

Abstract
The cell cytoskeleton is composed of microtubules, intermediate filaments, and actin, which provide a rigid support structure important for cell shape. However, it is also a dynamic signaling scaffold that receives and transmits complex mechanosensing stimuli that regulate normal physiological and aberrant pathophysiological processes. Studying cytoskeletal functions in its native state is inherently difficult due to its rigid and insoluble nature. This has severely limited detailed proteomic analyses of the complex protein networks that regulate the cytoskeleton. Here, we describe a purification method that enriches for the cytoskeleton and its associated proteins in their native state, which is also compatible with current mass spectrometry-based protein detection methods. This method can be used for biochemical, fluorescence and large-scale proteomic analyses of numerous cell types. Using this approach, 2346 proteins were identified in the cytoskeletal fraction of purified mouse embryonic fibroblasts, of which, 635 proteins were either known cytoskeleton proteins or cytoskeleton-interacting proteins. Functional annotation and network analyses using the Ingenuity Knowledge Database of the cytoskeletome revealed important nodes of interconnectivity surrounding well-established regulators of the actin cytoskeleton and focal adhesion complexes. This improved cytoskeleton purification method will aid our understanding of how the cytoskeleton controls normal and diseased cell functions.
Keywords: Cytoskeleton, actin scaffold, focal adhesion, proteomics
Introduction
The cytoskeleton is a filamentous subcellular network that is insoluble under many conventional lysis and detergent conditions. In addition, many cytoskeleton regulatory proteins bind with high affinity to the actin scaffold and focal adhesion complexes, making them highly insoluble [1–4]. This problem is further compounded by the inability of mass spectrometry techniques to detect low abundant regulatory cytoskeleton proteins, which can be masked by the multitude of cytoplasmic proteins typically present in whole cell extracts. Therefore, subtle changes in key protein and phosphoprotein signatures that associate with the cytoskeleton domains can be easily missed when using conventional whole cell extraction and protein detection methods. In fact, current isolation methods do not efficiently extract the cytoskeleton in its native state and, thus, can disrupt important interactions with binding partners [5–8]. These limitations have made it difficult to fully characterize the cytoskeletome [5–7]. Thus, there is a need for an improved enrichment method that allows for the biochemical purification of the cytoskeleton and its associated proteins in their native states that is compatible with modern quantitative mass spectrometry-based proteomic techniques.
Materials and methods
Cell culture
Low passage wild type mouse embryonic fibroblasts (MEFs) cells were cultured with Dulbecco’s modified Eagle’s medium (DMEM, GIBCO, Invitrogen) supplemented with 10% fetal bovine serum (FBS, Invitrogen) in 150 mm petri culture dishes. Cells were maintained at sub-confluency in a humidified atmosphere with 5% CO2 at 37°C with medium renewal at every 2 or 3 days.
Cytoskeleton extraction
Cells grown to 70% - 80% confluency in 150 mm were placed on ice. After removal of the culture medium, 5 mL of cold phosphate buffered saline (PBS) was used to wash the cells twice. Next, 5 mL of ice-cold cell lysis buffer (50 mM PIPES, 50 mM NaCl, 5% Glycerol, 0.1% NP-40, 0.1% Triton X-100 and 0.1% Tween 20) was added to the dish and kept on ice for 1.5 min. Lysates were collected and kept on ice for further use. The cells were further rinsed gently with 5 mL Tris-HCl buffer (50 mM Tris-HCl, pH 7.5) and incubated with 5 mL of Nuclease buffer [10 U/mL Benzoase nuclease (Sigma-Aldrich), 10 mM MgCl2 and 2 mM CaCl2 in 50 mM Tris-HCl buffer, pH 7.5] for 10 min at room temperature. After removal of the Nuclease buffer, aliquots of the previously collected lysates (in lysis buffer) were added to release and solubilize the DNA or RNA binding proteins for another 30 seconds on ice. Cytoskeletal proteins remaining bound to the dish were then rinsed using 5 ml of cold Tris-HCl buffer three times on ice, and solubilized/denatured in 500 μL of 1% SDS. The total protein concentration was determined using the BCA protein assay (Pierce). All the buffers used during the cytoskeleton extraction procedure contained protease (Roche protease inhibitor cocktail) and phosphatase inhibitors (5 mM NaF, 2 mM sodium vanadate and 10 mM β-glycerophosphate). For convenience, we have also developed in association with EMD Millipore scientists a cytoskeleton extraction kit using the extraction procedures and buffers described here. The kit is also compatible with traditional MS methods (ProteoExtract Cytoskeleton Enrichment Kit, #17-10195). Additional information and data is described in a White Paper found at EMD Millipore (http://www.millipore.com/techpublications/tech1/an4223en00).
Transient transfection, immunocytochemistry, and fluorescent microscopy
Cell morphology and protein localization during the extraction were monitored using a Nikon Ti-E wide-field fluorescent microscope. Wild type MEFs cells were cultured in 10 cm dishes to a 70% confluence and transiently transfected with plasmid combinations encoding either GFP-α-actinin/mCherry-actin or GFP-paxillin/mCherry-actin. The transfection cocktail was prepared as follows: 4 μg of each DNA in plasmid was mixed with 40 μL of Plus reagent (Invitrogen) and 500 μL serum-free DMEM medium, which was incubated at room temperature for 15 min; then 30 μL Lipofectamine (Invitrogen) diluted in 500 μL of serum-free DMEM medium was added and the mixture was incubated for an additional 15 min. The cells were rinsed twice with PBS, followed by incubation of the transfection cocktail in 4 mL of serum-free DMEM for 6 h. The media was then changed to complete media lacking antibiotics and the cells were incubated at 37°C overnight. The transfected cells were split and plated onto a fibronectin-coated glass-bottomed 4-well dish, which was incubated at 37°C for 6 hours before imaging.
For immunocytochemistry, the cells were fixed with 4 % paraformaldehyde. Actin was stained with Alexa Fluor 546-phalloidin (Molecular Probes) and α-tubulin or vimentin was counterstained with specific primary antibodies (Sigma-Aldrich and Cell Signaling Technology, respectively) and Alexa Fluor 488-conjugated secondary antibody. All imaging was done on an inverted microscope system (Eclipse Ti; Nikon and Olympus FV-1000) with a 60× oil objective lens (Supplementary Figure 2).
Western blot analysis
For western blot, equal amount of proteins (15 μg) from the cytoskeletal and soluble cell body fractions were loaded and separated by SDS-PAGE on a NuPage 4%–12% Bis-Tris gel (Invitrogen), blotted to nitrocellulose membrane (Immobilon, Whatman), probed and visualized using enhanced chemiluminscence (Thermo Scientific).
Gel separation and in-gel digestion
The cytoskeletal proteins (100 μg) were separated by SDS PAGE on a NuPage 4% - 12% Bis-Tris Gel in MOPS buffer (Invitrogen) at 180 V constant voltage for 50 min. After stained with Coomassie Blue, the gel was excise to 14 bands and cut into small cubes separately. Subsequently, the small gel pieces were reduced with 10 mM dithiothreitol (DTT) and alkylated with 55 mM iodoacetamide (IAA). Proteins in the gel pieces were digested in-gel with trypsin (Promega) at 37°C overnight; and the peptides were extracted from gels with 5% acetic acid in H2O and in CH3CN/H2O (1:1, v/v). The resulting peptide mixtures were dried and stored at −20°C for further analysis.
LC-MS/MS for Protein Identification
Online LC-MS/MS analysis was performed on an Agilent 1200 series pump and a LTQ-Orbitrap mass spectrometry system (Thermo Finnigan). A capillary with 50 mm was used for eluent splitting, and the flow rate of eluent for separation after splitting was adjusted to ~250 nL/min. The peptides were separated by a 15 cm long C18 separation column (75 μm i.d., packed with 5 μm C18 resin) with a 90-min linear gradient of 5% – 35% acetonitrile in 0.1% formic acid. The LTQ-Orbitrap instrument was operated at positive ion mode with spray voltage of 2.25 kV. The temperature of the ion transfer capillary was set at 200°C and the normalized collision energy was set to 35%. One micro-scan was used for each MS and MS/MS scan. All MS and MS/MS spectra were acquired in the data dependent mode. Survey full scan MS was acquired by LTQ-Orbitrap from m/z 400 to 1600; and 10 most intense ions were selected for MS/MS scan by collision induced dissociation (CID). The dynamic exclusion function was set as follows: repeat count 1, repeat duration 30 s, and exclusion duration 60 s. The cytoskeletal fraction and soluble fraction of LC/MS/MS were performed in triplicate analysis.
Database search and protein identification
All the MS/MS spectra were searched using SEQUEST (v 0.28, in Bioworks 3.3.1) against a composite sequence database containing common contaminations, the mouse IPI protein database (version 3.08) and its reversed complement with the following parameters: the maximum number of miss-cleavages for trypsin was set as two per peptide; Cysteine carbamidomethylation was set as a fixed modification, while Methionine oxidation was considered as a variable modification; the mass tolerances for MS and MS/MS were 5 ppm and 1 Da, respectively. Only proteins which were identified with more than 2 peptides were retained for further analysis. The final protein false discovery rate (FDR) was controlled as less than 1% and the peptide FDR was considered as less than 0.1%.
Bioinformatics
The functional classification and pathway analyses were performed using the Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems). Proteins enriched in either the cytoskeletal or soluble fractions were categorized based upon gene ontology and cellular localization. These data are presented in Figure 2. Protein interactome maps were generated by comparing the list of known actin or alpha-actinin interacting proteins curated by IPA with the list of proteins found in our cytoskeletal fraction, which are presented in supplemental figure 2.
Figure 2.

(A) Western blot analyses of the indicated proteins for total cell lysate, cytoskeleton fraction (Cyto), and soluble cytoplasmic fraction (Sol). Right, peptide count ratio of identified proteins in proteomic results. (B) SDS-PAGE and Coomassie blue staining of total cell lysate, cytoskeleton fraction, and the soluble cytoplasmic fraction. Note that the actin band (43kD) is enriched in the cytoskeleton fraction.
Results and discussion
The need to study the macromolecular composition and post-translational modifications of the cytoskeleton prompted us to develop a purification procedure that is compatible with current protein detection and mass spectrometry technologies (Figure 1). To develop this procedure, we used adherent MEF cells growing in standard culture media in the presence of serum. Under these conditions, MEF cells have a typical cytoskeleton composed of F-actin, vimentin, and microtubules (Supplementary Figure 1) [9]. To isolate the cytoskeleton, live MEFs were briefly treated with a cytoskeleton stabilization buffer containing protease and phosphatase inhibitors as well as a cocktail of low ionic strength detergents to remove soluble cytoplasmic proteins. Cells were then treated with a nuclear extraction buffer to remove contaminating DNA/RNA and their associated proteins. Under these conditions, the insoluble F-actin and intermediate filaments remain attached to the substratum, whereas the majority of microtubules are extracted (Supplementary Figure 1). The remaining cytoskeleton was then solubilized in 1% SDS and separated using SDS-PAGE. Gels were either transferred to nitrocellulose and western blotted for known cytoskeleton proteins or protein bands were cut from the gel, and enzymatically digested prior to high-resolution mass spectrometry analysis. To validate the efficiency of the cytoskeleton enrichment procedure, known cytoskeleton and focal adhesion proteins (α-actin, α-actinin, vinculin and Src) [10–12] and soluble cytoplasmic proteins (Erk2, gapdh, and α-tubulin) [13] were analyzed in the total cell lysate, soluble cytoplasmic extract, and cytoskeleton fractions by SDS-PAGE and Western blotting. As expected, α-actin, α-actinin, and Src were highly enriched in the cytoskeleton fraction, while the cytoplasmic Erk2, GAPDH, and α-tubulin were detected in the soluble fraction (Figure 2A). The presence of focal adhesion protein vinculin in both the soluble and cytoskeleton fraction is consistent with the known subcellular domains to which this protein localizes [14]. SDS-PAGE and Coomassie blue staining of the soluble and cytoskeleton fractions, also revealed a significantly different protein expression pattern, including actin (42kDa) which is significantly enriched the cytoskeleton fraction as expected (Figure 2B). These results demonstrate that the cytoskeleton extraction method removes cytoplasmic proteins, leaving behind the actin and intermediate filament cytoskeleton and their associated proteins.
Figure 1.

Flow diagram of the protocol used to isolate and prepare cytoskeleton proteins from wild type MEF cells for biochemical, microscopic and proteomic analyses.
To determine if the extraction procedure disrupted the native architecture of the cytoskeleton, which could perturb its protein scaffolding and signaling functions, we analyzed the morphological and molecular changes of the actin cytoskeleton and its scaffolding functions in the same live cell before, during and after the extraction procedure using fluorescence microscopy. MEF cells were engineered to co-express mCherry-actin and its binding partner GFP-α-actinin. In addition, MEF cells were engineered to co-express the focal adhesion marker protein GFP-paxillin along with the mCherry-actin reporter (Figure 3A and B). No significant differences in the architecture of the cytoskeleton and focal adhesions were observed, as the association of α-actinin and paxillin with the actin cytoskeleton remained robust and their spatial patterns were not altered during the extraction process (Figure 3A and B). It is also notable that the overall clarity of the fluorescence labeled cytoskeleton increased significantly as a result of the extraction process. This is likely due to removal of soluble cytoplasmic proteins. Thus, we conclude that the cytoskeleton retains its native state and its associated proteins during the extraction procedure.
Figure 3.

(A and B) Merged fluorescent micrographs of MEFs transiently transfected with mCherry-actin (red) and GFP-α-actinin (green) (A) or GFP-paxillin (green) (B) before (live), during, and after the cytoskeleton enrichment procedure (see Figure S1 for single wavelength images and the Supplemental Materials and Methods section for extraction buffer recipes). Importantly, there is a significant increase in signal:noise ratio and the clarity of the labeled cytoskeleton after extraction of the soluble fraction.
To globally profile the proteins that co-purify with the cytoskeleton fraction in comparison to the soluble fraction from MEF cells, we used SDS-PAGE separation followed by an in-gel digestion procedure coupled with high resolution protein detection mass spectrometry [15]. For protein identification, filters were optimized and set for each analysis separately to a final peptide and protein false discovery rate of less than 1%. Protein identification was considered only if a single protein was detected in two out of the three analyses with 2 or more matching peptides. A total of 2346 proteins from the cytoskeletal fraction and 2032 proteins from the soluble fraction were identified, respectively (Supplementary data). For comparison, proteins annotated from unique peptide sequences were arranged according to the percent amino acid sequence coverage across each protein. The total number of peptides identified for each protein is also shown for comparison since large molecular weight proteins are more likely to display more redundant peptide identifications (Supplementary data). In this regard, actin family members (43kD) display a high sequence coverage rate but a lower peptide count compared to a large 517kD protein like plectin, which binds to intermediate filaments. As expected, α-actinin, paxillin, and vinculin were reproducibly detected in the cytoskeleton fraction from numerous peptides with high sequence coverage, whereas GAPDH and tubulin were abundantly detected in the cytoplasmic fraction (Supplementary data).
Ingenuity Pathway Analysis (IPA) is a bioinformatics resource that facilitates the functional annotation and mapping of signaling networks involving a range of biomolecules as they relate to normal physiological and pathophysiological functions (www.ingenuity.com) [16]. Classifications of cytoskeletal fraction and soluble fraction are shown in Figure 4. Using this program, 42% of cytoskeleton-enriched proteins were annotated as cytoskeleton and focal adhesion-associated proteins [4]. In addition, 19% of the proteins were associated with membrane trafficking, which is well known to control cytoskeleton and integrin-mediated focal adhesion dynamics [17,18]. 11% of the proteins are functionally involved in signal transduction. Of these, Src and FAK are well-known effectors that control the cytoskeleton and focal adhesions [15,19]. In contrast, only 2% of the proteins were predicted to be involved in protein folding and degradation suggesting that this cellular machinery is primarily cytoplasmic and not strongly associated with the cytoskeleton scaffold. Proteins enriched in the soluble fraction were more broadly represented being associated with localization (19%), signaling (14%), transport (12%), and DNA/RNA processing (11%), whereas only a small portion were annotated to the cytoskeleton (8%).
Figure 4.
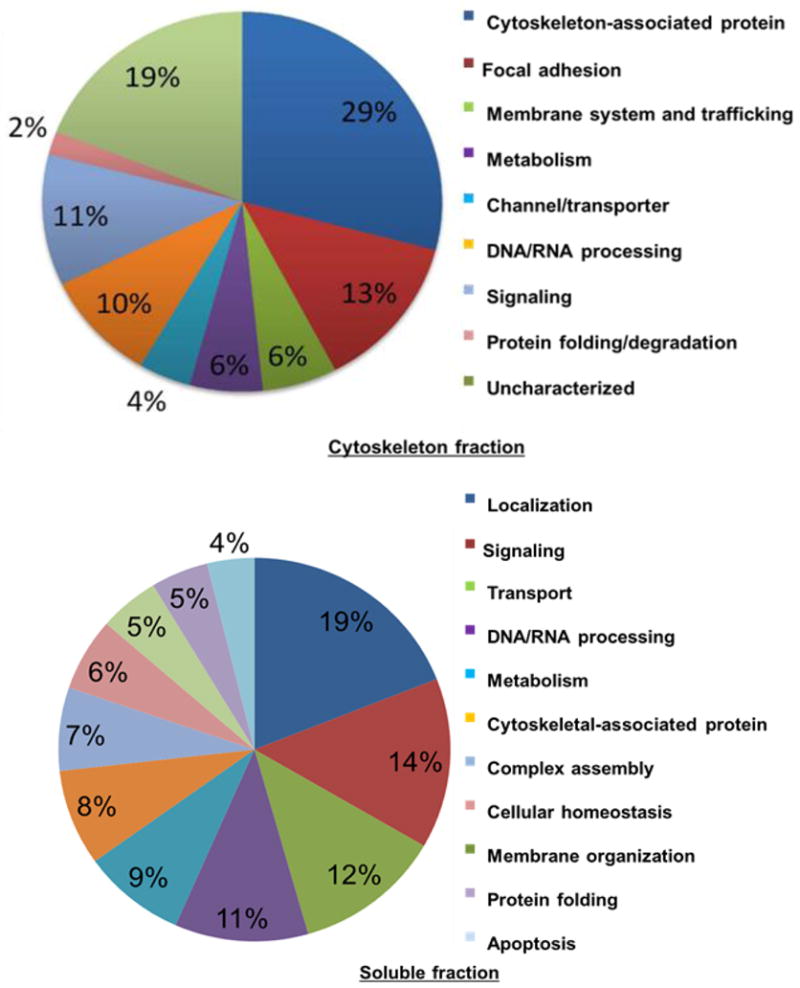
Pie chart showing the percentage of proteins from the reproducible list of soluble- (lower chart) and cytoskeleton-enriched (upper chart) proteins categorized according to their biological functions using Ingenuity Pathway Analysis (IPA) software.
Finally, using the IPA, we have conducted network analyses of the 2346 reproducibly identified cytoskeletome proteins. We manually created two unique interactomes from the cytoskeleton-enriched proteins. Supplementary Figure 3 (left) shows the cytoskeletome proteins that overlap with an Ingenuity-derived actin network, and Supplementary Figure 3 (right) shows the cytoskeletome proteins that overlap with an Ingenuity-derived α-actinin network. Overall, the interactomes support the above biochemical and mass spectrometry data demonstrating that the cytoskeleton extraction method enables the enrichment of protein complexes known to regulate cytoskeleton functions (e.g., vinculin, Src family kinases, talin, ZO-1, actins and α-actinins) [20]. For example, the α-actinin interactome shows that Src binds directly with α-actinin and catalytically acts on vinculin, talin, catenin and ezrin. These pathways have been previously shown to impact cell motility by regulating both actin and focal adhesion dynamics [21].
Conclusions
Together our data demonstrate that the cytoskeleton extraction procedure enriches for known cytoskeleton proteins in their native state, which can be detected by conventional fluorescence microscopy, western blotting, and mass spectrometry. Importantly, we have identified many new cytoskeleton-interacting proteins that may be the basis for new hypothesis-driven studies regarding the roles of these proteins in regulating the cytoskeleton. The improved cytoskeleton isolation technique will aid our understanding of the cytoskeleton and how it controls normal and diseased cell functions.
Supplementary Material
Identified proteins of cytoskeletal fraction and soluble fraction by LC/MS/MS.
*C/S: Cytoskeletal fraction/Soluble fraction.
*N/M: not matched; peptides identified exclusively in either the cytoskeletal or soluble fraction.
Figure 1. Fluorescent microscopy analysis of phalloidin (red) and α-tubulin (green) or vimentin (green) before and after cytoskeleton extraction.
Figure 2. Fluorescent microscopy analysis of mCherry-Actin and GFP-α-actinin and GFP-paxillin in MEF cells before, during and after being exposed to the cytoskeleton extraction procedure.
Figure 3.Ingenuity Pathway Analysis (IPA)-generated interactome map for actin network proteins present in the cytoskeletome (Left). Right, IPA-generated interactome map for α-actinin network proteins present in the cytoskeletome. Solid lines represent direct interactions, where the indicated proteins have been previously demonstrated to bind to one another. Dashed lines represent indirect interactions, where the indicated proteins do not directly bind to one another, but associate via other protein complexes.
Acknowledgments
We would like to thank members of the Klemke lab for helpful discussions and comments on this manuscript. This work was supported by the NIH-IRACDA (National Institutes of Health – Institutional Research and Academic Career Development Award) Postdoctoral Fellowship GM06852 (to J.A.K) and National Institutes of Health Grants CA097022 (to R.L.K.).
Abbreviations
- MEFs
mouse embryonic fibroblasts
- DMEM
Dulbecco’s modified Eagle’s minimum essential medium
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- DTT
dithiothreitol
- IAA
iodoacetamide
- CID
collision induced dissociation
- FDR
false discovery rate
- IPA
Ingenuity Pathway Analysis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Luna EJ, Hitt AL. CYTOSKELETON PLASMA-MEMBRANE INTERACTIONS. Science. 1992;258:955–63. doi: 10.1126/science.1439807. [DOI] [PubMed] [Google Scholar]
- 2.Cowin P, Burke B. Cytoskeleton-membrane interactions. Current Opinion in Cell Biology. 1996;8:56–65. doi: 10.1016/s0955-0674(96)80049-4. [DOI] [PubMed] [Google Scholar]
- 3.Chichili GR, Rodgers W. Cytoskeleton-membrane interactions in membrane raft structure. Cell Mol Life Sci. 2009;66:2319–28. doi: 10.1007/s00018-009-0022-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuo JC, Han XM, Hsiao CT, Yates JR, Waterman CM. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for beta-Pix in negative regulation of focal adhesion maturation. Nat Cell Biol. 2011;13:383–U109. doi: 10.1038/ncb2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nebl T, Pestonjamasp KN, Leszyk JD, Crowley JL, Oh SW, Luna EJ. Proteomic Analysis of a Detergent-resistant Membrane Skeleton from Neutrophil Plasma Membranes. Journal of Biological Chemistry. 2002;277:43399–409. doi: 10.1074/jbc.M205386200. [DOI] [PubMed] [Google Scholar]
- 6.Chen R, Jiang XN, Sun DG, Han GH, Wang FJ, Ye ML, et al. Glycoproteomics Analysis of Human Liver Tissue by Combination of Multiple Enzyme Digestion and Hydrazide Chemistry. Journal of Proteome Research. 2009;8:651–61. doi: 10.1021/pr8008012. [DOI] [PubMed] [Google Scholar]
- 7.Tomazella GG, daSilva I, Thome CH, Greene LJ, Koehler CJ, Thiede B, et al. Analysis of Detergent-Insoluble and Whole Cell Lysate Fractions of Resting Neutrophils Using High-Resolution Mass Spectrometry. Journal of Proteome Research. 2010;9:2030–6. doi: 10.1021/pr1000253. [DOI] [PubMed] [Google Scholar]
- 8.Xu P, Crawford M, Way M, Godovac-Zimmermann J, Segal AW, Radulovic M. Subproteome analysis of the neutrophil cytoskeleton. Proteomics. 2009;9:2037–49. doi: 10.1002/pmic.200800674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fabry B, Klemm AH, Kienle S, Schaffer TE, Goldmann WH. Focal Adhesion Kinase Stabilizes the Cytoskeleton. Biophys J. 2011;101:2131–8. doi: 10.1016/j.bpj.2011.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sjoblom B, Salmazo A, Djinovic-Carugo K. alpha-actinin structure and regulation. Cell Mol Life Sci. 2008;65:2688–701. doi: 10.1007/s00018-008-8080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamadi A, Deramaudt TB, Takeda K, Ronde P. Src activation and translocation from focal adhesions to membrane ruffles contribute to formation of new adhesion sites. Cell Mol Life Sci. 2009;66:324–38. doi: 10.1007/s00018-008-8424-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rudiger M. Vinculin and alpha-catenin: shared and unique functions in adherens junctions. Bioessays. 1998;20:733–40. doi: 10.1002/(SICI)1521-1878(199809)20:9<733::AID-BIES6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 13.Basuroy S, Seth A, Elias B, Naren AP, Rao R. MAR interacts with occludin and mediates EGF-induced prevention of tight junction disruption by hydrogen peroxide. Biochemical Journal. 2006;393:69–77. doi: 10.1042/BJ20050959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ziegler WH, Liddington RC, Critchley DR. The structure and regulation of vinculin. Trends Cell Biol. 2006;16:453–60. doi: 10.1016/j.tcb.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 15.Dong XL, Xiao YS, Jiang XN, Wang YS. Quantitative Proteomic Analysis Revealed Lovastatin-induced Perturbation of Cellular Pathways in HL-60 Cells. Journal of Proteome Research. 2011;10:5463–71. doi: 10.1021/pr200718p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gehlenborg N, O’Donoghue SI, Baliga NS, Goesmann A, Hibbs MA, Kitano H, et al. Visualization of omics data for systems biology. Nat Methods. 2010;7:S56–68. doi: 10.1038/nmeth.1436. [DOI] [PubMed] [Google Scholar]
- 17.Skalski M, Sharma N, Williams K, Kruspe A, Coppolino MG. SNARE-mediated membrane traffic is required for focal adhesion kinase signaling and Src-regulated focal adhesion turnover. Biochim Biophys Acta-Mol Cell Res. 2011;1813:148–58. doi: 10.1016/j.bbamcr.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 18.Kessels MM, Qualmann B. The syndapin protein family: linking membrane trafficking with the cytoskeleton. Journal of Cell Science. 2004;117:3077–86. doi: 10.1242/jcs.01290. [DOI] [PubMed] [Google Scholar]
- 19.Thamilselvan V, Basson MD. The role of the cytoskeleton in differentially regulating pressure-mediated effects on malignant colonocyte focal adhesion signaling and cell adhesion. Carcinogenesis. 2005;26:1687–97. doi: 10.1093/carcin/bgi135. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Kelber JA, Tran Cao HS, Cantin GT, Lin R, Wang W, et al. Pseudopodium-enriched atypical kinase 1 regulates the cytoskeleton and cancer progression [corrected] Proc Natl Acad Sci U S A. 2010;107:10920–5. doi: 10.1073/pnas.0914776107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takeda H, Nagafuchi A, Yonemura S, Tsukita S, Behrens J, Birchmeier W. V-src kinase shifts the cadherin-based cell adhesion from the strong to the weak state and beta catenin is not required for the shift. J Cell Biol. 1995;131:1839–47. doi: 10.1083/jcb.131.6.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Identified proteins of cytoskeletal fraction and soluble fraction by LC/MS/MS.
*C/S: Cytoskeletal fraction/Soluble fraction.
*N/M: not matched; peptides identified exclusively in either the cytoskeletal or soluble fraction.
Figure 1. Fluorescent microscopy analysis of phalloidin (red) and α-tubulin (green) or vimentin (green) before and after cytoskeleton extraction.
Figure 2. Fluorescent microscopy analysis of mCherry-Actin and GFP-α-actinin and GFP-paxillin in MEF cells before, during and after being exposed to the cytoskeleton extraction procedure.
Figure 3.Ingenuity Pathway Analysis (IPA)-generated interactome map for actin network proteins present in the cytoskeletome (Left). Right, IPA-generated interactome map for α-actinin network proteins present in the cytoskeletome. Solid lines represent direct interactions, where the indicated proteins have been previously demonstrated to bind to one another. Dashed lines represent indirect interactions, where the indicated proteins do not directly bind to one another, but associate via other protein complexes.