 g, 0.127 mmol, 29.3 % yield for 3 steps). 1H NMR (400 MHz, CDCl3, mixture of rotational isomers) δ 8.86–8.96 (m, 1H), 8.04–8.20 (m, 2H), 7.83 (d, J=7.9 Hz, 1H), 7.72 (t, J=7.6 Hz, 1H), 7.56 (t, J=7.5 Hz, 1H), 7.08 & 6.82 (br. s., 1H), 5.10–5.45 (m, 3H), 4.78 & 4.32–4.53 (m., 2H), 3.85–4.10 (m, 1H), 3.31–3.75 & 2.79 (m, 3H), 3.22 (dd, J=10.8, 17.5 Hz, 1H), 3.01 (dd, J=8.0, 17.4 Hz, 1H), 2.53 (m, 1H), 2.20–2.44 (m, 1H). HRMS (ESI-QTOF) m/z: calculated for C20H21BrFN4O4 [M+H]+ 479.0725; found 479.07199.
(2S,4R)-quinolin-3-ylmethyl 2-((((S)-3-bromo-4,5-dihydroisoxazol-5-yl)methyl)carbamoyl)-4-(prop-2-yn-1-yloxy)pyrrolidine-1-carboxylate (3)
NaH (60% oil suspension, 0.1402 g, 3.5 mmol, 1.5 eq.) was suspended in DMF (10 mL) and combined with commercially available (2S,4R)-1-tert-butyl 2-methyl 4-hydroxypyrrolidine-1,2-dicarboxylate (0.3959 g, 1.6mmol, 1 eq.) dissolved in DMF (2 mL). The resulting suspension was then stirred at −10°C for 30 minutes before propargyl bromide (1.80 mL, 20.4 mmol, 15 eq.) was added dropwise. The suspension was stirred for an hour at −10 °C and then stirred at room temperature for 15 hours. The solution was diluted with DCM (50 mL) and partitioned with brine (4 × 30 mL). The resultant organic phase was dried over sodium sulfate, evaporated under reduced pressure, and then purified by column chromatography (10%-40% EtOAc:Pentane) to yield 1-(tert-butyl) 2-methyl (2S, 4R)-4-(prop-2-yn-1-yloxy)pyrrolidine-1,2-dicarboxylate (0.093 g, 0.359 mmol, 25.3% yield).
tert-Butyl (2S, 4R)-4-methoxy-2-(prop-2-yn-1-yl)pyrrolidine-1-carboxylate (0.093 g, 0.359 mmol, 1 eq.) was dissolved in 5 mL of THF, combined with 1 M LiOH (0.34 mL, 0.34 mmol, 1 eq.), and stirred for an hour, before an equimolar amount of hydrochloric acid was added and the volatiles removed under reduced pressure. The aqueous solution was further acidified to pH 4 and then extracted with ethyl acetate (3 × 50 mL). The organic phases were combined, dried over sodium sulfate, and evaporated under reduced pressure to yield crude (2S, 4R)-1-(tert-butoxycarbonyl)-4-(prop-2-yn-1-yloxy)pyrrolidine-2-carboxylic acid (0.090 g, 0.334 mmol, 94 % yield).
This intermediate was elaborated using the amide coupling methodology employed for compound (2a). The crude product (0.044 g, 0.103 mmol, 30.6 % yield) from this step was combined with a second aliquot from a repeated preparation. The pooled tert-butyl (2S, 4R)-2-((((S)-3-bromo-4,5-dihydroisoxazol-5-yl)methyl)carbamoyl)-4-(prop-2-yn-1-yloxy)pyrrolidine-1-carboxylate (0.083 g, 0.193 mmol) was reacted with DCM/TFA as described above. The deprotected product was isolated as its TFA salt by evaporation of the volatiles and directly subjected to a coupling reaction with building block (5a) as described above (with an additional 0.5 eq. NMM). Preparative TLC (100% EtOAc) of the crude product furnished the title compound as a viscous oil (0.0438 g, 0.085 mmol, 44 % yield over two steps). 1H NMR (400 MHz, CDCl3, mixture of rotational isomers) δ 8.81–8.95 (m, 1H), 8.03–8.22 (m, 2H), 7.77–7.95 (m, 1H), 7.65–7.77 (m, 1H), 7.56 (t, J=7.9 Hz, 1H), 6.93 & 6.63 (2 br. s., 1H), 5.16–5.53 (m, 2H), 4.92 & 4.12 (2 s, 2H), 4.78 & 4.46 (2 br m, 1H), 4.26–4.43 (m, 2H), 3.55–3.90 (m, 2H), 3.33–3.54 (m, 1H), 3.13–3.31 (m, 1H), 2.96–3.12 (m, 1H), 2.64–2.85 (m, 1H), 2.34–2.48 (m, 1H), 2.24–2.34 (m, 1H), 1.98–2.20 (m, 1H). HRMS (ESI-QTOF) m/z: calculated for C23H24BrN4O5 [M+H]+ 515.0925; found 515.0921.
(S)-4-ethynylbenzyl 2-((((S)-3-bromo-4,5-dihydroisoxazol-5-yl)methyl)carbamoyl)pyrrolidine-1-carboxylate (4)
Compound (4) was prepared according to a previously published procedure (30)
(S)-(3-bromo-4,5-dihydroisoxazol-5-yl)methanamine (S-DHI)
The racemic DHI moiety was prepared by the [3+2] dipolar cycloaddition route reported in the literature (37). Deviating from the published procedure, we found that when we left out methylene chloride as a putative surfactant, we could filter dibromoformaldoxime as a shiny white solid from the reaction mixture in satisfactory yield (103 g, 64.9 %), obviating the need for an extraction and solvent evaporation step for this reactive and potentially dangerous intermediate. The S-DHI enantiomer was enriched from the crude racemic mixture by fractional crystallization with L-mandelic acid, inspired by a previously reported procedure (38). To this end, L-mandelic acid (42.4 g, 278 mmol) was dissolved in hot methanol (approx. 250 mL) and the crude product (99.7 g, 557 mmol combined from two cycloaddition preparations, individually furnishing 253 mmol in 49.8 % yield and 304 mmol in 55.4 % yield, respectively), was diluted with an equal volume of hot absolute ethanol. The two solutions were combined and a large amount of the mandelate salt crystallized upon cooling to room temperature. The solid was filtered off and recrystallized in batches from a mixture of methanol and ethanol furnishing the mandelate salt as long white needles. The desired compound could be obtained by suspending the crystals in water, followed by basifying the solution with 10% aqueous potassium carbonate to a pH of 11–12 and extracting the solution with methylene chloride (5x).
4-nitrophenyl (quinolin-3-ylmethyl) carbonate (5a)
Compound (5a) was prepared according to our previously published procedure (23)
Quinolin-3-ylmethyl 1H-imidazole-1-carboxylate (5b)
Commercial quinoline-3-carbaldehyde (15 g, 95 mmol) was reduced as previously described (23). The resulting quinolin-3-ylmethanol (15.2 g, 95 mmol) and di(1H-imidazol-1-yl)methanone (31.0 g, 191 mmol) were separately dissolved in 100 mL anhydrous acetonitrile each and combined while stirring at room temperature. Stirring was continued for another 30 minutes, while a large quantity of a white solid formed. The solid was filtered off, washed with little acetonitrile, and dried. Additional crops of white solid could be obtained by reducing the volume of the filtrate under reduced pressure and repeated filtration, furnishing quinolin-3-ylmethyl 1H-imidazole-1-carboxylate in excellent combined yield (22 g, 87 mmol, 91 % yield) and without the need for any purification. 1H NMR (500 MHz, CDCl3) δ 8.99 (d, J=2.20 Hz, 1H), 8.26 (d, J=1.71 Hz, 1H), 8.12–8.16 (m, 2H), 7.86 (d, J=8.06 Hz, 1H), 7.77 (ddd, J=1.22, 7.02, 8.36 Hz, 1H), 7.58–7.61 (m, 1H), 7.42 (s, 1H), 7.06 (s, 1H), 5.60 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 150.5, 148.4, 148.2, 137.0, 136.6, 130.8, 130.4, 129.3, 127.9, 127.33, 127.31, 126.7, 117.0, 67.4
5-biotinamidopentylamine hydrochloride (5-BP*HCl)
D-(+)-biotin (3.26 g, 13.35 mmol) and tert-butyl (5-aminopentyl)carbamate (2.7 g, 13.35 mmol), prepared by the method of Lee (39), and EDCI·HCl (2.94 g, 15.35 mmol) were suspended in acetonitrile (70 mL). Water (30 mL) was added, followed by NMM (2.70 g, 26.7 mmol). The mixture was stirred for 3 h, whereafter the volatiles were removed under reduced pressure, furnishing the crude product as a yellow oil. The oil was diluted with water and a first crop of the product could be filtered off. On standing for several days, a number of additional crops of product could be filtered off, accounting for a moderate additional yield. The crude product was loaded onto a silica gel column in ethyl acetate with methanol and eluted slowly with a gradient up to 15% methanol in ethyl acetate. Upon evaporation of the pooled fractions, tert-butyl 5-(biotinamidopentyl) carbamate (3.0 g, 7.00 mmol, 52.4 % yield) was obtained as a gel that slowly solidified under vacuum. An aliquot of the white solid (2.5 g, 5.83 mmol) was suspended in 15 mL of 4 M HCl in dioxane and stirred for 10 minutes. Then, 15 mL of methanol were added and residual suspended solids dissolved. Stirring was continued for another 10 minutes. The crude mixture was slowly added to 220 mL diethyl ether under vigorous stirring and stirring continued for 1 hour. During this time, a sticky oil had formed on the walls of the flask. The solvent was decanted off and the flask washed with additional ether. Then, the oily solid was taken up in methanol and the clear solution evaporated under reduced pressure, yielding the title compound as an off-white foam (1.3 g, 3.56 mmol, 61.1 % yield). 1H NMR (500 MHz, D2O) δ 4.53 (d, J=5.13 Hz, 1H), 4.35 (d, J=7.32 Hz, 1H), 3.27 (br. s., 1H obscured by residual methanol solvent signal), 3.12 (d, J=5.62 Hz, 2H), 2.81–2.98 (m, 3H), 2.71 (d, J=13.43 Hz, 1H), 2.08–2.26 (m, 2H), 1.15–1.79 (m, 12H). 13C NMR (126 MHz, D2O) δ 177.3, 165.8, 62.7, 60.9, 56.0, 40.3, 40.0, 39.6, 36.0, 28.53, 28.45, 28.3, 27.0, 25.8, 23.6.
In vitro transglutaminase assays
Human transglutaminase 2 (TG2) was expressed from plasmid pJLP4 in E. coli strain Rosetta 2(DE3) pLysS, and purified by sequential affinity and anion-exchange chromatography, as previously described (40). The other transglutaminases human TG1, TG3, Factor XIIIa as well as murine TG2 were also recombinantly expressed in E. coli, and chromatographically purified as will be described elsewhere (C.K. et al., unpublished data). The activity of recombinant transglutaminases was assessed using the glutamate dehydrogenase (GDH) coupled deamidation assay (40, 41) with Cbz-Gln-Gly or other appropriate peptides as acyl donor substrates in the absence of an amine-based acceptor substrate (C.K. et al., unpublished data). Inhibitor potencies were measured in this assay as previously described (42) and inhibition parameters kinh and Ki obtained by fitting the progress curves to an irreversible inhibition model (42, 43).
Hypoxia induced TG2 activation in mice
Chemical tools were formulated fresh and used the same day for each mouse experiment. ERW1041E was prepared in 20% DMSO (v/v) in water to 10mg/mL. 5-biotinamidopentylamine (5BP) was dissolved in PBS to 20mg/mL free amine equivalents. All chemical tools were then passed through a disposable 0.2 μm sterile polyvinylidenefluoride (PVDF) membrane filter prior to injection. C57BL/6 mice were obtained from Charles River Laboratories (Boston, Ma). The Tufts IACUC Committee approved all animal studies. Male C57BL/6 mice 6–8 weeks old (~20 grams body weight) were placed in normobaric hypoxic (FIO2 10.5%) chambers for 5–21 days or left in atmosphere as the normoxia-exposed animal controls. Thirty minutes prior to sacrifice, mice were injected with a TG2 substrate, 5BP, 100 mg/kg as an intraperitoneal (IP) injection. To ensure that 5BP incorporation depended on the TG2 activity, we contemporaneously administered 50 mg/kg IP of a selective TG2 inhibitor, ERW1041E. Mice were then sacrificed and heart and lung tissue dissected. While the heart was directly embedded in optimal cutting temperature (OCT), the left lung was inflated with 1mL OCT and frozen for sectioning. Frozen mouse tissue cryomolds were transferred to a Leica CM3050S cryostat already equilibrated to −20°C. Tissue blocks were removed from the cryomolds, cut into 8 micron thin sections, and transferred to room temperature superfrost plus microslides. Microslides were stored at −20°C post tissue transfer. Tissue mounted microslides were fixed in 4% (w/v) paraformaldehyde for 15 min at room temperature, thoroughly washed with PBS, and blocked with 5% (w/v) BSA in PBS supplemented with 0.1% (v/v) Tween-20 overnight at 4°C. Tissue mounted microslides were treated with primary antibody 1:500 (v/v) dilution of mouse anti-TG2 IgG MAb (TG100 + CUB7402 mix) in blocking buffer overnight at 4°C. Slides were washed three times with PBS supplemented with 0.1% (v/v) Tween-20 for 5 min at room temperature. Slides were then treated with secondary antibodies, goat anti-mouse (H-L) IgG Alexa Fluor 488 and Streptavidin Alexa Fluor 555 conjugates, diluted 1:1000 (v/v) in blocking buffer overnight at 4°C. Slides were thoroughly washed four times for 5 min each with PBS supplemented with 0.1% (v/v) Tween-20 and air dried in the dark at room temperature. Cover slips were then mounted using Vectashield mounting medium and sealed onto the slides using multiple coats of clear nail polish. Sealed slides were stored in the dark at 4°C, and imaged using a Zeiss LSM 510 Meta Confocal Microscope.
Fluorescent microscopy, image processing, and calculations
The amount of TG2 activity in mouse lung and heart tissues was assessed by fluorescent microscopy. All images were generated using a Zeiss LSM 510 Meta Confocal Microscope. Thin sectioned mouse tissues were imaged using a 10X EC Plan-Neofluar / 0.30 M27 dry lens. In general, all images were generated under the same microscope settings, where detector saturation was avoided, and were processed identically. For fluorescent microscopy processing of thin sectioned mouse tissue, separate laser channel images were generated independently in a multi-track mode to avoid fluorophore cross talk that could influence calculations. Images were processed using ImageJ (44). Each image was first split into separate red, green, and blue channels. Next, the intensity of each pixel in the split images was calculated. The intensity of red and green pixels were proportional to the amount of 5BP and TG2 at a given XY coordinate in the images, respectively, while the intensity of the blue pixels was negligible and ignored. The pixel intensities over the entire image were averaged for each channel, and the average 5BP pixel intensity (red) was divided by the average TG2 pixel intensity (green) to give a measure of TG2 activity for each image. TG2 activity measurements were normalized to those from control animal tissues.
Production of experimental pulmonary hypertension and measurement of hemodynamics in mice exposed to Su-5416 and hypoxia
Male C57BL/6 mice were obtained from Charles River Laboratories (Boston, MA). The Tufts IACUC Committee approved all animal studies. For hypoxic exposures, mice were placed in normobaric hypoxic (FIO2 10.5%) chambers for 3 weeks. Normoxia-exposed animals were used as controls. Where noted Su-5416 (Tocris Bioscience), mixed in carboxymethyl cellulose (0.5%), polysorbate 80 (0.4%), benzyl alcohol (0.9%) and NaCl (0.9%), was given weekly in 200 [L subcutaneous injections at a dose of 20 mg/kg to hypoxia exposed animals as previously reported in experimental models of pulmonary hypertension (12, 28). Food was changed twice weekly and water was given ad libitum.
Following three weeks of hypoxia, animals were removed to room air and anesthetized with ketamine/xylazine (50 mg/kg and and 8 mg/kg IP). Body weights (BW) were recorded. The right ventricular systolic pressure was measured by transthoracic insertion of a 25G needle connected to a liquid filled catheter and pressure transducer (Grass P23) into the right ventricle. Pressure wave forms were recorded (PowerLab AD Instruments, Colorado Springs, CO) for 5 min and an average systolic pressure was obtained. Following the pressure measurements the abdomen was opened and blood was collected through the inferior vena cava.
The animals were then euthanized by exsanguination. The chest was then opened; the pulmonary artery and trachea were cannulated; and the heart and lungs were removed en bloc. The right lung was ligated, removed and flash frozen. The left lung was fixed in 4% formalin at 23 cm H2O tracheal and 75 cm H2O pulmonary artery pressures. Following 30 min fixation the right lung was removed and further processed for later embedding. The heart was removed and the right ventricle (RV) was separated from the left ventricle and septum (LV + S). The weights of these tissues were recorded and the RV/LV + S and RV/BW ratios were calculated.
Supplementary Material
Acknowledgments
Funding Sources: This research was supported by grants from the NIH, R01 DK063158 to C.K. and R01 HL107713 to B.F.
NOMENCLATURE
- TG2
transglutaminase 2
- 5BP
5-biotinamidopentylamine
- ERW1041E
(S)-quinolin-3-ylmethyl-2-((((S)-3-bromo-4,5-dihydroisoxazol-5-yl)methyl)carbamoyl) pyrrolidine-1 carboxylate
- SU-5416
semaxinib, 3-[(3,5-Dimethyl-1H-pyrrol-2-yl)met-hylene]-1,3-dihydro-2H-indol-2-one
- BW
body weight
- RVSP
right ventricular systolic pressure
- RV
right ventricle
- LV
left ventricle
- LV + S
left ventricle plus septum
Footnotes
Author Contributions: Authors Thomas R. DiRaimondo, Cornelius Klock, and Rod Warburton contributed equally.
Supporting Information Available: The supplemental information provided illustrates the ostensibly elevated TG2 activity in mouse cardiac tissue regardless of hypoxia treatments and supplies NMR spectra of synthetic compounds.
References
- 1.Herve P, Drouet L, Dosquet C, Launay JM, Rain B, Simonneau G, Caen J, Duroux P. Primary pulmonary hypertension in a patient with a familial platelet storage pool disease: role of serotonin. Am J Med. 1990;89:117–20. doi: 10.1016/0002-9343(90)90111-p. [DOI] [PubMed] [Google Scholar]
- 2.Herve P, Launay JM, Scrobohaci ML, Brenot F, Simonneau G, Petitpretz P, Poubeau P, Cerrina J, Duroux P, Drouet L. Increased plasma serotonin in primary pulmonary hypertension. Am J Med. 1995;99:249–54. doi: 10.1016/s0002-9343(99)80156-9. [DOI] [PubMed] [Google Scholar]
- 3.Kereveur A, Callebert J, Humbert M, Herve P, Simonneau G, Launay JM, Drouet L. High plasma serotonin levels in primary pulmonary hypertension. Effect of long-term epoprostenol (prostacyclin) therapy. Arter Thromb Vasc Biol. 2000;20:2233–9. doi: 10.1161/01.atv.20.10.2233. [DOI] [PubMed] [Google Scholar]
- 4.Sato K, Webb S, Tucker A, Rabinovitch M, O’Brien RF, McMurtry IF, Stelzner TJ. Factors influencing the idiopathic development of pulmonary hypertension in the fawn hooded rat. Am Rev Respir Dis. 1992;145:793–7. doi: 10.1164/ajrccm/145.4_Pt_1.793. [DOI] [PubMed] [Google Scholar]
- 5.Dempsie Y, MacLean MR. Role of the serotonin transporter in pulmonary arterial hypertension. Expert Rev Clin Pharmacol. 2008;1:749–757. doi: 10.1586/17512433.1.6.749. [DOI] [PubMed] [Google Scholar]
- 6.Nemecek GM, Coughlin SR, Handley DA, Moskowitz MA. Stimulation of aortic smooth muscle cell mitogenesis by serotonin. Proc Natl Acad Sci USA. 1986;83:674–8. doi: 10.1073/pnas.83.3.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee SL, Wang WW, Lanzillo JJ, Fanburg BL. Serotonin produces both hyperplasia and hypertrophy of bovine pulmonary artery smooth muscle cells in culture. Am J Physiol. 1994;266:L46–52. doi: 10.1152/ajplung.1994.266.1.L46. [DOI] [PubMed] [Google Scholar]
- 8.Kahn AM, Bishara M, Cragoe EJ, Allen JC, Seidel CL, Navran SS, O’Neil RG, McCarty NA, Shelat H. Effects of serotonin on intracellular pH and contraction in vascular smooth muscle. Circ Res. 1992;71:1294–304. doi: 10.1161/01.res.71.6.1294. [DOI] [PubMed] [Google Scholar]
- 9.Morecroft I, Dempsie Y, Bader M, Walther DJ, Kotnik K, Loughlin L, Nilsen M, MacLean MR. Effect of tryptophan hydroxylase 1 deficiency on the development of hypoxia-induced pulmonary hypertension. Hypertension. 2007;49:232–6. doi: 10.1161/01.HYP.0000252210.58849.78. [DOI] [PubMed] [Google Scholar]
- 10.Izikki M, Hanoun N, Marcos E, Savale L, Barlier-Mur AM, Saurini F, Eddahibi S, Hamon M, Adnot S. Tryptophan hydroxylase 1 knockout and tryptophan hydroxylase 2 polymorphism: effects on hypoxic pulmonary hypertension in mice. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1045–52. doi: 10.1152/ajplung.00082.2007. [DOI] [PubMed] [Google Scholar]
- 11.Abid S, Houssaini A, Chevarin C, Marcos E, Tissot CM, Gary-Bobo G, Wan F, Mouraret N, Amsellem V, Dubois-Rande JL, Hamon M, Adnot S. Inhibition of gut- and lung-derived serotonin attenuates pulmonary hypertension in mice. Am J Physiol Lung Cell Mol Physiol. 2012;303:L500–8. doi: 10.1152/ajplung.00049.2012. [DOI] [PubMed] [Google Scholar]
- 12.Ciuclan L, Hussey MJ, Burton V, Good R, Duggan N, Beach S, Jones P, Fox R, Clay I, Bonneau O, Konstantinova I, Pearce A, Rowlands DJ, Jarai G, Westwick J, MacLean MR, Thomas M. Imatinib attenuates hypoxia-induced pulmonary arterial hypertension pathology via reduction in 5-hydroxytryptamine through inhibition of tryptophan hydroxylase 1 expression. Am J Respir Crit Care Med. 2013;187:78–89. doi: 10.1164/rccm.201206-1028OC. [DOI] [PubMed] [Google Scholar]
- 13.Eddahibi S, Hanoun N, Lanfumey L, Lesch KP, Raffestin B, Hamon M, Adnot S. Attenuated hypoxic pulmonary hypertension in mice lacking the 5-hydroxytryptamine transporter gene. J Clin Invest. 2000;105:1555–62. doi: 10.1172/JCI8678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guignabert C, Izikki M, Tu LI, Li Z, Zadigue P, Barlier-Mur AM, Hanoun N, Rodman D, Hamon M, Adnot S, Eddahibi S. Transgenic mice overexpressing the 5-hydroxytryptamine transporter gene in smooth muscle develop pulmonary hypertension. Circ Res. 2006;98:1323–30. doi: 10.1161/01.RES.0000222546.45372.a0. [DOI] [PubMed] [Google Scholar]
- 15.Walther DJ, Stahlberg S, Vowinckel J. Novel roles for biogenic monoamines: from monoamines in transglutaminase-mediated post-translational protein modification to monoaminylation deregulation diseases. FEBS J. 2011;278:4740–55. doi: 10.1111/j.1742-4658.2011.08347.x. [DOI] [PubMed] [Google Scholar]
- 16.Hummerich R, Thumfart JO, Findeisen P, Bartsch D, Schloss P. Transglutaminase-mediated transamidation of serotonin, dopamine and noradrenaline to fibronectin: evidence for a general mechanism of monoaminylation. FEBS Lett. 2012;586:3421–8. doi: 10.1016/j.febslet.2012.07.062. [DOI] [PubMed] [Google Scholar]
- 17.Song Y, Kirkpatrick LL, Schilling AB, Helseth DL, Chabot N, Keillor JW, Johnson GVW, Brady ST. Transglutaminase and polyamination of tubulin: posttranslational modification for stabilizing axonal microtubules. Neuron, Elsevier Inc. 2013;78:109–23. doi: 10.1016/j.neuron.2013.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Wei L, Laskin DL, Fanburg BL. Role of protein transamidation in serotonin-induced proliferation and migration of pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. 2011;44:548–55. doi: 10.1165/rcmb.2010-0078OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei L, Warburton RR, Preston IR, Roberts KE, Comhair Saa, Erzurum SC, Hill NS, Fanburg BL. Serotonylated fibronectin is elevated in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2012;302:L1273–9. doi: 10.1152/ajplung.00082.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klock C, Khosla C. Regulation of the activities of the mammalian transglutaminase family of enzymes. Protein Sci. 2012;21:1781–91. doi: 10.1002/pro.2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siegel M, Strnad P, Watts RE, Choi K, Jabri B, Omary MB, Khosla C. Extracellular transglutaminase 2 is catalytically inactive, but is transiently activated upon tissue injury. PLoS One. 2008;3:e1861. doi: 10.1371/journal.pone.0001861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dafik L, Albertelli M, Stamnaes J, Sollid LM, Khosla C. Activation and inhibition of transglutaminase 2 in mice. PLoS One. 2012;7:e30642. doi: 10.1371/journal.pone.0030642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watts RE, Siegel M, Khosla C. Structure-activity relationship analysis of the selective inhibition of transglutaminase 2 by dihydroisoxazoles. J Med Chem, American Chemical Society. 2006;49:7493–7501. doi: 10.1021/jm060839a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pinkas DM, Strop P, Brunger AT, Khosla C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007;5:e327. doi: 10.1371/journal.pbio.0050327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stamnaes J, Pinkas DM, Fleckenstein B, Khosla C, Sollid LM. Redox regulation of transglutaminase 2 activity. J Biol Chem. 2010;285:25402–9. doi: 10.1074/jbc.M109.097162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin X, Stamnaes J, Klock C, DiRaimondo TR, Sollid LM, Khosla C. Activation of extracellular transglutaminase 2 by thioredoxin. J Biol Chem. 2011;286:37866– 73. doi: 10.1074/jbc.M111.287490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DiRaimondo TR, Plugis NM, Jin X, Khosla C. Selective inhibition of extracellular thioredoxin by asymmetric disulfides. J Med Chem. 2013;56:1301–10. doi: 10.1021/jm301775s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ciuclan L, Bonneau O, Hussey M, Duggan N, Holmes AM, Good R, Stringer R, Jones P, Morrell NW, Jarai G, Walker C, Westwick J, Thomas M. A novel murine model of severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2011;184:1171–82. doi: 10.1164/rccm.201103-0412OC. [DOI] [PubMed] [Google Scholar]
- 29.Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4:140–56. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 30.Dafik L, Khosla C. Dihydroisoxazole Analogs for Labeling and Visualization of Catalytically Active Transglutaminase 2. Chem Biol, Elsevier Ltd. 2011;18:58–66. doi: 10.1016/j.chembiol.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Staab H. New Methods of Preparative Organic Chmistry IV. Syntheses Using Heterocyclic Amides (Azolides) Angew Chem, Int Ed IV. 1962:351–367. [Google Scholar]
- 32.D’Addona D, Bochet CG. Preparation of carbamates from amines and alcohols under mild conditions. Tetrahedron Lett. 2001;42:5227–5229. [Google Scholar]
- 33.Gerig JT, McLeod RS. Conformations of cis- and trans-4-fluoro-L-proline in aqueous solution. J Am Chem Soc. 1973;95:5725–9. doi: 10.1021/ja00798a046. [DOI] [PubMed] [Google Scholar]
- 34.Muszbek L, Yee VC, Hevessy Z. Blood coagulation factor XIII: structure and function. Thromb Res. 1999;94:271–305. doi: 10.1016/s0049-3848(99)00023-7. [DOI] [PubMed] [Google Scholar]
- 35.Mehta K. Mammalian transglutaminases: a family portrait. Prog Exp Tumor Res. 2005;38:1–18. doi: 10.1159/000084229. [DOI] [PubMed] [Google Scholar]
- 36.Glaser M, Robins EG. “Click labelling” in PET radiochemistry. J Label Compd Radiopharm. 2009;52:407–414. [Google Scholar]
- 37.Rohloff JC, Robinson J, III, Gardner JO. Bromonitrile oxide [3+2] cycloadditions in water. Tetrahedron Lett. 1992;33:3113–3116. [Google Scholar]
- 38.Castelhano AL, Billedeau R, Pliura DH, Bonaventura BJ, Krantz A. Synthesis, chemistry, and absolute configuration of novel transglutaminase inhibitors containing a 3-halo-4,5-dihydroisoxazole. Bioorg Chem. 1988;16:335–340. [Google Scholar]
- 39.Lee DW, Ha H, Lee WK. Selective Mono-BOC Protection of Diamines. Synth Commun. 2007;37:737–742. [Google Scholar]
- 40.Piper JL, Gray GM, Khosla C. High selectivity of human tissue transglutaminase for immunoactive gliadin peptides: implications for celiac sprue. Biochemistry. 2002;41:386–393. doi: 10.1021/bi011715x. [DOI] [PubMed] [Google Scholar]
- 41.Day N, Keillor JW. A continuous spectrophotometric linked enzyme assay for transglutaminase activity. Anal Biochem. 1999;274:141–4. doi: 10.1006/abio.1999.4255. [DOI] [PubMed] [Google Scholar]
- 42.Hausch F, Halttunen T, Maki M, Khosla C. Design, synthesis, and evaluation of gluten peptide analogs as selective inhibitors of human tissue transglutaminase. Chem Biol Elsevier. 2003;10:225–231. doi: 10.1016/s1074-5521(03)00045-0. [DOI] [PubMed] [Google Scholar]
- 43.Choi K, Siegel M, Piper JL, Yuan L, Cho E, Strnad P, Omary B, Rich KM, Khosla C. Chemistry and Biology of Dihydroisoxazole Derivatives: Selective Inhibitors of Human Transglutaminase 2. Chem Biol. 2005;12:469–475. doi: 10.1016/j.chembiol.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 44.Abramoff MD, Magalhaes PJ, Ram SJ. Image Processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.