

Summary
Acyl-carrier-protein (ACP) represents one of the most highly conserved proteins across all domains of life and is nature's way of transporting hydrocarbon-chains in vivo. Notably, type II ACPs serve as a crucial interaction hub within primary cellular metabolism1 by communicating transiently between partner enzymes of the numerous biosynthetic pathways2,3. However, the highly transient nature of such interactions and the inherent conformational mobility of ACP2 have stymied previous attempts to structurally visualize ACP tied to an overall catalytic cycle. This is essential to understanding a fundamental aspect of cellular metabolism leading to compounds that are not only useful to the cell, but are also of therapeutic value. For example, ACP is central to the biosynthesis of the lipid A (endotoxin) component of lipopolysaccharides (LPS) in Gram-negative microorganisms, which is required for their growth and survival4,5 and is an activator of the mammalian host's immune system6,7, thus emerging as an important therapeutic target8-10. During lipid A synthesis (Raetz Pathway), ACP shuttles acyl-intermediates linked to its prosthetic 4′-phosphopantetheine group (4′-PPT)2 among four acyltransferases, including LpxD11. Here we report the crystal structures of three forms of Escherichia coli ACP engaging LpxD, which represent stalled substrate and liberated products along the reaction coordinate. The structures reveal the intricate interactions at the interface that optimally position ACP for acyl-delivery and that directly involve the pantetheinyl group. Conformational differences among the stalled ACPs provide the molecular basis for the association-dissociation process. An unanticipated conformational shift of 4′-phosphopantetheine groups within the LpxD catalytic chamber reveals an unprecedented role of ACP in product release.
Although the paradigm for ACP association with protein partners is thought to be exceedingly transient, the LpxD acyltransferase in the Raetz Pathway (Supplementary Fig. 1a) binds ACP with very high affinity (Kd = 59 nM)12. LpxD transfers R-3-hydroxymyristoyl (β-OH-C14) acyl-chains that are delivered by ACP, to the amino-group of uridine diphosphate 3-O-(β-OH-C14)-α-D-glucosamine (UDP-acyl-GlcN)12 (Supplementary Fig. 1b). Moreover, LpxD follows an ordered sequential kinetic mechanism in which acyl-ACP binds first, and importantly, holo-ACP (acyl-chain has been liberated) dissociates last12. Given the canonical mode of ACP-mediated transfer often permits the rapid exchange between protein partners13, such pronounced association between ACP and LpxD is most unusual and instead suggests a ‘strong, transient’14 interaction that would require a yet to be identified ‘molecular trigger’ for dissociation. Thus, we reasoned that the crystallographic study of the ACP-LpxD complex offers a unique opportunity to gain the detailed molecular basis for ACP-based acyl-delivery and a deeper understanding of protein-protein communication more generally.
We present herein three X-ray co-crystal structures of ACP bound to LpxD, each of which captured a different form of the carrier-protein: intact-acyl-ACP, hydrolyzed-acyl-ACP, and holo-ACP (Fig. 1a and 1b, Supplementary Fig. 2, Supplementary Table 1). These structures were resolved to 2.1 Å, 2.9 Å, and 2.15 Å resolution, respectively (Supplementary Table 2). In each case, the triclinic unit cell contained two LpxD trimers with differing bound states of ACP (Supplementary Fig. 3). The overall B-values for all ACPs modeled in each structure are 2-3 fold higher than LpxD (Supplementary Fig. 4 and Supplementary Table 2) and, accordingly the observed electron density was weaker in some regions (Supplementary Fig. 3). Nonetheless, the placement of side-chains at the protein-protein interface, prosthetic groups, and acyl-chain(s) were apparent (Supplementary Fig. 2d-f and Supplementary Fig. 5). Our structures show the 4′-PPT (attached to Ser36) and its β-OH-C14 acyl-chains having vacated the canonical hydrophobic cavity extending through the core of ACP2,15-17 – all of which require considerable movement (Supplementary Fig. 6). The overall architecture of the LpxD trimer is similar to previously reported X-ray structures18,19 in that each monomer of LpxD can be subdivided into three domains (Fig. 1c and Supplementary Fig. 2): the N-terminal uridine-binding domain (UBD), which is tethered to the left-handed β-helix domain (LβH) that harbors the conserved catalytic His239 residue12, and a C-terminal domain (CTD).
Figure 1. Stalled ACPs bound to LpxD.
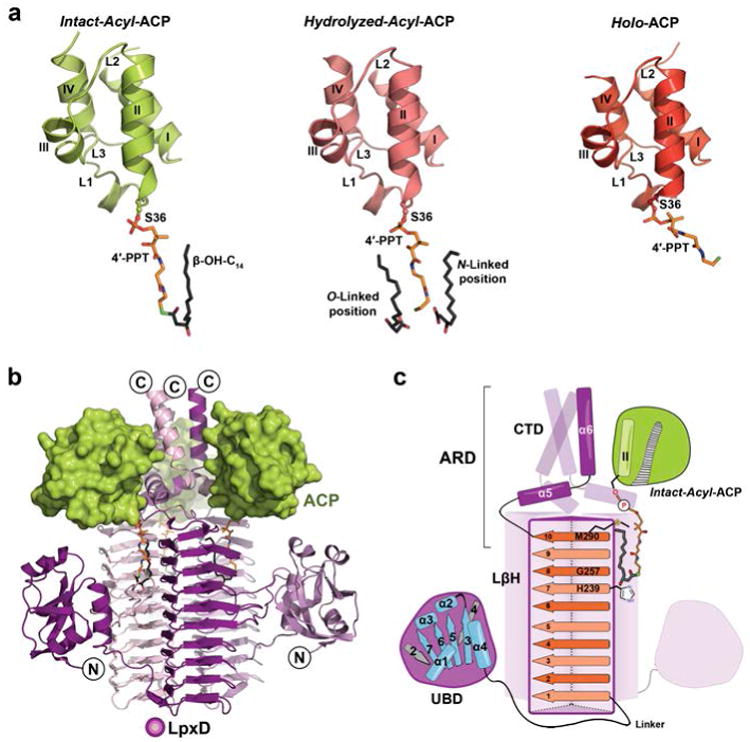
(a) The three forms of ACP: intact-acyl-ACP (green), hydrolyzed-acyl-ACP (salmon), and holo-ACP (red). ACP adopts a 4-helix bundle (I-IV) with several loop regions (L1-L3)28,29. The 4′-PPT arm and acyl-chains are shown as stick models and colored orange or dark gray, respectively, and by atom. (b) Overall architecture of the ACP-LpxD complex (intact-acyl-ACP shown). Three ACPs bind the carboxy-terminal end of the LpxD trimer (colored by chain purple/magenta/light pink). (c) Cartoon rendering showing the overall fold and interaction of acyl-ACP with LpxD. Highlighted dark purple is a single monomer of LpxD with subdomains indicated: uridine binding domain (UBD), left-handed β-helix domain (LβH), and C-terminal domain (CTD). ACP, interfacing with the ACP recognition domain (ARD), and its acyl-4′-PPT group are shown. The locations of the catalytic base (His239), oxyanion hole (Gly257), and molecular ‘hydrocarbon ruler’ (Met290) are indicated within the ten β-helical coils (orange strands) of a single LβH.
The structures reveal three molecules of the carrier-protein are localized to the C-terminal end of LpxD (Fig. 1b). Notably, we have identified the ACP recognition domain (ARD) (Fig. 1c), which is formed by the CTD and the last beta-coil of the LβH domain, providing the molecular basis for ACP association. This contrasts with a previous study that suggested the UBD domain as the likely ACP docking site due to its proximity to the catalytic cleft18. Although the analogous C-terminal region of the LpxA acyltransferase20 is found to adopt a completely different orientation from that of LpxD, it may serve a similar function in binding ACP (Supplementary Fig. 7).
By virtue of the complete engagement of ACP, three competent active sites are created (Fig. 2a). Each ACP-LpxD interface buries a surface area of ∼530 Å2 and is predominated by complementary electrostatic interactions (Fig. 2b). In addition, van der Waals contacts and extensive interaction with the prosthetic group contribute to the large binding footprint that explains the ‘strong, transient’ nature of these two protein partners. A combination of residues located on the ‘universal recognition helix’ (helix II)21, as well as portions of L1, L2, and helix-III of ACP provide the acidic surface that binds a pronounced basic patch on LpxD. This surface feature of ACP can be subdivided into two highly acidic regions, I and II, which include residues Glu30-Met44 and Ala45-Glu60, respectively. The complementary binding surface on LpxD involves residues from all three monomers (denoted by prime symbols) and forms a shallow groove between coiled-coils of the ARD into which helix-II packs (Supplementary Fig. 8).
Figure 2. Intermolecular interactions between ACP and LpxD.
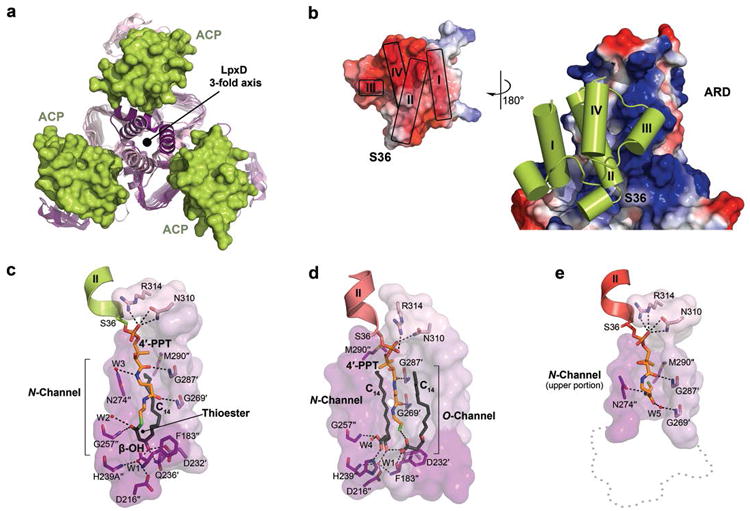
(a, b) Overview of the protein-protein interactions (intact-acyl-ACP depicted). (a) Top-down view of the ACP-LpxD complex showing three molecules of ACP bind per LpxD trimer. (b) Electrostatic surface representation of the ARD and ACP (inset), the potential contours were scaled to +79.2 (blue) and -79.2 (red) KbT e-1. (c, d, e) Detailed interactions between the LpxD reaction chamber and the bound acyl/4′-PPT groups of ACP. Hydrogen bonds are shown as black dashes. Molecular surfaces are of only those residues that contribute to interactions. (c)Intact-acyl-ACP complex. The β-OH-C14 acyl-chain delivered by ACP binds the hydrophobic N-channel and its terminal carbon-atoms pack near Met290″. (d)Hydrolyzed-acyl-ACP complex. An equivalent β-OH-C14 acyl-chain is shown bound to a newly identified hydrophobic channel (O-channel). (e)Holo-ACP complex. The 4′-PPT arm interacts at the far end of the N-channel, positioning the terminal thiol near Met290″. A ‘ghost outline’ of the catalytic cleft is indicated by a gray dotted line.
Within region I, Asp35, Ser36, Leu37, Asp38, Val40, Glu41, and Met44 are important for binding the N-terminal end of the recognition helix to the base of the ARD domain (Supplementary Fig. 8) and the interactions were notably present in all three structures. Region II of ACP interacts with the upper portion of the ARD domain, the details of which differ substantially among the three stalled ACP complexes (discussed below). Most of the residues within regions I and II are conserved among other type II carrier-proteins (Supplementary Fig. 2b) and have been implicated as key modulators of ACP association22,23. The most universal electrostatic interaction displayed across the intact-, hydrolyzed-, and holo-ACP structures is between Arg293 of LpxD and Asp35, Asp38, and Glu41 of ACP that flank Ser36. This coordination point is crucial for ACP association with LpxD as mutation of Arg293 to alanine results in a 23-fold increase in KM for acyl-ACP as compared to wild-type LpxD, with little effect on kcat12. These interactions orient ACP by positioning the pantetheinylated Ser36 residue toward the catalytic chamber of LpxD.
In addition to the protein-protein interactions, the prosthetic group and acyl-chain of intact-acyl-ACP extensively contact the surface of the partner enzyme (Supplementary Fig. 9). Inspection of the electron density indicates that the thioester scissile bond remained unbroken, which required a His239Ala LpxD variant (Supplementary Fig. 5a). The β-OH-C14 acyl-chain and the 4′-PPT arm adopt a horseshoe-like conformation, which in effect buries the acyl-chain between the prosthetic group and a pronounced hydrophobic channel (N-channel) formed between LpxD monomers (Fig. 2c). The 4′-phosphate moiety of 4′-PPT is directly coordinated by Asn310 and Arg314 of LpxD and may partially contribute to some level of specificity. The remainder of the 4′-PPT arm adopts a rather extended conformation, stretching over 14 Å, and interacts with residues primarily along the rim of the N-channel. This conformation places the thioester bond in proximity to the alanine-substituted His239 catalytic base and orients the carbonyl-oxygen of β-OH-C14 toward the amide nitrogen atom of Gly257″, corroborating its role in forming the oxyanion hole12,24.
Two features of LpxD specificity toward β-hydroxy-acyl-chains are explained by the intact-acyl-ACP structure. First, the terminal two carbon-atoms of β-OH-C14 pack against Met290″ located at the far end of the N-channel (Fig. 2c and Supplementary Fig. 9). Given that LpxD is highly specific for 14-carbon chain-lengths12, this supports the role of Met290 as a ‘hydrocarbon ruler’19. Second, LpxD is particularly selective for the β-hydroxyl group of the acyl-chain; its removal completely abrogates acyltransferase activity25. Our structures reveal an intricate hydrogen bond network between the β-hydroxyl group and Asp232′, Gln236′, and a critical water-mediated bridge with Asp216″ and the main-chain nitrogen atoms of Phe183″ and His239Ala″.
By contrast to the intact-acyl-ACP complex, the thioester bond is broken in the hydrolyzed-acyl-ACP structure (Fig. 2d, Supplementary Fig. 5b, and Supplementary Fig. 9). Notably, we discovered a second molecule of β-OH-C14 fatty acid bound to the LpxD surface (Supplementary Fig. 10), which reveals an additional hydrophobic channel (O-channel) that likely binds the acyl-chain that is ester-linked to the UDP-acyl-GlcN lipid substrate (Supplementary Fig. 1b). A superposition with the previously reported C. trachomatis LpxD in complex with UDP-GlcNAc18 illustrates the proximity of the carboxylate head group of the additional fatty acid to the anticipated binding locale of the 3-hydroxyl position of the GlcN ring (Supplementary Fig. 11).
What is striking about both the intact- and hydrolyzed-acyl-ACP structures is the conformational similarity between the 4′-PPT prosthetic groups that are fully extended (Fig. 2c and 2d), whereas the holo-ACP structure reveals an alternate conformation of its prosthetic group packing against the far end of the N-channel (Fig. 2e and Supplementary Fig. 9). Notably, in both intact- and hydrolyzed-acyl-ACP the pantetheinyl arms completely enclose the reaction chambers (Fig. 3a). Although this architecture likely stabilizes substrate binding within the hydrophobic N-channel, it raises a key question. Given a sequential ordered reaction mechanism, how does the di-acylated-GlcN product leave LpxD prior to holo-ACP if the reaction chamber is completely blocked? A structural comparison between intact- and holo-ACP sheds light on this matter by exposing a substantial movement that the 4′-PPT arm has undergone (Fig. 3b). Notably, the terminal thiol of the 4′-PPT has vacated the catalytic cleft and moves ∼15 Å to be situated near Met290. A sizable region of the reaction chamber closest to the catalytic cleft is now open to solvent, thereby giving the diacyl-GlcN product an opportunity to dissociate prior to the release of holo-ACP.
Figure 3. ACP conformations and reorganization of its prosthetic group.
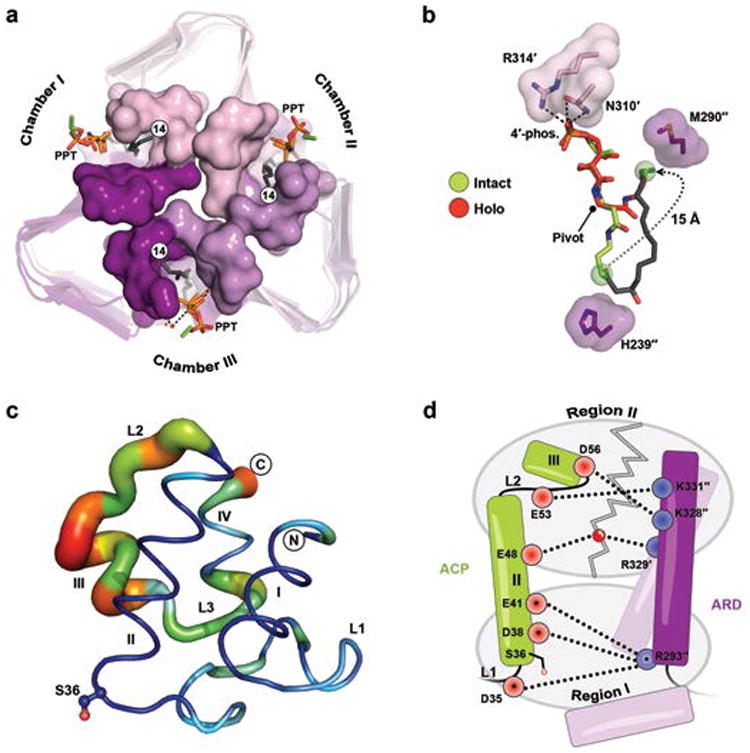
(a) Top-down view of LpxD (intact-acyl-ACP depicted) showing three reaction chambers enclosed by the 4′-PPT. (b) Structural comparison between the intact- (green) and holo-4′-PPT (red) prosthetic groups. The 4′-PPT rearrangement is indicated (gray dotted arrow). (c) Difference distance matrix calculated between fully modeled intact- (chain K) and holo-ACP (chain G). Deviations between like atoms are shown as a putty-sausage representation. Both the thickness and heatmap coloring indicate regions of least (thin, blue) to highest (thick, red) displacement. (d) Schematic summarizing differences in electrostatic interactions between ACP complexes. Residues are indicated as red (acidic) or blue (basic) circles and subdivided according to region I or II affiliation. Interactions made by intact-acyl-ACP alone are depicted (dashed lines) and those common among all complexes are represented with an asterisk. The gray zigzag indicates those electrostatic interactions that are broken in the hydrolyzed- and holo-ACP product complexes.
The remarkable difference in conformation displayed by the 4′-PPT arm, together with the ordered sequential mechanism of acyl-transfer, prompted us to propose that this ‘swing’ motion may in fact be involved in ‘triggering’ the release of lipid-product. Since every terminal thiol of holo-ACP was positioned ∼3.7 Å away from Met290 of LpxD, we envisaged the possibility of mutating this residue to a cysteine in an effort to induce a mixed-disulfide linkage post-catalysis. Biochemically, the Met290Cys mutation abrogates acyl-transfer to UDP-acyl-GlcN as compared to wild-type LpxD (Supplementary Fig. 12). This suggests that a covalent bond is formed between the cysteinyl and 4′-PPT thiols. Thus, we reasoned that the addition of reducing agent would rescue acyl-transfer. Accordingly, titration of DTT into the reaction mixture recovers activity of Met290Cys-LpxD to levels indistinguishable from that of wild-type enzyme (Supplementary Fig. 12). These data suggest that the observed 4′-PPT motion has functional relevance in the course of product release. Moreover, as typically exemplified with ‘strong, transient’ interactions14 this substantial movement of the pantetheine arm likely serves as the ‘molecular trigger’ that promotes the collapse of the ACP-partner complex.
Because our structures show different states of ACP stalled at the LpxD active site, an alignment of LpxD domains allows us to visualize the movements within ACP as it relates to the overall catalytic cycle (Fig. 3c), which to the best of our knowledge is unprecedented. Helix-I, helix-II, and portions of L1 remain relatively unchanged (RMSD of ∼1 Å) and are preserved for the purpose of remaining docked. The largest differences occur downstream of the recognition helix (Fig. 3c), including helices-III, -IV, L2 and L3 (RMSD of ∼3 Å) and are an indication of what interactions must break for dissociation. A closer inspection reveals that the intact-complex makes electrostatic interactions with the entire ARD interface that involves region II, whereas both the hydrolyzed- and holo-ACP structures do not (Fig. 3d). These data indicate that the more extensive interactions are important for molecular recognition during association; however, following acyl-transfer it is conformational changes in ACP that ultimately destabilize the protein-protein complex. In this context, one particular molecule of holo-ACP within the asymmetric unit is displaced from the others and possibly represents a different binding state revealing additional interactions which must break for dissociation (Supplementary Fig. 13).
The structures captured in this study begin to establish key molecular movements within ACP that initiate molecular recognition and a mechanism with which associations can be ultimately broken. In this context, we present a model for ACP-based synthesis of lipid A precursors catalyzed by LpxD (Fig. 4) and can begin to extract some general principles for these types of interactions. First, although there does not yet appear to be a consensus-sequence binding motif on the surface of partner proteins, both regions I and II of ACP seem to be frequently involved, with the majority of the small number of other ACP-based complexes determined revealing that helix-II, -III, L1 and L2 are consistently utilized22,23,26,27. Second, because these regions are broadly universal, exploiting specific residue contacts at the interface and fine-tuning the buried surface area at these regions likely dictate how ACP-based associations can be both specific and temporary. For instance, our structures reveal the first direct, electrostatic coordination with the 4′-phosphate of both acyl- and holo-ACP, which in addition to extensive protein-protein contacts provide the ‘strong, transient’ nature of the ACP-LpxD complex. Third, because we observe ACP tied to an entire catalytic cycle, a substantial conformational change downstream of the recognition helix-II may represent a more general communication mechanism for breaking ACP-partner complexes. Finally, the stalled ACPs captured herein have revealed an unprecedented role for carrier-proteins in product release, and the contribution of the pantetheinyl group in both the formation and dissociation of the ACP-partner complex. Perhaps motion of the pantetheinyl group in other ACP-based complexes provokes dissociation in a similar manner, especially when considering other ‘strong, transient’ protein partners.
Figure 4. Molecular basis for the ordered-sequential reaction mechanism and involvement of ACP in lipid-product release.

Acyl-ACP binds first (i) to free LpxD forming the (ii) binary complex. ACP associates with the ARD and the acyl-4′-PPT packs into the hydrophobic N-channel, thus (iii) enabling UDP-acyl-GlcN to bind next and initiate acyl-transfer. In the ternary product complex (iv) the 4′-PPT arm of hydrolyzed-acyl-ACP (transparent orange squiggle) completely encloses the reaction chamber, blocking UDP-diacyl-GlcN from leaving. By moving toward Met290 (dotted arrow), the 4′-PPT (dark orange squiggle) drives the (v) eventual release of UDP-diacyl-GlcN by opening up the catalytic chamber. This motion also ‘triggers’ (vi) conformational changes downstream of helix-II leading to holo-ACP dissociation.
Methods
Expression and purification of His6-LpxD
E. coli LpxD was over-expressed, and purified as previously reported12. Briefly, His6-LpxD was expressed in E. coli Rosetta/pLysS. The membrane-free fraction was loaded onto a 5 mL Ni-NTA (Qiagen, CA) column and eluted in one step with 200 mM imidazole. The His6-tag was left intact and the resulting LpxD was concentrated to ∼4-10 mL and loaded onto a High Load 26/60 Superdex 200 gel filtration column (GE, NJ) equilibrated with 10 mM Tris-HCl, pH 7.5, 500 mM NaCl, and 1 mM dithiothreitol (DTT). The protein peak had an elution profile consistent with that of the LpxD homotrimer. Fractions were pooled and concentrated to ∼40 mg mL-1 and stored in aliquots at −80 °C. The wild-type LpxD enzyme utilized in the LpxD assay was purified and stored in the absence of DTT.
Construction, expression and purification of His6-LpxD point mutants
Site-directed mutants designed to alter Met290 (Met290Cys), and His239 (His239Ala) were accomplished using the QuikChange PCR mutagenesis kit (Stratagene, CA). The LpxD variants were recombinantly expressed in C41(DE3) E. coli cells and purified in the same manner as described for wild-type His6-LpxD. The His239Ala LpxD was stored in 10 mM Tris-HCl, pH 7.5, 500 mM NaCl, and 1 mM TCEP, and Met290Cys LpxD was purified and stored in the absence of a reductant.
In vitro assay of LpxD
The LpxD catalyzed conversion of [α-32P]-UDP-3-acylGlcN to [α-32P]-UDP-2,3-diacylGlcN was monitored by thin layer chromatography (TLC) as previously reported25. The 20 μL assay mixture containing 40 mM HEPES (pH 7.4), 0.02 mg/mL BSA, 1.4 nM pure wild-type E. coli LpxD (or 4.2 nM Met290Cys LpxD), 6 μM R-3-hydroxymyristoyl-ACP (β-OH-C14-ACP), and 4 μM [α-32P]-UDP-3-O-(β-OH-C14)-GlcN (0.005-0.04 μCi/μL) was equilibrated at 30 °C, and the reaction was initiated by the addition of the enzyme. DTT (100 mM) was added to assess its effect on the catalysis of the wild-type and the mutant LpxD. A 1 μL aliquot of the reaction mixture was spotted onto a silica gel 60 plate (EMD Chemicals, Germany) at various time-points. After drying under a cold air stream, plates were developed with a chloroform/methanol/water/acetic acid solvent (25:15:4:2, v:v:v:v). The plates were dried and exposed overnight to a Molecular Dynamics PhosphorImager Screen. The conversion rate of the lipid substrate to product was measured using ImageQuant software.
Expression and purification of holo-ACP
The acp gene was amplified from E. coli W3110A genomic DNA using the ACP-forward and ACP-reverse primers (Supplementary Table 1) engineered to introduce downstream of the ACP coding region a spacer sequence, a ribosome binding site, followed by a translation spacer element (denoted PCR product A). The ACPS-forward and ACPS-reverse primers (Supplementary Table 1) were used to amplify acps gene from W3110A genomic DNA, which contained a spacer sequence, a ribosome binding site, and a translation spacer element upstream of the ACPS coding region (denoted PCR product B). The ACP-forward and ACPS-reverse primers were subsequently used to generate PCR product C, which contained acp and acps genes separated by the spacer sequence, ribosome binding site, and the translation spacer element. PCR product C was cloned into a pET16b* vector, which contained an engineered tobacco etch virus (TEV) protease cleavage site instead of Factor Xa, using the NdeI and XhoI restriction sites. E. coli DH5α competent cells were subsequently transformed with plasmid and transformants were selected at 37 °C on a LB-agar plate supplemented with 100 μg mL-1 ampicillin. The sequence of the plasmid encoding the N-terminally histidine-tagged ACP and non-tagged ACPS (pET16b*-AM) was confirmed by DNA sequencing.
ACP and ACPS were co-expressed using the pET16b*-AM vector in C41(DE3) E. coli cells and cultured at 37 °C in 1 L of LB broth supplemented with 100 μg mL-1 ampicillin. Upon reaching an OD600 of 0.6 the expression was induced for 6 hours with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at 30 °C. All the subsequent procedures were carried out at 4 °C. The cells were harvested, washed, resuspended and lysed in 20 mM HEPES, pH 8.0 containing 10% glycerol, 200 mM NaCl, and 2 mM DTT (Buffer A), supplemented with 20 mM imidazole. After removal of cell debris by centrifugation at 100,000 × g for 1 hour, the soluble fraction was loaded onto a Ni-NTA (Qiagen, CA) column equilibrated with Buffer A and 20 mM imidazole. The Ni-NTA column was washed with 10 column-volumes and His10-ACP was eluted in one step by Buffer A supplemented with 250 mM imidazole over the equivalent of 5 column-volumes. Although ACPS was not histidine-tagged, it co-purified with His10-ACP. The elution fraction was allowed to slowly rock with 1 mg of TEV protease and 2 mM EDTA overnight. Subsequently, the protein mixture was dialyzed against 20 mM MES, pH 6.0, and 2 mM DTT overnight. The TEV protease and residual His10-ACP were removed by passing the mixture through a 2 mL Ni-NTA column. The resulting flow-through fraction containing ACP was loaded onto a 5 mL QFF ion exchange column (GE, NJ). ACP eluted separately from ACPS by applying a linear gradient of 20 mM MES, pH 6.0, 0-500 mM NaCl, and 2 mM DTT over 50 column-volumes. The fractions corresponding to ACP were pooled together and dialyzed against 100 mM Tris-HCl, pH 7.1, and 200 mM NaCl (Buffer B) overnight. Holo-ACP was separated from residual apo-ACP by mixing the ACP sample with 5 mL of ThioPropyl Sepharose 6b resin (Sigma-Aldrich, MO) overnight. Apo-ACP, which lacks any free thiol group, did not bind to the resin, whereas, holo-ACP covalently attached to the resin via the terminal thiol group of its phosphopantetheine moiety. After washing the Thiopropyl Sepharose 6b with 25 mL of Buffer B, holo-ACP was eluted in 25 mL of Buffer B supplemented with 25 mM DTT. The complete removal of apo-ACP was confirmed by electrospray ionization mass spectrometry. The elution fraction was concentrated to ∼4-10 mL and loaded on a High Load 26/60 Superdex 200 gel filtration column equilibrated with 10 mM Tris-HCl, pH7.5, 200 mM NaCl, and 2 mM DTT. The relevant eluted fractions were concentrated to ∼20 mg mL-1 and stored in aliquots at −80 °C.
Production of β-OH-C14-ACP
Holo-ACP was charged enzymatically with R-3-hydroxymyristic acid (Santa Cruz Biotechnology, Inc.) by soluble acyl-ACP synthetase from Vibrio harveyi30. The soluble acyl-ACP synthetase (aasS) gene was synthesized by GenScript. The aasS gene was subcloned into pET-16b expression vector, overexpressed, and the His6-AasS was purified using nickel affinity chromatography as reported by Jiang et al.30. To generate β-OH-C14-ACP, 0.1 mM of holo-ACP was mixed with 0.001 mM AasS and 0.3 mM of the fatty acid (β-OH-C14) at room temperature for 3 hours in a buffer containing 100 mM Tris-HCl, pH 7.8, 10 mM ATP, and 10 mM MgCl2. To separate acyl-ACP from AasS, the reaction mixture was loaded onto a High Load 26/60 Superdex 200 gel filtration column equilibrated with 10 mM Tris-HCl, pH7.5, and 200 mM NaCl. The complete conversion of holo-ACP to β-OH-C14-ACP was confirmed by both electrospray ionization mass spectrometry and 2.5 M urea (19%) polyacrylamide (pH 9.5) gel electrophoresis31.
Crystallization and structure determination
Prior to crystallization, either acyl-ACP or holo-ACP was mixed with the wild-type or the catalytically inactive His239Ala LpxD in order to preform the protein-protein complex. Crystals of holo-ACP-LpxD were grown at 15 °C by mixing the protein solution with the precipitant (0.1 M MES pH 6.0, 0.2 M lithium sulfate, 20% PEG 4000) in ratios of 1:1 and 1:1.5. Crystals achieved full size in approximately 45 days. The crystals were transferred to a cryo-solution using a 50:50 ratio of paratone to mineral oil and immediately cryo-cooled to −180 °C in liquid nitrogen. Hydrolyzed-acyl-ACP-LpxD crystals were obtained by equilibrating the protein mixture against a well solution containing 0.1 M MES pH 6.5, 0.2 M ammonium sulfate, 20% PEG 8000 and incubating at 15 °C. The crystals were harvested on day 10 and cryo-cooled using a solution containing ammonium sulfate, 33% PEG 8000, 5 mM Tris-HCl pH 7.5, 190 mM NaCl, and 20% of the cryoprotectant ethylene glycol. In order to trap the intact-acyl-ACP-LpxD complex the His239Ala-LpxD mutant was purified in the presence of the reducing agent TCEP instead of DTT in an attempt to reduce hydrolysis of the thioester bond. His239Ala-LpxD and acyl-ACP were premixed using a 1:1 molar ratio prior to crystallization trials. The well solution and the cryoprotectant were the same as that described for the hydrolyzed-acyl-ACP structure, however, a ratio of 1.5:1 of protein to well solution was required.
The diffraction data were collected on the SER-CAT 22-BM and 22-ID beamlines at the Advanced Photon Source (APS) at Argonne National Laboratory. Data were processed using the HKL200032 software suite (Supplementary Table 2). Although the data for both intact- and holo-ACP complexes were processed to 2.1 and 2.13 Å, respectively, the hydrolyzed-acyl-ACP complex was trimmed to 2.9 Å due to the data completeness being unsatisfactory in higher resolution bins, i.e. well below 70%. This resulted in a higher signal to noise ratio in the 2.9 Å resolution bin as well as a much lower Rmerge value. The crystals of each complex belong to the P1 space group. The structures were solved by the molecular replacement method using the program PHASER within the PHENIX software suite33,34 and the previously determined E. coli LpxD structure (PDB 3EH0) as the search model. Two trimers of LpxD were observed in the triclinic unit cell for the intact-acyl-ACP, hydrolyzed-acyl-ACP, and holo-ACP co-crystal structures. In each case, however, ACP was intentionally omitted during the molecular replacement process, and instead was manually rebuilt into unbiased, contiguous Fo – Fc difference electron density by first rigid-fitting the E. coli apo-ACP coordinates (PDB 1T8K)35 into the resulting maps. The models were rebuilt using COOT36 and iterative structure refinement with restrained and TLS options was carried out using PHENIX33. For the lower resolution hydrolyzed-acyl-ACP structure hydrogens were included for refinement with automated optimization of X-ray/stereochemistry and ADP weights selected. Additionally, we utilized the high resolution E. coli apo-ACP structure (PDB 1T8K) as a reference model. The molecular coordinates and restraints of the 4′-PPT, β-OH-C14 -4′-PPT, and free β-OH-C14 fatty acid ligands were generated by using either the Dundee PRODRG2 Server37 or PHENIX Elbow33. Composite omit map and simulated annealing omit map calculations were conducted using CNS38. The protein-ligand interactions were identified by AREAIMOL calculations within the CCP4 suite39. The quality of the final models was validated using MOLPROBITY40. The data statistics are reported in Supplementary Table 2. Molecular figures were generated using PyMOL41.
A total of six ACP molecules were present in the intact-acyl-ACP structure (Supplementary Fig. 3). Both the hydrolyzed- and holo-ACP structures exhibited partially bound states of ACP within two or three LpxD active sites as interpreted by the lack of contiguous electron density for the majority of the ACP backbone. As a result, only a portion of ACP that includes Ser36 and its 4′-PPT prosthetic group were included in the final model: (i) hydrolyzed-acyl-ACP, residues 35-44 of chain L, residues 6-15, 27-53, and 62-73 of chain I, residues 1-15 and 27-73 of chain G, (ii) holo-ACP, residues 35-44 of chain H and chain L. The remaining LpxD active sites in the hydrolyzed- and holo-ACP structures contain fully modeled ACPs. In all three co-crystal structures, the electron density maps indicated that the N-terminal methionine of ACP was present and forms a key lattice contact with neighboring molecules of LpxD. Two additional residues (Ser-His) in the holo-ACP structure that remain from the TEV cleavage site could also be modeled.
The observed electron density for fully modeled intact-, hydrolyzed-, and holo-ACPs were weaker in some regions especially on the backside of the molecule which faces solvent; however, the placement of side-chains was apparent at the protein-protein interface (Supplementary Fig. 3). This implicates conformational heterogeneity throughout the lattice. Nonetheless, in all three co-crystal structures electron density was apparent in every active site for all atoms of the 4′-PPT group. In both the intact- and hydrolyzed-forms, pronounced electron density was also present for the β-OH-C14 acyl-chains located in the N-channel. In addition, electron density indicated that the hydrolyzed-acyl-ACP complex included two additional molecules of β-OH-C14 fatty acid bound to the hydrophobic O-channel, although the density was weaker toward the terminal carbon-atoms of the acyl-chains. To investigate the origin of this second fatty acid, we mixed acyl-ACP (12.86 mg mL-1) in a 1:2.25 v:v ratio with wild-type LpxD (26.38 mg mL-1) pre-incubated with 1 mM DTT in a solution that was consistent with the condition used for crystallization. The protein solution was incubated at 15 °C, and aliquots were taken at different time points and stored at –80 °C. Samples were run on a 2.5 M urea (19%) polyacrylamide (pH 9.5) gel31, which revealed that both DTT (a known phenomenon42) and LpxD enhance the cleavage of the thioester bond of acyl-ACP (Supplementary Fig. 10). This observation most likely explains why free β-OH-C14 fatty acid was available to bind the O-channel of LpxD.
Supplementary Material
Acknowledgments
We wish to acknowledge our co-author, Dr. Raetz, who shaped the lipid field with his curiosity and remarkable efforts, introducing many scientists to the field during his renowned career. The authors thank Drs. Richard Brennan and W. Todd Lowther for critical review of the manuscript. Finally, we thank Dr. Ziquiang Guan for the help with the mass spectrometry of ACP, Dr. Hak-Suk Chung and other members of Raetz laboratory, as well as Jonathan M. Burg for stimulating discussions. Crystal screening, data collection, and data processing were conducted in collaboration with the Duke Macromolecular X-ray Crystallography Shared Resource. Diffraction data were collected remotely at the Southeast Regional Collaborative Access Team 22-BM and 22-ID beamlines at the Advanced Photon Source, Argonne National Laboratory supported by the US Department of Energy, Office of Science, and the Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38. This work was supported by the NIH Grants GM-51310 and AI-055588 awarded to Drs. C. R. H. Raetz and P. Zhou.
Footnotes
Supplementary Information is available in the online version of the paper.
Author Contributions A.M., C.R.H.R., and C.W.P. designed research; A.M. performed all biochemical experiments under the guidance of C.R.H.R., P.Z. and C.W.P.; A.M. performed all protein expression, purification, and crystallization; A.M. and C.W.P. contributed to data collection, structure solution, and refinement; A.M., C.R.H.R. and C.W.P. analyzed and interpreted the structures; A.M. and C.W.P. made the figures and wrote the manuscript; A.M., C.R.H.R., P.Z. and C.W.P. discussed the results and commented on the manuscript.
Author Information Molecular coordinates and structure factors of intact–acyl-ACP, hydrolyzed-acyl-ACP, and holo-ACP in complex with LpxD have been deposited in the Protein Data Bank under accession codes 4IHF, 4IHG, and 4IHH, respectively. Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
References
- 1.Butland G, et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005;433:531–537. doi: 10.1038/nature03239. [DOI] [PubMed] [Google Scholar]
- 2.Byers DM, Gong H. Acyl carrier protein: structure-function relationships in a conserved multifunctional protein family. Biochem Cell Biol. 2007;85:649–662. doi: 10.1139/o07-109. [DOI] [PubMed] [Google Scholar]
- 3.White SW, Zheng J, Zhang YM, Rock The structural biology of type II fatty acid biosynthesis. Annu Rev Biochem. 2005;74:791–831. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- 4.Galloway SM, Raetz CRH. A mutant of Escherichia coli defective in the first step of endotoxin biosynthesis. J Biol Chem. 1990;265:6394–6402. [PubMed] [Google Scholar]
- 5.Belunis CJ, Clementz T, Carty SM, Raetz CRH. Inhibition of lipopolysaccharide biosynthesis and cell growth following inactivation of the kdtA gene in Escherichia coli. J Biol Chem. 1995;270:27646–27652. doi: 10.1074/jbc.270.46.27646. [DOI] [PubMed] [Google Scholar]
- 6.Poltorak A, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 7.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Onishi HR, et al. Antibacterial agents that inhibit lipid A biosynthesis. Science. 1996;274:980–982. doi: 10.1126/science.274.5289.980. [DOI] [PubMed] [Google Scholar]
- 9.Williams AH, Immormino RM, Gewirth DT, Raetz CRH. Structure of UDP-N-acetylglucosamine acyltransferase with a bound antibacterial pentadecapeptide. Proc Natl Acad Sci USA. 2006;103:10877–10882. doi: 10.1073/pnas.0604465103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jenkins RJ, Dotson GD. Dual targeting antibacterial peptide inhibitor of early lipid A biosynthesis. ACS Chem Biol. 2012;7:1170–1177. doi: 10.1021/cb300094a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raetz CRH, Reynolds CM, Trent MS, Bishop RE. Lipid A modification systems in gram-negative bacteria. Annu Rev Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartling CM, Raetz CRH. Steady-state kinetics and mechanism of LpxD, the N-acyltransferase of lipid A biosynthesis. Biochemistry. 2008;47:5290–5302. doi: 10.1021/bi800240r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leibundgut M, Jenni S, Frick C, Ban N. Structural basis for substrate delivery by acyl carrier protein in the yeast fatty acid synthase. Science. 2007;316:288–290. doi: 10.1126/science.1138249. [DOI] [PubMed] [Google Scholar]
- 14.Nooren IM, Thornton JM. Diversity of protein-protein interactions. EMBO J. 2003;22:3486–3492. doi: 10.1093/emboj/cdg359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan DI, Vogel HJ. Current understanding of fatty acid biosynthesis and the acyl carrier protein. Biochem J. 2010;430:1–19. doi: 10.1042/BJ20100462. [DOI] [PubMed] [Google Scholar]
- 16.Ploskon E, et al. Recognition of intermediate functionality by acyl carrier protein over a complete cycle of fatty acid biosynthesis. Chem Biol. 2010;17:776–785. doi: 10.1016/j.chembiol.2010.05.024. [DOI] [PubMed] [Google Scholar]
- 17.Roujeinikova A, et al. Structural studies of fatty acyl-(acyl carrier protein) thioesters reveal a hydrophobic binding cavity that can expand to fit longer substrates. J Mol Biol. 2007;365:135–145. doi: 10.1016/j.jmb.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 18.Buetow L, Smith TK, Dawson A, Fyffe S, Hunter WN. Structure and reactivity of LpxD, the N-acyltransferase of lipid A biosynthesis. Proc Natl Acad Sci USA. 2007;104:4321–4326. doi: 10.1073/pnas.0606356104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartling CM, Raetz CRH. Crystal structure and acyl chain selectivity of Escherichia coli LpxD, the N-acyltransferase of lipid A biosynthesis. Biochemistry. 2009;48 doi: 10.1021/bi901025v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raetz CRH, Roderick SL. A left-handed parallel beta helix in the structure of UDP-N-acetylglucosamine acyltransferase. Science. 1995;270:997–1000. doi: 10.1126/science.270.5238.997. [DOI] [PubMed] [Google Scholar]
- 21.Frederick AF, Kay LE, Prestegard JH. Location of Divalent Ion Sites in Acyl Carrier Protein Using Relaxation Perturbed 2d Nmr. FEBS Lett. 1988;238:43–48. doi: 10.1016/0014-5793(88)80222-9. [DOI] [PubMed] [Google Scholar]
- 22.Cryle MJ, Schlichting I. Structural insights from a P450 Carrier Protein complex reveal how specificity is achieved in the P450(BioI) ACP complex. Proc Natl Acad Sci USA. 2008;105:15696–15701. doi: 10.1073/pnas.0805983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parris KD, et al. Crystal structures of substrate binding to Bacillus subtilis holo-(acyl carrier protein) synthase reveal a novel trimeric arrangement of molecules resulting in three active sites. Structure. 2000;8:883–895. doi: 10.1016/s0969-2126(00)00178-7. [DOI] [PubMed] [Google Scholar]
- 24.Kraut DA, Carroll KS, Herschlag D. Challenges in enzyme mechanism and energetics. Annu Rev Biochem. 2003;72:517–571. doi: 10.1146/annurev.biochem.72.121801.161617. [DOI] [PubMed] [Google Scholar]
- 25.Kelly TM, Stachula SA, Raetz CR, Anderson MS. The firA gene of Escherichia coli encodes UDP-3-O-(R-3-hydroxymyristoyl)-glucosamine N-acyltransferase. The third step of endotoxin biosynthesis. J Biol Chem. 1993;268:19866–19874. [PubMed] [Google Scholar]
- 26.Agarwal V, Lin S, Lukk T, Nair SK, Cronan JE. Structure of the enzyme-acyl carrier protein (ACP) substrate gatekeeper complex required for biotin synthesis. Proc Natl Acad Sci USA. 2012;109:17406–17411. doi: 10.1073/pnas.1207028109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Babu M, et al. Structure of a SLC26 anion transporter STAS domain in complex with acyl carrier protein: implications for E. coli YchM in fatty acid metabolism. Structure. 2010;18:1450–1462. doi: 10.1016/j.str.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 28.Holak TA, Nilges M, Prestegard JH, Gronenborn AM, Clore GM. Three-dimensional structure of acyl carrier protein in solution determined by nuclear magnetic resonance and the combined use of dynamical simulated annealing and distance geometry. Eur J Biochem. 1988;175:9–15. doi: 10.1111/j.1432-1033.1988.tb14159.x. [DOI] [PubMed] [Google Scholar]
- 29.Roujeinikova A, et al. X-ray crystallographic studies on butyryl-ACP reveal flexibility of the structure around a putative acyl chain binding site. Structure. 2002;10:825–835. doi: 10.1016/s0969-2126(02)00775-x. [DOI] [PubMed] [Google Scholar]
- 30.Jiang Y, Chan CH, Cronan JE. The soluble acyl-acyl carrier protein synthetase of Vibrio harveyi B392 is a member of the medium chain acyl-CoA synthetase family. Biochemistry. 2006;45:10008–10019. doi: 10.1021/bi060842w. [DOI] [PubMed] [Google Scholar]
- 31.Rock CO, Cronan JE, Jr, Armitage IM. Molecular properties of acyl carrier protein derivatives. J Biol Chem. 1981;256:2669–2674. [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiu X, Janson CA. Structure of apo acyl carrier protein and a proposal to engineer protein crystallization through metal ions. Acta Crystallogr D. 2004;60:1545–1554. doi: 10.1107/S0907444904015422. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 38.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 39.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.3r1. 2010 [Google Scholar]
- 42.Stokes GB, Stumpf PK. Fat metabolism in higher plants. The nonenzymatic acylation of dithiothreitol by acyl coenzyme A. Arch Biochem Biophys. 1974;162:638–648. doi: 10.1016/0003-9861(74)90226-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.