

Abstract
The clinical management of cancer reflects a balance between treatment efficacy and toxicity. While typically, combination therapy improves response rate and time to progression compared with sequential monotherapy, it causes increased toxicity. Consequently, in cases of advanced cancer, emerging guidelines recommend sequential monotherapy, as a means to enhance quality of life. An alternative approach that could overcome nonspecific toxicity while retaining therapeutic efficacy, involves the combination of chemotherapy with targeted therapy. In the current study, we tested the hypothesis that combination therapy targeting survivin (BIRC5) and low-dose doxorubicin (Dox) will show enhanced therapeutic potential in the treatment of cancer, as compared to monotherapy with Dox. We demonstrate in both in vitro and in vivo models of breast cancer that combination therapy with a low dose of Dox and an anti-survivin siRNA nanodrug (MN-siBIRC5) is superior to mono-therapy with either low- or high-dose Dox alone. Importantly, therapeutic efficacy showed prominent sequence dependence. Induction of apoptosis was observed only when the cells were treated with Dox followed by MN-siBIRC5, whereas the reverse sequence abrogated the benefit of the drug combination. In vivo, confirmation of successful sequence dependent combination therapy was demonstrated in a murine xenografts model of breast cancer. Finally, to determine if the observed effect is not limited to breast cancer, we extended our studies to a murine xenograft model of pancreatic adenocarcinoma and found similar outcomes as shown for breast cancer.
Keywords: small interfering RNA, nanodrug, breast cancer, pancreatic cancer, doxorubicin
Introduction
Doxorubicin (Dox) is a common chemotherapeutic used in the treatment of a wide range of cancers including breast adenocarcinomas 1. Despite its excellent anti-tumor activity, Dox has a relatively low therapeutic index and its clinical usage is limited due to acute and chronic toxicities such as immunosupression and cardiotoxicity 2. In addition, resistance to this agent is common, representing a major obstacle to successful treatment 3. Therefore, reducing the cytotoxic effects of Dox while enhancing therapeutic efficacy and reducing or eliminating drug resistance is warranted.
One of the major factors responsible for chemoresistance, is the evolution of the tumor cells towards a phenotype that is resistant to apoptotic cell kill. On a molecular level, this phenotype manifests as the over-expression of antigens from the inhibitor of apoptosis (IAP) family. A key member of this family is survivin (encoded by the birc5 gene). Survivin, a multi-regulator of cell cycle and apoptosis 4,5 is over-expressed in all human cancers but demonstrates low expression in normal tissues 6. Its increased expression has been detected in 90% of primary breast cancers correlates with poor clinical outcomes. Furthermore, increased survivin levels have been shown to be significantly associated with negative hormone receptor status 7. Importantly, high levels of survivin have been detected in other cancers such as pancreatic cancer, where it correlates with both cellular proliferation and apoptosis 8 pointing to a possible ubiquitous role of this anti-apoptotic marker.
Considering the potential value of reducing or abolishing survivin expression as a means of overcoming chemoresistance, the process of RNA interference (RNAi) can prove valuable. Indeed, down-regulation of birc5 by RNAi demonstrated promise in acute lymphoblastic leukemia 9, lung, and cervical carcinoma in vitro 10, and breast cancer in vivo 11.
Still, the delivery of siRNA to solid tumors remains a challenging task because of poor uptake and low stability in vivo. To address this problem, we have previously established an image-guided in vivo siRNA delivery platform (MN-siRNA) that consists of superparamagnetic iron oxide nanoparticles (MN, detectable by magnetic resonance imaging, MRI), conjugated to siRNA (MN-siRNA) 12. We further improved this platform by designing a tumor-targeted version of this siRNA nanodrug (MN-siBIRC5) by functionalizing the nanoparticles with peptides (EPPT) specifically targeting the underglycosylated mucin 1 (uMUC-1) tumor antigen, and by attaching therapeutic synthetic siRNA targeting birc5 13. Using this nanodrug, we achieved efficient in vivo delivery of anti-survivin siRNA in murine breast cancer xenografts, resulting in moderate inhibition of tumor growth 13.
In the present study, we hypothesized that the observed moderate arrest of tumor growth could be significantly improved by using a combination therapeutic strategy that involves low-dose Dox and MN-siBIRC5 in murine xenograft models of breast (triple negative) cancer. Surprisingly, we found that the treatment protocol relied on strict sequence dependence of drug administration and could result in a highly significant inhibition of tumor growth. We also showed that the application of low-dose Dox in sequence-dependent combination with the anti-survivin siRNA nanodrug could overcome issues of morbidity and toxicity, and result in increased survival. Furthermore, we demonstrated the applicability of this approach to other types of cancer (pancreatic adenocarcinoma) attesting to the potential widespread utility of this approach. In both cases MRI was used to assure nanodrug delivery to the tumors.
Material and Methods
Nanodrug synthesis and characterization
The MN-siBIRC5 nanodrug was synthesized as described in 13. It consists of superparamagnetic iron oxide nanoparticles (for magnetic resonance imaging), conjugated to synthetic siRNA targeting the tumor-specific anti-apoptotic gene birc5. To ensure preferential tumor cell delivery of the nanodrug, it was also functionalized with peptides (EPPT) targeting the tumor specific antigen uMUC-1. uMUC-1 is overexpressed and underglycosylated on 90% of human breast malignancies and on over 50% of all human cancers 14. We have previously established the in vivo tumor-targeting properties of the MN-EPPT platform in a variety of adenocarcinoma models, including breast cancer 15–18. The parental MN (crosslinked dextran-coated superparamagnetic iron oxide nanoparticles) was synthesized as described in 16. The targeting EPPT peptide was coupled to MN to obtain the resultant MN-EPPT precursor probe using the heterobifunctional cross-linker, N-succinimidyl 3-(2-pyridyldithio) propionate (SPDP, Pierce Biotechnology, Rockford, IL). The peptide, EPPT, was attached to this linker via the sulfhydryl reactive pyridyl disulfide residue as described in 13.
Five-prime-sense thiol-modified birc5-targeting and scrambled siRNA duplexes were designed and synthesized by Dharmacon (Lafayette, CO). The siRNA oligos were then conjugated to MN-EPPT as described in 13 to obtain MN-siBIRC5 or MN-siSCR.
The conjugation efficiency and dissociation stoichiometry of siRNA from MN were characterized by performing gel electrophoresis of MN-siRNA, following incubation in the reducing buffer DTT as described 13.
Cell lines and culture
All of the studies were performed using either triple negative breast carcinoma BT-20 cells carrying mutated p53 or pancreatic adenocarcinomas CAPAN-2 cell lines (ATCC, Manassas, VA). Cells were cultured them in MEM (Na-pyruvate and non-essential amino acid added extra) and McCoy’s 5A media respectively supplemented with heat inactivated FBS (10%), 100U/ml penicillin and 100μ/ml streptomycin (Life Technology, Grand Island, NY). The cell lines were authenticated based on viability, recovery, growth, morphology, and isoenzymology by the supplier (ATCC). For studies on cytotoxicity, cells were treated with PBS, Dox (0.1μM) either alone or in sequential combination with the active or inactive nanodrug (50μg/ml Fe; 1.25nmoles/ml oligo): Dox⇒MN-siBIRC5: Dox alone for 24h and then MN-siBIRC5 for another 24 h; MN-siBIRC5⇒Dox: MN-siBIRC5 for 24h and then Dox for another 24 h.
Immunofluorescence
Immunostaining for cleaved PARP fragments was performed by fixing cells or tissue sections in 2% paraformaldehyde. Fixed cells were permeabilized and incubated with either anti-cleaved PARP antibody (Cell signaling, Danvers, MA) or anti-human survivin polyclonal antibody (R&D system, Minneapolis, MN) followed by Texas Red-conjugated goat anti-rabbit secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). Nuclear counterstaining with DAPI was performed. Immunostained cells were examined under a Nikon Eclipse 50i fluorescence microscope.
MTT assay
Cell proliferation was determined in BT-20 cells by MTT assay as previously described 19. For the IC50 studies, the cells were incubated with various concentrations of Dox (0–1μM), for 72 h at 37°C. The inhibitory concentration for 50% cell viability (IC50) was calculated based on the generated cell viability curves. For the combination treatment studies, the cells were incubated with PBS, Dox (0.1μM) either alone or in sequential combination with the active or inactive nanodrug (50μg/ml Fe; 1.25nmoles/ml oligo): Dox⇒MN-siBIRC5: Dox alone and then MN-siBIRC5; MN-siBIRC5⇒Dox: MN-siBIRC5 and then Dox. Results were derived from at least three independent sets of triplicate experiments.
Caspase-3 activity assay
To assess the effect of combination therapy on cell death, caspase activity was analyzed fluorometrically according to the manufacturer’s instructions (ENZO Life Sciences, Plymouth Meeting, PA).
For the detection of caspase-3 activity in tumors, tissues were homogenized in 1x lysis buffer and processed similarly to the cell lysate preparation.
Oil-Red O staining
Cells were fixed in formaldehyde (4%) and rinsed with 60% isopropanol before staining with filtered Oil-Red O (0.18%) in isopropanol/water. Finally cells were rinsed with tap water before being visualized in a phase contrast light microscope.
For quantitative estimation of Oil-Red O staining, cells were stained as above and the cell-bound Oil-Red O was serially extracted with isopropanol. In a parallel experiment, the number of cells was counted. To quantify the extracted Oil-Red O, absorbance at 510nm was determined using spectrophotometry.
Western Blotting
Cells were lysed in cell extraction buffer (Invitrogen, Camarillo, CA) containing 1mM PMSF and protease inhibitors cocktail (Sigma, St. Louis, MO). Western blot was performed according to a standard procedure using anti-human survivin polyclonal rabbit antibody (0.5μg/ml) (R&D Systems, Minneapolis, MN) followed by horseradish peroxidase-conjugated goat anti-rabbit secondary antibodies (Invitrogen, Camarillo, CA). Membranes were developed using ECL Plus Western Blotting detection reagents kit (GE Healthcare, Piscataway, NJ) according to the manufacturer’s specifications.
Cell cycle analysis
Cell cycle distribution was analyzed by fluorescence-activated cell-sorting (FACS). The cells (1 × 10 6) were trypsinized, washed once with PBS and fixed in 80% ethanol overnight at 4 ° C. The next day, fixed cells were washed with cold PBS and then resuspended in 1.0ml of DNA staining solution (50μg/ml propidium iodide, 0.5mg DNAse-free RNase A (Sigma) and 0.1% Triton X-100 in PBS) for 30 min at room temperature. The stained cells were analyzed using a FACScan laser flowcytometer (Becton Dickinson, San Jose, CA). The fraction of the G1, S and G2/M phase of cells on DNA histograms was analyzed using DNA content software.
Therapy studies
Five- to-6-week-old 30g female nu/nu mice (Massachusetts General Hospital Radiation Oncology breeding facilities) were used for in vivo animal experiments. Human triple negative breast cancer BT-20 or pancreatic adenocarcinoma cells CAPAN-2 were injected subcutaneously into the left flanks. Animals were used in experiments on day 10 after the inoculation, when tumors were 0.3<0.5 cm in diameter. Mice were randomly assigned to the various treatment groups (n = 5 mice per group). Mice were treated individually with either saline or Dox (2 or 10mg/kg body weight) (Sigma) via intraperitoneal (i.p.) injections. For combination treatment experiments, mice were injected with Dox (2mg/kg) i.p. followed 24h later by either MN-siBIRC5 (10mg/kg of Fe and 250nmole/kg of siRNA) or MN-siSCR, injected intravenously (i.v.). For reverse sequential testing, MN-siBIRC5 was injected first followed by Dox (2 mg/kg) 24 hrs later. Mice with BT-20 xenografts were treated once per week for 5 weeks and CAPAN-2 xenografts for 6 weeks. Mice were weighed, and tumor width and length were measured using calipers every week. Tumor volumes were calculated using the equation: V= (L × W2) × 0.5, where L is the length and W is the width of the tumor. Body weights were also monitored throughout the study. Mice were sacrificed when tumors reached the maximum allowable volume.
All animal experiments were performed in compliance with institutional guidelines and according to the animal protocol approved by the Institutional Animal Care and Use Committee at Massachusetts General Hospital.
In vivo MR and optical imaging
To test the tumor-specific nanodrug delivery, MR imaging was done before and 24hrs after nanodrug injections. Imaging was performed using a 9.4-T Bruker horizontal bore scanner equipped with ParaVision 3.0 software. The imaging protocol consisted of coronal T2-weighted spin echo (SE) pulse sequences with the following parameters: SE repetition time/echo time (TE) = 3,000/[8, 16, 24, 32, 40, 48, 56, 64]; field of view = 32 × 32 mm2; matrix size = 128 × 128 pixels; slice thickness = 0.5 mm; in-plane resolution = 250 × 250 μm2. Image reconstruction and analysis were performed using Marevisi 3.5 software.
T2 relaxation times were calculated by manually segmenting out the tumor on MR images. For T2 map analysis of relaxation times, the terminal slices were not included in order to avoid interference from partial volume effects.
In vivo gray scale photographic images were acquired immediately after each MR imaging session using a whole-body animal imaging system (IVIS Spectrum, Caliper Life Sciences, Hopkinton MA).
Statistical analysis
Data were expressed as either mean ± SD or SEM, where indicated. Statistical significance was determined by using Student’s t test. P value of <0.05 was considered statistically significant.
Results
The in vitro cytotoxicity of Dox plus MN-siBIRC5 combination treatment follows a sequence-dependent pattern in breast cancer cells
The triple negative (ER, PR and HER2) breast cancer cell line BT-20 was chosen in this study because it has a constitutive level of survivin expression. To identify the level of Dox that is associated with minimal growth inhibition in these cells for our combination therapy studies, we determined the IC50 of Dox using an in vitro cell proliferation assay (MTT assay). As shown in Suppl. Fig. 1, Dox inhibited cell proliferation in a dose dependent manner with an IC50 slightly below 0.7μM. To ensure minimal growth inhibition for our combination treatment studies, all subsequent experiments were carried out at doses of 0.1μM (IC10).
Next, we investigated whether combination therapy with the two drugs would be effective at inhibiting tumor cell proliferation and inducing cell death. BT-20 cells were exposed to low-dose Dox either alone or in sequential combination with MN-siBIRC5 or the inactive control MN-siSCR for 72h. We found that, when Dox was added together or 24h before the nanodrug, significant (p<0.001) enhancement in cellular growth inhibition was observed. In contrast, when the nanodrug was added prior to Dox there was no effect on cell viability (Fig. 1A). To test whether growth inhibition in BT-20 cells by sequential combination therapy reflects apoptosis, cells were evaluated by measuring caspase-3 activity and cleaved PARP, since both are crucial component of apoptotic cell death. Fig. 1B represents the results obtained for caspase-3 activity. Consistent with the cell viability data, significant (p<0.0005) level of increased caspase-3 activity (Fig. 1B) was observed only in sequential combination when Dox was added prior to the nanodrug. In contrast, either drug alone or the reverse sequential combination induced significantly less caspase-3 enzyme activity.
Fig. 1. In vitro pro-apoptotic effects of Dox and MN-siBIRC5 in breast adenocarcinoma (BT-20) cells.

(A) Effect of the drugs on cell proliferation, as evaluated by MTT assay. BT-20 cells were treated with PBS, Dox (0.1μM) or nanodrug either alone or in sequential combination (Dox-MN-siBIRC5: Dox alone for 24h and then MN-siBIRC5 for another 24 h; MN-siBIRC5-Dox: MN-siBIRC5 for 24h and then Dox for another 24 h). Results are expressed as mean ± SD and represent a summary of three independent experiments (n=3, two-tailed Student’s t-test, *P<0.001). (B) Effect of the drugs on caspase-3 activation. BT-20 cells were treated with PBS, Dox (0.1μM) or nanodrug either alone or in sequential combination (Dox-MN-siBIRC5: Dox alone for 24h and then MN-siBIRC5 for another 24 h; MN-siBIRC5-Dox: MN-siBIRC5 for 24h and then Dox for another 24 h). Caspase-3 activity was measured in cell lysates prepared 48h after additional incubation with the drugs. Caspase-3 activation was significantly higher in the Dox-MN-siBIRC5 treatment group relative to PBS. Data are expressed as mean ± SD and are representative of three independent experiments (n=3; two-tailed Student’s t-test, *P<0.05, **P<0.005). (C) Analysis of PARP cleavage. After exposing the cells to drug treatments (as above), immunofluorescence was performed to detect cleaved PARP fragments. Nuclei (DAPI, Blue) and cleaved PARP (Red) were visualized by fluorescence microscopy. PARP cleavage was most prominent in the Dox-MN-siBIRC5 treatment group.
Consistent with the above findings, a confirmatory immunofluorescence assay was performed to detect cleaved PARP (a substrate for caspase-3) in the cells. As expected, these experiments also revealed a successful sequential combination strategy with the addition of Dox prior to the nanodrug (Fig. 1C). Together these results indicated that the sequential combination treatment with Dox and MN-siBIRC5 induces cellular apoptosis via a pathway that involves caspase-3-mediated PARP cleavage.
Based on these findings we hypothesized that low-dose Dox induces phenotype associated with senescence without overt apoptosis. Consistent with this hypothesis, we observed morphological changes (Fig. 2A) and cytoplasmic lipid droplet deposition (Fig. 2B) associated with cellular stress. Indeed, cytoplasmic lipid droplet deposition has been implicated as a sign of mitochondrial dysfunction apparent prior to overt apoptosis 20. In addition, the prior literature suggests that low-dose Dox induces cell cycle arrest without cytoxocity 21. This observation was confirmed by our studies, in which cells treated with low-dose Dox were arrested in the G2 phase of the cell cycle (Fig. 2C).
Fig. 2. Effects of low-dose Dox on tumor cell phenotype.

(A) Morphological changes induced by treatment with low-Dox. Cell and nuclear enlargement were observed after 4 days of treatment with low-dose Dox. (B) Quantitation of lipid droplet synthesis as determined by Oil-Red O staining. Cells were treated with Dox as above and analyzed by spectrophotometry. Na-butyrate represents a positive control. Absorbance is expressed as OD per million of cells. Lipid droplet synthesis was significantly enhanced by low-dose Dox. Data are represented as means ± standard deviation (n = 3, two-tailed t-test, ***p<0.0005). (C) Cell cycle analysis. Cell cycle arrest in G2 was observed following treatment with low-dose Dox. (D) Survivin expression. BT-20 cells were treated with Dox (0.1μM) for the indicated times. Cell lysates were analyzed by western blot with anti-survivin antibody. Upregulation of survivin by Dox was observed as early as 16 hrs of treatment. (E) Survivin downregulation by MN-EPPT-siBIRC5. BT-20 cells were treated with Dox, MN-siBIRC5, or MN-siSCR for 48h. The lysate proteins were analyzed for survivin and beta actin expression by western blotting. Beta-actin was used as control.
Based on our observations, we also speculated that if low-dose Dox induces survivin expression, as shown in the literature 22, administration of Dox subsequent to survivin inhibition by MN-EPPT-siBIRC5 would annul the effect of the nanodrug. To determine this, we examined survivin expression in cells treated with low-dose Dox. A moderate/constitutive level of survivin was steadily expressed in BT-20 cells. However, a time-dependent increase in survivin protein was observed in cells treated with low-dose Dox (Fig. 2D), whereas addition of the nanodrug successfully inhibited expression of the antigen (Fig. 2E). These results suggested that low-dose Dox sensitized the cell to apoptosis by mediating cell cycle arrest. However, it also caused upregulation of survivin, inhibiting apoptosis. The subsequent depletion of survivin by the nanodrug abolished this anti-apoptotic effect, resulting in cell death.
The in vitro cytotoxicity of Dox plus MN-siBIRC5 combination treatment follows a sequence-dependent pattern in pancreatic cancer cells
Survivin is involved in the development various cancers including human pancreatic ductal carcinoma 23 and expressed in the majority of pancreatic cancer, where it correlates with proliferation and apoptosis 8. Therefore we examined whether sequential treatment with low-dose Dox and the anti-survivin nanodrug would have implications for cancer treatment that extend beyond breast cancer. Incubation of CAPAN-2 pancreatic adenocarcinoma cells first with Dox and then with the nanodrug demonstrated significant (p<0.0005) induction of caspase-3 activity (70% more than the PBS control) only when cells were exposed to sequential treatment (Fig. 3A). The increased levels of apoptosis were also confirmed by examining the levels of cleaved PARP fragments (Fig. 3B), demonstrating that the therapeutic efficacy of the combination treatment in pancreatic cancer cells relies on strict sequence dependence, similar to the results observed with breast cancer.
Fig. 3. In vitro pro-apoptotic effects of Dox and MN-siBIRC5 in pancreatic adenocarcinoma (CAPAN-2) cells.

(A) Effect of the combined drugs on caspase-3 activation. Cells were treated with PBS and Dox (0.1μM), either alone or in sequential combination with MN-siBIRC5 or MN-siSCR. For the sequential combination treatments, Dox was either added for 24h prior to MN-siBIRC5/MN-siSCR or in the reverse sequence. Caspase-3 activity was measured in cell lysates after 48h incubation. Caspase-3 activation was significantly higher in the Dox-MN-siBIRC5 treatment group relative to PBS. Data expressed as mean ± SD and are representative of three independent experiments (n=3; two-tailed Student t-test; ***P<0.0005). (B) Analysis of PARP cleavage. After exposing the cells to drug treatments (as above), immunofluorescence was done to detect cleaved PARP fragments. Nuclei (DAPI, Blue) and cleaved PARP 29 were visualized by fluorescence microscopy. PARP cleavage was most prominent in the Dox-MN-siBIRC5 treatment group.
The in vivo cytotoxicity of Dox plus MN-siBIRC5 combination therapy follows a sequence-dependent pattern in a murine xenograft model of breast cancer
To determine whether treatment of breast cancer xenografts with Dox followed by the MN-siBIRC5 nanodrug could inhibit tumor growth, mice subcutaneously implanted with human breast tumor cells (BT-20) were treated with low-dose Dox (2mg/kg) and high-dose Dox (10mg/kg) via intraperitoneal (i.p.) injection or in combination with MN-siBIRC5 (10mg/kg Fe; 250nmoles/kg siBIRC5) or MN-siSCR delivered intravenously (i.v.).
Delivery of the nanodrug to tumors was monitored by noninvasive imaging. The strong T2 magnetic susceptibility effects mediated by superparamagnetic iron oxide nanoparticles allows their visualization in tissue by magnetic resonance imaging (MRI). Namely, their presence in tissue is reflected by marked shortening of T2 relaxation times, resulting in a loss of signal (darkening) on MR images. Quantitative analysis of tumor T2 relaxation times revealed a more than 40% shortening of tumor T2 relaxation times 24 h after nanodrug injection, indicating the presence of MN-siBIRC5 in the tumor (Suppl. Fig. 2).
Consistent with our in vitro results, significant (p < 0.05) inhibition of tumor growth was observed only in the group in which Dox (2mg/kg) was administered prior to MN-siBIRC5 (Fig. 4A). The reverse application of drug treatment by injecting mice with MN-EPPT-siBIRC5 for 24h followed by Dox did not result in this effect (Fig. 4A). Treatment with a high dose of Dox (10mg/kg) displayed morbidity, whereas the low dose of Dox alone moderately inhibited primary tumor as compared to PBS alone (Fig. 4A). This indicated that the sequence of drug administration was essential to achieve maximum tumor growth inhibition. The advantage of the combination treatment was clearly seen when assessing animal survival. Whereas the high-dose Dox treatment clearly reduced tumor growth (Fig. 4A and B), it also led to considerable morbidity (Fig. 4C and D). Of all treatment groups, the mice subjected to the Dox-MN-siBIRC5 treatment sequence survived the longest (Fig. 4C) and displayed no signs of systemic toxicity, e.g. weight loss, as compared to PBS-treated controls (Fig. 4D).
Fig. 4. In vivo sequence-dependent therapy with low-dose Dox and MN-siBIRC5 in a murine xenograft model of breast adenocarcinoma.

(A) Mice bearing subcutaneous breast (BT-20) tumors were injected once a week for 5 weeks with 1) saline, 2) Dox (10mg/kg), 3) Dox (2mg/kg), 4) Dox (2mg/kg) followed 24h later by MN-siBIRC5 (10mg/kg Fe; 250nmoles/kg siBIRC5), 5) Dox (2mg/kg) followed 24h later by MN-siSCR (10mg/kg Fe; 250nmoles/kg siSCR), 6) MN-siBIRC5 for 24h followed by Dox (2mg/kg). The mean ± standard error of the mean (SEM, #44) of the relative tumor volumes is shown at the indicated time points (n = 5, two-tailed t-test, * p < 0.05). Tumor growth was significantly inhibited in the Dox-MN-siBIRC5 treatment group relative to the other groups. (B) Grayscale photographs reflecting tumor growth kinetics. Tumor growth was visibly inhibited in the Dox-MN-siBIRC5 treatment group relative to controls. (C) Survival of tumor-bearing mice during treatment. High-dose Dox, while inhibiting tumor growth, was associated with high mortality. (D) Body weight of tumor-bearing mice during treatment. High-dose Dox, while inhibiting tumor growth, was associated with high morbidity.
To further examine the reasons behind the tumor-growth reduction mediated by the sequential combination treatment, we analyzed tumor tissues for markers of apoptosis. We observed a significantly (p<0.0005) increased activity of caspase-3 enzyme in tumor extracts from mice treated with Dox 24h prior to MN-siBIRC5 (Fig. 5A), as compared to PBS treated samples. In concordance with the caspase-3 activation data, we also detected a higher level of cleaved PARP fragments (Fig. 5B) when Dox was administered prior to the nanodrug.
Fig. 5. Ex vivo analysis of tumor cell death following treatment with Dox and MN-siBIRC5 in BT-20 tumor xenografts.
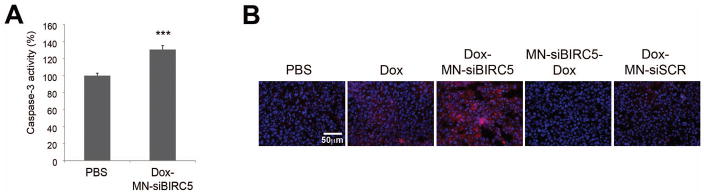
(A) Detection of caspase-3 activity in tumor lysates. Excised tumors were analyzed by caspase-3 enzyme assay. Data are represented as ± S.D. (n=8, two-tailed Student’s t-test, ***P<0.0005 compared to PBS control). Caspase-3 activation was significantly higher in the Dox-MN-siBIRC5 treatment group relative to PBS. (B) Detection of cleaved PARP fragments in tumor tissue sections by immunofluorescence. Nuclei stained with (DAPI, Blue) and cleaved PARP immunostained (Texas red, Red) were visualized by fluorescence microscopy. PARP cleavage was most prominent in the Dox-MN-siBIRC5 treatment group.
The in vivo cytotoxicity of Dox plus MN-siBIRC5 combination therapy follows a sequence-dependent pattern in a murine xenograft model of pancreatic cancer
To investigate the wide applicability of this approach we evaluated the antitumor effects of combination therapy on pancreatic cancer in vivo. Mice bearing a murine xenografts model of human pancreatic adenocarcinoma (CAPAN-2) were treated with PBS or Dox (2mg/kg) or in sequential combination of Dox and the nanodrug once a week for 7 weeks. The Dox treatment initially showed moderate inhibition of tumor growth, followed by tumor growth similar to the PBS control. The combination treatment showed inhibition of tumor growth by 68% compared to the PBS control mice on day 36 after treatment (p<0.05, Fig. 6A). Body weight was not significantly different between the treatment groups (data not shown), indicating the absence of systemic toxicity.
Fig. 6. In vivo sequence-dependent therapy with Dox and MN-siBIRC5 in a murine xenograft model of pancreatic adenocarcinoma.

(A) Mice bearing subcutaneous pancreatic (CAPAN-2) tumors were injected once a week for 6 weeks with saline, Dox (2mg/kg) alone and Dox followed 24h later by MN-siBIRC5 (10mg/kg Fe; 250nmoles/kg siBIRC5). The mean ± standard error of the mean (SEM) of the relative tumor volumes is shown at the indicated time points (n = 3, two-tailed Student t-test; P<0.05). Tumor growth was significantly inhibited in the Dox-MN-siBIRC5 treatment group relative to the other groups. (B) Detection of caspase-3 activity in tumor lysates. Excised tumors were analyzed by caspase-3 enzyme assay. Data are represented as ± S.D. (n=8, two-tailed Student’s t-test, **P<0.0005). Caspase-3 activation was significantly higher in the Dox-MN-siBIRC5 treatment group relative to PBS. (C) Detection of cleaved PARP in tumor tissue sections by immunofluorescence. Nuclei stained with (DAPI, Blue) and cleaved PARP immunostained (Texas red, Red) were visualized by fluorescence microscopy. PARP cleavage was most prominent in the Dox-MN-siBIRC5 treatment group.
Ex vivo analysis by caspase-3 activity and PARP cleavage in tissues clearly confirmed the induction of apoptosis by the sequence-dependent combination treatment (Fig. 6B and C). These results established that the sequence-dependent combination drug treatment with Dox and MN-siBIRC5 exerted a significant anti-tumor effect against pancreatic cancer in vivo and suggested that the observed anti-tumor effects may have a global relevance against adenocarcinoma.
Discussion
Cancer therapy often involves combination treatment protocols that rely on the synergism between different therapeutic agents. The goal is to combine drugs with different mechanisms of action to increase efficacy, while maintaining a favorable side effect and toxicity profile.
One example of a drug that is commonly used as a component of combination therapies is doxorubicin. Doxorubicin is one of the most widely used chemotherapeutic agents for the treatment of breast cancer. Although doxorubicin induces substantial antitumor activity and cytotoxicity 24, 25, it also leads to the development of resistance through altered bioavailability or inactivation and non-specific cytotoxicity 2. In the course of treatment with the drug, the delicate balance between drug sensitivity and resistance displayed by targeted tumor cells often gets lost. In addition, cumulative cardiotoxicity is a major limitation to the therapeutic use of doxorubicin and can lead to potentially fatal congestive heart failure 26, 27. Therefore, reducing the cytotoxic effects while enhancing therapeutic efficacy by developing combination therapy is warranted.
Here, we address this issue by examining the efficacy of a novel therapeutic strategy that combines a low-dose doxorubicin with a siRNA nanodrug that inhibits the anti-apoptotic gene birc5/survivin (MN-siBIRC5). This approach is selected as a means of increasing anticancer efficacy while limiting systemic toxicity, associated with high-dose doxorubicin monotherapy. Our results reveal that the nanodrug potentiates the anticancer activity of low doses of doxorubicin in a sequence dependent manner by direct activation of caspase-3 and PARP cleavage pathways, which leads to apoptosis and cell death. Namely, induction of apoptosis was observed only when the cells were treated with Dox followed by MN-siBIRC5, whereas the reverse sequence abrogated the benefit of the drug combination. This observation is likely pertinent to a scenario in which drug resistance is not established and could be applied as a means of avoiding its emergence. In a situation in which drug resistance is present, pre-treatment with a Survivin inhibitor followed by a high-dose of Dox may be necessary as evidenced by the recent literature 28.
The findings described here have practical implications for cancer therapy. Our data provide compelling evidence that sequence dependent combination treatment using low-dose doxorubicin and an anti-survivin agent is efficacious against both breast and pancreatic cancer. Considering the disadvantages of standard chemotherapy, e.g. non-specific delivery, toxicity to healthy tissues, and the possibility for resistance to it, we envision a future in which chemotherapy is complemented by sequence dependent molecularly targeted therapies. The studies presented here have evolved in response to this vision and illustrate the promise of alternative nano-therapeutic approaches with fewer adverse side effects in comparison to currently available chemotherapeutic regimens.
Supplementary Material
Determination of 50% inhibition of cell proliferation (IC50) manifested by Dox in BT-20 cells. Dox (0–1μM) was added to BT-20 cells for 72h and cell proliferation was analyzed by MTT assay. PBS treated cells served as controls. An IC50 of 0.5 μM was determined.
MR imaging of subcutaneous mammary tumors representing color-coded pre-and post contrast T2 maps. At 24h after tail vein injection of the nanodrug there was a decrease of signal intensity (T2 shortening) associated with the tumors (outlined), which indicates accumulation of MN-siBIRC5 in the tumor. Error bars denote mean ± SD (n = 3, two-tailed Student t-test, **P<0.005).
Novelty and Impact.
We show for the first time that sequence-dependent therapy with low-dose doxorubicin and an anti-survivin siRNA nanodrug can have a profound effect on primary tumor growth, while avoiding the issues of toxicity and morbidity associated with standard chemotherapy. This finding can form the basis of more effective clinical therapeutic interventions.
Acknowledgments
We thank Pamela Pantazopoulos for help with the in-vitro and ex-vivo studies.
Grant Support:
This work was supported in part under Grants R00CA129070 and R01CA16346101A1 from the National Cancer Institute and a Research Grant from the Breast Cancer Alliance.
Footnotes
Conflict of Interest Statement: The authors have no conflict of interest to disclose.
References
- 1.Chew HK. Medical management of breast cancer: today and tomorrow. Cancer Biother Radiopharm. 2002;17:137–49. doi: 10.1089/108497802753773766. [DOI] [PubMed] [Google Scholar]
- 2.Hardenbergh PH, Recht A, Gollamudi S, Come SE, Hayes DF, Shulman LN, O’Neill A, Gelman RS, Silver B, Harris JR. Treatment-related toxicity from a randomized trial of the sequencing of doxorubicin and radiation therapy in patients treated for early stage breast cancer. Int J Radiat Oncol Biol Phys. 1999;45:69–72. doi: 10.1016/s0360-3016(99)00127-3. [DOI] [PubMed] [Google Scholar]
- 3.Smith L, Watson MB, O’Kane SL, Drew PJ, Lind MJ, Cawkwell L. The analysis of doxorubicin resistance in human breast cancer cells using antibody microarrays. Mol Cancer Ther. 2006;5:2115–20. doi: 10.1158/1535-7163.MCT-06-0190. [DOI] [PubMed] [Google Scholar]
- 4.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–21. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 5.Pennati M, Folini M, Zaffaroni N. Targeting survivin in cancer therapy: fulfilled promises and open questions. Carcinogenesis. 2007;28:1133–9. doi: 10.1093/carcin/bgm047. [DOI] [PubMed] [Google Scholar]
- 6.Fukuda S, Pelus LM. Survivin, a cancer target with an emerging role in normal adult tissues. Mol Cancer Ther. 2006;5:1087–98. doi: 10.1158/1535-7163.MCT-05-0375. [DOI] [PubMed] [Google Scholar]
- 7.Ryan BM, Konecny GE, Kahlert S, Wang HJ, Untch M, Meng G, Pegram MD, Podratz KC, Crown J, Slamon DJ, Duffy MJ. Survivin expression in breast cancer predicts clinical outcome and is associated with HER2, VEGF, urokinase plasminogen activator and PAI-1. Ann Oncol. 2006;17:597–604. doi: 10.1093/annonc/mdj121. [DOI] [PubMed] [Google Scholar]
- 8.Sarela AI, Verbeke CS, Ramsdale J, Davies CL, Markham AF, Guillou PJ. Expression of survivin, a novel inhibitor of apoptosis and cell cycle regulatory protein, in pancreatic adenocarcinoma. Br J Cancer. 2002;86:886–92. doi: 10.1038/sj.bjc.6600133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morrison DJ, Hogan LE, Condos G, Bhatla T, Germino N, Moskowitz NP, Lee L, Bhojwani D, Horton TM, Belitskaya-Levy I, Greenberger LM, Horak ID, et al. Endogenous knockdown of survivin improves chemotherapeutic response in ALL models. Leukemia. 2012;26:271–9. doi: 10.1038/leu.2011.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trabulo S, Cardoso AM, Santos-Ferreira T, Cardoso AL, Simoes S, Pedroso de Lima MC. Survivin silencing as a promising strategy to enhance the sensitivity of cancer cells to chemotherapeutic agents. Mol Pharm. 2011;8:1120–31. doi: 10.1021/mp100426e. [DOI] [PubMed] [Google Scholar]
- 11.Li Z, Yin PH, Yang SS, Li QY, Chang T, Fang L, Shi LX, Fang GE. Recombinant attenuated Salmonella typhimurium carrying a plasmid co-expressing ENDO-VEGI151 and survivin siRNA inhibits the growth of breast cancer in vivo. Mol Med Rep. 2013;7:1215–22. doi: 10.3892/mmr.2013.1308. [DOI] [PubMed] [Google Scholar]
- 12.Medarova Z, Pham W, Farrar C, Petkova V, Moore A. In vivo imaging of siRNA delivery and silencing in tumors. Nat Med. 2007;13:372–7. doi: 10.1038/nm1486. [DOI] [PubMed] [Google Scholar]
- 13.Kumar M, Yigit M, Dai G, Moore A, Medarova Z. Image-guided breast tumor therapy using a small interfering RNA nanodrug. Cancer Res. 2010;70:7553–61. doi: 10.1158/0008-5472.CAN-10-2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayes DF, Mesa-Tejada R, Papsidero LD, Croghan GA, Korzun AH, Norton L, Wood W, Strauchen JA, Grimes M, Weiss RB, et al. Prediction of prognosis in primary breast cancer by detection of a high molecular weight mucin-like antigen using monoclonal antibodies DF3, F36/22, and CU18: a Cancer and Leukemia Group B study. J Clin Oncol. 1991;9:1113–23. doi: 10.1200/JCO.1991.9.7.1113. [DOI] [PubMed] [Google Scholar]
- 15.Moore A, Medarova Z, Potthast A, Dai G. In vivo targeting of underglycosylated MUC-1 tumor antigen using a multimodal imaging probe. Cancer Res. 2004;64:1821–7. doi: 10.1158/0008-5472.can-03-3230. [DOI] [PubMed] [Google Scholar]
- 16.Medarova Z, Pham W, Kim Y, Dai G, Moore A. In vivo imaging of tumor response to therapy using a dual-modality imaging strategy. Int J Cancer. 2006;118:2796–802. doi: 10.1002/ijc.21672. [DOI] [PubMed] [Google Scholar]
- 17.Medarova Z, Rashkovetsky L, Pantazopoulos P, Moore A. Multiparametric monitoring of tumor response to chemotherapy by noninvasive imaging. Cancer Res. 2009;69:1182–9. doi: 10.1158/0008-5472.CAN-08-2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh SK, Uchida M, Yoo B, Ross AW, Gendler SJ, Gong J, Moore A, Medarova Z. Targeted imaging of breast tumor progression and therapeutic response in a human uMUC-1 expressing transgenic mouse model. Int J Cancer. 2013;132:1860–7. doi: 10.1002/ijc.27872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matito C, Mastorakou F, Centelles JJ, Torres JL, Cascante M. Antiproliferative effect of antioxidant polyphenols from grape in murine Hepa-1c1c7. Eur J Nutr. 2003;42:43–9. doi: 10.1007/s00394-003-0398-2. [DOI] [PubMed] [Google Scholar]
- 20.Boren J, Brindle KM. Apoptosis-induced mitochondrial dysfunction causes cytoplasmic lipid droplet formation. Cell Death Differ. 2012;19:1561–70. doi: 10.1038/cdd.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blagosklonny MV, Robey R, Bates S, Fojo T. Pretreatment with DNA-damaging agents permits selective killing of checkpoint-deficient cells by microtubule-active drugs. J Clin Invest. 2000;105:533–9. doi: 10.1172/JCI8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aliabadi HM, Mahdipoor P, Uludag H. Polymeric delivery of siRNA for dual silencing of Mcl-1 and P-glycoprotein and apoptosis induction in drug-resistant breast cancer cells. Cancer Gene Ther. 2013;20:169–77. doi: 10.1038/cgt.2013.8. [DOI] [PubMed] [Google Scholar]
- 23.Satoh K, Kaneko K, Hirota M, Masamune A, Satoh A, Shimosegawa T. Expression of survivin is correlated with cancer cell apoptosis and is involved in the development of human pancreatic duct cell tumors. Cancer. 2001;92:271–8. doi: 10.1002/1097-0142(20010715)92:2<271::aid-cncr1319>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 24.Gamen S, Anel A, Perez-Galan P, Lasierra P, Johnson D, Pineiro A, Naval J. Doxorubicin treatment activates a Z-VAD-sensitive caspase, which causes deltapsim loss, caspase-9 activity, and apoptosis in Jurkat cells. Exp Cell Res. 2000;258:223–35. doi: 10.1006/excr.2000.4924. [DOI] [PubMed] [Google Scholar]
- 25.Lu Y, Tatsuka M, Takebe H, Yagi T. Involvement of cyclin-dependent kinases in doxorubicin-induced apoptosis in human tumor cells. Mol Carcinog. 2000;29:1–7. [PubMed] [Google Scholar]
- 26.Hershman DL, Shao T. Anthracycline cardiotoxicity after breast cancer treatment. Oncology (Williston Park) 2009;23:227–34. [PubMed] [Google Scholar]
- 27.Barrett-Lee PJ, Dixon JM, Farrell C, Jones A, Leonard R, Murray N, Palmieri C, Plummer CJ, Stanley A, Verrill MW. Expert opinion on the use of anthracyclines in patients with advanced breast cancer at cardiac risk. Ann Oncol. 2009;20:816–27. doi: 10.1093/annonc/mdn728. [DOI] [PubMed] [Google Scholar]
- 28.Yin Q, Shen J, Chen L, Zhang Z, Gu W, Li Y. Overcoming multidrug resistance by co-delivery of Mdr-1 and survivin-targeting RNA with reduction-responsible cationic poly(beta-amino esters) Biomaterials. 2012;33:6495–506. doi: 10.1016/j.biomaterials.2012.05.039. [DOI] [PubMed] [Google Scholar]
- 29.Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, Weinstein CL, Hruban RH, Yeo CJ, Kern SE. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994;8:27–32. doi: 10.1038/ng0994-27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Determination of 50% inhibition of cell proliferation (IC50) manifested by Dox in BT-20 cells. Dox (0–1μM) was added to BT-20 cells for 72h and cell proliferation was analyzed by MTT assay. PBS treated cells served as controls. An IC50 of 0.5 μM was determined.
MR imaging of subcutaneous mammary tumors representing color-coded pre-and post contrast T2 maps. At 24h after tail vein injection of the nanodrug there was a decrease of signal intensity (T2 shortening) associated with the tumors (outlined), which indicates accumulation of MN-siBIRC5 in the tumor. Error bars denote mean ± SD (n = 3, two-tailed Student t-test, **P<0.005).