

Summary
PDZ domain interactions are involved in signaling and trafficking pathways that coordinate crucial cellular processes. Alignment-based PDZ binding motifs identify the few most favorable residues at certain positions along the peptide backbone. However, sequences that bind the CAL (CFTR-Associated Ligand) PDZ domain reveal only a degenerate motif that overpredicts the true number of high affinity interactors. Here, we combine extended peptide-array motif analysis with biochemical techniques to show that non-motif ‘modulator’ residues influence CAL binding. The crystallographic structures of 13 new CAL:peptide complexes reveal defined, but accommodating stereochemical environments at non-motif positions, which are reflected in modulator preferences uncovered by multi-sequence substitutional arrays. These preferences facilitate the identification of new high-affinity CAL binding sequences and differentially affect CAL and NHERF PDZ binding. As a result, they also help determine the specificity of a PDZ domain network that regulates the trafficking of CFTR at the apical membrane.
Introduction
If each residue is uniquely specified, a decapeptide can encode more than 1013 distinct sequences, providing a versatile and specific mechanism to encode protein:protein interactions. Indeed, domains recognizing short linear motif (SLiM) peptides are found throughout the human genome, including the abundant family of PDZ domains first recognized in the proteins PSD-95, Dlg, and ZO-1 (Davey et al., 2012; Harris and Lim, 2001; Lee and Zheng, 2010; Nourry et al., 2003). PDZ target interactions are governed by a variety of constraints, including local concentration, multidentate binding interfaces, and interactions with additional proteins (Luck et al, 2012). Nevertheless, the recognition of a generally C-terminal peptide by the PDZ binding cleft is the core of the interaction, conferring critical target specificity. As a result, the underlying sequence:affinity relationships are essential to understand connectivity in PDZ-mediated protein networks.
To identify the binding preferences of individual domains, early studies compared the sequences of known binding partners and highlighted the importance of the amino acids at the P0 (extreme C-terminus) and P−2 positions (e.g., Songyang et al., 1997). High-throughput screens of phage-display and peptide-array libraries have since revealed more complex ‘motifs’ involving varying combinations of up to seven C-terminal residues, consistent with stereochemical interactions observed in individual complexes (Doyle et al., 1996; Laura et al., 2002; Schultz et al., 1998; Skelton et al., 2003; Stiffler et al., 2007; Tonikian et al., 2008). Furthermore, although few motifs have been investigated beyond the P−6 position, biochemical and structural studies have identified affinity contributions and stereochemical contacts extending as far as the P−10 position (reviewed in Luck et al., 2012).
Even with extended motifs, PDZ binding motifs on average constrain fewer than four of the potential interacting residues and often accommodate similar residues at a given position (Davey et al., 2012). As a result, PDZ domains are frequently promiscuous, binding multiple target proteins and sharing targets with other PDZ domains. This is illustrated by a set of PDZ proteins that regulate the intracellular trafficking and localization of the cystic fibrosis transmembrane conductance regulator (CFTR): the CFTR-Associated Ligand (CAL) and the NHE3 Regulatory Factor proteins NHERF1 and NHERF2 (Cheng et al., 2002; Guerra et al., 2005; Wolde et al., 2007). Consistent with shared target specificity, sequence alignments revealed overlapping motifs at the P0 and P−2 positions, and sequence optimization in the four C-terminal residues led to only 10-fold selectivity for the CAL PDZ (CALP) domain (Vouilleme et al., 2010). Using a peptide-array approach to extend the sequence iteratively towards the N-terminus, we engineered a CALP inhibitor (iCAL36; sequence ANSRWPTSII) with 170-fold selectivity (Cushing et al., 2010; Vouilleme et al., 2010).
Here we characterize the interaction of the CAL PDZ domain with these N-terminal (‘upstream’, i.e., P−4 to P−9) binding determinants and with the P−1 and P−3 side chains. Although not captured by sequence alignment techniques, they act as binding ‘modulators’ for each domain, exerting a modest influence individually, but a powerful impact collectively. Crystallographic analysis of 13 new CALP:peptide complexes spanning a wide range of affinities reveals an accommodating pharmacophore binding model. In-depth stereochemical and substitutional array analysis provides clues to the identification of preferences that remain hidden from alignment motifs. These modulatory effects facilitate the identification of high-affinity binding sequences and contribute to inhibitor specificity in the context of CFTR trafficking.
Results
Sequence alignments identify only core binding motifs for CAL and NHERF
Based on the demonstrated contributions of upstream residues to the selectivity of our engineered iCAL36 peptide, we attempted to identify extended binding motifs for the CAL and NHERF PDZ domains. We performed WebLogo analysis of all 10 C-terminal positions in peptides identified by screening a 6223HumLib array (Cushing et al., 2008) with CALP or with the NHERF1 and NHERF2 PDZ1 and PDZ2 domains (N1P1, N1P2, N2P2, and N2P2, respectively). However, the results (Figures 1A and S1A) did not highlight any significant upstream preferences outside the previously examined core residues P0 to P−3 (Roberts et al., 2012; Vouilleme et al., 2010).
Figure 1. Sequence determinants of CAL PDZ binding and CAL/NHERF specificity.
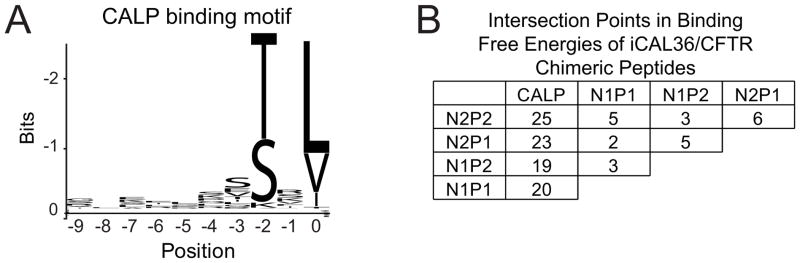
(A) WebLogo analysis (Crooks et al., 2004) of the top 100 sequences that bound CALP in a 6223HumLib peptide array experiment reveals clear preferences at the P0 and P−2 positions, similar to the short, degenerate binding motifs seen with the NHERF PDZ domains (Figure S1A). The C-terminal residue (P0) position is labeled 0; adjacent residues are −1, −2, etc.
(B) For each PDZ domain, free energies of binding (kcal/mol) were calculated from the binding affinities determined by FP for 10 different iCAL36/CFTR chimeric peptide sequences (Table 1). The number of intersection points in pairwise free-energy plots (Figure S2) were tabulated to reveal rank-order exchanges as a function of sequence.
Weak secondary CALP preferences are observed at a few positions (e.g., S>E>V at P−3; Figure 1A). To test the robustness of such signatures, we calculated motifs using a varying number of input sequences, and observed that both the rank order and stereochemical nature of the residues changed. For example, at P−3, the top 10 CALP-binding sequences yielded the pattern E>S>T, whereas the top 50 sequences yielded S>I>Q (Figure S1B). Furthermore, while the top 10 CALP-binding sequences show modest signals, e.g., for Arg at P−1 or Ser at P−7, these grow progressively weaker or shift with the addition of more sequences (Figures 1A and S1B). To evaluate the possibility of correlated binding requirements across multiple ligand positions, we reprobed our top 100 sequences using the MUltiple Specificity Identifier (MUSI) program, which uses both single and multiple position weight matrices to determine specificity patterns (Kim et al., 2011). Overall, the results again confirm the highly degenerate CALP binding motif originally seen at the P0 and P−2 positions (Figure S1C).
Similar patterns are observed for the NHERF domains (Figure S1A). In some cases, secondary preferences seen at the P−3 position (e.g., S>E>T for N1P2 and E>S>D for N2P2) are slightly stronger than those observed for CALP. At least in the case of the N2P2 domain, the presence of acidic residues is broadly consistent with a P−3 preference for Asp reported in earlier phage-display studies (Tonikian et al., 2008). However, as for CALP, no strong signatures were observed upstream of P−3.
Chimeric peptides reveal positive and negative affinity modulators
Although our experience suggested that residues outside the P0/P−2 motif positions significantly enhanced binding selectivity of iCAL36 for CAL vs. NHERF domains, we had not focused on CALP affinity per se, which changed relatively little in the later stages of iCAL36 sequence engineering (Vouilleme et al., 2010). Given the lack of upstream binding motifs, we decided to explore the free-energy contributions of non-motif residues, using nested sets of chimeric peptides based on reference sequences with both high (iCAL36; Ki = 23 μM) and low (CFTR; Ki = 420 μM) affinity for the CALP domain (Amacher et al., 2013). Using fluorescence polarization (FP) competition experiments, we determined Ki values for each of the chimeric peptides binding to CALP (Table 1). CALP has modest preferences for an -9ANSR-6 sequence over -9TEEE-6 (iCAL36VQDTRL vs. CFTR), Trp over Val at P−5 (iCAL36QDTRL vs. iCAL36VQDTRL), and Gln over Pro at P−4 (iCAL36 vs. iCAL36Q-4). A positive CALP preference for a P−1 Arg is also suggested by the 5-fold higher affinity of the iCAL36TRL sequenced compared to iCAL36L and the approximate equivalence of the Ser/Thr motif at P−2 (Vouilleme et al., 2010). However, the most significant effect is seen at P−3. Replacing -5WPT-3 with -5VQD-3 in iCAL36VQD-3 dramatically reduces CALP affinity (Ki > 1 mM). Since neither the P−5 nor P−4 substitutions account for such a large effect, we synthesized a CFTR peptide with a single substitution at P−3. CFTRT-3 (TEEEVQTTRL; Ki = 18 μM) binds 23-fold more tightly than the native CFTR sequence (Table 1).
Table 1.
FP binding affinities of CALP and NHERF PDZ domains for iCAL36/CFTR chimeric peptides.
| Ki (μM) | |||||||
|---|---|---|---|---|---|---|---|
| Peptidea | Sequenceb | CALP | CALP E317A | N1P1 | N1P2 | N2P1 | N2P2 |
| iCAL36TRL | ANSRWPTTRL | 4.5 ± 0.8 | 2.2 ± 1.2 | 8.0 ± 1.4 | 110 ± 10 | 8.7 ± 3.1 | 16.5 ± 3.9 |
| iCAL36Q-4 | ANSRWQTSII | 14.8 ± 3.5 | NDd | 280 ± 100 | >1000 | 300 ± 100 | 450 ± 150 |
| CFTRT-3 | TEEEVQTTRL | 18 ± 5.5 | 15.1 ± 2.0 | 0.76 ± 0.51 | 4.6 ± 1.6 | 1.3 ± 0.4 | 0.45 ± 0.25 |
| iCAL36c | ANSRWPTSII | 22.6 ± 8.0 | 15.1 ± 2.1 | >1000 | >1000 | >1000 | >1000 |
| iCAL36Lc | ANSRWPTSIL | 23.6 ± 2.2 | NDd | 230 ± 100 | >1000 | 76.8 ± 1.6 | 530 ± 120 |
| iCAL36QDTRL | ANSRWQDTRL | 56.2 ± 5.6 | 16.2 ± 3.5 | 1.5 ± 0.2 | 6.7 ± 2.4 | 3.1 ± 1.4 | 1.1 ± 0.6 |
| iCAL36VQDTRL | ANSRVQDTRL | 230 ± 30 | 140 ± 40 | 2.5 ± 0.3 | 6.5 ± 2.3 | 3.8 ± 1.4 | 0.94 ± 0.42 |
| CFTRc | TEEEVQDTRL | 420 ± 80 | 64.7 ± 1.8 | 0.47 ± 0.2 | 1.8 ± 0.5 | 0.83 ± 0.33 | 0.07 ± 0.05 |
| CFTRIc | TEEEVQDTRI | 490 ± 120 | NDd | 10.7 ± 2.8 | 39.8 ± 1.0 | 19.1 ± 2.9 | 2.5 ± 0.43 |
| iCAL36VQD-3 | ANSRVQDSII | >1000 | 770 ± 50 | >1000 | >1000 | >1000 | 420 ± 120 |
Substitutions noted in subscript; the final substituted position is C-terminal unless numbered.
Bold indicates residue(s) from the CFTR sequence; non-bold indicates residues from iCAL36.
Values previously reported in Amacher et al., 2013.
ND indicates that value was not measured.
To test the hypothesis that non-motif residues also impact NHERF1 and NHERF2 affinities, we measured the binding constants of the chimeric peptides for each of the four PDZ domains (N1P1, N1P2, N2P2, and N2P2, respectively) in FP displacement assays (Table 1). The NHERF PDZ domains bind the CFTR and iCAL36 peptides with high affinity for CFTR (Ki <2 μM) and low affinity for iCAL36 (Ki >1 mM) (Amacher et al., 2013). Among these domains, substitutions at P−1 (iCAL36TRL vs. iCAL36L), P−3 (CFTRT-3), P−4 (iCAL36Q-4), and P−6-P−9 (CFTR vs. iCAL36VQDTRL) each affect individual Ki values.
Finally, to explore the potential of these non-motif differences to contribute to PDZ inhibitor selectivity, we developed an approach to identify substitutions that switched rank order of affinity. We first plotted the binding free energies for the series of peptides along a vertical axis for each domain. When placed side-by-side for a given pair of PDZ domains, with straight line connections between the values for each peptide, rank-order switches appear as points of intersection (Figure S2). Compared to each NHERF PDZ domain, the CALP affinities exhibit 19–25 pairwise affinity crossovers (Figure 1B) that presumably account for the engineered selectivity of iCAL36. Thus, although the CAL and NHERF binding motifs fail to highlight preferences at P−1, and P−3 through P−9, these residues are capable of ‘modulating’ both the affinity and selectivity of peptides for these PDZ domains. Furthermore, despite high sequence identity (58–72%) between the NHERF PDZ domains, pairwise comparisons reveal 2–6 crossovers (Figure 1B), suggesting potential non-motif contributions to selectivity even in this tightly related cluster.
All six C-terminal positions form side chain-dependent interactions
Despite the absence of motif signatures at four of the six residues from P0 to P−5, each residue directly contacts the surface of the CALP binding site, as illustrated in Figure 2A for one of the two protomers in our previously published CALP:iCAL36 structure (Amacher et al., 2013). Corresponding contact surfaces on the peptide are highlighted by comparison of the solvent-accessible surface of the bound peptide in the absence (Figure 2C) and presence of the CALP domain (Figure 2D).
Figure 2. Six peptide positions interact with the CALP binding cleft.

(A) The van der Waals surface of the CALP protein is shown together with the bound iCAL36 peptide (stick figure), with individual residues labeled and colored by position. The CALP surface (cyan) is colored at contact sites according to the closest peptide residue: P0 Ile (red), P−1 Ile (purple), P−2 Ser (gray), P−3 Thr (orange), P−4 Pro (yellow), P−5 Trp (blue), P−6 Arg (green), P−7 Ser (black), P−8 Asn (pink), and P−9 Ala (forest green).
(B) The peptide side-chain interaction index (IPDZ) is shown as a function of residue position for the peptides interacting with the A- (gray), and B- (black) CALP protomers of the asymmetric unit.
(C–D) The van der Waals surface of the bound conformation of the iCAL36 peptide is shown in the same perspective as in panel A (left) and following rotation by 135° around the vertical axis (right). The surface is colored by residue as in (A). To illustrate the surface contacted by CALP, individual atoms with IPDZ values > 0.5 are colored white in (D), involving residues P0-P−5.
The non-motif contacts also represent potentially significant stereochemical constraints. The fraction of solvent-accessible surface area (SASA) buried serves as an interaction index (IPDZ) that gauges the extent of side-chain engagement at each position along the backbone (Figure 2B). At the non-motif position P−5, IPDZ values (0.87 and 0.63 for the A and B protomers, respectively) are comparable to those for the motif residue P−2 (0.73 and 0.64). The other side chains exhibit IPDZ values ranging from 0.26 to 0.60, and even for residues with lower side-chain IPDZ values, several side-chain atoms are occluded at least 50% upon CALP docking (atoms colored white in Figure 2D).
Of course, contact mapping cannot by itself distinguish between favorable and unfavorable binding relationships. It also does not capture long-range (e.g., electrostatic) interactions or shifts in the free energy of the unbound state. Nevertheless, the observed steric interactions are consistent with evidence that non-motif residues can impact CALP affinity.
Defined CALP binding sites interact with each residue position
Given the characteristically shallow peptide-binding pocket of the CALP domain, we wished to determine whether our substituted peptides adopt significantly variable conformations, which could complicate stereochemical analysis and obscure modulator preferences in sequence alignments. To address this point, we crystallized and solved the structures of CALP bound to four chimeric iCAL36/CFTR peptides, selected to cover a range of affinities: iCAL36TRL (Ki = 4.5 μM), iCAL36Q-4 (Ki = 14.8 μM), iCAL36QDTRL (Ki = 56.2 μM), and iCAL36VQD-3 (Ki >1 mM),. To further explore sequence and conformational space, we also crystallized CALP bound to two additional low-affinity binding partners with unrelated sequences. HPV E6 viral oncoprotein C-terminal peptides have been studied in complex with other PDZ domains by NMR and X-ray crystallography (Charbonnier et al., 2011; Liu et al., 2007; Mischo et al., 2013; Zhang et al., 2007). Here, we generated CALP complexes with the decameric E6 C-terminal peptides from HPV16 (SSRTRRETQL) and HPV18 (RLQRRRETQV) (Jeong et al., 2007; Pim et al., 2012), with Ki values of 340 ± 70 μM and 490 ± 20 μM, respectively.
Structure determination and refinement protocols were designed to avoid phase bias (Table S1; Figures S3–4). Final data and refinement statistics are shown in Table 2. In general, the iCAL-based structures are very similar. When the PDZ backbone atoms of each structure are aligned to the CALP:iCAL36 B-protomer, they exhibit an average RMSD value of 0.34 ± 0.09 Å, with a maximum of only 0.52 Å (B-protomer of CALP:iCAL36TRL). Visually, the superposition of these CALP domains (Figure 3A) confirms that the framework of the binding cleft is largely independent of ligand sequence.
Table 2.
Data collection and refinement statistics for CALP co-crystals with iCAL36/CFTR chimeric peptides.
| iCAL36Q-4 | iCAL36TRL | iCAL36VQD-3 | iCAL36QDTRL | iCAL36QDTRL CALP-E317A |
HPV16 E6 | HPV18 E6 | |
|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
| Data Collection | |||||||
| Space Group (Number) | P 21 21 21 (19) | P 1 (1) | P 21 (4) | P 63 2 2 (182) | P 63 2 2 (182) | P 21 (4) | P 21 (4) |
| Unit cell dimensions | |||||||
| a,b,c (Å) | 36.0,47.7,98 | 30.8,50.2,55.2 | 36.3,48.8,54.9 | 61.6,61.6,97.6 | 61.8,61.8,97.4 | 33.1,48.4,52 | 33.2,48.0,52.9 |
| α,β,γ (°) | 90,90,90 | 68.8,75.8,87.9 | 90,92.8,90 | 90,90,120 | 90,90,120 | 90,101.6,90 | 90,102.0,90 |
| Resolutiona (Å) | 19.9–1.48 (1.58–1.48) | 19.3–1.75 (1.79–1.7) | 19.6–1.90 (2.03–1.9) | 19.7–1.50 (1.58–1.5) | 19.1–1.80 (1.85–1.8) | 19.4–1.80 (1.90–1.8) | 19.3–1.34 (1.42–1.34) |
| Rsymb (%) | 5.9 (45.0) | 4.5 (37.2) | 6.5 (36.0) | 4.9 (61.5) | 11.0 (61.7) | 5.9 (52.3) | 3.8 (26.5) |
| I/σI | 34.60 (6.93) | 15.48 (2.60) | 15.58 (3.59) | 49.46 (6.32) | 26.01 (6.53) | 20.37 (3.16) | 36.69 (7.53) |
| Completeness (%) | 99.7 (99.2) | 94.8 (91.1) | 99.6 (99.5) | 99.9 (100.0) | 99.9 (100.0) | 99.4 (98.1) | 94.2 (77.8) |
| Refinement | |||||||
| Total # of reflections | 28,816 | 30,585 | 15,162 | 18,179 | 10,780 | 14,953 | 34,482 |
| Reflections in test set | 1,460 | 1,493 | 763 | 920 | 560 | 758 | 1,729 |
| Rworkd/Rfreee | 18.1/20.9 | 17.2 /22.1 | 17.9/22.7 | 19.7/22.1 | 18.1/22.1 | 18.3/22.3 | 17.2/18.1 |
| Number of atoms: | |||||||
| Protein | 1467 | 2927 | 1426 | 774f | 772 f | 1431 | 1507 |
| Water | 180 | 267 | 141 | 69 | 69 | 147 | 258 |
| Ramachandran plotg (%) | 96.8/3.2/0/0 | 95.6/4.4/0/0 | 98.0/2.0/0/0 | 97.3/2.7/0/0 | 100/0/0/0 | 96.6/3.4/0/0 | 97.4/2.6/0/0 |
| Bav (Å2) | |||||||
| Protein | 13.27 | 18.96 | 22.38 | 19.36 | 16.32 | 23.40 | 9.86 |
| Solvent | 24.62 | 28.73 | 31.27 | 30.03 | 27.43 | 31.13 | 23.10 |
| Bond length RMSD (Å) | 0.006 | 0.006 | 0.007 | 0.007 | 0.008 | 0.007 | 0.007 |
| Bond angle RMSD (°) | 1.078 | 1.006 | 1.097 | 1.169 | 1.203 | 1.064 | 1.167 |
Values in parentheses are for data in the highest-resolution shell.
Rsym = ΣhΣi |I(h) − Ii(h)| / ΣhΣi Ii(h), where Ii(h) and I(h) values are the i-th and mean measurements of the intensity of reflection h.
SigAno = |F(+) − F(−)| / σ
Rwork = Σ ||Fobs|h − |Fcalc||h / Σ |Fobs|h, h ∈ {working set}
Rfree is calculated as Rwork for the reflections h ∈ {test set}
including sulfocysteine at position 319 (see Figure S4)
Core/allowed/generously allowed/disallowed
Figure 3. Peptide residues lie in distinct CALP binding sites.
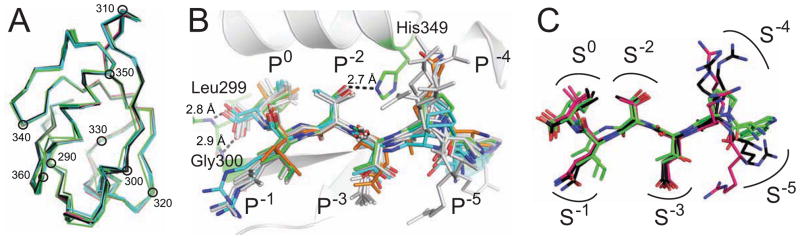
(A) Cα traces are shown for CALP protomers from each of four different space-groups (Table S1), following superposition of main-chain atoms on the B-protomer of CALP:iCAL36. Traces are colored by space group: P212121 (black), P1 (green), P21 (cyan), and P6322 (pink). Every 10th residue is indicated with a black circle and residue number. The tight clustering reveals no gross conformational changes in the PDZ domain based on lattice packing, peptide sequence, or affinity. See also Figure S3.
(B) Following superposition of PDZ main-chain atoms, chimeric iCAL36/CFTR peptides (stick figures) bind CALP (cartoon, gray) in similar conformations, despite sequence and affinity differences (Table 1). Characteristic PDZ hydrogen bonds are shown as dashed lines, including carboxylate interactions with residues Gly300 and Leu299 and a P−2 interaction with His349 (stick figures). iCAL36 (green carbons) and the highest (iCAL36TRL; cyan) and lowest (iCAL36VQD-3; orange) affinity peptides are highlighted for reference. Other peptides (gray) include iCAL36QDTRL, iCAL36Q-4, iCAL36L, HPV16 E6, and HPV18 E6. See also Figure S8A–G.
(C) Following superposition of PDZ main-chain atoms, bound peptide conformations are shown for C-terminal peptides derived from the HPV16 (black) and HPV18 (pink) E6 proteins together with iCAL36 (green). Binding sites are shown schematically for each position (S0, S−1, etc.). Non-carbon atoms are colored by element (red = O, blue = N)
This domain superposition also reveals strong conformational similarity of the peptides. We first compared the complexes formed by derivatives of the iCAL sequence, including the chimeric peptides described above, as well as earlier structures of iCAL36 and iCAL36L (Amacher et al., 2013). Despite widely varying binding affinities, all peptide co-complexes reflect canonical Class I PDZ domain-target interactions: the P0 residue with the carboxylate-binding loop, the P−2 residue with His349, and conserved β-sheet interactions (Figure 3B). Pairwise comparisons of the peptides reveal Cα variations less than 1.1 Å for the P0 to P−3 positions, and up to 4.4 Å for the P−4 and P−5 positions. These latter variations may partially reflect conformational changes due to differential crystal packing (Figure S3), but the side chains at each position still contact relatively similar surfaces on CALP. Thus, even the substitution of half of the residues in the peptide has only a limited effect on the stereochemical environment experienced by the side chain at a given position along the backbone.
Comparison of iCAL36 with the HPV E6 peptides also revealed similar peptide backbone conformations, despite very low sequence similarity (Figure 3C). Thus, in all of our structures, the side chains at a given peptide position are oriented similarly, contacting a common CALP local environment. As a result, we can extend previous models (e.g., Kang et al., 2003) to define a pharmacophore model in which positions P0 to P−5 each interact with cognate sites S0 to S−5 (Figure 3C). According to this model, each CALP site exhibits a characteristic set of stereochemical constraints that favor the binding of a corresponding subset of complementary amino acid(s). If the constraints are stringent, they may be present in enough sequences to appear as part of a motif. If they are more accommodating, they may not, but may still contribute to affinity as modulators.
Positional SubAna analysis confirms multiple modulatory residues
As a further test of the idea that each peptide side chain contacts a defined stereochemical environment relatively independent of sequence context, we performed substitutional analysis (SubAna) using peptide arrays derived from a variety of starting sequences. Each SubAna array contains a single peptide sequence with all 20 natural L-amino acids individually substituted at each position along the chain (Boisguerin et al., 2007). We chose to use SubAnas based on 10 starting sequences that encompassed a number of different CALP-binding attributes (sequences described in detail in the supplemental information, Figure S5).
Except for the artificial iCAL36 sequence, these peptides are derived from endogenous proteins, include known interacting partners (Cheng et al., 2002; Wente et al., 2005), cover a wide range of affinities for CALP (Cushing et al., 2008), and provide a variety of binding motif residue combinations (S/T at the P−2 position, I/L/V at the P0 position) for comparison. Furthermore, while the amount of protein bound is measured at a single concentration under non-equilibrium conditions, we can qualitatively compare trends across arrays (Boisguerin et al., 2011). In order to compare positional preferences across the full set of peptides, we separated each SubAna array into six strips, corresponding to the residues from P0 to P−5, and then aligned all of the strips for P0, all of the strips for P−1, etc. The resulting collage (Figure 4) shows the variability of side-chain preference(s) at a given position. Consistent selective preferences at the P0 and P−2 positions reflect their status as motif residues, largely independent of peptide context.
Figure 4. Position-specific peptide arrays reveal modulator preferences for CALP.

SubAna arrays of 10 peptides are clustered by residue position, with each row labeled according to the background sequence of the source array (see key for sequence information). The WT residues are listed to the right of each row. Residues in the initial sequence are substituted by all 20 L-amino acids, as indicated at top (see text). The motif positions P0 and P−2 reveal clear preferences for only a few residues. Positive modulator preferences are seen as consistently stronger binding than WT following substitution of e.g., Arg at P−1 or Trp at P−5, independent of the background peptide sequence. Negative preferences are also seen as consistently weaker binding compared to WT, e.g., following substitution of Gly at P−1 or Asp at P−3. See also Figure S5.
At the other positions, a more heterogeneous picture emerges, consistent with the lack of motif constraints observed previously (Figure 1B). A residue may be favored for certain sequences, and disfavored for others. For example, a P−4 Phe residue appears to be strongly favored for CFTR (row 7) and other sequences, while disfavored for VIPR2 (row 9). However, despite this variability, certain trends are observed that apply to most or all sequences. For example, our positional arrays confirm positive preferences for a P−1 Arg and for a P−5 Trp residue. They also reveal negative preferences, including P−3 Asp (Figure 4), consistent with the affinity difference observed between CFTR and CFTRT-3 (Table 1). CALP also appears to disfavor a Gly or Pro at P−1 and Pro at P−3.
Single substitutions at P−5 modulate binding affinity
To investigate the stereochemistry of preferential accommodation of a modulator residue, we focused on the impact of single residue substitutions in iCAL36 at the P−5 position. Competition FP experiments confirm a range of affinities for P−5-substituted peptides (Table 3). The Leu- and Tyr-substituted peptides bind CALP with Ki ≤100 μM, whereas the Ala- and His-substituted peptides each show ≥10-fold losses in affinity. A P−5 His side chain results in the weakest affinity of the set, at 450 μM, comparable to that of CALP:CFTR binding.
Table 3.
CALP binding affinities for P−5-substituted iCAL36 peptides.
| Ki (ìM) | Contact Surface Areac (Å2) | |||
|---|---|---|---|---|
|
| ||||
| Peptide | Sequencea | CALP | Peptide | P−5 Position |
|
| ||||
| iCAL36b | ANSRWPTSII | 22.6 ± 8.0 | 533.4 | 109.5 |
| L-iCAL36 | ANSRLPTSII | 68 ± 20 | 492.2 | 73.9 |
| Y-iCAL36 | ANSRYPTSII | 85.3 ± 1.5 | 518.2 | 99.6 |
| F-iCAL36 | ANSRFPTSII | 110 ± 20 | 506.9 | 86.6 |
| V-iCAL36 | ANSRVPTSII | 190 ± 40 | 485.8 | 66.4 |
| A-iCAL36 | ANSRAPTSII | 275 ± 80 | 461.9 | 42.5 |
| H-iCAL36 | ANSRHPTSII | 450 ± 10 | 505.5 | 79.2 |
Substitutions at the P−5 position are indicated in bold.
Values previously reported in Amacher et al., 2013.
Contact surface area for each P−5 position includes all atoms of the residue.
All 6 peptides were crystallized in complex with CALP. Structures were determined using the methods described above for the chimeric peptide complexes (Table S1 and supplemental methods), with data and refinement statistics reported in Table 4. For structural comparisons, we aligned the main-chain atoms of the P−5-substituted peptide:CALP complexes, using CALP:iCAL36 as a reference. Since the superposition of chimeric peptides had shown slight variations at the C-terminal core (positions P0 to P−3), we first compared the positions of the Cα atoms in these residues. In this region, the RMSD for the P−5-substituted peptides is less than the coordinate error (Figure 5A). There is a larger degree of variability at the P−4 and P−5 positions (≤3 Å), but the side chains remain clustered, again suggesting that modulations in binding affinity at the P−5 residue are due to local binding effects, not to large-scale alterations of residue interactions (Figure 5A).
Table 4.
Data collection and refinement statistics for CALP co-crystals with P−5-substituted iCAL36 peptides.
| A-iCAL36
|
L-iCAL36
|
V-iCAL36
|
H-iCAL36
|
F-iCAL36
|
Y-iCAL36
|
|
|---|---|---|---|---|---|---|
| Data Collection | ||||||
| Space Group | P 21 21 21 | P 21 21 21 | P 21 21 21 | P 21 21 21 | P 21 21 21 | P 21 21 21 |
| Unit cell dimensions | ||||||
| a,b,c (Å) | 36.7,47.8,97.6 | 36.2,47.6,97.3 | 36.7,47.9,97.8 | 36.2,47.8,98.1 | 36.2,48.1,97.6 | 36.2,47.8,97.5 |
| α,β, γ (°) | 90,90,90 | 90,90,90 | 90,90,90 | 90,90,90 | 90,90,90 | 90,90,90 |
| Resolutiona (Å) | 18.5–1.14 (1.19–1.14) | 34.0–1.2 (1.24–1.2) | 19.7–1.46 (1.54–1.46) | 19.5–1.47 (1.58–1.47) | 19.6–1.2 (1.26–1.2) | 18.5–1.09 (1.15–1.09) |
| Rsymb (%) | 11.6 (39.3) | 6.3 (51.5) | 6.8 (50.4) | 9.3 (50.6) | 8.4 (47.1) | 7.5 (34.5) |
| I/σI | 10.14 (3.99) | 20.98 (4.24) | 16.71 (3.05) | 21.17 (6.39) | 13.28 (3.86) | 15.57 (4.98) |
| Completeness (%) | 99.4 (99.9) | 99.9 (99.9) | 98.1 (88.7) | 98.3 (97.2) | 98.3 (95.7) | 98.2 (92.4) |
| Refinement | ||||||
| Total # of reflections | 63,011 | 53,398 | 30,171 | 28,702 | 53,211 | 70,084 |
| Reflections in the test set | 3,144 | 2,674 | 1,516 | 1,446 | 2,658 | 3,525 |
| Rworkd/Rfreee | 18.5/19.5 | 18.9/20.1 | 17.9/19.5 | 18.0/20.7 | 18.5/18.8 | 18.2/18.8 |
| Number of atoms: | ||||||
| Protein | 1502 | 1483 | 1472 | 1428 | 1522 | 1520 |
| Water | 236 | 230 | 180 | 170 | 227 | 220 |
| Ramachandran plotf (%) | 94.8/3.4/0/0 | 96.6/3.4/0/0 | 97.3/2.7/0/0 | 96.6/3.4/0/0 | 97.4/2.6/0/0 | 97.3/2.7/0/0 |
| Bav (Å2) | ||||||
| Protein | 12.35 | 12.27 | 14.85 | 9.77 | 12.20 | 11.95 |
| Solvent | 25.17 | 23.39 | 27.11 | 21.72 | 24.24 | 23.52 |
| Bond length RMSD (Å) | 0.005 | 0.006 | 0.006 | 0.006 | 0.006 | 0.007 |
| Bond angle RMSD (°) | 1.108 | 1.102 | 1. 043 | 1.063 | 1.107 | 1.073 |
Values in parentheses are for data in the highest-resolution shell.
Rsym = ΣhΣi |I(h) − Ii(h)| / ΣhΣi Ii(h), where Ii(h) and I(h) values are the i-th and mean measurements of the intensity of reflection h.
SigAno = |F(+) − F(−)| / σ
Rwork = Σ ||Fobs|h − |Fcalc|| h / Σ |Fobs| h, h∈ {working set}
Rfree is calculated as Rwork for the reflections h∈ {test set}
Core/allowed/generously allowed/disallowed
Figure 5. Stereochemical analysis of modulator preferences.

(A) The clustering of CALP-bound P−5-substituted iCAL36 peptides (stick figures) is shown following main-chain superposition of the CALP B-protomers. Peptides are colored by sequence: iCAL36 (W; green carbons), F- (red), Y- (black), H- (yellow), A- (cyan), L- (orange), and V- (purple). RMSD offsets at the P0-P−3 residues were within coordinate error, while modest variations were observed at the P−4 and P−5 positions (black arrow). The largest difference is 3.2 Å, between the Cα of the L- and Cα of the A-substituted peptides.
(B) Each substituted P−5 side chain (colored stick figure) is shown, as docked against its CALP binding interface (surface, cartoon and stick figure). The residues that contact the P−5 residue (His309, Gly310, Val311) are labeled in the CALP:F-iCAL36 structure. Each panel is labeled with the P−5 residue identity. See also Figure S6.
(C) The electrostatic potential surface of WT CALP (left) reveals a highly polarized distribution on either side of the peptide binding cleft. The electrostatic potential surface of CALP-E317A (right) shows a reduced, although still negative, interaction surface for the P−3 Asp residue of iCAL36QDTRL (insets). See also Figure S7.
(D) Electrostatic interactions are shown for P−1 Arg (left) and P−3 Asp (right) residues of the iCAL36QDTRL peptide (stick figure, yellow carbons) bound to CALP (gray Cα trace). At the top, nearby charged CALP residues are shown (stick figures, green carbons). The P−1 Arg forms a hydrogen bond (dashed line) with Gln314. The closets charged residue for the P−3 Asp is Glu317 (d > 5 Å). In the lower panels, the corresponding electrostatic potential surfaces are shown (rendered at 10 kBT/e) underscoring the negative electrostatic environment of both the P−1 and the P−3 side chains.
As seen for the chimeric peptides (Figure 3C), all P−5 residues interact with S−5, comprised of the same His309-Gly310-Val311 hydrophobic ledge that interacts with the P−5 Trp in iCAL36 (Amacher et al., 2013). However, despite this shared environment (Figure 5B; Figure S6), there is no obvious correlation of the observed range of affinities with stereochemical parameters, including electrostatic interactions or buried surface area metrics. For example, although the highest affinity peptide (Trp) is associated with the largest contact surface area, the second highest-affinity peptide (Leu) has a smaller contact surface than the significantly weaker His, Phe, or Tyr peptides (Table 3). However, more sophisticated computational approaches have shown promise in capturing CALP affinity determinants: the K* algorithm used energy minimization of conformational ensembles to estimate partition functions for the free and bound partners. Interestingly, 8 of the top 11 K*-designed sequences contained a P−1 Arg, including kCAL01 (WQVTRV), which has the highest affinity observed to date for a hexameric sequence (Roberts et al., 2012).
Electrostatic potential contributes to modulator preferences at P−1 and P−3
Our SubAna comparisons (Figure 4) also revealed prominent modulatory preferences at P−1 (favoring Arg) and P−3 (disfavoring Asp), both of which involve charged residues. As a result, we investigated the electrostatic environment of the corresponding binding sites. Rendered at 10 kBT/e, the CALP van der Waals surface potential reveals distinct positive and negative surfaces on opposite sides of the peptide-binding cleft (Figure 5C, Figure S7A). In the CALP:iCAL36QDTRL structure, both the P−1 Arg and the P−3 Asp residues point towards a ridge of negative potential, comprised of the β2, β3, α1, and β4 secondary structure elements, as well as connecting loops. This electrostatic environment could thus account for both of these modulator preferences (Figure 5D).
Specifically, the P−1 Arg is hydrogen-bonded with Gln322, facilitating a favorable interaction between the positively charged arginine guanidino group and an area of negative potential (Figure 5D). In contrast, the negatively charged P−3 Asp side-chain carboxylate is positioned directly adjacent to the negative potential surface, creating a Coulombic repulsion, even though the nearest charged residue is >5 Å away. In order to test the role of this side chain in determining modulator preferences, we expressed and purified a CALP-E317A mutant. Unfortunately, a crystal structure of the mutant (Table 2, Figure S7B–C and supplemental information available online) showed that the region of negative electrostatic potential was only modestly attenuated relative to wild-type (Figure 5C, insets). Correspondingly small (<7-fold) effects were observed on peptide binding affinities (Table 1).
However, a clearer picture is provided by analysis of the NHERF PDZ domains, which do not share the charge distribution of CALP. For example, in N1P1, the residue corresponding to CALP-Glu317 is Leu41, a nonpolar residue. Consistently, all four NHERF1/2 PDZ domains make favorable hydrogen bonds with the P−3 Asp, which is in a positive environment (Figure S7D–G). Indeed, in contrast to the dramatic increase in binding affinity for CALP upon substitution of the P−3 Asp to Thr, CFTRT-3 affinity was ~2–6-fold weaker than that of CFTR for each of the NHERF1/2 PDZ domains (Table 1), confirming their modest preference for the P−3 Asp.
Using modulator preferences to identify new high affinity sequences for CAL
Lastly, we wished to evaluate the ability of modulator preferences to improve our classification of peptide affinities for the CALP domain. The human deca-C-terminome, the set of C-terminal decapeptides for all 84,888 protein isoforms listed in the UniProt database (The UniProt Consortium, 2012), contains a total of 57,632 unique sequences. 2,197 of these sequences conform to the CALP binding motif (Figure 6A). However, CALP only weakly binds some of the peptide sequences that satisfy its motif, including iCAL36VQD-3 and the CFTR C-terminus (Table 1), and others previously tested (Cushing et al., 2008; Roberts et al., 2012).
Figure 6. Impact of binding motif and modulator residues on PDZ binding specificity.

(A) Evaluation of the number of unique C-terminal decamers in the human proteome that satisfy the CALP binding motif (red residues in example sequences listed at right).
(B) A schematic pharmacophore model of the series of side chain-binding sites along the CAL PDZ cleft is shown, some of which are identified as binding motif residues by high-throughput analysis (Figure 1A). Positive (green) and negative (red) preferences are shown at non-motif sites, together with the frequencies of these residues in the pool of potential CALP binders. FP displacement titrations confirm the expected affinities for representative peptides that contain positive (left panel, green) and negative (right, red) modulator residues, suggesting that modulators can significantly restrict the number of binding partners by adjusting the overall affinity balance of the PDZ:target interaction. See also Figure S8H.
To detect candidate high affinity sequences, we applied the positive modulator preferences at P−1 and P−5. Of the 2197 motif sequences, 141 and 36 contain an Arg and a Trp side chain, respectively (Figure 6B). Only four sequences contain both of our positive modulator preferences. Although they may not be physiological partners, all have CALP affinities better than or comparable to previously reported endogenous CALP ligands (Cushing et al., 2008): insulin-like growth factor-binding protein 3 receptor (TM219_HUMAN; RESHWSRTRL; Ki = 29 ± 8 μM), semaphorin 4G isoform 3 (SEM4G_HUMAN; PHSPWSFSRV; Ki = 46 ± 14 μM), histone H4 transcription factor (HINFP_HUMAN; CGCSWFATRV; Ki = 46 ± 24 μM), and proteasome subunit beta type-9 (B4DZW2_HUMAN; GFETWEGSRL; Ki = 120 ± 60 μM). Moreover, the proteasomal sequence contains a P−3 Gly residue (underlined), which has a negative influence in the positional arrays (Figure 4) and thus may account for its somewhat lower affinity. Likewise, endogenous targets containing the strongest negative modulator (Asp at P−3) are among the weakest CALP interactors (Figure 6B). Taken together, our data confirm the value of modulator preferences in assessing the affinity of candidate CALP-binding sequences.
Discussion
As illustrated by the weak CALP binding of C-terminal peptides from the known endogenous interactors CFTR and β-catenin (Cushing et al., 2008), there is abundant evidence that SLiM peptide affinity is not always the sole determinant of in vivo interactions, which also depend on molecular and cellular context (Luck et al., 2012). Nevertheless, PDZ:peptide binding remains a key element of specificity. Thus, deciphering the relationship between peptide sequence and PDZ affinity is a core requirement for understanding the networks of interactions formed by these ubiquitous protein-protein interaction domains. Over the past 15 years, studies have identified binding motifs and contacts from the C-terminus to the P−10 position (Appleton et al., 2006; Luck et al., 2012; e.g., Skelton et al., 2003; Smith and Kortemme, 2010; Thomas et al., 2008).
However, recent evidence suggests that PDZ sequence:affinity relationships can involve either more or fewer residues than indicated by motifs defined by alignments of high-affinity sequences. For example, while the Lin-7 homolog A (Lin7A) PDZ domain exhibits a highly selective motif with up to 6 determinants (Tonikian et al., 2008), fewer than four of these motif constraints are satisfied in any single Lin7A target sequence (The UniProt Consortium, 2012), and no annotated human C-terminal sequence matches the complete Lin7A PDZ binding motif. In contrast, although the binding motif for the CAL PDZ domain is highly degenerate (Figure 1B, Figure S1A), many motif-compatible sequences actually have negligible affinity for the domain (Roberts et al., 2012).
Overall, it appears that PDZ domains achieve sufficient binding affinity by harnessing strong free energy contributions at a few anchor positions, supplemented by additional stereochemical interactions that may involve either additional motif constraints or non-motif ‘modulator’ residues. Structural comparisons suggest that such modulator preferences reflect relatively loose pharmacophore sites, each of which engages the side chain at a particular position (Figure 3C). In fact, although only a limited number of other PDZ domains have been co-crystallized with more than one peptide ligand (e.g., Babault et al., 2011; Grembecka et al., 2006; Kang et al., 2003; Nomme et al., 2011), as for CALP, superpositions in these cases also suggest that each domain imposes a characteristic binding conformation on its ligands, and can thus define a set of domain-specific sequence preferences (Figure 3B, Figure S8).
Whether these preferences are readily captured in alignment motifs depends on a number of factors, including the stringency of the preference itself, as well as the nature of the experiment, limitations of the technique (e.g., number of sequences tested), and the baseline affinity of the domain for the peptide backbone. In many cases, preferences are subtle and lead only to marginal enrichment relative to random sequences. For example, our P−5 residue analysis suggests that all 7 substituted amino acids (Trp/Leu/Tyr/Phe/Val/Ala/His) could have been found in peptides that were included in the binding motif analysis, as the substituted iCAL peptides all bind CALP with affinity greater than that of CFTR, a top 100 sequence hit. This variability may reflect the attenuated specificity of surface-exposed stereochemical interactions (e.g., Serrano et al., 1990). Whatever the source of accommodation, alignments are unlikely to achieve sufficient enrichment to create an obvious motif constraint. Negative contributions are particularly difficult to detect robustly by alignment, since the expected observation frequency of a “neutral” residue is its already low rate of natural occurrence, associated with a correspondingly high level of random variability.
Our data (Figure 4) suggest that multi-sequence substitution analysis represents an effective extension of mutagenetic approaches (e.g., Wiedemann et al., 2004) to highlight preferences that are insufficiently robust or sequence-independent to establish clear motif signatures. SubAna arrays can also readily detect negative contributions (e.g., Asp at P−3) that have important implications for the biological specificity of PDZ interactions. The P−3 Asp side chain at the C-terminus of CFTR significantly limits its affinity for CAL, which targets CFTR to the lysosome, but not for NHERF1 and NHERF2, which functionally stabilize CFTR at the apical membrane (Table 1). Thus, the CFTR sequence at this position specifically favors interactions with PDZ proteins that promote endocytic recycling over degradation (Cheng et al., 2002).
This example underscores the potential for modulator residues to differentially affect interactions with PDZ domains that share overlapping motifs. Recent work by a number of groups suggests that PDZ interactions are a finely tuned product of evolution operating both on the PDZ domains (Ernst et al., 2009; Kaneko et al., 2010; McLaughlin et al., 2012) and on the target C-terminal sequences themselves (Kim et al., 2012). If degenerate two- or three-residue motifs were sufficient to determine target affinity for PDZ domains such as CAL or NHERF, such evolution would be limited by a very small set of combinatorial possibilities, as would the engineering of highly selective sequences. While individual modulator residues may contribute only modestly to target affinity, collectively they have the potential to significantly differentiate PDZ specificity profiles. As a result, characterizing these previously cryptic binding determinants may be critical both for a fuller understanding of PDZ domain interaction networks and for the development of specific peptide inhibitors of individual nodes.
Experimental Procedures
Protein Expression and Purification
The CALP E317A mutant was generated using the Quikchange Lightning (Agilent Technologies) protocol. The WT and mutant CALP (Amacher et al., 2011) and NHERF PDZ domains (Cushing et al., 2008) were expressed and purified as previously described. For fluorescence polarization (FP) experiments, proteins were dialyzed overnight into storage buffer (150 mM NaCl; 0.02% [w/v] sodium azide, 25 mM sodium phosphate pH 8.0, 0.2 mM tris[2-carboxyethyl]phosphine [TCEP], 5% [v/v] glycerol). For CALP crystallization, the protein was stored in GF2 buffer (25 mM Tris, 150 mM sodium chloride, 0.02% [w/v] sodium azide, 0.1 mM TCEP, 0.1 mM ATP, 5% [v/v] glycerol) (Amacher et al., 2011), prior to dialysis into crystallization buffer. Peptides for co-crystallization and FP experiments were synthesized and purified by the Tufts Core Peptide Facility.
Crystallization and structure determination
Detailed methods for crystallization and determination of each co-crystal structure are described in supplemental information (Table S1). Complexes were crystallized, data collected, and structures determined as previously described (Amacher et al., 2011; Amacher et al., 2013). Coordinates and structure factors have been deposited in the Protein Data Bank, with accession codes 4JOE, 4JOF, 4JOG, 4JOH, 4JOJ, 4JOK, 4JOP, 4JOR, 4K6Y, 4K72, 4K75, 4K76, and 4K78 (Table S1).
Structure analysis
Electrostatic potential surface calculations and structure alignments were performed in PYMOL (DeLano, 2008). The contact surface of each peptide residue was visualized using CHIMERA (Pettersen et al., 2004). SASA, including contact, was calculated using AREAIMOL (Lee and Richards, 1971), part of the CCP4 suite of programs (Collaborative Computational Project, 1994). Contact surface area for each peptide residue atom was calculated as [SASA]COMPLEX – [SASA]ALONE. The Interaction Index (IPDZ) is defined as the fractional change in solvent accessible surface area upon ligand binding, using the equation:
Fluorescence Polarization (FP)
FP assays were performed as previously described (Cushing et al., 2008; Vouilleme et al., 2010). Triplicate experiments were performed, using separate protein and reporter stock solutions in independent peptide titrations. The Ki values were determined using a SOLVER-based least-squares fitting algorithm in EXCEL, as described previously (Amacher et al., 2013; Cushing et al., 2008; Vouilleme et al., 2010). The following reporter peptides were used: F*-iCAL36 (F*-ANSRWPTSII) for CALP and E317A CALP; F*-CFTR6 (F*-VQDTRL) for N1P1 and N1P2; and F*-CFTR10 (F*-TEEEVQDTRL) for N2P1 and N2P2. F* corresponds to a fluorescein group linked to the peptide N-terminus via an amino-hexanoic acid linker. The concentrations used for each PDZ domain were ~1.5 × Kd, where the Kd values of each PDZ domain for their respective reporter peptides were: CALP (Kd=0.97 μM), E317A CALP (0.61 μM), N1P1 (0.37 μM), N1P2 (1.08 μM), N2P1 (0.32 μM), and N2P2 (0.23 μM).
Peptide Arrays
The full set of 6223HumLib sequences is described elsewhere (Roberts et al., 2012; supplemental material). Position-specific SubAna arrays were generated by juxtaposing data for a given peptide position from a series of SubAna arrays based on distinct peptide sequences. Individual SubAna arrays were synthesized and binding analyzed as previously described (Boisguerin et al., 2007; Vouilleme et al., 2010).
Proteome analysis of sequences
The Python scripting language (van Rossum, 2009) was used to map all occurrences of the CALP binding motif and the frequency distribution of individual modulator preferences in the set of all decameric C-terminal peptides present in the FASTA file provided by UniProt Knowledgebase release 2012_10 (The UniProt Consortium, 2012).
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R01-DK075309 and R01-HG004499, training grants T32-GM008704 to JFA and T32-DK007301 to PRC, and IDeA awards P20-RR018787 and P20-GM103413 to the Dartmouth Lung Biology COBRE), the Cystic Fibrosis Foundation RDP (STANTO11R0), a Hitchcock Foundation PPG, and by the Deutsche Forschungsgemeinschaft (DFG grant VO 885/3-2). We thank the entire Madden lab and Dr. James Fraser (UCSF) for advice and discussions, as well as Dr. Karl Griswold and Dr. Tom Scanlon (Dartmouth) for insight on rendering electrostatic potential maps. We would also like to thank Edwin Lazo, Dr. Vivian Stojanoff and Dr. Jean Jakoncic (NSLS) for assistance with data collection and analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amacher JF, Cushing PR, Bahl CD, Beck T, Madden DR. Stereochemical Determinants of C-terminal Specificity in PDZ Peptide-binding Domains: A novel contribution of the carboxylate-binding loop. J Biol Chem. 2013;288:5114–5126. doi: 10.1074/jbc.M112.401588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amacher JF, Cushing PR, Weiner JA, Madden DR. Crystallization and preliminary diffraction analysis of the CAL PDZ domain in complex with a selective peptide inhibitor. Acta Crystallogr. 2011;F67:600–603. doi: 10.1107/S1744309111009985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleton BA, Zhang Y, Wu P, Yin JP, Hunziker W, Skelton NJ, Sidhu SS, Wiesmann C. Comparative structural analysis of the Erbin PDZ domain and the first PDZ domain of ZO-1. Insights into determinants of PDZ domain specificity. J Biol Chem. 2006;281:22312–22320. doi: 10.1074/jbc.M602901200. [DOI] [PubMed] [Google Scholar]
- Babault N, Cordier F, Lafage M, Cockburn J, Haouz A, Prehaud C, Rey FA, Delepierre M, Buc H, Lafon M, Wolff N. Peptides Targeting the PDZ Domain of PTPN4 Are Efficient Inducers of Glioblastoma Cell Death. Structure. 2011;19:1518–1524. doi: 10.1016/j.str.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Boisguerin P, Ay B, Radziwill G, Fritz RD, Moelling K, Volkmer R. Characterization of a putative phosphorylation switch: adaptation of SPOT synthesis to analyze PDZ domain regulation mechanisms. Chembiochem. 2007;8:2302–2307. doi: 10.1002/cbic.200700518. [DOI] [PubMed] [Google Scholar]
- Boisguerin P, Madden DR, Volkmer R. Target-oriented peptide arrays in a palliative approach to cystic fibrosis (CF) In: Tschesche H, editor. Methods in Protein Biochemistry. Berlin: de Gruyter; 2011. pp. 249–269. [Google Scholar]
- Charbonnier S, Nomine Y, Ramirez J, Luck K, Chapelle A, Stote RH, Trave G, Kieffer B, Atkinson RA. The structural and dynamic response of MAGI-1 PDZ1 with noncanonical domain boundaries to the binding of human papillomavirus E6. J Mol Biol. 2011;406:745–763. doi: 10.1016/j.jmb.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Cheng J, Moyer BD, Milewski M, Loffing J, Ikeda M, Mickle JE, Cutting GR, Li M, Stanton BA, Guggino WB. A Golgi-associated PDZ domain protein modulates cystic fibrosis transmembrane regulator plasma membrane expression. J Biol Chem. 2002;277:3520–3529. doi: 10.1074/jbc.M110177200. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Consortium, The UniProt. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushing PR, Fellows A, Villone D, Boisguerin P, Madden DR. The relative binding affinities of PDZ partners for CFTR: a biochemical basis for efficient endocytic recycling. Biochemistry. 2008;47:10084–10098. doi: 10.1021/bi8003928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushing PR, Vouilleme L, Pellegrini M, Boisguerin P, Madden DR. A stabilizing influence: CAL PDZ inhibition extends the half-life of DeltaF508-CFTR. Angew Chem Int Ed Engl. 2010;49:9907–9911. doi: 10.1002/anie.201005585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey NE, Van Roey K, Weatheritt RJ, Toedt G, Uyar B, Altenberg B, Budd A, Diella F, Dinkel H, Gibson TJ. Attributes of short linear motifs. Mol Biosyst. 2012;8:268–281. doi: 10.1039/c1mb05231d. [DOI] [PubMed] [Google Scholar]
- DeLano W. The PyMOL molecular graphics system. DeLano Scientific LLC; Palo Alto, CA: 2008. The PyMOL molecular graphics system. [Google Scholar]
- Doyle DA, Lee A, Lewis J, Kim E, Sheng M, MacKinnon R. Crystal structures of a complexed and peptide-free membrane protein-binding domain: molecular basis of peptide recognition by PDZ. Cell. 1996;85:1067–1076. doi: 10.1016/s0092-8674(00)81307-0. [DOI] [PubMed] [Google Scholar]
- Ernst A, Sazinsky SL, Hui S, Currell B, Dharsee M, Seshagiri S, Bader GD, Sidhu SS. Rapid evolution of functional complexity in a domain family. Sci Signal. 2009;2:ra50. doi: 10.1126/scisignal.2000416. [DOI] [PubMed] [Google Scholar]
- Grembecka J, Cierpicki T, Devedjiev Y, Derewenda U, Kang BS, Bushweller JH, Derewenda ZS. The binding of the PDZ tandem of syntenin to target proteins. Biochemistry. 2006;45:3674–3683. doi: 10.1021/bi052225y. [DOI] [PubMed] [Google Scholar]
- Guerra L, Fanelli T, Favia M, Riccardi SM, Busco G, Cardone RA, Carrabino S, Weinman EJ, Reshkin SJ, Conese M, Casavola V. Na+/H+ exchanger regulatory factor isoform 1 overexpression modulates cystic fibrosis transmembrane conductance regulator (CFTR) expression and activity in human airway 16HBE14o- cells and rescues DeltaF508 CFTR functional expression in cystic fibrosis cells. J Biol Chem. 2005;280:40925–40933. doi: 10.1074/jbc.M505103200. [DOI] [PubMed] [Google Scholar]
- Harris BZ, Lim WA. Mechanism and role of PDZ domains in signaling complex assembly. J Cell Sci. 2001;114:3219–3231. doi: 10.1242/jcs.114.18.3219. [DOI] [PubMed] [Google Scholar]
- Jeong KW, Kim HZ, Kim S, Kim YS, Choe J. Human papillomavirus type 16 E6 protein interacts with cystic fibrosis transmembrane regulator-associated ligand and promotes E6-associated protein-mediated ubiquitination and proteasomal degradation. Oncogene. 2007;26:487–499. doi: 10.1038/sj.onc.1209837. [DOI] [PubMed] [Google Scholar]
- Kaneko T, Sidhu SS, Li SS. Evolving specificity from variability for protein interaction domains. Trends Biochem Sci. 2010;36:183–190. doi: 10.1016/j.tibs.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Kang BS, Cooper DR, Devedjiev Y, Derewenda U, Derewenda ZS. Molecular roots of degenerate specificity in syntenin’s PDZ2 domain: reassessment of the PDZ recognition paradigm. Structure. 2003;11:845–853. doi: 10.1016/s0969-2126(03)00125-4. [DOI] [PubMed] [Google Scholar]
- Kim J, Kim I, Yang JS, Shin YE, Hwang J, Park S, Choi YS, Kim S. Rewiring of PDZ domain-ligand interaction network contributed to eukaryotic evolution. PLoS Genet. 2012;8:e1002510. doi: 10.1371/journal.pgen.1002510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Tyndel MS, Huang H, Sidhu SS, Bader GD, Gfeller D, Kim PM. MUSI: an integrated system for identifying multiple specificity from very large peptide or nucleic acid data sets. Nucleic Acids Res. 2011;40:e47. doi: 10.1093/nar/gkr1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laura RP, Witt AS, Held HA, Gerstner R, Deshayes K, Koehler MF, Kosik KS, Sidhu SS, Lasky LA. The Erbin PDZ domain binds with high affinity and specificity to the carboxyl termini of delta-catenin and ARVCF. J Biol Chem. 2002;277:12906–12914. doi: 10.1074/jbc.M200818200. [DOI] [PubMed] [Google Scholar]
- Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Zheng JJ. PDZ domains and their binding partners: structure, specificity, and modification. Cell Commun Signal. 2010;8:8. doi: 10.1186/1478-811X-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Henry GD, Hegde RS, Baleja JD. Solution structure of the hDlg/SAP97 PDZ2 domain and its mechanism of interaction with HPV-18 papillomavirus E6 protein. Biochemistry. 2007;46:10864–10874. doi: 10.1021/bi700879k. [DOI] [PubMed] [Google Scholar]
- Luck K, Charbonnier S, Trave G. The emerging contribution of sequence context to the specificity of protein interactions mediated by PDZ domains. FEBS Lett. 2012;586:2648–2661. doi: 10.1016/j.febslet.2012.03.056. [DOI] [PubMed] [Google Scholar]
- McLaughlin RN, Jr, Poelwijk FJ, Raman A, Gosal WS, Ranganathan R. The spatial architecture of protein function and adaptation. Nature. 2012;491:138–142. doi: 10.1038/nature11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischo A, Ohlenschlager O, Hortschansky P, Ramachandran R, Gorlach M. Structural insights into a wildtype domain of the oncoprotein E6 and its interaction with a PDZ domain. PLoS One. 2013;8:e62584. doi: 10.1371/journal.pone.0062584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomme J, Fanning AS, Caffrey M, Lye MF, Anderson JM, Lavie A. The Src homology 3 domain is required for junctional adhesion molecule binding to the third PDZ domain of the scaffolding protein ZO-1. J Biol Chem. 2011;286:43352–43360. doi: 10.1074/jbc.M111.304089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nourry C, Grant SG, Borg JP. PDZ domain proteins: plug and play! Sci STKE. 2003;2003:RE7. doi: 10.1126/stke.2003.179.re7. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pim D, Bergant M, Boon SS, Ganti K, Kranjec C, Massimi P, Subbaiah VK, Thomas M, Tomaic V, Banks L. Human papillomaviruses and the specificity of PDZ domain targeting. FEBS J. 2012;279:3530–3537. doi: 10.1111/j.1742-4658.2012.08709.x. [DOI] [PubMed] [Google Scholar]
- Roberts KE, Cushing PR, Boisguerin P, Madden DR, Donald BR. Computational Design of a PDZ Domain Peptide Inhibitor that Rescues CFTR Activity. PLoS Comput Biol. 2012;8:e1002477. doi: 10.1371/journal.pcbi.1002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Hoffmuller U, Krause G, Ashurst J, Macias MJ, Schmieder P, Schneider-Mergener J, Oschkinat H. Specific interactions between the syntrophin PDZ domain and voltage-gated sodium channels. Nat Struct Biol. 1998;5:19–24. doi: 10.1038/nsb0198-19. [DOI] [PubMed] [Google Scholar]
- Serrano L, Horovitz A, Avron B, Bycroft M, Fersht AR. Estimating the contribution of engineered surface electrostatic interactions to protein stability by using double-mutant cycles. Biochemistry. 1990;29:9343–9352. doi: 10.1021/bi00492a006. [DOI] [PubMed] [Google Scholar]
- Skelton NJ, Koehler MF, Zobel K, Wong WL, Yeh S, Pisabarro MT, Yin JP, Lasky LA, Sidhu SS. Origins of PDZ domain ligand specificity. Structure determination and mutagenesis of the Erbin PDZ domain. J Biol Chem. 2003;278:7645–7654. doi: 10.1074/jbc.M209751200. [DOI] [PubMed] [Google Scholar]
- Smith CA, Kortemme T. Structure-based prediction of the peptide sequence space recognized by natural and synthetic PDZ domains. J Mol Biol. 2010;402:460–474. doi: 10.1016/j.jmb.2010.07.032. [DOI] [PubMed] [Google Scholar]
- Stiffler MA, Chen JR, Grantcharova VP, Lei Y, Fuchs D, Allen JE, Zaslavskaia LA, MacBeath G. PDZ domain binding selectivity is optimized across the mouse proteome. Science. 2007;317:364–369. doi: 10.1126/science.1144592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, Dasgupta J, Zhang Y, Chen X, Banks L. Analysis of specificity determinants in the interactions of different HPV E6 proteins with their PDZ domain-containing substrates. Virology. 2008;376:371–378. doi: 10.1016/j.virol.2008.03.021. [DOI] [PubMed] [Google Scholar]
- Tonikian R, Zhang Y, Sazinsky SL, Currell B, Yeh JH, Reva B, Held HA, Appleton BA, Evangelista M, Wu Y, et al. A specificity map for the PDZ domain family. PLoS Biol. 2008;6:e239. doi: 10.1371/journal.pbio.0060239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rossum G. In: The Python Language Reference. Drake FL Jr, editor. Python Software Foundation; 2009. [Google Scholar]
- Vouilleme L, Cushing PR, Volkmer R, Madden DR, Boisguerin P. Engineering peptide inhibitors to overcome PDZ binding promiscuity. Angew Chem Int Ed Engl. 2010;49:9912–9916. doi: 10.1002/anie.201005575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wente W, Stroh T, Beaudet A, Richter D, Kreienkamp HJ. Interactions with PDZ domain proteins PIST/GOPC and PDZK1 regulate intracellular sorting of the somatostatin receptor subtype 5. J Biol Chem. 2005;280:32419–32425. doi: 10.1074/jbc.M507198200. [DOI] [PubMed] [Google Scholar]
- Wiedemann U, Boisguerin P, Leben R, Leitner D, Krause G, Moelling K, Volkmer-Engert R, Oschkinat H. Quantification of PDZ domain specificity, prediction of ligand affinity and rational design of super-binding peptides. J Mol Biol. 2004;343:703–718. doi: 10.1016/j.jmb.2004.08.064. [DOI] [PubMed] [Google Scholar]
- Wolde M, Fellows A, Cheng J, Kivenson A, Coutermarsh B, Talebian L, Karlson K, Piserchio A, Mierke DF, Stanton BA, et al. Targeting CAL as a negative regulator of DeltaF508-CFTR cell-surface expression: an RNA interference and structure-based mutagenetic approach. J Biol Chem. 2007;282:8099–8109. doi: 10.1074/jbc.M611049200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Dasgupta J, Ma RZ, Banks L, Thomas M, Chen XS. Structures of a human papillomavirus (HPV) E6 polypeptide bound to MAGUK proteins: mechanisms of targeting tumor suppressors by a high-risk HPV oncoprotein. J Virol. 2007;81:3618–3626. doi: 10.1128/JVI.02044-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.