

Abstract
Toll-like receptors (TLR) are innate immune receptors typically activated by microbial-associated molecular patterns (MAMPs) during infection or damage-associated molecular patterns (DAMPs) as a result of tissue injury. Recent findings suggest that TLR2 and TLR4 signaling play important roles in developmental and adult neuroplasticity, and in learning and memory. In addition, activation of TLR2 and TLR4 worsens ischemic injury to the heart and brain in animal models of myocardial infarction and stroke. TLR activation is also implicated in thermoregulation and fever in response to infection. However, it is not known whether TLRs participate in the regulation of the sympathetic and/or parasympathetic components of the autonomic nervous system (ANS). Here we provide evidence that TLR2 and TLR4 influence autonomic regulation of heart rate (HR) body temperature and energy metabolism in mice. We show that mice lacking TLR2 or TLR4 exhibit reduced basal HR, which results from an increase of parasympathetic tone. In addition, thermoregulatory responses to stress are altered in TLR2−/− and TLR4−/− mice, and brown fat-dependent thermoregulation is altered in TLR4−/− mice. Moreover, TLR2−/− and TLR4−/− mice consume less food and exhibit a greater mass compared to wild type mice. Collectively, our findings suggest important roles for TLR2 and TLR4 in the ANS regulation of cardiovascular function, thermoregulation, and energy metabolism.
Keywords: Innate Immunity, Toll-like receptors, TLR2, TLR4, Autonomic nervous system, ANS, heart rate, stress
Introduction
Toll-like receptors (TLR) comprise a family of 10–12 innate immune receptors in mammals. TLRs are typically activated during infection by microbial-associated molecular patterns (MAMPs; to be distinguished from the former term, pathogen associated molecular patterns, PAMPs, as it is acknowledged now that molecular patterns on non-pathogenic bacteria also activate TLRs) (Takeda and Akira, 2004). TLRs are also activated by damage-associated molecular patterns (DAMPs), a set of endogenous TLR ligands released in body tissues as a result of tissue injury (Yu et al., 2010). TLRs are expressed in a variety of immune-related cell types, as well as non-immune cells such as intestinal epithelial cells (Marques and Boneca, 2011), endothelial cells (Garrafa et al., 2011) as well as cells of the central nervous system (CNS) including microglia, astrocytes, oligodendrocytes and neurons (Bsibsi et al., 2002; Tang et al., 2008). TLRs and TLR-related signaling are increasingly implicated in developmental and adult neuroplasticity, including the regulation of neural progenitor cell proliferation (Lathia et al., 2008; Okun et al., 2010b; Rolls et al., 2007; Shechter et al., 2008), axonal guidance (Cameron et al., 2007; Ma et al., 2007; Ma et al., 2006), learning and memory (Okun et al., 2012; Okun et al., 2010a), and metabolism (Shechter et al., 2013).
Via counterbalancing actions of sympathetic and parasympathetic neurons, the autonomic nervous system (ANS) regulates body systems critical for many basic functions of the organism including heart rate (HR), blood pressure, thermoregulatory thermogenesis and energy metabolism (Cannon and Nedergaard, 2004) in both health and disease. In thermoregulatory thermogenesis, the parasympathetic innervation of brown adipose tissue (BAT) only appears in two minor BAT depots, but not in the major interscapular BAT depot. BAT non-shivering thermogenesis is mostly triggered by the release of norepinephrine from its sympathetic nerve terminals, stimulating β3-adrenoceptors (Bartness et al., 2010). Sympathetic control of brown adipose tissue is also essential for the cold acclimation-recruited norepinephrine-mediated thermogenesis (Cannon and Nedergaard, 2004). Activation of either TLR2 or TLR4 is implicated in thermoregulation and fever in response to infection (Romanovsky et al., 2006). Activation of members of the TLR family of receptors explains how secreted products of different microbes can evoke the same biological response, fever. Fever-inducing substances or pyrogens, such as Lipopolysaccharides (LPS) and cell wall components of Gram-negative bacteria such as E. coli, are recognized by TLR4 (Poltorak et al., 1998). Similarly to TLR4 activation, Gram-positive bacterial MAMPs such as peptidoglycan, lipoteichoic acid, as well as products from mycobacteria, yeast or fungi that stimulate TLR2 (Akira, 2003) can evoke fever (Hubschle et al., 2006; Nakagawa et al., 2002). Although much less studied, stimulation of TLR3 using polyinosine–polycytidylic acid (poly (I:C)), a synthetic analogue of double-stranded RNA from viruses (Alexopoulou et al., 2001) is also capable of inducing fever (Nakagawa et al., 2002). Moreover, it is likely that most ligands that activate TLRs are pyrogens. However, it is not known whether this process is mediated by regulation of the sympathetic and/or parasympathetic components of the ANS.
Through its regulation via the parasympathetic/sympathetic controls, HR is an established indicator for numerous pathological conditions. Increasing autonomic parasympathetic control of HR is tightly linked to higher survival rates in patients with heart disease (Bigger et al., 1992). Utilizing rodents for the study of the different controls of HR is an important step toward understanding the contribution of genes affecting these traits in pathologies related to the ANS. Heart failure occurs when there is an inability of the heart to pump enough blood to meet the requirements of the body’s metabolizing tissues. Ischemic cardiomyopathy and hypertension-induced cardiac hypertrophy are the most common causes of heart failure (Brown et al., 2005; Gradman and Alfayoumi, 2006). Heart failure is associated with chronic inflammation (Anker and von Haehling, 2004) and up-regulation of TLRs in cardiac muscle (Birks et al., 2004; Frantz et al., 1999). TLR2 or TLR4 deficiency, however, prevents increase in myocyte and cardiac size following pressure overload (Favre et al., 2007; Ha et al., 2005). The acute activation of TLR signaling may be beneficial in the short term, but ongoing tissue damage which results in chronic activation of TLRs can lead to heart failure (Topkara et al., 2011). Both TLR antagonists and agonists have been shown to be protective in heart failure. Eritoran, a TLR4 inhibitor, has been shown to reduce cardiac hypertrophy in a mouse model of aortic constriction by inhibiting a TLR4-mediated inflammatory response (Ehrentraut et al., 2011a; Ehrentraut et al., 2011b). Thus, patients with pressure overload induced heart failure may benefit from the inhibition of TLR4 signaling. However, the pharmacodynamics of eritoran requires administration by continuous infusion that would not be practical for the treatment of patients with chronic heart failure. Heart failure is associated with the activation of the sympathetic nervous system (Francis, 1989; Meredith et al., 1993) and this association contributes to increased mortality rates (Eichhorn and Bristow, 1996; Goldsmith, 1999). In addition, cardiovascular disease may lead to an inflammatory process within the brain and also play a key role in activation of the sympathetic nervous system (Zhang et al., 2010). Additional evidence link TLRs to both inflammatory processes and sympathetic control. In a mouse model of ischemia-induced heart failure, angiotensin II receptors in the brain stem contribute to the central sympathetic response through TLR4 and MyD88-mediated inflammatory response. In addition, intra-cerebroventricular infusion of the angiotensin receptor blocker Losartan reduces both inflammatory and sympathetic responses (Ogawa et al., 2011). Fluvastatin, an HMG-CoA reductase inhibitor, reduces the inflammatory response in patients with chronic heart failure with an inhibitory effect on monocyte TLR4-mediated immune response (Foldes et al., 2008; Navi et al., 2013).
While both the cardio-vascular and thermoregulatory response are attributes of the ANS, and TLR2 and TLR4 are both intimately linked to a myriad of heart and cardiovascular related pathologies as well as thermoregulatory alterations following inflammation, the mechanism that underlies these effects is not clear. We thus asked whether there is a link between TLR signaling and ANS regulation of HR, thermoregulation, and/or energy metabolism.
Here, we show that: (1) mice lacking TLR2 or TLR4 exhibit reduced HR; (2) thermoregulatory responses to stress are altered in TLR2−/− and TLR4−/− mice; (3) brown fat-dependent thermoregulation is altered in TLR4−/− mice; and (4) mice lacking either TLR2 or TLR4 consume less food and maintain a greater mass compared to WT mice. The above suggests that TLR2 and TLR4 influence energy intake and metabolism and help regulate key aspects of the ANS.
Materials and Methods
Animals
Young adult male congenic TLR4−/− mice (B6.B10ScN-Tlr4lps-del/JthJ) (n = 20), TLR2−/− mice (B6.129-Tlr2tm1Kir/J) (n = 20) and genetically matched background WT mice (B6.B10) (n = 20) at identical ages were purchased from Jackson Laboratories (Bar Harbor, ME, USA). All experiments were completed using mice starting at 2 months old and lasted until mice were 5 months old. Animal care and experimental procedures followed NIH guidelines and were approved by the National Institute on Aging Animal Care and Use Committee. The different experimental paradigms conducted in this study are chronologically depicted in Scheme 1.
Scheme 1.

Chronological indication of the different experimental paradigms conducted in this study.
Drugs
The following drugs were used in this study: atropine methyl-nitrate, a blood-brain barrier-impenetrant competitive antagonist of muscarinic acetylcholine receptor types M1, M2, M3, M4 and M5 (MP Biomedicals; 2 mg/kg, (Griffioen et al., 2013)); the beta-1 adrenergic receptor antagonist atenolol a (MP Biomedicals; 2 mg/kg, (Griffioen et al., 2013)); the beta-3 adrenergic receptor agonist CL316243 (Tocris Bioscience; 1 mg/kg, (Fu et al., 2008)); and the beta-3 adrenergic receptor antagonist SR59230A hydrochloride (Tocris Bioscience; 0.5 mg/kg, (Bexis and Docherty, 2009)). All drugs were injected intraperitoneally, and the maximal injected volume of the drugs was 200 μl, diluted in phosphate buffered saline.
Telemetry
A telemetry system was used to continuously monitor physiological and behavioral parameters of mice in their home cages as described previously (Griffioen et al., 2011). Briefly, a transmitter, TA10ETA-F20 (Data Sciences International, St Paul, MN, USA), which monitors electrocardiogram (ECG), core body temperature, and general activity, was surgically implanted in each of the mice. Two biopotential leads were routed subcutaneously lateral to midline of the chest and secured to chest muscles with silk sutures (Ethicon). Telemetry data were continuously recorded in 2.5 min bins, every 10 min. A total of 30 mice (of the 60 mice used in this study) were subjected to telemetry analysis. To this end, mice (n=10 per group) were implanted with transmitters at 2 months of age and allowed to recover for a month before beginning recording.
Restraint Stress
Immobilization stress was induced using a 50 ml polypropylene plastic tube, perforated throughout the length of the tube to allow unhindered breathing. Physiological variables were recorded for 30 min before the stress session, during the 1 h stress period, and for 1–2 h after the stress. Following the 1 h stress period, mice were immediately returned to their cages. All stress sessions were performed between 1100 and 1300 h.
Cold Water Swim Stress
Baseline recording was conducted for 30 minutes prior to the swim stress session. Temperature was maintained at 20°C, and exposure time to cold water was 15 minutes, during which telemetric measurements were continuously recorded. Following the stress session, mice were allowed to recover in their home cage, during which telemetric recordings were continuously acquired.
Plasma corticosterone measurements
To measure baseline corticosterone levels, blood samples were drawn from mice during their activity time. Blood was gathered using a retro-orbital bleeding technique, using a heparinized micro-hematocrit capillary tube (Fisher Scientific; Pittsburgh, PA). Blood samples were then centrifuged at 12,500 rpm for 12 min at 4° and plasma supernatant was removed and stored at −80°. Plasma corticosterone concentrations were quantified using an RIA kit (MP Biomedicals; Solon, OH) according to manufacturer’s instructions.
Metabolic analyses
A Comprehensive Lab Animal Monitoring System (CLAMS; Columbus Instruments, Columbus, OH) was used to evaluate general animal activity, food consumption, water intake, O2 consumption, CO2 production, and energy expenditure. Activity (counts) was measured on both the x and z axis using infrared beams, and the number of beam breaks was determined during a 48 hour period. Food intake (g) was measured by recording the difference in the scale measurement of the center feeder from one time point to another. The accumulated drink volume (ml) was measured using a volumetric drinking monitor, which detects the lick number and volume consumed. The Respiratory Exchange Ratio (RER) was also calculated using the ratio of VCO2 to VO2 (which changes depending on the energy source the animal uses). Energy expenditure was calculated using the gas exchange data [energy expenditure = (3.815 +1.232 *RER) *VO2] and expressed as kJ/kg/h. When carbohydrates are the only substrate being oxidized, the RER will be ~1.0, and it will be ~0.7 when only fatty acids are oxidized. The animals were continuously monitored for 48 hours, which included 2 complete light/dark cycles.
NMR analysis
The assessment was acquired by nuclear magnetic resonance (NMR) using the Minispec LF90 (Bruker Optics, Billerica, MA) and measurements of lean and fat were performed in whole, live, awake restrained mice.
Data and statistical analysis
Data were analyzed by two-way repeated measures ANOVA with Bonferroni’s post-hoc test and Student’s t-test for individual comparisons between groups using Prizm 5 (GraphPad software, Inc., San Diego, CA, USA). Results are expressed as mean ± SE and significance was set at p < 0.05.
Results
TLR2−/− and TLR4−/− mice have altered autonomic control of HR
It is unknown whether TLRs regulate HR. We therefore determined whether mice lacking either TLR2 (TLR2−/− mice) or TLR4 (TLR4−/− mice) exhibit altered HR. Mice were implanted with telemetry transmitters that monitor HR, home cage activity and core body temperature. Both TLR2−/− and TLR4−/− mice exhibited significantly lower baseline HR in both light and dark cycles compared to WT mice (light phase: WT: 489±7.2, TLR4: 468±4.9, TLR2: 462±6.3; dark phase: WT: 568±9.4, TLR4: 542±4.9, TLR2: 539±4.5; F2,27=7.71, P<0.0022, Figure 1A, right panel), suggesting that TLR2 and TLR4 deficiency contributes to baseline HR. To further determine whether parasympathetic and/or sympathetic activity to the heart is altered in TLR2−/− and TLR4−/− mice, we measured HR in the presence of antagonists of parasympathetic cholinergic (atropine) or sympathetic beta-adrenergic (atenolol) receptors. TLR2−/− but not TLR4−/− mice exhibited a significantly augmented HR elevation in response to atropine compared to WT mice (TLR2: 172.4±21.2; WT: 105.7±15.4, P<0.05, t-Test, Figure 1B). This suggests that TLR2−/− mice have enhanced parasympathetic activity to the heart. Atenolol decreased HR to a similar degree in WT, TLR2−/− and TLR4−/− mice (−60.9±23.1, −52.7±20.3, −36.07±16.1 respectively, Figure 1C), suggesting that sympathetic control of HR is unchanged in TLR2−/− and TLR4−/− mice. The intrinsic HR, determined by ganglionic blockade with hexamethonium bromide, was not different in TLR2−/− or TLR4−/− mice compared with WT mice (596±30.4, 572±30.3 and 580±30.2 respectively, Figure 1D), indicating that the observed differences in basal HR result from altered autonomic activity to the heart, and not the intrinsic beat rate.
Figure 1. Mice lacking TLR2 or TLR4 exhibit a reduced resting heart rate: evidence that TLR2 and TLR4 signaling reduce parasympathetic tone.

(A) Left panel: Baseline HR of TLR2−/−, TLR4−/− mice and their respective WT mice were measured in mice implanted with transmitters and HR was recorded during 2 consecutive days. Right panel: average baseline HR from both light and dark cycles during 2 consecutive days. (B) Atropine sulfate was injected intraperitonealy to mice and HR was measured for 1 hour, during which TLR2−/− mice exhibited increased change in HR. (C) Atenolol was injected intraperitonealy to mice and HR was measured for 1 hour, during which no significant change in HR was noted. (D) Hexamethonium was injected intraperitonealy to mice and HR was measured for 1 hour, during which all mouse strains exhibited similar intrinsic HR.
The effect of acute restraint stress on HR in TLR2−/− and TLR4−/− mice
To test whether TLR2 or TLR4 deficiency also alters HR responses to stress, we exposed mice to restraint stress. Although TLR2−/− and TLR4−/− mice had a lower basal HR, all mice exhibited similar elevation in HR during restraint stress (Figure 2A). However, when normalized to baseline, the change in HR in TLR2−/− and TLR4−/− mice during the stress period was significantly augmented compared to WT mice (F2,162=12.05, P<0.0001, Figure 2B). This suggests that although TLR2−/− and TLR4−/− mice have lower basal HRs, their HR during restraint stress reaches a level similar to that of WT mice.
Figure 2. Evidence for subtle effects of TLR2 and TLR4 signaling on the cardiovascular response to acute restraint stress.
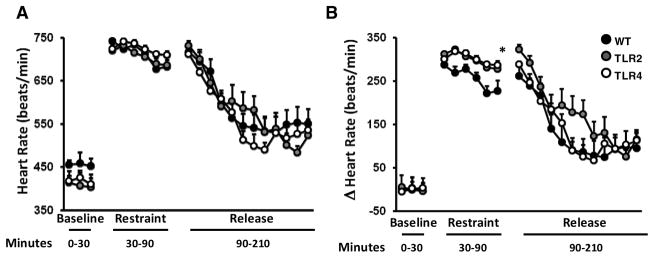
TLR2−/−, TLR4−/− and their respective WT mice, were implanted with transmitters, and subsequently subjected to a 30-minute restraint stress. (A) HR was measured prior, during and following stress. (B) Change in HR compared to baseline HR.
Thermoregulatory responses to stress are altered in TLR2−/− and TLR4−/− mice
To test whether autonomic control of body temperature is altered in TLR2−/− and TLR4−/− mice, we measured body temperature during a 48-hour period in their home cage and during and after exposure to stressors. Baseline core body temperature was lower in TLR2−/− mice compared with TLR4−/− and WT mice during the day (35.73±0.068, 35.79±0.027 and 35.955±0.065 respectively, F2,54=3.6, P=0.0341, Figure 3A) and specifically during the first third of the light cycle (35.865±0.078, 36.153±0.058 and 36.246±0.062 respectively, F2,162=7,53, P=0.0007, Figure 3B). However, during restraint stress, TLR2−/− and TLR4−/− mice exhibit delayed thermoregulation compared with WT mice (F2,162=37.94, P<0.0001, Figure 3C). Further, when normalized to baseline body temperature, the change in body temperature during restraint stress was higher in TLR2−/− mice (F2,162=37.94, P<0.0001, Figure 3D). Further, these effects were not correlated to altered activity levels (Figure 3E).
Figure 3. TLR2−/− and TLR4−/− mice exhibit altered thermoregulation during acute restraint stress.

TLR2−/−, TLR4−/− and their respective WT mice were implanted with transmitters and core body temperature and activity were recorded during 2 consecutive days (A) Total averages of baseline core body temperature in the light and dark cycles (B) Core body temperature during light (5am–9am, 9am–1pm and 1pm–5pm) and dark cycles (5pm–9pm, 9pm–1am and 1am–5am) (C) baseline core body temperature was recorded prior, during and after acute restraint stress (D) change in temperature compared with baseline temperature (D) activity levels prior to, during and after restraint stress.
In mice, heat production predominantly arises from activation of brown fat. We asked whether the activation of brown fat by the ANS is altered in TLR2−/− and TLR4−/− mice. To do this, we used antagonists to the receptors responsible for heat production in brown fat including cholinergic receptors located on the stellate ganglion neurons that innervate brown fat (atropine), and adrenergic receptors located on brown fat cells (SR59230A). Atropine treatment resulted in a small and significant increase of body temperature in TLR2−/− and TLR4−/− but not in WT mice (0.41±0.15°C, 0.39±0.1°C and −0.081±0.17°C respectively, P=0.004, t-Test, Figure 4A). The latter finding suggested that TLR2 and TLR4 signaling might enhance parasympathetic stimulation of thermogenesis. Expectedly, attenuating sympathetic control using atenolol had no effect on core body temperature, as beta-1 adrenergic receptors are not expressed on brown fat (Zhao et al., 1998) (−0.035±0.19°C, 0.015±0.12°C and −0.117±0.12°C in WT, TLR2−/− and TLR4−/− mice respectively, Figure 4B). Activation of beta-3 adrenergic receptors with the agonist CL316243, significantly increased core body temperature in WT (36.89±0.06 °C vs. 35.6±0.05 °C, P<0.05, t-Test), TLR2−/− (36.99±0.08 °C vs. 35.59±0.11 °C, t-Test) and TLR4−/− (36.37±0.1 °C vs. 35.71±0.12 °C, t-Test) (Figure 4C, left panel). However, when normalized to baseline, the change in body temperature was significantly lower in TLR4−/− mice compared with TLR2−/− or WT mice (0.66±0.14°C, 1.4±0.07°C and 1.29±0.09°C respectively, P=0.003, t-Test, Figure 4C, right panel). In contrast, antagonism of beta-3 receptors using SR59230A did not result in a significant change in body temperature compared to baseline temperature either WT, TLR2−/− or TLR4−/− mice (0.37±0.15°C, 0.16±0.16°C and 0.02±0.12°C respectively, Figure 4D). Although no effect was observed for SR59230A in these experiments, it should be noted that the beta-3 adrenergic receptors are also expressed on tissues other than brown fat and that the specificity of this antagonist has been questioned (Ootsuka et al., 2011). These results suggest that beta-3 receptor-dependent thermoregulation by brown adipose tissue is altered in TLR4−/− but not in TLR2−/− mice.
Figure 4. Evidence for differential effects of developmental deficiency in TLR2 and TLR4 on thermoregulation.

(A) TLR2−/−, TLR4−/− and WT mice were implanted with transmitters and core body temperature was recorded prior to and after mice were injected intraperitoneally with (A) atropine methyl nitrate, (B) Atenolol, (C) CL316243 or (D) SR59230A.
Because we observed alterations in both agonist- and stress-induced thermoregulation in TLR2−/− and TLR4−/− mice, we next assessed thermoregulation under a stressful condition that involves a change in temperature. For this experiment, we utilized the cold-swim stress paradigm. During a cold swim stress, TLR2−/− and TLR4−/− mice maintain their body temperature at a higher temperature than WT mice, and their recovery to baseline temperature occurs faster (F2,1512=32.8, P<0.0001, Figure 5A), and inhibiting beta-3 adrenergic receptors using a beta-3 blocker does not significantly change this effect (Figure 5B). None of these effects was correlated to altered activity levels during cold swim stress or during recovery (Figure 5C).
Figure 5. TLR2 and TLR4 influence body temperature responses to cold water swim stress; effect of a beta-3 receptor antagonist.

(A) TLR2−/−, TLR4−/− and WT mice implanted with transmitters and were recorded for core body temperature prior to, during and after exposure to cold swim stress (B) with a beta-3 antagonist. (C) Activity levels prior to, during and after cold swim stress.
Evidence that TLR2 and TLR4 regulate energy metabolism
To understand whether the baseline ANS alterations we observed in mice lacking TLR2 or TLR4 have functional implications with regards to energy metabolism, we analyzed various metabolic parameters in TLR2−/−, TLR4−/− and WT mice. TLR2−/−, but not TLR4−/− mice were less active than WT mice (F1,198=9.78, P<0.0058, Figure 6A). This effect was due to lower activity during the dark cycle (TLR2: 6.72±0.97; WT: 11.86±1.6, F2,54=4.39, P=0.0172, Figure 6B), and this was correlated with reduced baseline corticosterone levels in TLR2−/− mice but not TLR4−/− mice compared with WT mice (TLR2−/−: 37±5.9; WT: 67±11, P=0.0168, T-test, Figure 6C).
Figure 6. TLR2 deficiency reduces activity levels during the dark cycle; Correlation with resting circulating corticosterone levels.
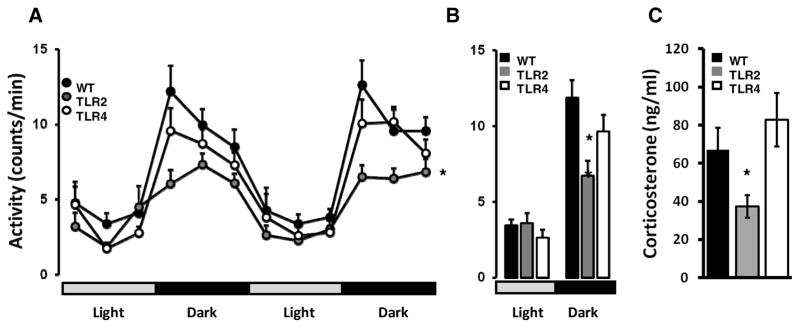
Mice implanted with transmitters were recorded during 2 consecutive dark/light cycles for activity in their home cage. (A) TLR2−/− and TLR4−/− mice exhibited reduced activity levels (B) average activity levels during all 48 hours shown that TLR2−/− mice are less active than TLR4−/− or WT mice during the night cycle (C) resting baseline circulating corticosterone levels in TLR2−/−, TLR4−/− and WT mice during the night cycle. * P<0.05.
Although the body weight of both TLR2−/− and TLR4−/− mice was higher than WT mice (F2,108=43.57, P<0.0001, Figure 7A), we could not attribute this to higher muscle mass, fat mass or the ratio between the two parameters (Figure 7B–D). Interestingly, despite having a higher body weight, both TLR2−/− and TLR4−/− mice exhibited reduced food intake compared to WT mice (F2,3045=3.87, P=0.0371, Figure 7E), whereas TLR4−/− mice exhibit a trend towards lower water consumption compared with WT mice (F2,3045=3.27, P=0.0581, Figure 7F).
Figure 7. Deficiency for TLR2 and TLR4 affect food and water consumption while not affecting muscle or fat mass.

(A) TLR2−/−, TLR4−/− and WT mice were monitored for body weight during the study. Mice were subjected to NMR spectrometry analysis of body composition, and measured for muscles content, fat content and fat:muscle ratio. Both TLR2−/− mice and TLR4−/− mice show no difference in (B) muscle mass (C) fat mass or (D) muscle:fat ratio. TLR2−/−, TLR4−/− and WT mice were placed in metabolic cages and continuous monitoring of feeding behavior during 48 hours was conducted. Cumulative (E) food and (F) water intake were recorded. TLR2−/− mice and TLR4−/− mice exhibited reduced eating and drinking compared with WT mice. * P<0.05.
As both thermoregulation, activity levels and food intake were altered in TLR2−/− and TLR4−/− mice compared to WT mice, we next assessed the metabolic status of these mice using a CLAMS system, which continuously measures O2 consumption, CO2 production, heat production and total energy expenditure. The Respiratory exchange ratio (RER) was calculated, and indicates the main source of fuel being metabolized by the mice. An RER of 0.85-0.7 indicates a combination of fatty acids and carbohydrates being metabolized, as opposed to an RER value of 1 or above which indicates the use of carbohydrates, or an RER of <0.7, which indicates the use of fatty acids being used as a predominant fuel source. TLR2−/− mice exhibited a significantly lower RER than WT mice (F2,3045=3.65, P=0.043, Figure 8A), suggesting that these animals utilized mainly fatty acids instead of carbohydrates as their major energy source. Additionally, TLR2−/− mice produced significantly less CO2 than WT and TLR4−/− mice (F2,3045=12.22, P=0.0003, Figure 8B), even though they consumed similar amounts of O2 (Figure 8C). There was no notable difference in heat production among the three mouse strains (Figure 8D). The total energy expenditure was lower in TLR2−/− mice (F2,3045=10.46, P=0.0007, Figure 8E), which is potentially attributed to their decreased food intake and reduced activity levels.
Figure 8. Evidence that developmental TLR2 and TLR4 deficiency affects energy metabolism.

TLR2−/− mice, TLR4−/− mice and WT mice (n=10 per group) were individually placed in metabolic cages and analyzed during 48 hours for (A) Respiratory exchange ratio (RER) (B) Cumulative CO2 production. TLR2−/− mice exhibited reduced CO2 production compared with WT mice. (C) Cumulative O2 consumption (D) Heat production. (E) Energy expenditure. TLR2−/− mice exhibited reduced energy expenditure compared with WT control mice. * P<0.05.
Discussion
TLRs are best known for their fundamental roles in innate immunity, namely, the identifying microbes and promoting an efficient immune response towards invading pathogen (Takeda and Akira, 2004). However, recent findings suggest that mammalian TLRs also possess developmental roles, as well as physiological and metabolic roles in adults. For example, TLR5-deficient mice exhibit hyperphagia and develop hallmark features of metabolic syndrome, including hyperlipidemia, hypertension, insulin resistance and increased adiposity (Vijay-Kumar et al., 2010). Developmental deficiency or activation of TLRs 3 and 4 have been implicated in altering multiple aspects of cognitive learning and memory (Okun et al., 2012; Okun et al., 2010a; Okun et al., 2011), and recently TLR2 was implicated in regulation of metabolism (Shechter et al., 2013). We therefore hypothesized that a developmental TLR deficiency may play important roles in additional non-immune aspects of the nervous system such as the ANS. Herein, we describe novel findings in mice lacking TLR2 or TLR4 providing evidence that TLRs play important roles in the regulation of autonomic control of HR, body temperature and energy metabolism under physiological, non-pathological conditions. Despite a large body of evidence linking TLRs to cardiovascular pathologies (Hofmann et al., 2011; Vallejo, 2011), it was previously unknown whether TLR signaling regulate HR. TLR2−/− and TLR4−/− mice exhibit reduced basal HR during both the dark and light cycles, suggesting that TLR2 and TLR4 expression promotes an elevated HR in a circadian cycle-independent manner. Consistent with the latter possibility, atropine treatment resulted in a greater elevation of HR in TLR2−/− but not TLR4−/− mice, which indicates a reduced parasympathetic tone in these mice. While a stronger parasympathetic tone in TLR2−/− mice certainly contributes to the reduced HR levels in these mice, it is possible also that the reduced activity levels during night cycle contributes to the lower HR levels in TLR2−/− mice. The lower daytime HR levels, cannot be explained in a similar way, as activity levels during day time are similar between the different TLR strains and WT mice. A possible mechanism for the lower activity of TLR2−/− mice during the night cycle could be the lower levels of circulating corticosterone in these mice.
Although the HRs of TLR2−/− and TLR4−/− mice were reduced throughout the circadian cycle under non-stress home cage conditions, the maximum HRs of these TLR mutant mice attained during restraint stress were essentially identical to the maximum stress-induced HR of WT mice. The higher change in HR during stress (stress-induced HR normalized to baseline HR), suggests a greater dynamic control of HR in mice lacking TLR2 or TLR4. By suppressing parasympathetic activity, TLR2 and to a lesser extent, TLR4 signaling may therefore increase cardiovascular risk. Indeed, it was recently reported that TLR2−/− mice are less likely to suffer a fatal arrhythmia in an experimental model of myocardial infarction (Mersmann et al., 2010). Moreover, TLR4 signaling contributes to an elevated blood pressure in spontaneously hypertensive rats (Bomfim et al., 2012), which may result from TLR4-mediated reduction in parasympathetic tone. An alternative explanation, however, is that perhaps TLR signaling is adaptive in times of stress and restricts HR and body temperature elevation.
Beta-3 receptors are expressed in brown adipose tissue cells where their activation stimulates thermogenesis and lipolysis (Chartoumpekis et al., 2011). Under non-stressful conditions, there were no discernible differences in body temperature among TLR2−/− or TLR4−/− mice and WT mice during the light or dark cycles of the day. However, TLR2−/− mice exhibited greater elevations of body temperature during restraint stress compared to wild type mice. It is surprising that a lack of TLR signaling resulted in a greater increase in body temperature considering that typical TLR activation by microbial-derived ligands such as LPS in the case of TLR4 or peptidoglycan in the case of TLR2, causes a pyrogenic fever response which increases temperature. However, MAMPs-derived activation of TLR2 and TLR4 is thought to activate signaling pathways different from those that are active following DAMPs-derived TLR activation (Okun et al., 2011).
Interestingly, atropine had no significant effect on body temperature in wild type mice, but increased body temperature in TLR2−/− and TLR4−/− mice. The latter finding might be explained by increased parasympathetic activity in mice lacking TLR2 or TLR4, and is consistent with the HR data suggesting that TLR2 and TLR4 signaling reduce parasympathetic activity. The magnitude of the elevation of body temperature in response to activation of noradrenergic receptors was significantly attenuated in TLR4−/− mice, suggesting that TLR4 signaling mediates, in part, the thermogenic response to sympathetic neuron activation. Restoration of body temperature after swimming in cold was faster in TLR2−/− and TLR4−/− mice compared to WT mice, suggesting that TLR2 and TLR4 signaling can exacerbate hyperthermia under conditions where heat generation by brown fat cells is inhibited.
Compared to WT mice, TLR2−/− and TLR4−/− mice maintained higher body weights. The higher body weight in TLR2−/− and TLR4−/− mice appear at odds with their reduced food and water intake compared to WT mice, and suggest that TLR2−/− and TLR4−/− mice are more efficient at utilizing nutrients. The reason for this is unclear, but at least in TLR2−/− mice, this could be due to a better utilization of both carbohydrates and fat sources compared to WT mice, as indicated by a lower RER. While the mechanism for this is unclear, it is possible that alterations in gut microbiota in TLR2−/− and TLR4−/− mice compared with WT mice, which have been reported previously (Caricilli et al., 2011; Ubeda et al., 2012) play a role in this selectivity toward higher carbohydrate utilization.
Considering the lower daily food intake by TLR2−/− and TLR4−/− mice, it is possible that the alterations in HR in TLR2−/− and TLR4−/− mice might simply be due to the lower daily food intake of these mice, compared to WT mice. Further experiments are warranted to specifically clarify this issue. Moreover, Because TLR2−/− and TLR4−/− mice lack TLR expression throughout development, it is possible that certain aspects of the ANS are modulated by TLR2 and TLR4 during embryonic development or prior to weaning. Thus, it will be important to determine in subsequent studies, what is the relative contributions of developmental effects and altered function to ANS-related phenotypes of these mice documented in the present study. Of high relevance, a recent study (Shechter et al., 2013) assessed age-induced obesity in TLR2 deficient mice. Shechter and colleagues suggest that hypothalamic TLR2 serves as an endogenous protective mechanism designed to maintain energy homeostasis and to compensate for metabolic alterations. While our study strengthens this understanding, certain inconsistencies remain, such as the lower food consumption by TLR2−/− mice in our study compared to a higher food consumption indicated in the study by Shecther et al. The different observation reported by the different studies that show roles for TLR2 in regulation of metabolism probably stem from altered gut microbiota, itself a factor determined by diet composition, housing conditions and genetic background. Although relatively complicated and expensive to address, understanding the complex interplay between gut microbiota, TLR2 expression and the regulation of metabolism is of essence in order to understand the exact role of TLR2 in particular and TLRs in general in regulating metabolism.
To the best of our knowledge, no experimental data exists on the effects of SNPs in either TLR2 or TLR4 on the ANS in humans. Thus, the current study indicates the possibility that SNPs in TLR2 or TLR4 may affect properties of ANS regulation, such as HR or metabolism. Emerging evidence indicate that TLR signaling plays roles in both the development and adult function of the CNS. For example, TLR2 (Okun et al., 2010b) and TLR3 (Lathia et al., 2008) inhibit the proliferation of neural progenitor cells in the developing brain, TLR2 and TLR4 differentially regulate adult hippocampal neurogenesis (Rolls et al., 2007), and TLR3 signaling negatively regulates memory retention in adult mice (Okun et al., 2010a). In addition, a recent study in which WT and TLR4−/− mice were subjected to a battery of behavioral tests provided evidence that TLR4 signaling can influence brain development. In this study, TLR4 affected spatial reference memory acquisition and memory retention, and motor performance, whereas acute TLR4 signaling in the brain modifies anxiety-related behaviors (Okun et al., 2012). The signaling pathways by which TLRs affect neuronal plasticity remain to be established, but may involve regulation of the activities of transcription factors such as cyclic AMP response element-binding protein and nuclear factor kappa b (Okun et al., 2012; Okun et al., 2010a; Wu et al., 2012). Our findings suggesting novel roles for TLR2 and TLR4 in regulating the ANS, pave the way for further studies aimed at elucidating the underlying cellular and molecular mechanisms. Specifically, we hypothesize that activating/antagonizing the parasympathetic branch of the ANS in mice deficient in TLRs such as TLR2 and TLR4 may have a detrimental/beneficial effect on mice under stress. Further, understanding whether SNPs in TLR2 and TLR4 have implications on autonomic properties of the CNS may help medically assess whether individuals with such SNPs are more prone to sympathetic/parasympathetic regulation impairments, narrowing down the possible mechanism(s) responsible for the impairments in these patients.
Acknowledgments
The authors’ work was supported by the Intramural Research Program of the National Institute on Aging, as well as by Bar Ilan University, The Gonda Multidisciplinary Brain Research Center.
Footnotes
Conflict of Interest Statement:
All authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akira S. Mammalian Toll-like receptors. Current opinion in immunology. 2003;15:5–11. doi: 10.1016/s0952-7915(02)00013-4. [DOI] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Anker SD, von Haehling S. Inflammatory mediators in chronic heart failure: an overview. Heart. 2004;90:464–470. doi: 10.1136/hrt.2002.007005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartness TJ, Vaughan CH, Song CK. Sympathetic and sensory innervation of brown adipose tissue. Int J Obes (Lond) 2010;34(Suppl 1):S36–42. doi: 10.1038/ijo.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bexis S, Docherty JR. Role of alpha 1- and beta 3-adrenoceptors in the modulation by SR59230A of the effects of MDMA on body temperature in the mouse. British journal of pharmacology. 2009;158:259–266. doi: 10.1111/j.1476-5381.2009.00186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigger JT, Jr, Fleiss JL, Steinman RC, Rolnitzky LM, Kleiger RE, Rottman JN. Frequency domain measures of heart period variability and mortality after myocardial infarction. Circulation. 1992;85:164–171. doi: 10.1161/01.cir.85.1.164. [DOI] [PubMed] [Google Scholar]
- Birks EJ, Felkin LE, Banner NR, Khaghani A, Barton PJ, Yacoub MH. Increased toll-like receptor 4 in the myocardium of patients requiring left ventricular assist devices. The Journal of heart and lung transplantation: the official publication of the International Society for Heart Transplantation. 2004;23:228–235. doi: 10.1016/S1053-2498(03)00106-2. [DOI] [PubMed] [Google Scholar]
- Bomfim GF, Dos Santos RA, Oliveira MA, Giachini FR, Akamine EH, Tostes RC, Fortes ZB, Webb RC, Carvalho MH. Toll-like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin Sci (Lond) 2012;122:535–543. doi: 10.1042/CS20110523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–687. doi: 10.1146/annurev.pharmtox.45.120403.095802. [DOI] [PubMed] [Google Scholar]
- Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. Journal of neuropathology and experimental neurology. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- Cameron JS, Alexopoulou L, Sloane JA, DiBernardo AB, Ma Y, Kosaras B, Flavell R, Strittmatter SM, Volpe J, Sidman R, Vartanian T. Toll-like receptor 3 is a potent negative regulator of axonal growth in mammals. J Neurosci. 2007;27:13033–13041. doi: 10.1523/JNEUROSCI.4290-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiological reviews. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- Caricilli AM, Picardi PK, de Abreu LL, Ueno M, Prada PO, Ropelle ER, Hirabara SM, Castoldi A, Vieira P, Camara NO, Curi R, Carvalheira JB, Saad MJ. Gut microbiota is a key modulator of insulin resistance in TLR 2 knockout mice. PLoS Biol. 2011;9:e1001212. doi: 10.1371/journal.pbio.1001212. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Chartoumpekis DV, Habeos IG, Ziros PG, Psyrogiannis AI, Kyriazopoulou VE, Papavassiliou AG. Brown adipose tissue responds to cold and adrenergic stimulation by induction of FGF21. Mol Med. 2011;17:736–740. doi: 10.2119/molmed.2011.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrentraut H, Weber C, Ehrentraut S, Schwederski M, Boehm O, Knuefermann P, Meyer R, Baumgarten G. The toll-like receptor 4-antagonist eritoran reduces murine cardiac hypertrophy. European journal of heart failure. 2011a;13:602–610. doi: 10.1093/eurjhf/hfr035. [DOI] [PubMed] [Google Scholar]
- Ehrentraut S, Lohner R, Schwederski M, Ehrentraut H, Boehm O, Noga S, Langhoff P, Baumgarten G, Meyer R, Knuefermann P. In vivo Toll-like receptor 4 antagonism restores cardiac function during endotoxemia. Shock. 2011b;36:613–620. doi: 10.1097/SHK.0b013e318235805f. [DOI] [PubMed] [Google Scholar]
- Eichhorn EJ, Bristow MR. Medical therapy can improve the biological properties of the chronically failing heart. A new era in the treatment of heart failure. Circulation. 1996;94:2285–2296. doi: 10.1161/01.cir.94.9.2285. [DOI] [PubMed] [Google Scholar]
- Favre J, Musette P, Douin-Echinard V, Laude K, Henry JP, Arnal JF, Thuillez C, Richard V. Toll-like receptors 2-deficient mice are protected against postischemic coronary endothelial dysfunction. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:1064–1071. doi: 10.1161/ATVBAHA.107.140723. [DOI] [PubMed] [Google Scholar]
- Foldes G, von Haehling S, Okonko DO, Jankowska EA, Poole-Wilson PA, Anker SD. Fluvastatin reduces increased blood monocyte Toll-like receptor 4 expression in whole blood from patients with chronic heart failure. International journal of cardiology. 2008;124:80–85. doi: 10.1016/j.ijcard.2006.12.024. [DOI] [PubMed] [Google Scholar]
- Francis GS. The relationship of the sympathetic nervous system and the renin-angiotensin system in congestive heart failure. American heart journal. 1989;118:642–648. doi: 10.1016/0002-8703(89)90291-3. [DOI] [PubMed] [Google Scholar]
- Frantz S, Kobzik L, Kim YD, Fukazawa R, Medzhitov R, Lee RT, Kelly RA. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J Clin Invest. 1999;104:271–280. doi: 10.1172/JCI6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Isobe K, Zeng Q, Suzukawa K, Takekoshi K, Kawakami Y. The effects of beta(3)-adrenoceptor agonist CL-316,243 on adiponectin, adiponectin receptors and tumor necrosis factor-alpha expressions in adipose tissues of obese diabetic KKAy mice. European journal of pharmacology. 2008;584:202–206. doi: 10.1016/j.ejphar.2008.01.028. [DOI] [PubMed] [Google Scholar]
- Garrafa E, Imberti L, Tiberio G, Prandini A, Giulini SM, Caimi L. Heterogeneous expression of toll-like receptors in lymphatic endothelial cells derived from different tissues. Immunology and cell biology. 2011;89:475–481. doi: 10.1038/icb.2010.111. [DOI] [PubMed] [Google Scholar]
- Goldsmith SR. Angiotensin II and sympathoactivation in heart failure. J Card Fail. 1999;5:139–145. doi: 10.1016/s1071-9164(99)90036-2. [DOI] [PubMed] [Google Scholar]
- Gradman AH, Alfayoumi F. From left ventricular hypertrophy to congestive heart failure: management of hypertensive heart disease. Prog Cardiovasc Dis. 2006;48:326–341. doi: 10.1016/j.pcad.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Griffioen KJ, Rothman SM, Ladenheim B, Wan R, Vranis N, Hutchison E, Okun E, Cadet JL, Mattson MP. Dietary energy intake modifies brainstem autonomic dysfunction caused by mutant alpha-synuclein. Neurobiology of aging. 2013;34:928–935. doi: 10.1016/j.neurobiolaging.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffioen KJ, Wan R, Okun E, Wang X, Lovett-Barr MR, Li Y, Mughal MR, Mendelowitz D, Mattson MP. GLP-1 receptor stimulation depresses heart rate variability and inhibits neurotransmission to cardiac vagal neurons. Cardiovascular research. 2011;89:72–78. doi: 10.1093/cvr/cvq271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha T, Li Y, Hua F, Ma J, Gao X, Kelley J, Zhao A, Haddad GE, Williams DL, William Browder I, Kao RL, Li C. Reduced cardiac hypertrophy in toll-like receptor 4-deficient mice following pressure overload. Cardiovascular research. 2005;68:224–234. doi: 10.1016/j.cardiores.2005.05.025. [DOI] [PubMed] [Google Scholar]
- Hofmann U, Ertl G, Frantz S. Toll-like receptors as potential therapeutic targets in cardiac dysfunction. Expert opinion on therapeutic targets. 2011;15:753–765. doi: 10.1517/14728222.2011.566560. [DOI] [PubMed] [Google Scholar]
- Hubschle T, Mutze J, Muhlradt PF, Korte S, Gerstberger R, Roth J. Pyrexia, anorexia, adipsia, and depressed motor activity in rats during systemic inflammation induced by the Toll-like receptors-2 and -6 agonists MALP-2 and FSL-1. American journal of physiology Regulatory, integrative and comparative physiology. 2006;290:R180–187. doi: 10.1152/ajpregu.00579.2005. [DOI] [PubMed] [Google Scholar]
- Lathia JD, Okun E, Tang SC, Griffioen K, Cheng A, Mughal MR, Laryea G, Selvaraj PK, ffrench-Constant C, Magnus T, Arumugam TV, Mattson MP. Toll-like receptor 3 is a negative regulator of embryonic neural progenitor cell proliferation. J Neurosci. 2008;28:13978–13984. doi: 10.1523/JNEUROSCI.2140-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Haynes RL, Sidman RL, Vartanian T. TLR8: an innate immune receptor in brain, neurons and axons. Cell Cycle. 2007;6:2859–2868. doi: 10.4161/cc.6.23.5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Li J, Chiu I, Wang Y, Sloane JA, Lu J, Kosaras B, Sidman RL, Volpe JJ, Vartanian T. Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. J Cell Biol. 2006;175:209–215. doi: 10.1083/jcb.200606016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques R, Boneca IG. Expression and functional importance of innate immune receptors by intestinal epithelial cells. Cellular and molecular life sciences: CMLS. 2011;68:3661–3673. doi: 10.1007/s00018-011-0829-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith IT, Eisenhofer G, Lambert GW, Dewar EM, Jennings GL, Esler MD. Cardiac sympathetic nervous activity in congestive heart failure. Evidence for increased neuronal norepinephrine release and preserved neuronal uptake. Circulation. 1993;88:136–145. doi: 10.1161/01.cir.88.1.136. [DOI] [PubMed] [Google Scholar]
- Mersmann J, Berkels R, Zacharowski P, Tran N, Koch A, Iekushi K, Dimmeler S, Granja TF, Boehm O, Claycomb WC, Zacharowski K. Preconditioning by toll-like receptor 2 agonist Pam3CSK4 reduces CXCL1-dependent leukocyte recruitment in murine myocardial ischemia/reperfusion injury. Crit Care Med. 2010;38:903–909. doi: 10.1097/CCM.0b013e3181ce50e6. [DOI] [PubMed] [Google Scholar]
- Nakagawa Y, Maeda H, Murai T. Evaluation of the in vitro pyrogen test system based on proinflammatory cytokine release from human monocytes: comparison with a human whole blood culture test system and with the rabbit pyrogen test. Clinical and diagnostic laboratory immunology. 2002;9:588–597. doi: 10.1128/CDLI.9.3.588-597.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navi A, Patel H, Shaw S, Baker D, Tsui J. Therapeutic role of toll-like receptor modification in cardiovascular dysfunction. Vascular pharmacology. 2013;58:231–239. doi: 10.1016/j.vph.2012.10.001. [DOI] [PubMed] [Google Scholar]
- Ogawa K, Hirooka Y, Kishi T, Sunagawa K. Brain AT1 receptor activates the sympathetic nervous system through toll-like receptor 4 in mice with heart failure. Journal of cardiovascular pharmacology. 2011;58:543–549. doi: 10.1097/FJC.0b013e31822e6b40. [DOI] [PubMed] [Google Scholar]
- Okun E, Barak B, Saada-Madar R, Rothman SM, Griffioen KJ, Roberts N, Castro K, Mughal MR, Pita MA, Stranahan AM, Arumugam TV, Mattson MP. Evidence for a developmental role for TLR4 in learning and memory. PLoS One. 2012;7:e47522. doi: 10.1371/journal.pone.0047522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun E, Griffioen K, Barak B, Roberts NJ, Castro K, Pita MA, Cheng A, Mughal MR, Wan R, Ashery U, Mattson MP. Toll-like receptor 3 inhibits memory retention and constrains adult hippocampal neurogenesis. Proc Natl Acad Sci U S A. 2010a;107:15625–15630. doi: 10.1073/pnas.1005807107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun E, Griffioen KJ, Mattson MP. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 2011;34:269–281. doi: 10.1016/j.tins.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun E, Griffioen KJ, Son TG, Lee JH, Roberts NJ, Mughal MR, Hutchison E, Cheng A, Arumugam TV, Lathia JD, van Praag H, Mattson MP. TLR2 activation inhibits embryonic neural progenitor cell proliferation. J Neurochem. 2010b;114:462–474. doi: 10.1111/j.1471-4159.2010.06778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ootsuka Y, Kulasekara K, de Menezes RC, Blessing WW. SR59230A, a beta-3 adrenoceptor antagonist, inhibits ultradian brown adipose tissue thermogenesis and interrupts associated episodic brain and body heating. American journal of physiology Regulatory, integrative and comparative physiology. 2011;301:R987–994. doi: 10.1152/ajpregu.00085.2011. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, Schwartz M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol. 2007;9:1081–1088. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- Romanovsky AA, Steiner AA, Matsumura K. Cells that trigger fever. Cell Cycle. 2006;5:2195–2197. doi: 10.4161/cc.5.19.3321. [DOI] [PubMed] [Google Scholar]
- Shechter R, London A, Kuperman Y, Ronen A, Rolls A, Chen A, Schwartz M. Hypothalamic neuronal toll-like receptor 2 protects against age-induced obesity. Sci Rep. 2013;3:1254. doi: 10.1038/srep01254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, Ronen A, Rolls A, London A, Bakalash S, Young MJ, Schwartz M. Toll-like receptor 4 restricts retinal progenitor cell proliferation. J Cell Biol. 2008;183:393–400. doi: 10.1083/jcb.200804010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Tang SC, Lathia JD, Selvaraj PK, Jo DG, Mughal MR, Cheng A, Siler DA, Markesbery WR, Arumugam TV, Mattson MP. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid beta-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Experimental neurology. 2008;213:114–121. doi: 10.1016/j.expneurol.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topkara VK, Evans S, Zhang W, Epelman S, Staloch L, Barger PM, Mann DL. Therapeutic targeting of innate immunity in the failing heart. J Mol Cell Cardiol. 2011;51:594–599. doi: 10.1016/j.yjmcc.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubeda C, Lipuma L, Gobourne A, Viale A, Leiner I, Equinda M, Khanin R, Pamer EG. Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J Exp Med. 2012;209:1445–1456. doi: 10.1084/jem.20120504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo JG. Role of toll-like receptors in cardiovascular diseases. Clin Sci (Lond) 2011;121:1–10. doi: 10.1042/CS20100539. [DOI] [PubMed] [Google Scholar]
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Li W, Zhou C, Lu F, Gao T, Liu Y, Cao J, Zhang Y. Ketamine inhibits lipopolysaccharide-induced astrocytes activation by suppressing TLR4/NF-kB pathway. Cell Physiol Biochem. 2012;30:609–617. doi: 10.1159/000341442. [DOI] [PubMed] [Google Scholar]
- Yu L, Wang L, Chen S. Endogenous toll-like receptor ligands and their biological significance. Journal of cellular and molecular medicine. 2010;14:2592–2603. doi: 10.1111/j.1582-4934.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Yuan X, Jin PF, Hou JF, Wang W, Wei YJ, Hu S. Alteration of parasympathetic/sympathetic ratio in the infarcted myocardium after Schwann cell transplantation modified electrophysiological function of heart: a novel antiarrhythmic therapy. Circulation. 2010;122:S193–200. doi: 10.1161/CIRCULATIONAHA.109.922740. [DOI] [PubMed] [Google Scholar]
- Zhao J, Cannon B, Nedergaard J. Thermogenesis is beta3- but not beta1-adrenergically mediated in rat brown fat cells, even after cold acclimation. The American journal of physiology. 1998;275:R2002–2011. doi: 10.1152/ajpregu.1998.275.6.R2002. [DOI] [PubMed] [Google Scholar]