

Abstract
The discovery that survivin, a small anti-apoptotic protein, is involved in chemoresistance, opens a new scenario to overcome the drug resistance in cancer. It was shown that siRNA can efficiently inhibit the expression of survivin in cancer cells. However, the clinical use of siRNA is still hampered by an unfavorable pharmacokinetic profile. To address this problem, earlier we developed a novel system to deliver siRNA into cancer cells. Namely, we reversibly modified the survivin siRNA with a phosphothioethanol (PE) portion via a reducible disulfide bond and incorporated the resulting siRNA-S-S-PE conjugate into nanosized polyethyelene glycol2000-phosphatidyl ethanolamine (PEG2000-PE)-based polymeric micelles (PM), obtaining survivin siRNA PM. The activity of these nanopreparations was evaluated by survivin protein down-regulation, tumor cell growth inhibition, and chemosensitization of the treated tumor cells to paclitaxel (PXL). We found a significant decrease of cell viability and down-regulation of survivin protein levels after treatment with survivin siRNA PM in several cancer cell lines. In addition, the down-regulation of survivin by treating cells with survivin siRNA PM, elicited a significant sensitization of the cells to PXL, in both sensitive and resistant cancer cell lines. Finally, we demonstrate successful co-delivery of PXL and survivin siRNA in the same PM leading to superior therapeutic activity compared to their sequential administration. Our results support the use of this new platform for the treatment of the most aggressive tumors.
1. Introduction
Survivin, the smallest member of the inhibitors of apoptosis (IAP) family, has gained much attention in recent years as a promising new target in cancer therapy due to its differential expression in tumours compared to normal tissues [1]. Survivin plays an important role in the negative regulation of apoptosis as well as in cell division [2,3]. Moreover, survivin expression in malignant tissues has been correlated with drug resistance [4]. Accordingly, inhibition of survivin has been of clear interest for cancer therapy. In the last years, many researchers have proposed various ways to counteract survivin activity in cancer cells with the aim to inhibit the tumor growth potential and to sensitize the tumor cells to chemotherapeutic agents. RNA interference (RNAi) offers an attractive and powerful approach to efficiently inhibit survivin expression in cancer cells [5]. A. Carvalho et al. [6] were the first to use siRNA to suppress survivin levels in HeLa cells, showing a specific depletion of survivin for at least 60 h after the transfection with a specific siRNA. Seth et al. have demonstrated the in vivo silencing of survivin and a significant dose-dependent decrease of tumor volumes after intravesical instillation of liposomes containing survivin siRNA in an animal model of bladder cancer [7]. Despite all the potential of siRNA in cancer treatment, selective inhibition of an over-expressed gene via RNAi requires an effective delivery strategy that ameliorates the significant issues associated with its pharmacokinetic profile. In particular, the poor stability in biological fluids and the low cellular uptake impaired siRNA direct use in clinical trials. In the literature, several approaches for siRNA delivery in vitro and in vivo, such as viral vectors [8,9], hydrodynamic injection [10], cationic polymer and lipids [11,12] are reported. However, only few have demonstrated clinical applicability due to toxicity and poor stability in biological fluids. Therefore, the transition of siRNA-based approach to the clinical setting requires the development of a suitable delivery system.
We previously reported bio-reductive PM for siRNA delivery based on siRNA conjugated to PE via a disulfide linkage [13]. This strategy is based on the dramatically higher concentration of reductases in the tumor microenvironment over normal tissues [14] as well as glutathione inside cancer cells [15]. Chemical conjugation of siRNA and incorporation of siRNA conjugate into PM offers the advantage of protecting the siRNA from degradation in vitro by a facile reaction and at the same time, the cleavable disulfide bonds linked to the siRNA, allow to liberate it free when inside the cell for target-specific gene silencing. Thus, the conjugated siRNA can be incorporated via the PE moiety into a non toxic delivery system, such as PEG2000-PE-based PM [16], to become stable in physiological conditions and able accumulate in the areas with an abnormal vascularization, i.e. tumors, via the enhanced permeability and retention (EPR) effect.
Here, we formulated nanosized PEG2000-PE PM for anti-survivin siRNA delivery. In vitro cytotoxicity and survivin protein levels studies revealed the ability of survivin siRNA PM to inhibit efficiently the cellular growth and to down-regulate the survivin in different cancer cell lines. In a second phase, we investigated the potential of combination therapy with survivin siRNA and a chemotherapeutic agent, PXL. PXL exhibits its anticancer activity by promoting tubulin polymerization and stabilizing microtubules, which results in mitotic G2/M arrest and apoptosis [17]. The clinical effectiveness of PXL, an agent widely used in clinic for the treatment of several tumors, is often hampered by acquired drug resistance [18]. Since sensitization to PXL by survivin down-regulation has been reported [19–21] we evaluated co-treatments with PXL and anti survivin siRNA. Cells were either subjected to survivin siRNA before PXL treatment or treated with PM simultaneously encapsulating PXL and survivin siRNA. Our results suggest that the developed survivin siRNA PM greatly sensitize the cells to PXL treatment and the simultaneous delivery of survivin siRNA and PXL, by using PM, significantly enhances the tumor response to PXL in resistant cancer cell lines.
2. Materials and Methods
Survivin siRNA with the following sense sequence 5’-GCAUUCGUCCGGUUGCGCUdTdT-3’ and a scrambled siRNA with the following sense sequence 5’-AUGAACUUCAGGGUCAGCUdTdT-3’ have been used. Both siRNAs modified at the 3′-end of the sense strand with N-succinimidyl 3-(2-pyridyldithio)propionate (SPDP) group were purchased from Thermo Scientific Dharmacon (Pittsburgh PA, USA). The paclitaxel (PXL) was purchased from LC Laboratories (Woburn MA, United States). The 1,2-dipalmitoyl-sn-glycero-3-phosphothioethanol (PE-SH, MW 731) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(poly(ethylene glycol))-2000] (PEG2000-PE) were from Avanti Polar Lipids (Alabaster, AL). The d-Salt dextran desalting column was from Pierce (Rockford, IL, USA). Acetonitrile (HPLC grade), analytical grade chloroform (CH3Cl), DMSO, methanol (MeOH) and Triton X-100 were supplied by Sigma Aldrich (Saint Louis, MO). The human total survivin immunoassay, Surveyor IC, was purchased from R&D System (Minneapolis, MN). RNase/DNase-free water was obtained from MP Biomedicals (Solon, OH), the phosphate saline buffer (PBS) 10× solution and bovine serum albumin (Fraction V) were from Fisher Scientific (Fair Lawn, NJ). β-tubulin antibody (G-8) was from Santa Cruz Biotechnology (Dallas, Texas, USA). Fluorescein (FITC)-conjugated AffiniPure Donkey Anti-Mouse IgG (H+L) was provided by Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). Hoechst 33342 trihydrochloride, trihydrate, was purchased from Molecular Probes (Eugene, Oregon, USA). Vecta Shield mounting medium for fluorescence, H-1000, was from Vector Laboratories, Inc. (Burlingame, CA).
2.1 Cell Culture
Human ovarian cancer cell line (A2780) and human breast cancer cell line (MDA-MB231) were cultured in DMEM medium, containing 10% fetal bovine serum (FBS), 100 U/mL penicillin G sodium and 100 mg/mL streptomycin sulfate (complete medium), in a humidified atmosphere of 95% air 5% CO2 at 37 °C. Human ovarian cancer cell line sensitive (SKOV3) and multi drug resistant (MDR) (SKOV3-tr) were grown in the complete RPMI1640 medium. The SKOV3-tr cells have been widely characterized and are known to overexpress the MDR-1 gene (Duan Z et al 1999). All the cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Trypan Blue solution and trypsin were from CellGro (Kansas City, MO).
2.2 Synthesis of survivin siRNA-S–S-PE Conjugate
The survivin siRNA-S–S-PE conjugate was synthesized as previously described by T. Musacchio et al. [13]. Briefly, an aqueous solution of the SPDP-activated siRNA (20 nmol in 120 µL of RNase/DNase-free water), was added dropwise to a solution of PE-SH (2 µmol) in DMSO and CHCl3 (total volume of organic solvents 350 µL). The reaction was carried out for 48 h at room temperature with continuous stirring. The un-reacted reagents were removed by desalting column. The collected samples containing the survivin siRNA-S-S-PE conjugate were freeze-dried for overnight. After freeze-drying, the survivin siRNA-S-S-PE conjugate was hydrated with PBS pH 7.4 at a final siRNA concentration of 20 nmol/ml and ultracentrifuged for 1 min at 14.5 × 1000 rpm to further remove mixed solvents and/or PE-SH. The survivin siRNA-S–S-PE conjugate was stored at −20 °C. The conjugation efficiency and the amount of survivin siRNA-S–S-PE conjugate was determined, after purification, by absorbance at 260 nm using a Nanodrop (2000c Spectrophotometers, Thermo Scientific). Scrambled siRNA was modified following the same protocol.
2.3 Incorporation of siRNA-S–S-PE in PE-PEG2000 Micelles
The PEG2000-PE micelles containing siRNA-S-S-PE were prepared by hydration of a thin polymeric film [16]. In particular, PEG2000-PE was dissolved in chloroform (20 mg/ml) and the resulting solution was added to a 50 ml round-bottom flask. The organic solvent was removed under reduced pressure by a rotary evaporator under nitrogen atmosphere, followed by freeze-drying. Then, the polymeric film was hydrated with 1 ml of survivin siRNA-S-S-PE in phosphate buffer at pH 7.4 at different PEG2000-PE/siRNA-S-S-PE weight ratio (1:200, 1:500, 1:750). The resulting dispersion was gently vortexed to form mixed micelles, so-called survivin siRNA PM. PEG2000-PE-based PM containing scrambled siRNA-S-S-PE and plain PM were prepared similarly. Each formulation was prepared in triplicate.
2.4 Co-encapsulation of PXL and survivin siRNA-S–S-PE in PEG2000-PE PM
PXL was incorporated in survivin siRNA PM as follow. Briefly, an organic solution of PXL in methanol (1 mg/mL) was added to the PEG2000-PE mixture in chloroform. The initial loading of PXL into micelles was 1% w/w. The incorporation of survivin siRNA-S–S-PE into these micelles was determined as reported above. The encapsulation efficiency of PXL in PM was determined as reported by T. Musacchio et al. [22].
2.5 Characterization of PM
The mean diameter of PM containing survivin siRNA-S-S-PE alone or in combination with PXL, was determined at 20°C by the dynamic light scattering (DLS) using a Zeta Plus Instrument (Brookhaven Instrument Co., Holtsville, NY). Briefly, each sample was diluted in deionizer/filtered water and analyzed with detector at 90° angle. As a measure of the particle size distribution, polydispersity index (P.I) was used. For each batch, mean diameter and size distribution were the mean of three measures. For each formulation, the mean diameter and P.I. were calculated as the mean of three different batches.
2.6 siRNA-S–S-PE Conjugate Incorporation in PEG2000-PE PM
The quantitative analysis of siRNA-S-S-PE in PM was performed by the size-exclusion high-performance liquid chromatography (SEC-HPLC). For the analysis, the HPLC system (D-7000 HPLC, Hitachi, Japan) equipped with a Shodex protein KW-804 column (Showa Denko, Japan) and an UV detection at 280 nm, was used. The mobile phase was composed of 50 mM NaCl and 50 mM Tris-HCl (pH 8.0) and the flow rate at 1.0 mL/min. The siRNA-S-S-PE loading efficiency into PM was evaluated by ratio of the area under the peaks at the same retention time (tr ca. 10 min) of survivin siRNA-S-S-PE not encapsulated in PM and the survivin siRNA-S-S-PE initially added to the PM. As a control, plain PM were also analyzed. To confirm the data collected by SEC-HPLC, we analyzed the same samples also by the reverse phase HPLC (RP-HPLC). The RP-HPLC analysis was carried out as reported by T. Musacchio, et al. [13]. Then, we evaluated the loading efficiency of PXL in survivin siRNA PM. The quantitative analysis of PXL was determined by RP-HPLC as reported by T. Musacchio et al. [22], using a XBridge column (4.6mm×250mm, Waters, Milliford, USA). The mobile phase consisted of water and acetonitrile with volume ratio 60:40, the elution was performed at a rate of 1.0 ml/min, and PXL was detected from injected sample (50µL) at 227nm.
2.7 Cell viability assay
A2780, MDA-MB 231, SKOV3 and SKOV3-tr cells were seeded at a density of 3 × 103 cells/well in 96-well culture plates for 24 h. After 24 h, the cells were treated with various concentrations of survivin siRNA-S–S-PE free or in PM, scrambled siRNA-S-S-PE in PM and plain PM, in serum-contained media. The final concentration of siRNA-S-S-PE was in the range of 200 to 17.6 nM. After 6 h, the medium was replaced with fresh medium, and the cells were incubated until the 48-hour-time point was reached. Cells without treatment were used as control. The cell viability was determined by Cell Titer Blue assay following manufacturer’s protocol. The experiments were done in triplicate on three different sample preparations.
2.8 Survivin Protein Assay
A2780, MDA-MB231, SKOV3 and SKOV3-tr cells (3×104 cells per well) were seeded into 48-well plates. Cells were treated with survivin siRNA PM at a final concentration of 200 nM in serum-containing medium for 6 h. Cells were washed once with fresh medium and maintained in fresh medium until 48 h. Cells were rinsed three times with PBS and treated with 200 µl of cold lysis buffer for 30 minutes on ice (R&D system). Cell lysates were collected, vortexed, and incubated on ice for other 15 minutes twice. Cell debris was removed by centrifugation at 2000g for 5 min, and protein concentrations were determined by BCA assay after 6-fold dilution in PBS. Samples were added into captured antibody pre-coated 96-well plates, and human survivin was assayed by ELISA after 6-fold dilution in assay buffer (R&D system).
2.9 Chemosensitization Study
MDA-MB231, SKOV3 and SKOV3-tr cells (3×103 per well) were seeded into 96-well plates. Cells were treated with survivin siRNA PM at the concentration of 200 nM for 6 h. After 6 h, the medium was replaced with fresh medium, and the cells were incubated for another 48 h. Cells were then exposed to different concentrations of PXL for 24 h. Cell viability was detected by the Cell Titer Blue assay.
2.10 Immunohistochemical staining
SKOV3-tr cells (2×104 per coverslip) were seeded and grown on glass coverslips coated with 1% gelatin cross-linked with 0.5% glutaraldehyde in a 12-well plate. After 24 h, cells were treated with 200 nM of survivin siRNA PM for 6 h. After 6 h, the medium was replaced with fresh medium, and the cells were incubated for another 48 h. Then, cells were exposed to 40 nM of free PXL for 24 h. As controls, free PXL (40 nM), survivin siRNA PM, and untreated cells were used. Cells were washed three times with PBS and then were fixed with 4% paraformaldehyde. Fixed cells were made permeable with 0.2% Triton X-100 in PBS and incubated with a monoclonal antibody against β tubulin (1:10) for 1 h in 1% bovine serum albumin/PBS. After washing, cells were incubated with a secondary FITC-labeled mouse-immunoglobulin G targeting antibody (1:100) for 1 h in 1% bovine serum albumin/PBS. After washing, cells were incubated with Hoechst 33342 (5 µM) for nuclear staining. The coverslips were mounted on glass slides with Fluoromount-G (Fisher Scientific, Waltham, MA) medium and sealed using a nail lacquer. The slides were observed with a Zeiss LSM 700 inverted confocal microscope (Carl Zeiss Co. Ltd., Jena, Germany) equipped with a 63×, 1.4-numerical aperture plan-apochromat oil-immersion objective. The images were analyzed using the ImageJ version 1.42 software (NIH, Bethesda, MD).
2.11 In vitro cytotoxicity of PM co-loaded with the combination of survivin siRNA-S-S-PE and PXL
SKOV3-tr cells (3×103 cells/well) were seeded in 96-well plates. After 24 h the cells were treated with different concentrations of survivin siRNA /PXL PM for 6 h in serum contained media. After 6 h, the medium was replaced with fresh medium, and the cells were incubated for another 72 h. Same concentrations of free PXL, survivin siRNA PM, and scrambled siRNA PM were used as controls. The cell viability was assessed by the Cell Titer Blue assay.
2.12 Statistical analysis
For comparison of several groups, one-way ANOVA with Bonferroni correction was performed using GraphPad Prism version 5.0 software (GraphPad Software, Inc, San Diego, CA). All numerical data are expressed as mean ± SD, n = 3 or 4, from 3 different experiments. Any p values less than 0.05 was considered statistically significant.
3. Results
3.1 Synthesis and characterization of survivin siRNA PM
In this study, we proposed a new strategy to stabilize and deliver siRNA against survivin into cancer cells. As the first step, we synthesized a survivin siRNA conjugated with a phospholipid (PE-SH) with the disulfide linkage at the 3′-end of the modified siRNA SPDP sense strand. The yield of the conjugation reaction between siRNA and PE was ~90%. Survivin siRNA-S-S-PE conjugate was then incorporated in PM. We have optimized the weight ratio (siRNA conjugate to PEG2000-PE to the micelle-forming component) required to obtain PM with narrow size distribution and high siRNA-S-S-PE incorporation efficiency. More specifically, we prepared PM at a survivin siRNA-S-S-PE/PEG2000-PE weight ratio of 1:200, 1:500 and 1:750, respectively. As reported in Table I, at the ratios tested, the PM were characterized with mean diameter of ~20 nm and a narrow size distribution, with a P.I. ≤ 0.2. The incorporation efficiency of survivin siRNA-S-S-PE in PM was determined by SEC-HPLC. After injection of survivin siRNA PM, SEC-HPLC analysis demonstrated that the peak at ca. 8 min corresponded to PM containing survivin siRNA, while the peak at ca. 10 min corresponded to free siRNA-S-S-PE (Figure 1). Plain PM analyzed at the same concentration as survivin siRNA PM showed the same peak as PM containing survivin siRNA at ca. 8 min. Therefore, we calculated the survivin siRNA incorporation efficiency by the ratio of free survivin siRNA-S-S-PE, not incorporated in PM, and survivin siRNA-S-S-PE initially added to micelle-forming components.
Table I.
Physical characteristics of survivin siRNA PM.
| Formulations (weight ratio) |
Mean Diameter (nm ± SD) |
P.I. ± SD | Survivin siRNA incorporation efficiency (% ± SD) |
|---|---|---|---|
| Survivin siRNA PM (1:200) | 18.7 ± 3.5 | 0.191 ± 0.08 | 26.0 ± 5.0 |
| Survivin siRNA PM (1:500) | 18.3 ± 2.0 | 0.189 ± 0.06 | 31.0 ± 3.8 |
| Survivin siRNA PM (1:750) | 21.5 ± 3.3 | 0.160 ± 0.05 | 52.0 ± 1.6 |
Figure 1.
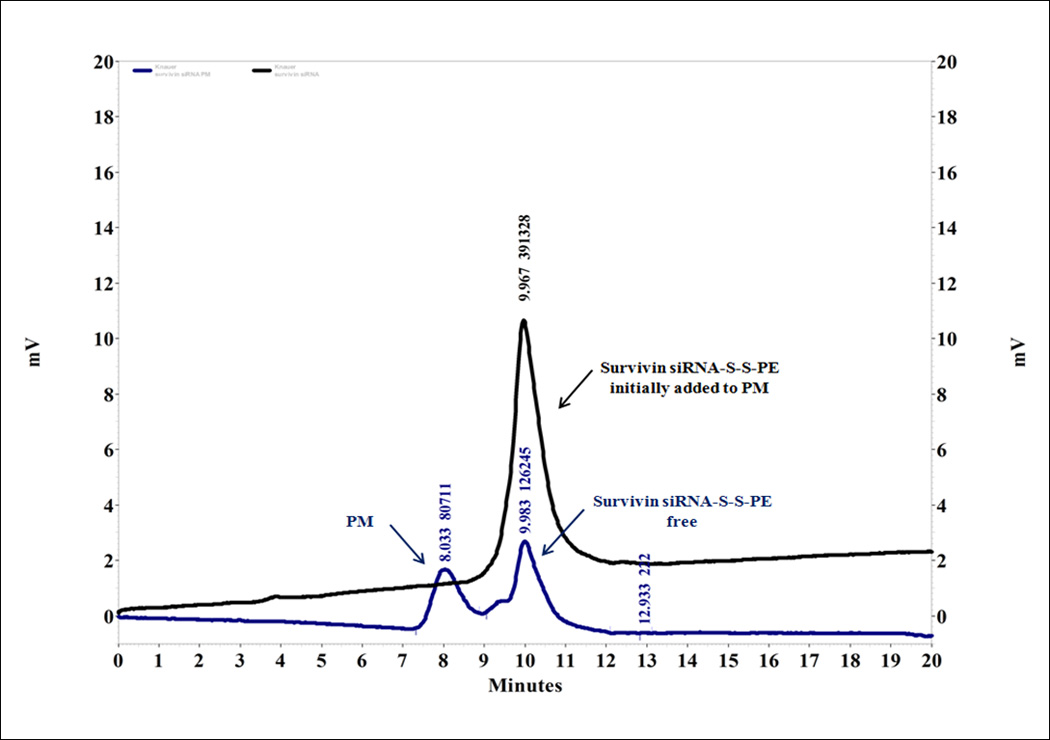
Incorporation efficiency of the modified siRNA into PM determined by SEC. The degree of the incorporation efficiency was measured by ratio of the area under the peak of free survivin siRNA-S-S-PE, not incorporated in PM, and the siRNA-S-S-PE initially added to PM at the same retention time (tr ca. 10 min). As a reference, plain PM were used.
As shown in the Table I, the highest loading efficiency, about 50%, was reached only with PM prepared with the highest amount of polymer, namely with a weight ratio of 1:750. These data were confirmed by RP-HPLC using previously described methods [13] (data not shown). This formulation was selected for the subsequent studies.
In the following step, the co-encapsulation of survivin siRNA-S-S-PE and PXL in PM was investigated. In particular, the attention was focused on the effect of PXL on the physical characteristics of PM, i.e. size and siRNA-S-S-PE loading efficiency. As reported in the Table II, when PXL was added to survivin siRNA PM, no change in the size and in the loading efficiency was observed. In particular, survivin siRNA/PXL PM had a mean diameter of about 22 nm with narrow size distribution (PI< 0.2). Chromatographic analysis of non-encapsulated survivin siRNA-S-S-PE and PXL showed an encapsulation efficiency of about 50% and 70%, respectively.
Table II.
Physical characteristics of PM co-loaded with survivin siRNA-S-S-PE and PXL.
| Formulations | Mean Diameter (nm ± SD) |
P.I. ± SD | Survivin siRNA incorporation efficiency (% ± SD) |
PXL incorporation efficiency (% ± SD) |
|---|---|---|---|---|
| Survivin siRNA PM | 21.5 ± 3.3 | 0.160 ± 0.05 | 50.0 ± 1.0 | - |
| Survivin siRNA/PXL PM | 25.0 ± 3.6 | 0.190 ± 0.07 | 51.0 ± 1.5 | 69.9 ± 2.5 |
3.2 Effect of survivin siRNA PM on the viability of cancer cell lines
The effect of survivin siRNA-S-S-PE incorporated in PM on the growth of different human cancer cell lines, namely breast (MDA-MB231), ovarian (A2780, SKOV3), and paclitaxel-resistant ovarian cell (SKOV3-tr), was investigated by the Cell Titer Blue assay. In the Figure 2, the cell viability (%) after treatment with 200 nM of survivin siRNA-S-S-PE in PM in the different cell lines analyzed after 48 h, is reported. In all sensitive cell lines, when considering survivin siRNA-S-S-PE incorporated in PM, we found a significant (p<0.001 survivin siRNA PM versus the other treatments) antiproliferative effect. The highest cell growth inhibition was observed in ovarian cancer cells, especially in A2780, reaching a reduction of cell viability of about 70%. No toxicity of PM or micelles prepared with irrelevant siRNA was observed indicating the effect was mediated by RNAi toxicity, which corresponds to earlier mentioned data [16]. In PXL-resistant SKOV3 tr cells however, no significant reduction of the cell viability was found under the action of siRNA-containing micelles.
Figure 2.
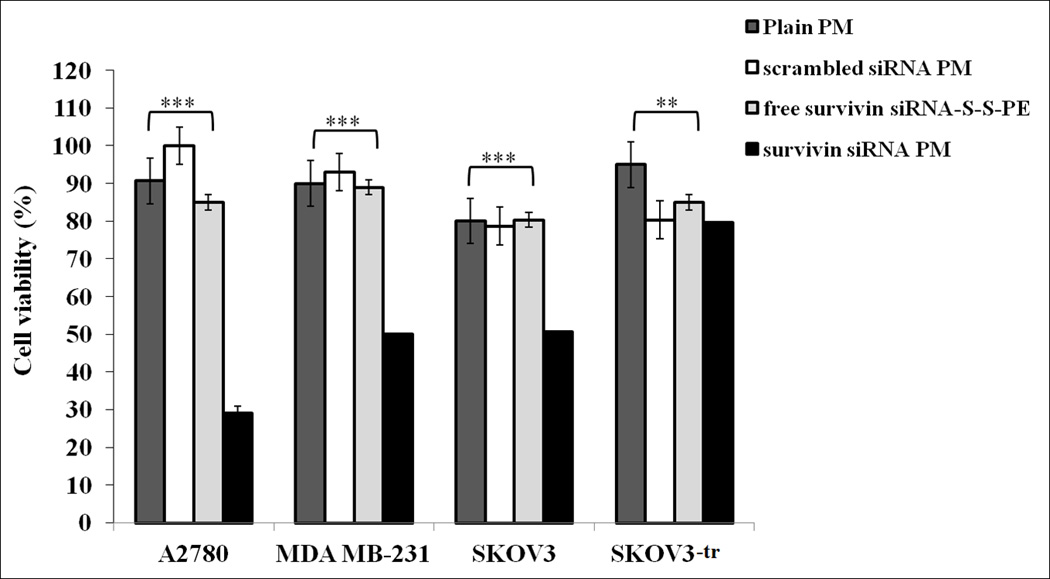
Viability of different cancer cells. Cells were treated with survivin siRNA PM at a final siRNA concentration of 200 nM in the serum-containing medium for 6 h. Cell viability in the presence of survivin siRNA PM, free survivin siRNA-S-S-PE conjugate, PM containing scrambled siRNA-S-S-PE, and plain PM was followed by Cell Titer Blue assay after 48 h of incubation. **p < 0.01, and ***p < 0.001 values were obtained by comparing survivin siRNA PM to all the other treatments. Results were obtained from three independent experiments in triplicate (n = 9). Mean ± SD.
3.3 Survivin protein levels
Survivin protein levels in the cells treated with survivin siRNA-S-S-PE in PM were then evaluated by ELISA. As illustrated in Figure 3, after treatment with PM containing survivin siRNA-S-S-PE, a significant down-regulation of survivin, by about 30% (p<0.01 survivin siRNA PM versus the other treatments), was observed in all the cell lines. Noteworthy, a significant down-regulation (by about 30%) of survivin levels was observed also in survivin overexpressing SKOV3-tr cells. In cells treated with free survivin siRNA-S-S-PE, and with PM containing scrambled siRNA-S-S-PE, no significant decrease of survivin level was observed.
Figure 3.
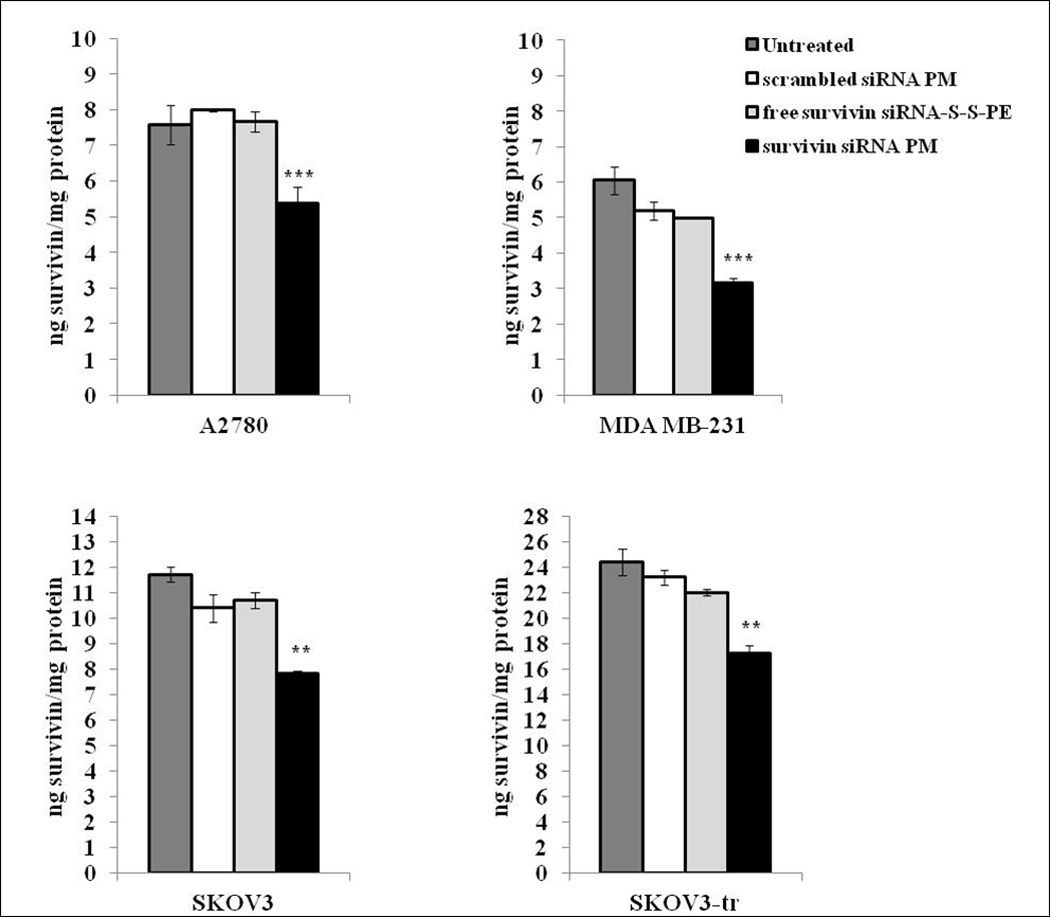
Survivin protein levels in A2780, MDA-MB231, SKOV3, and SKOV3-tr cells after different treatments. Survivin siRNA was quantified 48 h after the treatment by the ELISA method. The data are expressed as ng of survivin protein per mg of protein. Data = mean ± SD (n =3). **p < 0.01, and ***p < 0.001 values were obtained by comparing survivin siRNA PM to all the other treatments. Results were obtained from three independent experiments in triplicate (n = 9). Mean ± SD.
3.4 Chemosensitization study
In order to evaluate whether the down-regulation of survivin resulting from the treatment with survivin siRNA PM is able to sensitize cells to PXL, chemosensitization studies were performed. In particular, survivin siRNA PM pre-treated MDA-MB-231, SKOV3 and SKOV3-tr cells were exposed to increasing concentrations of PXL, and the resultant cell viabilities are summarized in Figures 4 a, b and c, respectively. As detailed in Figure 4, in all the cell lines tested, no toxicity was detected with free PXL at a dose range from 40 nM to 3.5 nM after 24 h of treatment. Increasing the incubation time to 72 h lead to significant toxicity in PXL-sensitive cell lines whereas none was detected in SKOV3-tr, using the same range of concentrations (data not shown). Confirming our initial hypothesis, the down-regulation of survivin by pre-treatment of the cells with survivin siRNA PM, strongly sensitized the cells to PXL. In particular, a significant increase of the PXL cytotoxicity was observed after only 24 h of cell exposure to the drug (Figure 4). These results were confirmed by the immunofluorescence analysis.
Figure 4.
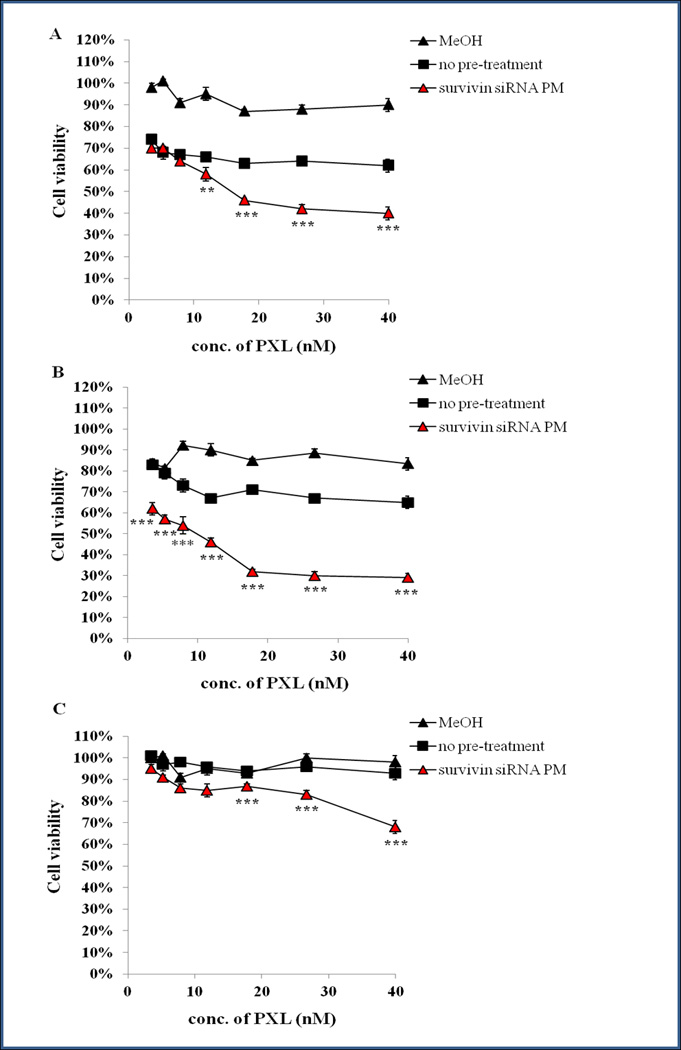
Chemosensitization of MDA MB-231(A), SKOV3 (B), and SKOV3-tr (C) cells mediated by the pre-treatment with survivin siRNA PM. Cells were incubated with survivin siRNA PM (range of siRNA concentrations from 200 to 17.5 nM) for 6 h. Forty eight hours later, cells were challenged with various concentrations of PXL. The viability of cells was measured 24 h later by the CTB assay. Data = mean ± SD (n =3).**p < 0.01, and ***p < 0.001 values were obtained by comparing survivin siRNA PM pre-treated cells (survivin siRNA PM) to PXL no pre treated cells (no pre-treatment).
Figure 5 shows the influence of the pre-treatment with survivin siRNA PM on the PXL activity in microtubules stabilization in the resistant cancer cell line, SKOV3-tr. In untreated cells, in cells treated for 24 h with 40 nM of PXL and cells treated with survivin siRNA PM for 48 h (Figure 5 A,B,C, respectively), microtubules appear “healthy” with elongated and fibrillary extensions from around the nucleus to the cell periphery. In contrast, the down regulation of survivin mediated by pretreatment with survivin siRNA PM for 48 h, sensitize SKOV3-tr cells to PXL action on microtubule organization (Figure 5D). Here, microtubules appear disassembled and characterized by a loss of fibrillar extensions as confirmed by the dense organization of microtubule network around the nucleus. The nuclei begin to fragment, reflective of cell death. These results confirm that survivin down-regulation enhances the PXL activity in microtubule destabilization.
Figure 5. Effect of PXL on microtubule stabilization after survivin down-regulation in SKOV3-tr cells.
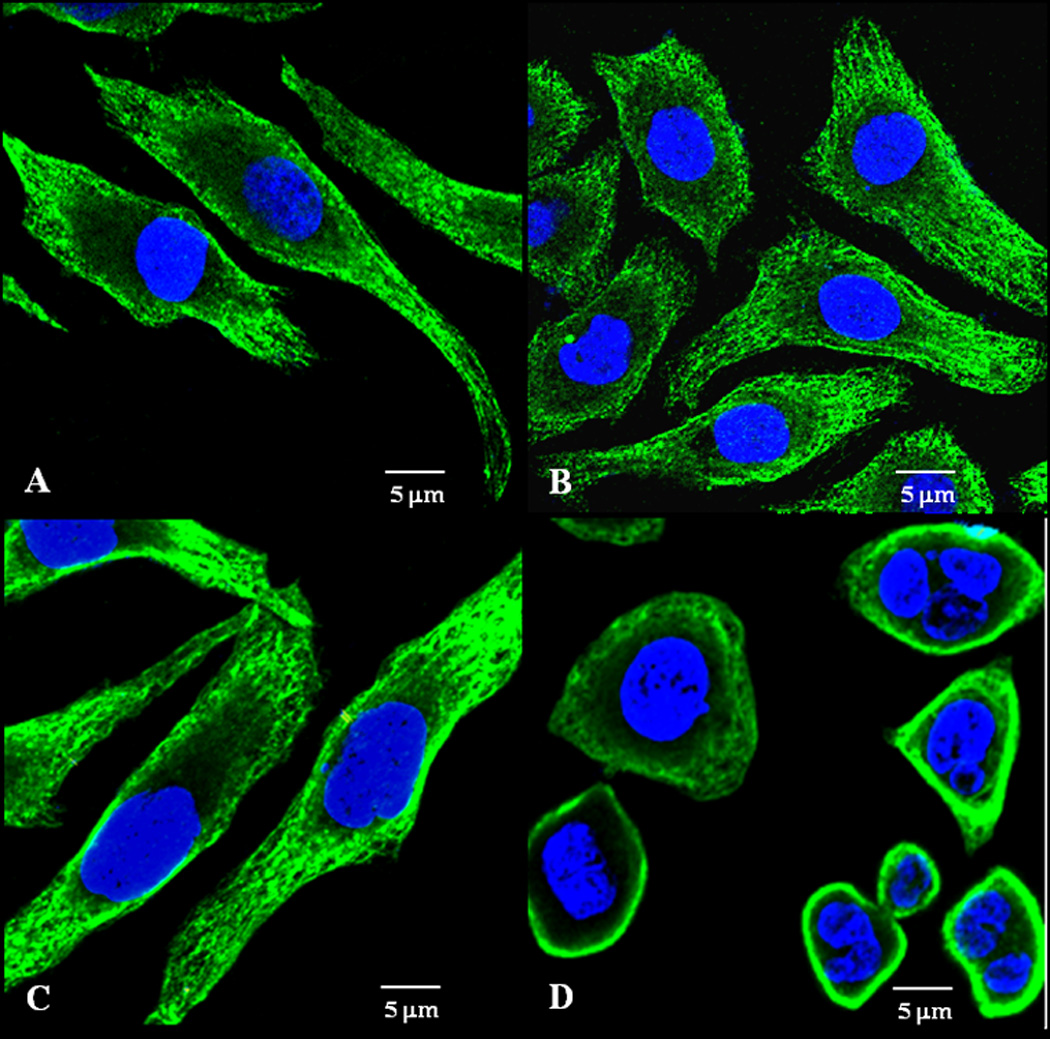
SKOV3-tr cells were pre-treated with 200 nM survivin siRNA PM for 48 h followed by the treatment with 40 nM of PXL for 24 h. Cells were then stained for β-tubulin (green). The nuclei (blue) were stained with DAPI. A–D: Representative images of three independent experiments showing organization of microtubules. Untreated cells (A); cells treated with free PXL for 24h (B); cells treated with survivin siRNA PM for 72 h (C); cells pre-treated with survivin siRNA PM for 48 h followed by treatment with PXL for 24 h (D). Data = mean ± SD (n =3).
3.5 Multifunctional therapy: survivin siRNA-S-S-PE and PXL co-encapsulated in PM
Finally, we investigated the effect of survivin siRNA-S-S-PE and PXL co-loaded in PM on the viability of a PXL-resistant cancer cell line, SKOV3-tr. For this purpose, we studied the effect of increasing concentrations of survivin siRNA-S-S-PE and PXL co-encapsulated in PM on the cell viability. In Figure 6, the viability of SKOV3-tr cells (%) 72 h after the treatment with free PXL, survivin siRNA PM and survivin siRNA/PXL PM, is shown. After the treatment with free PXL, no significant inhibition of the cell growth was observed. On the contrary, the simultaneous delivery of PXL and survivin siRNA-S-S-PE by PM to SKOV3-tr cells lead to a significant inhibition of cell growth compared to all other treatments (p<0.001 survivin siRNA/PXL PM versus the other treatments).
Figure 6.
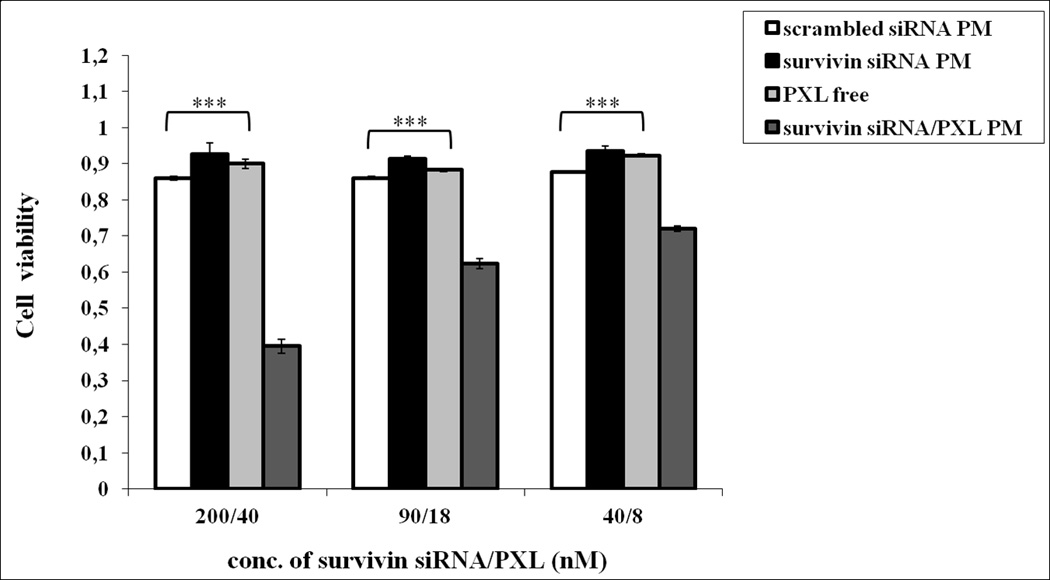
The viability of SKOV3-tr cancer cells. The cells were treated with the different formulations at 37°C for 6 h. After 72 h, the cell viability was measured by the Cell Titer Blue assay. Data = mean ± SD (n =3). ***p < 0.001 values were obtained by comparing each treatment to survivin siRNA/PXL PM treated cells.
4. Discussion
Survivin, a member of the IAP family, is considered a challenging target for cancer therapy. Survivin, not usually detected in normal adult tissue, is over-expressed in malignant tumors where plays a key role in hampering apoptosis, promoting cell proliferation, mitosis and angiogenesis [23]. Moreover, there is evidence that survivin up-regulation may be a predictive factor in determining drug-resistance. Interestingly, it has been demonstrated that survivin inhibition reduces tumor growth potential and markedly enhances tumor cell response to anticancer agents, such as PXL, etoposide, cisplatin [24], as well as to immunotherapy and ionizing radiation [25,26]. The resounding demonstrations of the efficacy of survivin inhibition in pre-clinical experiments has strongly incited to develop new strategies to target survivin in human tumors. Recently, siRNA directed against survivin, has been proposed as valid approach to specifically suppress survivin in cancer cells. In fact, it was shown to sensitize and strengthen the tumor response to chemotherapeutic drugs [27]. However, the major obstacle to therapeutic RNAi is the disadvantageous pharmacokinetic profile, which strongly hampers achieving effective concentrations in intracellular site of action. Therefore, delivery strategies that stabilize siRNA and enhance intracellular uptake in vivo need to be developed.
A new method addressed to stabilize and deliver the siRNA was recently proposed by us. In particular, a siRNA “reversibly” conjugated to a phospholipid by a disulfide linkage was synthesized [13]. The resulting modified siRNA was incorporated in nano-sized PEG2000-PE PM via its hydrophobic lipid moiety. As a result, the stability of PM-incorporated siRNA against nucleolytic degradation was dramatically increased, and, at the same time, the system easily released free unmodified siRNA in reducing conditions (similar to those inside cancer cells with characteristic high glutathione concentration). PEG2000-PE micelles has gained in the last years much attention due to their high stability in vivo, the possibility to efficiently encapsulate poor soluble drugs, such as PXL [16], and the ability to be accumulate in tumors via the EPR effect. In the present work, a siRNA against survivin was modified with a PE moiety and then incorporated in nanosized PM in order to deliver the survivin siRNA in cancer cells.
The survivin siRNA-S-S-PE was synthesized with a 90% of reaction yield. PM containing survivin siRNA optimized in terms of polymer concentration and weight ratio between polymer and survivin siRNA-S-S-PE demonstrated a mean size of about 20 nm and a narrow size distribution. The survivin siRNA was efficiently incorporated in PM only when a higher amount of PE-PEG2000 was used. In particular, the use of a survivin siRNA/PEG2000-PE weight ratio of 1:750 allowed reaching the encapsulation efficiency of about 50% (Figure 1). The hydrophobic interaction between the PE moiety of siRNA-S-S-PE and that of the PEGylated lipid serves as the driving force underlying lipid-modified siRNA firm incorporation into PM.
Cell culture experiments showed a significant cytotoxic effect of survivin siRNA when delivered by PM. In particular, the treatment with survivin siRNA PM resulted in a significant decrease in cell viability in sensitive cancer cells and a significant down-regulation of survivin protein levels, which was well in line with some previous observations [28]. It is noteworthy that in a resistant cancer cell line (SKOV3-tr), although little cytotoxic effect was observed, a significant down-regulation of survivin was still achieved following treatment with survivin siRNA PM. In all the cases, the treatment with the scrumbled siRNA conjugate in PM and plain PM did not yield any noticeable effect (Figures 2 and 3). Thus, PM proposed in this study are able to deliver the anti-survivin siRNA into the cells, as demonstrated by the significant inhibition of survivin expression. It is important that survivin siRNA PM demonstrated a silencing activity even in presence of serum. Free siRNA or siRNA delivered by commercial transfection agents, such as Lipofectamine, has no activity in presence of serum [29], Evidently, the PEG shell layer of PM sterically shields the siRNA in PM from accessibility of serum proteins, thereby protecting it from aggregation and enzymatic degradation.
As the next step, we have investigated the potential of combination therapy with survivin siRNA and a chemotherapeutic agent, PXL. PXL, one of the broad spectrum anticancer agents, was selected as a model anticancer drug, due to its efficacy often hampered by acquiring drug resistance by cancer cells [30]. On the other hand, there are different lines of evidence indicating that survivin inhibition enhances the antitumor activity of PXL [20,21]. With this in mind, we investigated the potential of the survivin down-regulation by survivin siRNA PM on the chemosensitization of cancer cells to PXL. With this in mind, we have studied the effect of cell pre-treatment survivin siRNA on the PXL chemosensitization of sensitive and resistant cancer cells. In sensitive cancer cells (MDA-MB231 and SKOV3), the pre-treatment with survivin siRNA for 48 h before exposing the cells to PXL, elicited a significant improvement in the citotoxicity of the drug. In particular, a significant cytotoxic effect of PXL was observed at earlier times, namely after only 24 h of PXL treatment. In the same condition, but without the pre-treatment with survivin siRNA PM, PXL showed a cytotoxic effect only after 72 h of treatment. It is noteworthy that in the human paclitaxel-resistant cancer cells, SKOV3-tr, even after 72 h no any significant cytotoxicity was observed following treatment with PXT alone. The pretreatment of SKOV3-tr with survivin siRNA PM and the consequent surviving down-regulation lead to a significant cell sensitization to PXL (Figure 4).
To confirm the key role of the survivin down-regulation on PXL sensitization in SKOV3 tr cell line, we have studied the microtubules organization by immunostaining the cells treated for β-tubulin. Survivin has been shown to bind polymerized microtubules in vitro where, presumably, it stabilizes the mitotic spindle [31]. J. Tran et al. [32] demonstrated a role of survivin in maintaining the integrity of the microtubule network of a human endothelial cell (HUVECs). In particular, they showed that in HUVECs cells, retrovirally infected with wild-type survivin, the over-expression of survivin could preserve the microtubule integrity of the cells treated with PXL. Our results, suggest that survivin siRNA PM mediated survivin down regulation, could counteract the PXL chemoresistance in SKOV3-tr cell lines (Figure 5). In fact, in the absence of the pre-treatment with survivin siRNA PM, no changes on the microtubules morphology were observed after exposure to PXL. The microtubules appeared like in “healthy” cells, with elongated and spread out tubulin filaments. Interestingly, in survivin siRNA PM pre-treated cells, the exposure to only 40 nM of PXL for 24 h, yielded significant changes in microtubules morphology. In particular, SKOV3-tr cells displayed a significant lack of microtubule organization reflective of cell death. Our data, therefore, support previous works demonstrating that survivin over-expression can negatively influence the sensitivity of cancer cell lines to chemotherapeutic agents, such as PXL.
Finally, we have also investigated if the developed PM could be used for co-encapsulation of survivin siRNA and PXL in mixed PM, in order to deliver simultaneously the two agents in cancer cells. With this in mind, we have evaluated if the presence of PXL could affect the properties of survivin siRNA PM in terms of size distribution and survivin siRNA encapsulation efficiency. When PXL was added to survivin siRNA PM, no changes in the size and in the encapsulation efficiency was observed. The mixed PM co-loading the two agents demonstrated a narrow size distribution and an incorporation efficiency of survivin siRNA and PXL of about 50% and 70%, respectively. Then, we investigated the in vitro activity of the multifunctional PM in the PXL-resistant cancer cell line. The results revealed that the simultaneous delivery of PXL and survivin siRNA in the cells lead to a significantly enhanced cytotoxicity (Figure 6). In particular, after 72 h of treatment with PM containing the two agents combined, a strong cytotoxic effect was observed. On the contrary, the treatment with free PXL did not elicit any significant effect. In accordance with previous findings [19] and the data observed by immunohistochemical analysis, survivin inhibition can strongly influence the sensitivity of SKOV3-tr cells to PXL.
On the whole, these results demonstrated that the survivin siRNA-S-S-PE PM developed in this work look like a promising tool to inhibit survivin expression in cancer cells. Moreover, due to the possibility to co-encapsulate lipophilic drugs, such as PXT, this system could be also used to overcome multidrug resistance in the treatment of chemoresistant tumors.
5. Conclusions
In this study, a novel self-assembling nano-system for the siRNA delivery in cancer therapy was developed. The developed system efficiently delivered survivin siRNA into different cancer cells, leading to a significantly down-regulated survivin expression. Interestingly, cells treated with the survivin siRNA PM, but not free siRNA, sensitized cells to PXL treatment. In addition, this system proposes a new platform for stimuli-sensitive co-delivery of chemotherapeutic drugs and siRNA for multifunctional therapy. Indeed, the use of PM co-delivering PXL and survivin siRNA simultaneously was able to reverse drug resistance in an aggressive cancer cell line. These results support further studies able to verify the in vivo activity of the developed delivery system.
Acknowledgments
This work was supported by the NIH grant U54 CA151881. The authors alone are responsible for the content and writing of this article.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
None
References
- 1.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 1997;3:917–921. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 2.Altieri DC. Validating survivin as a cancer therapeutic target. Nat. Rev. Cancer. 2003;3:46–54. doi: 10.1038/nrc968. [DOI] [PubMed] [Google Scholar]
- 3.Yang D, Welm A, Bishop JM. Cell division and cell survival in the absence of survivin. Proc. Natl. Acad. Sci. U.S.A. 2004;101:15100–15105. doi: 10.1073/pnas.0406665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu J, Tan M, Huang WC, Li P, Guo H, Tseng LM, Su XH, Yang WT, Treekitkarnmongkol W, Andreeff M, Symmans F, Yu D. Mitotic deregulation by survivin in ErbB2-overexpressing breast cancer cells contributes to Taxol resistance. Clin. Cancer Res. 2009;15:1326–1334. doi: 10.1158/1078-0432.CCR-08-0954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 6.Carvalho A, Carmena M, Sambade C, Earnshaw WC, Wheatley SP. Survivin is required for stable checkpoint activation in taxol-treated HeLa cells. J. Cell Sci. 2003;116:2987–2998. doi: 10.1242/jcs.00612. [DOI] [PubMed] [Google Scholar]
- 7.Seth S, Matsui Y, Fosnaugh K, Liu Y, Vaish N, Adami R, Harvie P, Johns R, Severson G, Brown T, Takagi A, Bell S, Chen Y, Chen F, Zhu T, Fam R, Maciagiewicz I, Kwang E, McCutcheon M, Farber K, Charmley P, Houston ME, Jr, So A, Templin MV, Polisky B. RNAi-based therapeutics targeting survivin and PLK1 for treatment of bladder cancer. Mol. Ther. 2011;19:928–935. doi: 10.1038/mt.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomanin R, Scarpa M. Why do we need new gene therapy viral vectors? Characteristics, limitations and future perspectives of viral vector transduction. Curr. Gene Ther. 2004;4:357–372. doi: 10.2174/1566523043346011. [DOI] [PubMed] [Google Scholar]
- 9.Devroe E, Silver PA. Therapeutic potential of retroviral RNAi vectors. Expert Opin. Biol. Ther. 2004;4:319–327. doi: 10.1517/14712598.4.3.319. [DOI] [PubMed] [Google Scholar]
- 10.Lewis DL, Wolff JA. Systemic siRNA delivery via hydrodynamic intravascular injection. Adv. Drug Deliv. Rev. 2007;59:115–123. doi: 10.1016/j.addr.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Hong K, Zheng W, Baker A, Papahadjopoulos D. Stabilization of cationic liposome-plasmid DNA complexes by polyamines and poly(ethylene glycol)-phospholipid conjugates for efficient in vivo gene delivery. FEBS Let. 1997;400:223–237. doi: 10.1016/s0014-5793(96)01397-x. [DOI] [PubMed] [Google Scholar]
- 12.Cardoso AL, Simões S, de Almeida LP, Pelisek J, Culmsee C, Wagner E, Pedroso de Lima MC. siRNA delivery by a transferrin-associated lipid-based vector: a non-viral strategy to mediate gene silencing. J. Gene Med. 2007;9:170–183. doi: 10.1002/jgm.1006. [DOI] [PubMed] [Google Scholar]
- 13.Musacchio T, Vaze O, D’Souza G, Torchilin VP. Effective Stabilization and Delivery of siRNA: Reversible siRNA-Phospholipid Conjugate in Nanosized Mixed Polymeric Micelles. Bioconjugate Chem. 2010;21:1530–1536. doi: 10.1021/bc100199c. [DOI] [PubMed] [Google Scholar]
- 14.Balendiran GK, Dabur R, Fraser D. The role of glutathione in cancer. Cell Biochem. Funct. 2004;22:343–352. doi: 10.1002/cbf.1149. [DOI] [PubMed] [Google Scholar]
- 15.Estrela JM, Ortega A, Obrador E. Glutathione in cancer biology and therapy. Crit. Rev. Clin. Lab. Sci. 2006;43:143–181. doi: 10.1080/10408360500523878. [DOI] [PubMed] [Google Scholar]
- 16.Sawant RR, Torchilin VP. Polymeric micelles: polyethylene glycol-phosphatidylethanolamine (PEG-PE)-based micelles as an example. Methods Mol. Biol. 2010;624:131–149. doi: 10.1007/978-1-60761-609-2_9. [DOI] [PubMed] [Google Scholar]
- 17.Gallagher BM., Jr Microtubule-stabilizing natural products as promising cancer therapeutics. Curr. Med. Chem. 2007;14:2959–2967. doi: 10.2174/092986707782794014. [DOI] [PubMed] [Google Scholar]
- 18.Singla AK, Garg A, Aggarwal D. Paclitaxel and its formulations. Int. J. Pharm. 2002;235:179–192. doi: 10.1016/s0378-5173(01)00986-3. [DOI] [PubMed] [Google Scholar]
- 19.Shen J, Yin Q, Chen L, Zhang Z, Li Y. Co-delivery of paclitaxel and survivin shRNA by pluronic P85-PEI/TPGS complex nanoparticles to overcome drug resistance in lung cancer. Biomaterials. 2012;33:8613–8624. doi: 10.1016/j.biomaterials.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 20.Shen J, Sun H, Xu P, Yin Q, Zhang Z, Wang S, Yu H, Li Y. Simultaneous inhibition of metastasis and growth of breast cancer by co-delivery of twist shRNA and paclitaxel using pluronic P85-PEI/TPGS complex nanoparticles. Biomaterials. 2013;34:1581–1590. doi: 10.1016/j.biomaterials.2012.10.057. [DOI] [PubMed] [Google Scholar]
- 21.Hu Q, Li W, Hua X, Hua Q, Shen J, Jin X, Zhou J, Tang G, Chu PK. Synergistic treatment of ovarian cancer by co-delivery of survivin shRNA and paclitaxel via supramolecular micellar assembly. Biomaterials. 2012;33:6580–6591. doi: 10.1016/j.biomaterials.2012.05.060. [DOI] [PubMed] [Google Scholar]
- 22.Musacchio T, Laquintana V, Latrofa A, Trapani G, Torchilin VP. PEG-PE micelles loaded with paclitaxel and surface-modified by a PBR-ligand: synergistic anticancer effect. Mol. Pharm. 2009;6:468–479. doi: 10.1021/mp800158c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat. Rev, Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- 24.Zaffaroni N, Daidone MG. Survivin expression and resistance to anticancer treatments: perspectives for new therapeutic interventions. Drug Resist. Updat. 2002;5:65–72. doi: 10.1016/s1368-7646(02)00049-3. [DOI] [PubMed] [Google Scholar]
- 25.Kanwar JR, Shen WP, Kanwar RK, Berg RW, Krissansen GW. Effects of survivin antagonists on growth of established tumors and B7-1 immunogene therapy. J. Natl. Cancer Inst. 2001;93:1541–1552. doi: 10.1093/jnci/93.20.1541. [DOI] [PubMed] [Google Scholar]
- 26.Sah NK, Munshi A, Hobbs M, Carter BZ, Andreeff M, Meyn RE. Effect of downregulation of survivin expression on radiosensitivity of human epidermoid carcinoma cells. Int. J. Radiat. Oncol. Biol. Phys. 2006;66:852–859. doi: 10.1016/j.ijrobp.2006.06.049. [DOI] [PubMed] [Google Scholar]
- 27.Shapira A, Livney YD, Broxterman HJ, Assaraf YG. Nanomedicine for targeted cancer therapy: towards the overcoming of drug resistance. Drug Resist. Updat. 2011;14:150–163. doi: 10.1016/j.drup.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 28.Li SD, Huang L. Targeted Delivery of Antisense Oligodeoxynucleotide and Small Interference RNA into Lung Cancer Cells. Mol. Pharm. 2006;3:579–588. doi: 10.1021/mp060039w. [DOI] [PubMed] [Google Scholar]
- 29.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat. Rev. Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Podolski-Renić A, Andelković T, Banković J, Tanić N, Ruždijić S, Pešić M. The role of paclitaxel in the development and treatment of multidrug resistant cancer cell lines. Biomed. Pharmacother. 2011;65:345–353. doi: 10.1016/j.biopha.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 31.Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 32.Tran J, Master Z, Yu JL, Rak J, Dumont DJ, Kerbel RS. A role for survivin in chemoresistance of endothelial cells mediated by VEGF. Proc. Natl. Acad. Sci. U.S.A. 2002;99:4349–4354. doi: 10.1073/pnas.072586399. [DOI] [PMC free article] [PubMed] [Google Scholar]