

Abstract
Introduction
Autoantibodies including anti-human protein S antibody (anti-hPS Ab) and anti-human protein C antibody (anti-hPC Ab) can be detected in patients with autoimmune diseases with hypercoagulability. The objective of the present study was to determine the effects and molecular pathways of these autoantibodies on tissue factor (TF) expression in human coronary artery endothelial cells (HCAECs).
Materials and Methods
HCAECs were treated with anti-hPS Ab or anti-hPC Ab for 3 hours. TF expression was measured by real-time PCR and Western blot. TF-mediated procoagulant activity was determined by a commercial kit. MAPK phosphorylation was analyzed by Bio-Plex luminex immunoassay and Western blot. The potential proteins interacting with anti-hPS Ab were studied by immunoprecipitation, mass spectrometry and in vitro pull-down assay.
Results
Anti-hPS Ab, but not anti-hPC Ab, specifically induced TF expression and TF-mediated procoagulant activity in HCAECs in a concentration-dependent manner. This effect was confirmed in human umbilical endothelial cells (HUVECs). ERK1/2 phosphorylation was induced by anti-hPS Ab treatment, while inhibition of ERK1/2 by U0216 partially blocked anti-hPS Ab-induced TF upregulation (P<0.05). In addition, anti-hPS Ab specifically cross-interacted with platelet phosphofructokinase (PFKP) in HCAECs. Anti-hPS Ab was able to directly inhibit PFKP activities in HCAECs. Furthermore, silencing of PFKP by PFKP shRNA resulted in TF upregulation in HCAECs, while activation of PFKP by fructose-6-phosphate partially blocked the effect of anti-hPS Ab on TF upregulation (P<0.05).
Conclusions
Anti-hPS Ab induces TF expression through a direct interaction with PFKP and ERK1/2 activation in HCAECs. Anti-hPS Ab may directly contribute to vascular thrombosis in the patient with autoimmune disorders.
Keywords: Autoantibody, anti-human protein S antibody, endothelial cell, tissue factor, platelet phosphofructokinase, ERK1/2
Introduction
Protein S is a vitamin K-dependent plasma protein that is mainly synthesized by both hepatocytes and megakaryocytes and serves as a cofactor for the anticoagulant reaction catalyzed by activated protein C [1]. Deficiency of protein S is associated with an increased risk of vascular thrombosis [2]. Acquired deficiency of protein S has been reported in systemic autoimmune disorders [3-7], and many other diseases including HIV infection [8,9], estrogen treatment [10] and malignancies [11]. Anti-protein S autoantibodies are present in a large proportion of patients with acquired protein S deficiency in the antiphospholipid syndrome (APS) and systemic lupus erythematosus (SLE) [12-15]. The patient with infection of chickenpox generated an autoantibody directed against protein S, leading to the thromoembolic complication [16,17]. The autoantibodies directly against a combination of phospholipids with prothrombin, protein C or protein S have been proposed to play a critical role in the vascular thrombosis [13,18]. In addition, these autoantibodies may directly regulate expressions and/or activities of many coagulation factors including tissue factor (TF) in the vascular system.
TF is a transmembrane protein constitutively expressed in many cell types outside of the vasculature, but is not normally expressed on endothelial cells or peripheral blood cells. However, in response to stimulation with certain agents, endothelial cells and monocytes express TF via transcription activation [19,20]. Increased TF activity has been implicated as a mechanism of thrombosis in a number of thrombotic conditions including atherosclerosis [21, 22]. There is growing evidence that autoantibodies could stimulate cellular TF expression in patients with systemic autoimmune disease [23-25]. However, it is not known how these autoantibodies regulate TF expression. Autoantibodies may cross-react with different autoantigens, which share certain structural features [26-29].
Since autoantibody against human protein S is clinically associated with increased thrombosis [13], we hypothesized that anti-human protein S antibody (anti-hPS Ab) may contribute thrombosis through specific molecular pathways. In this study, we have demonstrated that anti-hPS Ab, but not anti-human protein C antibody (hPS Ab), specifically increases TF expression through direct interaction with platelet phosphofructokinase (PFKP) in human coronary endothelial cells (HCAECs). PFKP is a key regulatory enzyme of glycolysis in most mammalian tissues. It is a new function for PFKP mediating TF expression. This study provides a new molecular mechanism of autoantibodies contributing vascular thrombosis.
Materials and Methods
Chemicals and reagents
TNF-α, anti-hPS Ab (polyclonal), anti-hPC Ab, anti-human IgG Ab (anti-hIgG Ab), monoclonal mouse anti-human β-actin Ab, and fructose-6-phosphate (F-6-P) were purchased from Sigma (St. Louis, MO, USA). Anti-hPS Ab (monoclonal) and a tissue factor human chromogenic activity assay kit were obtained from Abcam (Cambridge, MA, USA). Monoclonal anti-human TF Ab, IMUBIND TF ELISA Kit and human protein S (as a part of the Protein S - IMUCLONE™ Free Protein S ELISA kit) were purchased from American Diagnostica Inc (Stamford, CT, USA). Anti-phospho- and total-ERK1/2 antibodies were purchased form Cell Signaling (Danvers, MA, USA). PFKP recombinant protein with GST and mouse anti-PFKP Ab were purchased from Novus Biologicals Inc (Littleton, CO, USA).
Cell cultures
HCAECs and human umbilical endothelial cells (HUVECs) were purchased from Cambrex (Walkersville, MD, USA) at passage three. Cells were cultured in serum starvation medium (EBM-2 supplemented with 0.5% FBS, CA-1000, heparin, and ascorbic acid, and no growth factors added) for 12-16 hours. The cells were treated with anti-hPS Ab or anti-hIgG Ab for an indicated amount (0-100 μg/mL) and time point (0-7 hours). TNF-α treatment (5 ng/mL) was used as a positive control.
Real time PCR
Total RNA form HCAECs was extracted, and cDNA was synthesized. Quantitative real-time PCR was performed using iQ SYBR Green Supermix Kit (BioRad) following the manufacturer's instruction. The PCR primer sequences are: TF forward: 5′-GTGATTCCCTCCCGAACAGTT-3′; reverse: 5′-CTGGCCCATACACTCTACCG-3′; β-actin forward: 5′-CTGGAACGGTGAAGGTGACA-3′, reverse: 5′-AAGGGACTTCCTGTAACAATGCA-3’. GAPDH forward: 5′-CGTGCCGCCTGGAGAAACC-3′, reverse: 5′-TGGAAGAGTGGGAGTTGCTGTTG-3′. PFKP forward: 5′-GGGGATGCTCAAGGTATGAAC-3′, reverse: 5′-TCGGCCTCTGCGATGTTTG-3′. iCycler software was used to analyze the calibration curve by plotting the threshold cycle (Ct) versus the logarithm of the number of copies for each calibrator. The relative amount of mRNA for each gene was normalized based on that of the house-keeping gene β-actin or GAPDH calculated as [2(Ct[β-actin or GAPDH] - Ct[gene of interest]) ].
Western blot
Cells were lysed in a Cell Lysis buffer (Sigma). The total protein concentration was determined with the protein assay kit (Bio-Rad). Twenty five micrograms of protein samples were separated on 10% SDS-PAGE gels, which were then transferred to the nitrocellulose membrane. The membrane was blocked in 5% nonfat powdered milk. The membrane was incubated with the primary antibody at 4°C overnight, followed by incubation with HRP conjugated secondary Ab and detected with SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL, USA).
TF protein level and activity
Serum-starved HCAECs were treated with different concentrations of anti-hPS Ab (20, 40 or 100 μg/mL). Isotype control IgG, heat-inactivated anti-hPS Ab, and TNF-a were included as controls. Cellular TF protein levels and TF-mediated procoagulant activity were measured by commercial IMUBIND Tissue Factor ELISA Kit and Tissue Factor Human Chromogenic Activity Assay Kit, respectively, according to manufacturer's instructions.
Bioplex luminex immunoassay
Serum-starved HCAECs were treated with anti-hPS Ab (20 μg/mL) and the cell lysates were harvested at different time points (0, 10, 20, 40, 60 and 120 minutes) with Bio-Plex Cell Lysis Kit. The phosphorylation levels of MAPKs were analyzed by Bio-Plex phosphoprotein and total target protein assays (Bio-Rad) according to manufacturer's instructions.
Immunoprecipitation and protein sequencing
The proteins of HCAECs were extracted with Mem-PER Eukaryotic Membrane Protein Extraction Reagent Kit (Pierce). HCAEC protein lysates were incubated with rabbit anti-hPS Ab for 2 hours at 4°C. Protein G sepharose was then added. The suspension was mixed for 1 hour at 4°C and then subjected to centrifugation (12,000 × g) for 2 minutes. The supernatant was discarded and the Laemmli sample buffer (BioRad) was added to the beads and boiled for 5 minutes, and then subjected to centrifugation for 5 minutes. The resultant supernatant fluid was run in SDS-PAGE. The gel was stained with 0.5% Coomassie Blue R for 30 minutes and destained with 5% acetic acid and 10% methanol. The protein band was cut out and sent to the Protein Chemistry Core Lab at the Baylor College of Medicine for protein sequencing. The protein was digested by trypsin and sequenced with MALDI-TOF instrument.
PFKP activity
HCAECs were homogenized in 50 mM Tris-HCl buffer (pH 8.0) containing 50 mM-potassium fluoride and 2.5 mM-EDTA with a homogenizer. The homogenate was centrifuged for 15 minutes (at 10,000g) and the supernatant was collected for PFKP activity assay. PFKP crude extract was incubated with Ab for 15 minutes at room temperature before starting PFKP activity assay. PFKP activity assay was performed in 50 μL reaction mixture containing 33 mM Tris/HCl buffer, 50 mM KCl, 5 mM MgCl2, 1 mM DTT (dithiothreitol), 20 mM fructose-6-P, 2.4 mM DPNH, 2.0 mM ATP, 0.36 units of aldolase, 15 units of Triose phosphate isomerase, 5.1 units of glycerol phosphate dehydrogenase, 10 μL of the PFKP homogenate and 1 μL of Ab. The assay was run in a spectrophotomoter at room temperature. PFKP activity was represented as percent loss of DPNH in the reaction at A340.
PFKP shRNA silencing
The 29mer shRNA constructs against PFKP and control construct plasmid were purchased from Origen Technologies, Inc (Rockville, MD). The sequences of the two PFKP shRNAs are AAGTACAAGGCCAGCTATGACGTGTCGGA and CCACAGGATGCTCGCCATCTATGATGGCT. The shRNA constructs and control construct were transfected into Phoenix Ampho packaging cells (Orbigen, San Diego, CA, USA) to produce recombinant retroviruses. HCAECs were then infected by the viral supernatant and selected in growth medium containing 1 μg/mL puromycin for 1 week. All uninfected cells were killed by puromycin. The shRNA silencing effects in stably transfected HCAECs were verified by measuring PFKP mRNA abundance.
Statistical analysis
Data are presented as mean ± SEM. Statistical significance was determined by one-way analysis of variance (ANOVA) or Student's t-test (two tailed). A value of P < 0.05 was considered significant.
Results
Anti-hPS Ab specifically induces TF expression and TF-mediated procoagulant activity in HCAECs and HUVECs
To determine the specific effects of anti-hPS Ab on TF expression in HCAECs, we performed real-time PCR, ELISA and Western blot to measure TF mRNA and protein levels, respectively. Both TF mRNA and protein levels in HCAECs were increased in a concentration-dependent manner in response to anti-hPS Ab treatment (Fig. 1A, and Fig. 2A, 2B). As a negative control, anti-hIgG Ab had no effects on TF mRNA and protein expression. The same concentrations of anti-hPC Ab did not show any significant effect on TF expression in HCAECs (Fig. 1B). The time course analysis of anti-hPS Ab-induced TF mRNA expression in HCAECs was shown in Fig. 1C. Treatment with anti-hPS Ab resulted in a 2.5-fold increase in TF mRNA levels that reached the maximal at 3 hours. Commercial source of anti-hPS Ab (polyclonal, Sigma) was used to generate above data. In order to establish such a unique role of anti-hPS-Ab in TF expression, we used another anti-hPS Ab (monoclonal) from different source (Abcam) for assaying the TF-mediated procoagulant activity in both HCAECs and HUVECs. As expected, anti-hPS Ab (monoclonal) significantly increased TF-mediated procoagulant activity in both HCAECs and HUVECs in a concentration dependent manner (Fig. 2C and 2D). Western blot confirmed that anti-hPS Ab (monoclonal) also specifically increased TF protein levels in HUVECs (Fig. 2E). These data indicate that anti-hPS Ab, but not anti-hPC Ab, could mimic the effects of patient autoantibodies in terms of inducing TF expression.
Fig. 1.
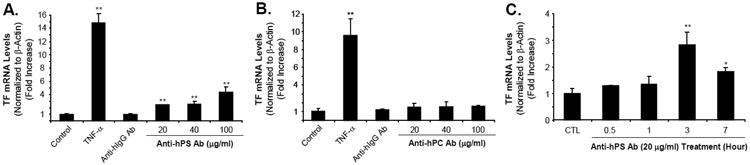
Effect of anti-hPS Ab on TF mRNA levels in HCAECs. (A). Concentration-dependent response of anti-hPS Ab (polyclonal, Sigma) in TF mRNA expression. (B). Concentration-dependent response of anti-hPC Ab in TF mRNA expression. HCAECs were serum-starved for 12-16 hours, and then treated with 20-100 μg/mL anti-hPS Ab or anti-hPC Ab, 100 μg/mL anti-hIgG Ab or 5 ng/mL TNF-α for 3 hours. (C). Time course of anti-hPS Ab-induced TF mRNA expression. HCAECs were serum-starved for 12-16 hours, and then treated with 20 μg/mL anti-hPS Ab for indicated time points. TF mRNA expression was measured by real-time PCR. The relative amount of TF mRNA was normalized to β-actin. Data shown are the mean + SEM of triplicate determinations. **P < 0.01 versus untreated controls.
Fig. 2.
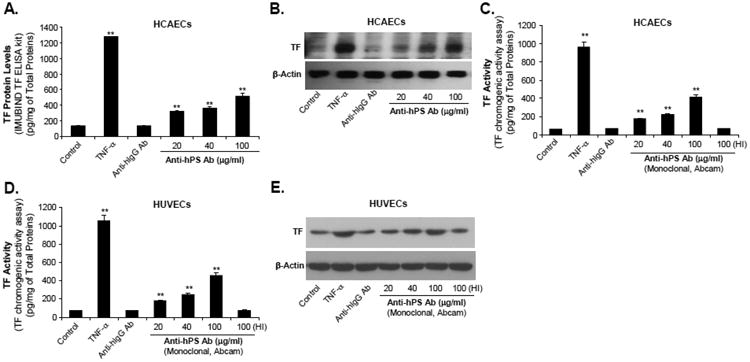
Effect of anti-hPS Ab on TF protein levels as well as TF-mediated procoagulant activity in HCAECs and HUVECs. (A). Concentration-dependent response of anti-hPS Ab-induced TF protein levels (ELISA) in HCAECs. Serum-starved HCAECs were treated with 20-100 μg/mL anti-hPS Ab (polyclonal, Sigma), 100 μg/mL anti-hIgG Ab or 5 ng/mL TNF-a for 3 hours. The cellular TF protein levels were measured by IMUBIND Tissue Factor ELISA Kit. (B). Concentration dependent response of anti-hPS Ab-induced TF protein levels (Western blot) in HCAECs. Serum-starved HCAECs were treated with 20-100 μg/mL anti-hPS Ab, 100 μg/mL anti-hIgG Ab or 5 ng/mL TNF-α for 3 hours. TF protein expression was detected by Western blot. β-actin was used as a loading control. (C). Effect of anti-hPS Ab (monoclonal, Abcam) on TF-mediated procoagulant activity in HCAECs. Serum-starved HCAECs were treated with 20-100 μg/mL anti-hPS Ab, 40 μg/mL anti-hIgG Ab, 5 ng/mL TNF-a or heat-inactivated anti-hPS Ab (100 ng/mL) for 3 hours. The TF activity levels were measured by a tissue factor human chromogenic activity assay kit (Abcam). (D). Effect of anti-hPS Ab (monoclonal, Abcam) on TF-mediated procoagulant activity in HUVECs. (E). Effect of anti-hPS Ab (monoclonal, Abcam) on TF expression (Western blot) in HUVECs. Data shown are the mean + SEM of triplicate determinations. **P < 0.01 versus untreated controls.
ERK1/2 activation is involved in anti-hPS Ab-induced TF expression in HCAECs
Since MAPKs are able to mediate TF expression in human endothelial cells [30-32], we determined whether MAPKs are also involved in anti-hPS Ab-induced TF expression. The activation status of 3 major MAPKs (ERK2, JNK and p38) was analyzed by Bio-Plex luminex immunoassay. Anti-hPS Ab treatment (20 µg/mL) for 40 minutes substantially increased the phosphorylation of ERK2 in HCAECs by 5.5-fold compared with untreated cells (at 0 minute). However, there were no changes of phosphorylation in JNK and p38 in response to anti-hPS Ab treatment (Fig. 3A). Meanwhile, Western blot also revealed a significant increase of phosphorylation of ERK1/2 in HCAECs at 40 minutes of anti-hPS Ab treatment compared with untreated cells at 0 minute (Fig. 3B). Moreover, anti-hPS Ab-induced increase of TF mRNA and protein levels was effectively blocked by co-incubation of anti-hPS Ab with the specific ERK1/2 inhibitor U0216 (P < 0.05, Fig. 4A and 4B). As a control, anti-human TF Ab was included in the study and did not show any effect on TF expression. Thus, ERK1/2 activation is involved in anti-hPS Ab-induced TF expression in HCAECs.
Fig. 3.
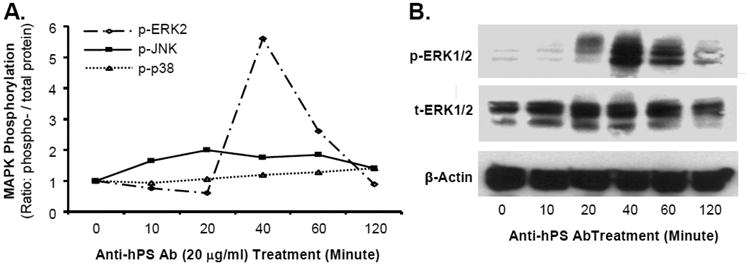
Effect of anti-hPS Ab on MAPK phosphorylation in HCAECs. (A). The activation status of MAPKs (ERK2, JNK and p38) was analyzed by Bio-Plex immunoassay. Serum-starved HCAECs were treated with anti-hPS Ab (20 μg/mL) and the cell lysates were harvested at different time points with Bio-Plex Cell Lysis Kit. The phosphoprotein and total proteins of MAPKs were analyzed by a Luminex 100TM analyzer and Bio-Plex Manager software (BioRad). (B). Phosphorylation of ERK1/2 was detected by Western blot. Serum-starved HCAECs were treated with anti-PS Ab (20 μg/mL) for different time points. The phosphorylated and total ERK1/2 proteins were detected by Western blot. β-actin was used as a loading control. Representative results from 3 experiments are shown.
Fig. 4.
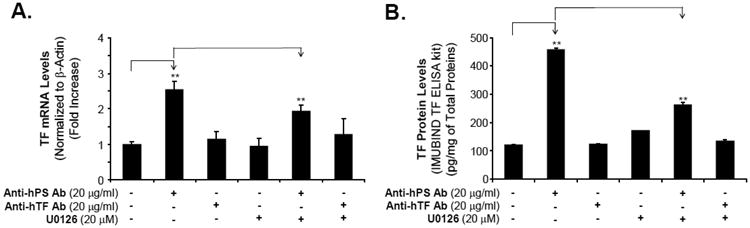
Effect of ERK1/2 inhibitor on anti-hPS Ab-induced TF expression and activity in HCAECs. (A). Effect of ERK1/2 inhibitor on anti-hPS Ab-induced TF mRNA expression in HCAECs. Serum-starved HCAECs we incubated with anti-hPS Ab or anti-hTF Ab, with the presence or absence of ERK1/2 inhibitor (U0126) for 3 hours. TF mRNA expression was measured by real-time PCR (BioRad). The relative amount of TF mRNA was normalized to β-actin. Data shown are the mean + SEM of triplicate determinations. (B). Effect of MEK1/2 inhibitor on anti-hPS Ab-induced TF protein activity in HCAECs. Serum-starved HCAECs were incubated with anti-hPS Ab or anti-hTF Ab, with the presence or absence of ERK1/2 inhibitor (U0126) for 3 hours. The cellular TF activity was measured by IMUBIND Tissue Factor ELISA Kit. Data shown are the mean + SEM of duplicate determinations. **P < 0.01 versus untreated controls. #P < 0.01 versus anti-hPS Ab group.
Anti-hPS Ab specifically interacts with PFKP in HCAECs
Since autoantibodies may cross-react with a wide spectrum of antigens, we performed immunoprecipitation and protein sequencing to identify the potential anti-hPS Ab-binding protein in HCAECs. The protein sequencing results showed that the anti-hPS Ab-binding protein is PFKP. To confirm that anti-hPS Ab could directly bind to PFKP, we performed immunoprecipitation assay using purified PFKP-GST and anti-hPS Ab, and we found that anti-hPS Ab immunoprecipitated the PFKP, while the negative control Abs (anti-hPC Ab and anti-hIG Ab) had no such effects (Fig. 5A). Furthermore, we found that the binding activity between PFKP-GST and anti-hPS Ab was effectively abolished by adding an excess amount of hPS (Fig. 5B), suggesting that hPS might block the domain on anti-hPS Ab. hPS used in the current study is a part of the Protein S - IMUCLONE™ Free Protein S ELISA kit; and it is a highly purified free protein with a full biological activity. hPS interacting with anti-hPS Ab competitively inhibited PFKP-GDT interacting with anti-hPS Ab. These findings demonstrate the cross-reactivity between anti-hPS Ab and PFKP.
Fig. 5.
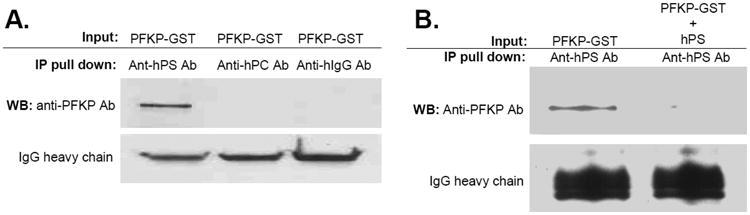
Specific interaction between anti-hPS Ab and PFKP. (A). Immunoprecipitation assay. Purified recombinant human PFKP-GST (0.5 μg) was mixed with anti-hPS Ab (2 μg) or control Abs (anti-hPC Ab and anti-hIgG Ab) (2 μg) at 4°C for 2 hours, followed by incubation with protein A agarose beads for additional 1 hour. Bound proteins were subjected to SDS-PAGE and immunoblotted with anti-PFKP Ab. (B). Human protein S abolished the binding activity between PFKP-GST and anti-hPS Ab. Purified recombinant human PFKP-GST (0.5 μg) was mixed with anti-hPS Ab (2 μg) and human protein S (20 μg) at 4°C for 2 hours, followed by incubation with protein A agarose beads for additional 1 hour. Bound proteins were subjected to SDS-PAGE and immunoblotted with anti-PFKP Ab. Representative results from 3 experiments are shown.
PFKP is directly involved in anti-hPS Ab-induced TF expression in HCAECs
Since anti-hPS Ab could bind with PFKP, we performed PFKP activity assay to observe the effects of anti-hPS Ab on PFKP activity. Fig. 6A showed that anti-hPS Ab significantly reduced the PFKP activity compared with control groups. To study the effects of PFKP levels on TF expression, we knocked down the expression of PFKP around 30% in HCAECs by PFKP shRNA (Fig. 6B). In the PFKP shRNA transfected HCAECs, the TF mRNA expression was significantly increased for more than 80% compared with control shRNA vector transfected cells (Fig. 6C). These data suggest that anti-hPS Ab might increase TF expression through inhibiting PFKP activity in HCAECs. To further confirm this concept, we incubated HCAECs with anti-hPS Ab in the presence or absence of fructose-6-phosphate, which is a PFKP activator. As shown in Fig. 6D, fructose-6-phosphate partially abolished the effect of anti-hPS Ab on TF mRNA expression (P < 0.01), which indicate that PFKP could mediate the effects of anti-hPS Ab on TF expression.
Fig. 6.
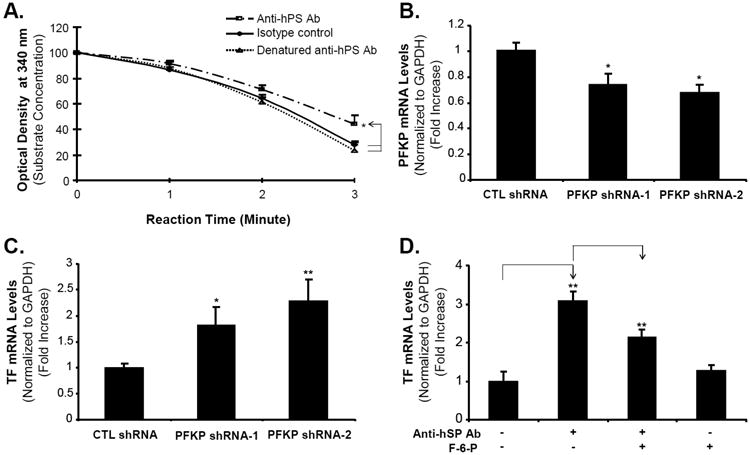
Role of PFKP in anti-hPS Ab-induced TF expression in HCAECs. (A). PFKP activity assay. The PFKP crude extract from HCAECs was incubated with anti-hPS Ab, heat denatured-anti-hPS Ab or isotype control Ab for 15 minutes, and then the PFKP activity was analyzed in the reaction mixture for 3 minutes at room temperature. The PFKP activity was represented as percent loss of DPNH in the reaction at A340. (B). PFKP mRNA expression in stably transfected HCAECs. The 29mer shRNA constructs against PFKP and control construct plasmid were transfected into Phoenix Ampho packaging cells to produce recombinant retroviruses. HCAECs were then infected by the viral supernatants and selected in growth medium containing 1 μg/mL puromycin for 1 week. The PFKP mRNA expression in stably transfected HCAECs was measured by real-time PCR. (C). TF mRNA expression in stably transfected HCAECs. The TF mRNA expression in stably transfected HCAECs was measured by real-time PCR. (D). Fructose-6-phosphate blocked the effects of anti-hPS Ab. Serum-starved HCAECs were incubated with anti-hPS Ab in the presence or absence of PFKP activator (fructose-6-phosphate) for 3 hours. TF mRNA expression was measured by real-time PCR. The relative amount of specific gene mRNA was normalized to GAPDH. Data shown are the mean + SEM of triplicate determinations. *P < 0.05 and **P < 0.01 versus untreated controls. #P < 0.01 versus anti-hPS Ab group.
Discussion
In the current study, we have found that anti-hPS Ab specifically induces TF expression through a direct interaction with PFKP in HCAECs and ERK1/2 activation. Previous studies have shown that anti-hPS Abs were present in a large proportion of patients with acquired protein S deficiency, who are positive for the lupus anticoagulant or positive for HIV infection, by measuring their binding activities to hPS or specific peptides [4,8]. However, concentrations (ng/mL) of anti-hPS Abs in human plasma or serum are currently not available, although we know that the concentration of PS in normal plasma is ∼25 μg/ml [33]; and the mean level of serum IgG is 111.8 mg/ml [34]. It has been proposed that autoantibodies might directly contribute to high thrombosis events in the patients [18]. There is growing evidence that increased TF activity on vascular endothelial cells or monocytes is an important mechanism of hypercoagulability in APS and SLE [35-40]. TF is the physiological initiator of normal coagulation as well as clotting observed in thrombotic disease. Autoantibodies and/or immune complexes circulating in APS patients appear to enhance the expression of TF activity on monocytes and endothelial cells [36-38,41]. Our data have shown that TF mRNA and protein expression levels as well as TF-mediated procoagulant activity in human endothelial cells were significantly increased by anti-hPS Ab treatment in a concentration-dependent manner. The time course analysis has shown that stimulation with anti-hPS Ab resulted in a 2.5-fold increase in TF mRNA levels in endothelial cells and the maximal stimulation was at 3 hours. These data suggest that anti-hPS Ab-induced TF expression on vascular endothelial cells may contribute to hypercoagulability in the patients with protein S deficiency.
MAPKs are activated by bacterial lipopolysaccharide and other signaling molecules, and are critical in signaling pathways mediating cellular activation [42]. In our current study, we found that anti-hPS Ab transiently increased ERK2 phosphorylation levels, but not p38 and JNK, and peak response was at 40 minutes after treatment of anti-hPS Ab by both Bio-Plex luminex immunoassay and Western blot analyses. Furthermore, we found ERK2 phosphorylation had a functional significance because the specific ERK1/2 inhibitor U0216 significantly inhibited the promoting effect of anti-hPS Ab on TF mRNA and protein expression. These data indicate that the activation of the ERK signaling pathway is essential for anti-hPS Ab-induced TF expression. Vega-Ostertag et al. reported that the phosphorylation of p38 MAPK was involved in the up-regulation of TF on endothelial cells by antiphospholipid antibodies [30]. López-Pedrera et al. showed that antiphospholipid antibodies induces TF expression in monocytes isolated from APS patients by activating ERK1/2 and p38 [43]. Several reports described the signal transduction pathways activating TF expression in endothelial cells, including ERK1/2 and transcription factors AP-1, NFκB, early growth response factor 1 (EGR-1), and nuclear factor of activated T cells (NFAT) [44-46]. It is possible that anti-hPS antibody may activate these transcription factors mentioned above in these studies.
PFKP catalyzes the irreversible conversion of fructose-6-phosphate to fructose-1,6-bisphosphate and is a key regulatory enzyme in glycolysis [47,48]. Our immunoprecipitation experiments and enzymatic assays have shown that anti-hPS Ab could cross-react with PFKP, reducing PFKP activity. The consequence of PFKP downregulation was to increase TF expression in HCAECs. Furthermore, the PFKP activator fructose-6-phosphate significantly attenuated the effects of anti-hPS Ab on TF mRNA expression, which confirms the participation of PFKP in anti-hPS Ab stimulating TF expression. To the best of our knowledge, our current study is the first report that PFKP is involved in autoantibody-induced TF expression, which suggests a novel molecular mechanism in autoimmune disease.
In conclusions, our results have shown that anti-hPS Ab, but not anti-hPC Ab, induces TF expression as well as TF-mediated procoagulant activity through reducing PFKP function and increasing ERK1/2 activity in human endothelial cells, which might help designing new therapies for autoantibody-triggered thrombosis in patients with autoimmune disorders. Further studies need to be done to determine the interaction mechanisms between anti-hPS Ab and PFKP, as well as the anti-hPS Ab-related transcription factors in the endothelial cells.
Acknowledgments
Sources of Funding: This work was partially supported by research and training grants from the National Institutes of Health (HL083471 to C Chen), and by the Michael E. DeBakey Department of Surgery, Baylor College of Medicine, Houston, Texas, USA. Dr. Dan Liao and Dr. Zhengdong Liang were supported by a NIH training grant (T32HL083774).
Footnotes
Conflict of Interest Statement: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Walker FJ. Protein S and the regulation of activated protein C. Semin Thromb Hemost. 1984;10:131–8. doi: 10.1055/s-2007-1004415. [DOI] [PubMed] [Google Scholar]
- 2.Bereczky Z, Kovács KB, Muszbek L. Protein C and protein S deficiencies: similarities and differences between two brothers playing in the same game. Clin Chem Lab Med. 2010;48(Suppl 1):S53–66. doi: 10.1515/CCLM.2010.369. [DOI] [PubMed] [Google Scholar]
- 3.D'Angelo A, Della Valle P, Crippa L, Pattarini E, Grimaldi LM, Vigano D'Angelo S. Brief report: autoimmune protein S deficiency in a boy with severe thromboembolic disease. N Engl J Med. 1993;328:1753–7. doi: 10.1056/NEJM199306173282405. [DOI] [PubMed] [Google Scholar]
- 4.Walker FJ. Regulation of activated protein C by protein S. The role of phospholipid in factor Va inactivation. J Biol Chem. 1981;256:11128–31. [PubMed] [Google Scholar]
- 5.Suh CH, Hilliard B, Li S, Merrill JT, Cohen PL. TAM receptor ligands in lupus: protein S but not Gas6 levels reflect disease activity in systemic lupus erythematosus. Arthritis Res Ther. 2010;12:R146. doi: 10.1186/ar3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ginsberg JS, Demers C, Brill-Edwards P, Bona R, Johnston M, Wong A, Denburg JA. Acquired free protein S deficiency is associated with antiphospholipid antibodies and increased thrombin generation in patients with systemic lupus erythematosus. Am J Med. 1995;98:379–83. doi: 10.1016/S0002-9343(99)80317-9. [DOI] [PubMed] [Google Scholar]
- 7.Crowther MA, Johnston M, Weitz J, Ginsberg JS. Free protein S deficiency may be found in patients with antiphospholipid antibodies who do not have systemic lupus erythematosus. Thromb Haemost. 1996;76:689–91. [PubMed] [Google Scholar]
- 8.Sorice M, Griggi T, Arcieri P, Circella A, d'Agostino F, Ranieri M, et al. Protein S and HIV infection. The role of anticardiolipin and anti-protein S antibodies. Thromb Res. 1994;73:165–75. doi: 10.1016/0049-3848(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 9.Kiser KL, Badowski ME. Risk factors for venous thromboembolism in patients with human immunodeficiency virus infection. Pharmacotherapy. 2010;30:1292–302. doi: 10.1592/phco.30.12.1292. [DOI] [PubMed] [Google Scholar]
- 10.van Ommen CH, Fijnvandraat K, Vulsma T, Delemarre-Van De Waal HA, Peters M. Acquired protein S deficiency caused by estrogen treatment of tall stature. J Pediatr. 1999;135:477–81. doi: 10.1016/s0022-3476(99)70171-x. [DOI] [PubMed] [Google Scholar]
- 11.Ideguchi H, Ohno S, Ueda A, Ishigatsubo Y. Catastrophic antiphospholipid syndrome associated with malignancies (case report and review of the literature) Lupus. 2007;16:59–64. doi: 10.1177/0961203306073166. [DOI] [PubMed] [Google Scholar]
- 12.Sorice M, Griggi T, Circella A, Lenti L, Arcieri P, Domenico di Nucci G, Mariani G. Protein S antibodies in acquired protein S deficiencies. Blood. 1994;83:2383–4. [PubMed] [Google Scholar]
- 13.Erkan D, Zhang HW, Shriky RC, Merrill JT. Dual antibody reactivity to beta2-glycoprotein I and protein S: increased association with thrombotic events in the antiphospholipid syndrome. Lupus. 2002;11:215–20. doi: 10.1191/0961203302lu178oa. [DOI] [PubMed] [Google Scholar]
- 14.Guermazi S, Regnault V, Gorgi Y, Ayed K, Lecompte T, Dellagi K. Further evidence for the presence of anti-protein S autoantibodies in patients with systemic lupus erythematosus. Blood Coagul Fibrinolysis. 2000;11:491–8. doi: 10.1097/00001721-200007000-00012. [DOI] [PubMed] [Google Scholar]
- 15.Regnault V, Boehlen F, Ozsahin H, Wahl D, de Groot PG, Lecompte T, de Moerloose P. Anti-protein S antibodies following a varicella infection: detection, characterization and influence on thrombin generation. J Thromb Haemost. 2005;3:1243–9. doi: 10.1111/j.1538-7836.2005.01270.x. [DOI] [PubMed] [Google Scholar]
- 16.Phillips WG, Marsden JR, Hill FG. Purpura fulminans due to protein S deficiency following chickenpox. Br J Dermatol. 1992;127:30–2. doi: 10.1111/j.1365-2133.1992.tb14821.x. [DOI] [PubMed] [Google Scholar]
- 17.D'Angelo A, Della Valle P, Crippa L, Pattarini E, Grimaldi LM, Viganò D'Angelo S. Brief report: autoimmune protein S deficiency in a boy with severe thromboembolic disease. N Engl J Med. 1993;328:1753–7. doi: 10.1056/NEJM199306173282405. [DOI] [PubMed] [Google Scholar]
- 18.Oosting JD, Derksen RH, Bobbink IW, Hackeng TM, Bouma BN, de Groot PG. Antiphospholipid antibodies directed against a combination of phospholipids with prothrombin, protein C, or protein S: an explanation for their pathogenic mechanism? Blood. 1993;81:2618–25. [PubMed] [Google Scholar]
- 19.Gregory SA, Morrissey JH, Edgington TS. Regulation of tissue factor gene expression in the monocyte procoagulant response to endotoxin. Mol Cell Biol. 1989;9:2752–5. doi: 10.1128/mcb.9.6.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen VM, Hogg PJ. Encryption and decryption of tissue factor. J Thromb Haemost. 2013;11(Suppl 1):277–84. doi: 10.1111/jth.12228. [DOI] [PubMed] [Google Scholar]
- 21.Muhlfelder TW, Teodorescu V, Rand J, Rosman A, Niemetz J. Human atheromatous plaque extracts induce tissue factor activity (TFa) in monocytes and also express constitutive TFa. Thromb Haemost. 1999;81:146–50. [PubMed] [Google Scholar]
- 22.Winckers K, ten Cate H, Hackeng TM. The role of tissue factor pathway inhibitor in atherosclerosis and arterial thrombosis. Blood Rev. 2013;27:119–32. doi: 10.1016/j.blre.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 23.Amengual O, Atsumi T, Khamashta MA, Hughes GR. The role of the tissue factor pathway in the hypercoagulable state in patients with the antiphospholipid syndrome. Thromb Haemost. 1998;79:276–81. [PubMed] [Google Scholar]
- 24.Reverter JC, Tassies D, Font J, Khamashta MA, Ichikawa K, Cervera R, et al. Effects of human monoclonal anticardiolipin antibodies on platelet function and on tissue factor expression on monocytes. Arthritis Rheum. 1998;41:1420–7. doi: 10.1002/1529-0131(199808)41:8<1420::AID-ART11>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 25.Motoki Y, Nojima J, Yanagihara M, Tsuneoka H, Matsui T, Yamamoto M, Ichihara K. Anti-phospholipid antibodies contribute to arteriosclerosis in patients with systemic lupus erythematosus through induction of tissue factor expression and cytokine production from peripheral blood mononuclear cells. Thromb Res. 2012;130:667–73. doi: 10.1016/j.thromres.2011.11.048. [DOI] [PubMed] [Google Scholar]
- 26.Jarjour WN, Minota S, Roubey RA, Mimura T, Winfield JB. Autoantibodies to nucleolin cross-react with histone H1 in systemic lupus erythematosus. Mol Biol Rep. 1992;16:263–6. doi: 10.1007/BF00419666. [DOI] [PubMed] [Google Scholar]
- 27.Ruf J, Ferrand M, Durand-Gorde JM, De Micco C, Carayon P. Significance of thyroglobulin antibodies cross-reactive with thyroperoxidase (TGPO antibodies) in individual patients and immunized mice. Clin Exp Immunol. 1993;92:65–72. doi: 10.1111/j.1365-2249.1993.tb05949.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun KH, Hong CC, Tang SJ, Sun GH, Liu WT, Han SH, Yu CL. Anti-dsDNA autoantibody cross-reacts with the C-terminal hydrophobic cluster region containing phenylalanines in the acidic ribosomal phosphoprotein P1 to exert a cytostatic effect on the cells. Biochem Biophys Res Commun. 1999;263:334–9. doi: 10.1006/bbrc.1999.1305. [DOI] [PubMed] [Google Scholar]
- 29.DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med. 2001;7:1189–93. doi: 10.1038/nm1101-1189. [DOI] [PubMed] [Google Scholar]
- 30.Vega-Ostertag M, Casper K, Swerlick R, Ferrara D, Harris EN, Pierangeli SS. Involvement of p38 MAPK in the up-regulation of tissue factor on endothelial cells by antiphospholipid antibodies. Arthritis Rheum. 2005;52:1545–54. doi: 10.1002/art.21009. [DOI] [PubMed] [Google Scholar]
- 31.Maruyama K, Morishita E, Yuno T, Sekiya A, Asakura H, Ohtake S, Yachie A. Carbon monoxide (CO)-releasing molecule-derived CO regulates tissue factor and plasminogen activator inhibitor type 1 in human endothelial cells. Thromb Res. 2012;130:e188–93. doi: 10.1016/j.thromres.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 32.Chen Y, Wang J, Yao Y, Yuan W, Kong M, Lin Y, et al. CRP regulates the expression and activity of tissue factor as well as tissue factor pathway inhibitor via NF-κB and ERK 1/2 MAPK pathway. FEBS Letters. 2009;583:2811–18. doi: 10.1016/j.febslet.2009.07.037. [DOI] [PubMed] [Google Scholar]
- 33.Griffin JH, Gruber A, Fernández JA. Reevaluation of total, free, and bound protein S and C4b-binding protein levels in plasma anticoagulated with citrate or hirudin. Blood. 1992;79:3203–11. [PubMed] [Google Scholar]
- 34.Gonzalez-Quintela A, Alende R, Gude F, Campos J, Rey J, Meijide LM, et al. Serum levels of immunoglobulins (IgG, IgA, IgM) in a general adult population and their relationship with alcohol consumption, smoking and common metabolic abnormalities. Clin Exp Immunol. 2008;151:42–50. doi: 10.1111/j.1365-2249.2007.03545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greaves M. Antiphospholipid antibodies and thrombosis. Lancet. 1999;354:1031. doi: 10.1016/S0140-6736(05)76636-8. [DOI] [PubMed] [Google Scholar]
- 36.Roubey RA. Tissue factor pathway and the antiphospholipid syndrome. J Autoimmun. 2000;15:217–20. doi: 10.1006/jaut.2000.0397. [DOI] [PubMed] [Google Scholar]
- 37.Zhou H. Dilazep and dipyridamole inhibit tissue factor expression on monocytes induced by IgG from patients with antiphospholipid syndrome. Acta Pharmacol Sin. 2004;25:1366–71. [PubMed] [Google Scholar]
- 38.Teruel R, Pérez-Sánchez C, Corral J, Herranz MT, Pérez-Andreu V, Saiz E, et al. Identification of miRNAs as potential modulators of tissue factor expression in patients with systemic lupus erythematosus and antiphospholipid syndrome. J Thromb Haemost. 2011;9:1985–92. doi: 10.1111/j.1538-7836.2011.04451.x. [DOI] [PubMed] [Google Scholar]
- 39.Motoki Y, Nojima J, Yanagihara M, Tsuneoka H, Matsui T, Yamamoto M, Ichihara K. Anti-phospholipid antibodies contribute to arteriosclerosis in patients with systemic lupus erythematosus through induction of tissue factor expression and cytokine production from peripheral blood mononuclear cells. Thromb Res. 2012;130:667–73. doi: 10.1016/j.thromres.2011.11.048. [DOI] [PubMed] [Google Scholar]
- 40.Adams MJ, Palatinus AA, Harvey AM, Khalafallah AA. Impaired control of the tissue factor pathway of blood coagulation in systemic lupus erythematosus. Lupus. 2011;20:1474–83. doi: 10.1177/0961203311418267. [DOI] [PubMed] [Google Scholar]
- 41.Kornberg A, Blank M, Kaufman S, Shoenfeld Y. Induction of tissue factor-like activity in monocytes by anti-cardiolipin antibodies. J Immunol. 1994;153:1328–32. [PubMed] [Google Scholar]
- 42.Bohgaki M, Atsumi T, Yamashita Y, Yasuda S, Sakai Y, Furusaki A, et al. The p38 mitogen-activated protein kinase (MAPK) pathway mediates induction of the tissue factor gene in monocytes stimulated with human monoclonal anti-beta2Glycoprotein I antibodies. Int Immunol. 2004;16:1633–41. doi: 10.1093/intimm/dxh166. [DOI] [PubMed] [Google Scholar]
- 43.López-Pedrera C, Buendía P, Cuadrado MJ, Siendones E, Aguirre MA, Barbarroja N, et al. Antiphospholipid antibodies from patients with the antiphospholipid syndrome induce monocyte tissue factor expression through the simultaneous activation of NF-kappaB/Rel proteins via the p38 mitogen-activated protein kinase pathway, and of the MEK-1/ERK pathway. Arthritis Rheum. 2006;54:301–11. doi: 10.1002/art.21549. [DOI] [PubMed] [Google Scholar]
- 44.Moll T, Czyz M, Holzmüller H, Hofer-Warbinek R, Wagner E, Winkler H, et al. Regulation of the tissue factor promoter in endothelial cells. Binding of NF kappa B-, AP-1-, and Sp1-like transcription factors. J Biol Chem. 1995;270:3849–57. doi: 10.1074/jbc.270.8.3849. [DOI] [PubMed] [Google Scholar]
- 45.Bavendiek U, Libby P, Kilbride M, Reynolds R, Mackman N, Schönbeck U. Induction of tissue factor expression in human endothelial cells by CD40 ligand is mediated via activator protein 1, nuclear factor kappa B, and Egr-1. J Biol Chem. 2002;277:25032–9. doi: 10.1074/jbc.M204003200. [DOI] [PubMed] [Google Scholar]
- 46.Bochkov VN, Mechtcheriakova D, Lucerna M, Huber J, Malli R, Graier WF, et al. Oxidized phospholipids stimulate tissue factor expression in human endothelial cells via activation ofERK/EGR-1 and Ca(++)/NFAT. Blood. 2002;99:199–206. doi: 10.1182/blood.v99.1.199. [DOI] [PubMed] [Google Scholar]
- 47.Uyeda K. Phosphofructokinase. Adv Enzymol Relat Areas Mol Biol. 1979;48:193–244. doi: 10.1002/9780470122938.ch4. [DOI] [PubMed] [Google Scholar]
- 48.Foe LG, Kemp RG. Isolation and characterization of phosphofructokinase C from rabbit brain. J Biol Chem. 1979;260:726–30. [PubMed] [Google Scholar]