

Abstract
Osteocytes comprise the overwhelming majority of cells in bone and are its only true “permanent” resident cell population. In recent years, conceptual and technological advances on many fronts have helped to clarify the role osteocytes play in skeletal metabolism and the mechanisms they use to perform them. The osteocyte is now recognized as a major orchestrator of skeletal activity, capable of sensing and integrating mechanical and chemical signals from their environment to regulate both bone formation and resorption. Recent studies have established that the mechanisms osteocytes use to sense stimuli and regulate effector cells (e.g. osteoblasts and osteoclasts) are directly coupled to the environment they inhabit – entombed within the mineralized matrix of bone and connected to each other in multicellular networks. Communication within these networks is both direct (via cell-cell contacts at gap junctions) and indirect (via paracrine signaling by secreted signals). Moreover, the movement of paracrine signals is dependent on movement of both solutes and fluid through the space immediately surrounding the osteocytes (i.e. the Lacunar-Canalicular System, LCS). Finally, recent studies have also shown that the regulatory capabilities of osteocytes extend beyond bone to include a role in endocrine control of systemic phosphate metabolism. This review will discuss how a highly productive combination of experimental and theoretical approaches has managed to unearth these unique features of osteocytes and bring to light novel insights into the regulatory mechanisms operating in bone.
Keywords: Osteocytes, Biomechanics, Mechanotransduction, Intercellular communication
1. Introduction: Cellular Effectors and Controllers in Bone Remodeling
Osteocytes are the most abundant cell type in bone, and arguably the least well understood. The technical advances that greatly accelerated research into osteoblast and osteoclast biology in the 1970’s and 1980’s, such as the identification of cell type-specific markers, development of techniques for bone cell isolation and culture, and the establishment of phenotypically stable cell lines [1–3], have only lately proved fruitful for osteocytes. Until the last decade, osteoblasts and osteoclasts – the effector cells that carry out bone formation and resorption – remained the focus of research aimed at understanding bone modeling and remodeling. While those studies yielded a wealth of information, it has become increasingly appreciated that control of these processes is concerned less with regulating the activities of the effector cells than with determining when and where they are produced in the first place. Both osteoclasts and osteoblasts, after all, are formed from their respective progenitor cells only “on demand” when required to re-shape (“model”) bone in response to altered mechanical demands or to remove and replace (“remodel”) bone that has outlived its usefulness. The cells that process the cues and trigger the initiation of bone resorption or formation must therefore be present in bone before the effector cells are formed. Consistent with this recognition, a growing body of evidence now supports the concept that the osteocyte is the principal cell type responsible for integrating the mechanical and chemical signals that govern modeling and remodeling and control the onset of both bone resorption and formation. In addition, more recent studies demonstrate that osteocytes exert regulatory influences beyond the borders of bone by participating in endocrine pathways that regulate phosphate metabolism.
This review will summarize the data implicating osteocytes as major regulatory cells in bone, and will focus particularly on the way their unique local environment inside the mineralized matrix of bone permits them to play that role. We will first consider some general features of the osteocytes, their environment, and the implications of that environment for osteocyte function. Next we will address the osteocytes as mechanosensors and the potential mechanisms available for detecting the mechanical status of bone. Third, we will consider potential mechanisms of osteocyte communication with each other, and with other bone cell types (osteoblasts, osteoclasts and bone lining cells) in how they relate to the control of bone formation and resorption. Finally, we will discuss recent evidence implicating osteocytes as participants in an endocrine pathway governing systemic phosphate metabolism.
2. Osteocytes and their pericellular space: the constraints of living in a cave
Osteocytes are former osteoblasts that become surrounded by unmineralized matrix (osteoid) during bone formation; they make up 90–95% of the adult bone cell population. The complex phenotypic transition from osteoblast to osteocyte has been reviewed recently [1]. A major feature of the transition, however, is a dramatic shift in both shape and function from a cuboidal cell specialized for active secretion of extracellular matrix to a dendritic cell with a small cell body and numerous long, slender processes that connect to their neighbors – both nearby osteocytes and cells on the surface of bone, i.e. osteoblasts and bone lining cells [2–4]. The network of interconnected osteocytes is visible as the lacunae (holes in bone matrix occupied by osteocyte cell bodies) and canaliculi (channels in bone that surrounding the osteocyte processes) that constitute perhaps the most distinctive morphological feature of bone tissue at the microscopic level. Formation of osteocyte processes begins before the bone matrix mineralizes [3]. The specific mechanisms have yet to be fully elucidated in vivo. However, they appear to be dynamic and regulated [5]. Osteocytes in vitro, for example, can elongate their cell processes by secreting E11/gp38 dendritic proteins in response to fluid shear stress [6]. Matrix metalloproteinases are also required to complete the extension of processes through bone matrix. Holmbeck and colleagues showed that transgenic mice lacking membrane type matrix metalloproteinase 1 (MT1-MMP, or MMP14) form osteocyte processes, but never develop pericellular space around them [7]. Inuoe et al. also demonstrated that MMP-2 is essential for the formation of osteocyte canaliculi and networks in calvarial bone [8].
Once the osteoid mineralizes, the osteocytes are trapped in place and remain buried for the rest of their lives. However, residing in such a restrictive environment does not appear to prevent some of them from living long lives. Osteocytes in interstitial lamellae and other regions of bone that are not subject to wholesale turnover by remodeling often survive throughout the life of the organism. For example, inner ear bones (bones that do not undergo remodeling) of 95 year old humans lost about half of their osteocytes, indicating that the surviving osteocytes in the bone were approximately as old as the individual. [9]. By contrast, the same study showed that about 30% of osteocyte lacunae were empty in rib bones – a bone that undergoes remodeling throughout life.
Osteocytes also experience other constraints as a result of their site of residence. First, direct contact of osteocytes with other cells is limited to the tips of their processes and does not occur along extended cell-cell borders, as in tissues where cells are closely packed in two or three dimensions. As a result, the number of cell contact-mediated reception sites is proportional to the number of processes. Moreover, signals received at the tip of a process must travel a considerable distance to reach the bulk of cellular machinery in the cell body. In this respect, signal transmission among osteocytes may resemble that within dendritic trees of nerve cells. (Interestingly, osteocyte processes also contact the mineralized matrix surrounding them at occasional points [10, 11], a feature that will be discussed further in Section 3). Second, only a narrow pericellular fluid space separates osteocyte cell bodies and processes from a mineralized bone matrix that is sparingly permeable to fluid and solutes. The pericellular space within the lacunar-canalicular system (LCS) of an osteocyte network is, however, continuous with the periosteal, intracortical and endocortical envelopes, since the osteocytes also contact lining cells or osteoblasts on bone surfaces [12]. As such it constitutes the principal, if not sole conduit through which nutrients and wastes can move between osteocytes and the rest of the body. In addition, fluid movement through the LCS is a means by which osteocytes sense global tissue mechanical loading as detailed in Section 3. Briefly, both osteocyte cell bodies and cell processes are subject to fluid flows and forces generated by loading, and several mechanisms have been proposed to explain the ability of osteocytes to sense them. In summary, however, the movement of fluids and solutes through the pericellular space of the LCS appears to impact virtually every aspect of osteocyte function.
The pericellular space between the osteocyte cell body and the lacunar wall is approximately 0.5–1 μm in width [13], while the membranes of osteocyte processes are separated from the canalicular walls by an “annular” space in the range of 50–100 nm [13]. These dimensions were determined from ultrastructural studies carried out under conditions used to study cartilage to prevent cell shrinkage and proteoglycan collapse. The composition of the pericellular fluid has not been precisely defined. However, it likely resembles that in articular cartilage, in terms of its ionic composition and may well contain a diverse array of macromolecules, some of which are the products of osteocytes. Osteocytes synthesize several bone matrix noncollagenous proteins including osteopontin (OPN), osteocalcin (OCN) and dentin matrix protein 1 (DMP1), as well as proteoglycans and hyaluronic acid (HA) [14, 15]. Osteocytes have also been reported to produce several cytokines (discussed in a later section). One function of macromolecules within the LCS is to maintain the patency of the fluid space, a process that appears to require active cellular participation since osteocyte lacunae and canaliculi mineralize after osteocytes die [16]. OPN and DMP1 have the capacity to bind calcium and to inhibit mineralization, and so are likely candidates for this role [17–19]. Perlecan also may contribute to maintaining open LCS channels, since mice lacking this proteoglycan were recently found to exhibit unusually narrow canaliculi [14].
The organization of proteoglycans within the LCS is a matter of particular interest. Experience with cartilage had demonstrated that traditional means of processing for electron microscopy led to the collapse of GAG chains in matrix proteoglycans and thus produced highly inaccurate representations of the space occupied by the proteoglycans [10]. A consequence of the use of structure-preserving tissue processing methods in the study by You et al. [13] was the novel demonstration that osteocyte cell bodies were surrounded by a rather disorganized pericellular matrix and that osteocyte processes in canaliculi exhibited discrete fibrous connections to the canalicular walls termed “tethers” at regularly spaced intervals. The presence of tethering elements (if not their number and molecular identity) had been predicted previously on theoretical grounds as a means to explain the mechanoresponsiveness of osteocytes (discussed in greater detail in Section 3) [13]. While the molecular identity of the tethers has yet to be established, proteoglycans are likely to be a crucial element. In fact, perlecan is a reasonable candidate since the length of its core protein is similar to that of the annular space [20].
The presence of tethers, proteoglycans and other macromolecules within the LCS suggests that movement of both fluids and solutes would be restricted to some extent, and this has been verified experimentally. The permeability of the LCS to fluorescein, a low molecular weight dye, was found to be similar to that of articular cartilage [21]. Solute transport through the LCS occurs both by diffusion and convection. Diffusion depends on solute concentration gradients, while convection results because the fluid itself moves through the pericellular space. Since fluid is incompressible, deformation of bone under mechanical load forces it to flow (like squeezing a sponge), as demonstrated by Piekarski and Munro [22]. Major sources of mechanical load driving convective fluid flow through the LCS (in both directions) are muscular action and reaction forces due to physical activity [22, 23]. The permeability characteristics of the bone LCS have been interrogated extensively by Fritton, Knothe-Tate, Wang and other investigators, primarily using fluorescent solutes of differing molecular dimensions [19, 24–26]. Their results indicated that solute movement is highly dependent on size; small solutes under 1 kDa or roughly 1 nm diameter (e.g. steroid hormones, glucose, ATP, NO, CO2, and O2) move easily by diffusion in the fluid space [19, 21, 24, 27, 28], while larger solutes in the range of 10–70 kDa or up to 6 nm diameter (e.g. growth factors, hormones and cytokines like parathyroid hormone (9.5 kDa), sclerostin (24 kDa) and RANKL (35 kDa) require convective transport [19, 24, 29–31]. Molecules greater than 70 kDa (such as monoclonal antibodies) were effectively excluded from the LCS [19]. From the example molecules identified above, it suggests that potential osteocyte signaling pathways utilizing secreted messengers of various sizes will exhibit different dependencies on fluid movement through the LCS. It also follows that their movements will depend on the composition/organization of the pericellular matrix. How pericellular matrix composition and organization are regulated on a day-to-day basis, and whether they vary between cortical and cancellous bone, or with changes during aging and disease, are not known.
3. Osteocytes as Mechanosensors
Bone has historically been recognized as an exquisitely mechanosensitive tissue, as exemplified by the long histories of manipulating bone structure by the application of forces in orthopaedics and orthodontics well before the studies of Julius Wolff and Rudolf Virchow in the 19th century [32]. Equally longstanding, however, is the mystery of how bone recognizes and responds to mechanical loading. Even as osteocytes became recognized as the critical sensors of mechanical loading in bone over the last decade, investigators of mechanobiology at the cellular level have been faced with a multitude of choices to explain the mechanisms that might account for their mechanosensitivity. Virtually all cells are intrinsically sensitive to mechanical stresses produced by a range of stimuli including substrate deformation, fluid shear or changes in hydrostatic pressure, and osteocytes experience all of these stimuli to some degree. During normal physiological loading (e.g. walking), interstitial fluid pressure causes flow into and out of the LCS and producing shear stresses along osteocyte membranes [22, 23]. Changes in interstitial hydrostatic pressure also result [33, 34], as does deformation of bone matrix itself [35], though the rigidity of the tissue indicates that such deformations are very small [36, 37].
Physiological tissue strain experienced by bone ranges from 400–3000 microstrain (με, 0.04–0.3%) [36, 37], with the vast majority of in vivo loading occurring at the low end of this range [37]. However, mechanical strains that elicit biological responses from osteocytes in vitro range from 5000–10,000 με (5–10%) [38, 39], similar to all connective tissue cells in vitro. Moreover, bone typically breaks at approximately 1% strain [40]. Thus it is unrealistic for the healthy osteocytes to be experiencing such high strains in vivo. These findings imply that osteocytes must experience something very different from mechanical strain in the cellular level in situ. Recent evidence from both in vivo and in vitro studies now appears to support the concept that mechanical loading-induced fluid flow in the LCS is likely the predominant force that osteocytes recognize and respond to by regulating bone modeling and/or remodeling activity [38].
Nearly two decades ago, Weinbaum et al. recognized that the fluid shear stress experienced by osteocyte processes was of the same order of magnitude as that experienced by endothelial cells [23]. This suggests that fluid shear stress generated from mechanical loading-induced fluid flow might be one of the major mechanical stimuli that act on osteocytes. Indeed, numerous in vitro studies utilizing isolated primary osteocytes or established osteocytic cell lines (principally the mouse cell line MLO-Y4) have shown that fluid flow induces osteocytes to produce several physiologically relevant secondary messengers including ATP [41], nitric oxide [42], Ca2+ [43], and prostaglandins [44]. Because the forces generated by flow are directly dependent on fluid flow rates, their magnitudes differ dramatically between the small caliber canaliculi and the much larger lacunae. Fluid moving in the narrow canalicular space between the osteocyte process and bony wall (radial dimension of 50–100 nm [13]) generate much higher fluid shear stresses than in a larger pericellular space in lacunae (0.5 – 1 μm [13]) where fluid velocities are lower [29, 45] (like water flowing from a narrow garden hose versus water in a swimming pool). As a result, potential sensory mechanisms in osteocyte cell processes and cell bodies would necessarily be different.
With additional discoveries about the osteocytic pericellular environment and attachments over the last decade came further understanding of the effect of fluid flow on the other forces that acted on the osteocytic cell membrane, and to novel insights into the likely nature of cellular mechanotransduction in situ. Weinbaum, Schaffler and colleagues posited that “tethering” of osteocyte process membranes to canalicular walls would produce both drag forces and radially-directed strains in the cell process cell membrane in response to interstitial fluid flow [46, 47]. Critically, they predicted these membrane strains to be much greater than the applied tissue-level strains. Several key pieces of experimental evidence support this model (i) Ultrastructural studies showed that osteocyte processes contain a regular array of bundled microfilaments typical of the actin filaments observed in cell processes (Figure 1A), and this actin bundled within osteocyte cell processes is highly rigid compared to the cell body [13] (ii) Additional electron microscopic evidence identified transverse tethering elements spaced regularly along the osteocyte processes and linking them with the canalicular walls through the pericellular space [13]. Recent data suggest that perlecan is a likely candidate for these tethers [14] (Figure 1B). iii) Immunohistochemical evidence also indicates that osteocyte processes contain CD44 [15], a transmembrane protein whose extracellular domain is a receptor for hyaluronic acid and whose intracellular region links to actin cytoskeleton [48]. Certain cell-associated proteoglycans (i.e. syndecan, heparan sulfate) have core proteins that span the plasma membrane and exhibit intracellular signal transducing capability in endothelial cells [49, 50], but their relevance to osteocytes is unknown. Intriguingly, in vitro evidence by Reilly et al. indicates that cell-associated proteoglycans may have a specific role in osteocyte mechanotransduction. Enzymatic treatment of osteocytes in vitro to remove cell surface-associated proteoglycans eliminated the increase of PGE2 production in response to fluid flow, but did not abolish calcium signaling [51].
Figure 1.

Schematic representation of key microstructural/ultrastructural features of osteocytes implicated in mechanosensing. A) Transmission electron (TEM) photomicrograph of an osteocyte process containing bundled F-actin (large black structures indicated by white arrow). Actin bundles in osteocyte processes increase their stiffness and limits membrane deformation.* B) TEM photomicrograph of proteoglycan tethering elements (black arrows) bridging osteocyte cell process to bony canalicular wall. Tethering proteoglycans such as perlecans are spaced approximately every 40 nm along the osteocyte process. These proteoglycan tethers exert a resistance to mechanical loading induced fluid flow, and the resulting drag force may be sensed at the cell process membrane. C) Fluorescent immunohistochemical (IHC) staining showing that β1 integrins (white arrows) are located only on osteocyte cell bodies.** D) TEM photomicrographs demonstrating the discrete protrusions from the canalicular wall (matrix hillock protrusions), which contact osteocyte processes; comparable direct bone-to membrane-attachment sites are not seen in osteocyte lacunae. E) Fluorescent IHC staining for β3 integrins (white arrows) are present in a punctate pattern along osteocyte processes. β3 integrins have shown to have a similar periodicity and spacing pattern to matrix hillock protrusions, and are absent from cell bodies. The localized strains at these adhesion sites caused by fluid flow-induced membrane deformations would be one to two orders of magnitude larger than whole-tissue strains (i.e. “local strain amplification”). *** [* Figure from You et al. [13] reprinted with permission; ** and ***: Figures from McNamara et al. [10], reprinted with permission.]
In addition to transverse tethers, osteocyte processes also attach directly to canalicular walls. McNamara et al. found discrete “hillocks” protruding from canalicular walls and forming close contacts with osteocyte process membranes [10, 52]. Kamioka et al. verified this finding by 3-D high voltage electron microscopy, though their images suggested a more crest-like protrusion from the structures along the osteocyte processes (Figure 1D)[11]. The molecular nature of the direct attachments between the cellular process and bony wall has not been definitively established. However, several lines of evidence indicate that the attachments are mediated by β3 integrins. McNamara et al. demonstrated by immunohistochemistry that β3 integrins in bone are found at discrete points between osteocyte cell bodies, suggesting a location on osteocyte processes (Figure 1E). In contrast, β1 integrins were found only on the osteocyte cell bodies (Figure 1C) [10]. Osteopontin, a bone matrix protein that interacts with β3 integrins via its Arg-Gly-Asp (RGD) recognition sequence and with mineral crystals via acidic amino acids and multiple phosphorylation sites, has been identified on canalicular walls and seems the likely ligand for β3 integrins in osteocyte processes [53]. Osteopontin has also been considered a major ligand for β3 integrins in osteoclasts, mediating their binding to bone surfaces during resorption [54]. β3 integrins have also demonstrated the ability to transduce mechanical signals in several cell types [55, 56] including osteocytic cells in vitro [57].
To investigate how the presence of direct integrin attachments along the osteocyte process would contribute to Weinbaum’s original strain amplification theory, Wang et al. mathematically modeled the mechanical effects of direct contacts between osteocyte processes and canalicular walls via rigid “spot-welds”, in combination with flexible proteoglycan tethers. They found that membrane strains at the integrin attachments were an order of magnitude larger than the radial strains from tethering elements predicted from previous mathematical models, and two orders of magnitude greater than whole-tissue strains [52]. In other words, focal attachments between the cell process membrane and the bony wall provided additional sites with even greater membrane strain amplification. Because of this strain amplification complex, tiny forces (1–10 pN) can produce membrane strains of 5000–10,000 με at the attachment sites, which are the levels of membrane strain known to trigger calcium influx and initiate biological signals in a wide range of connective tissues [39].
As noted previously, the osteocyte processes are much more rigid than the cell body due to their tightly cross-linked actin bundles, a feature which predicts increased mechanosensitivity of these processes compared to the cell bodies in response to fluid stress [58]. Wu et al. recently confirmed this regional difference in mechanosensitivity within a cell using a novel fluid stimulus probe. They found that loads of 1–2.3 pN applied to osteocytic processes in vitro evoked calcium signaling while similar loads applied to all bodies caused no response. This combination of mathematical prediction and experimental findings suggest that the osteocyte processes are substantially more sensitive to mechanical forces/deformation than the cell body.
The observation that β1 and β3 integrins appear to localize on discrete regions of osteocytes that differ in mechanosensitivity suggests that the two integrins also have distinct mechanical functions. This is an intriguing finding since β1 integrins, which comprise the largest and most diverse integrin family, have also been shown to transduce mechanical signals in a wide range of cell types [59, 60]. However, mice with a conditional knockout of β1 integrins in osteoblast-lineage cells displayed no skeletal differences from their wild type counterparts unless they were challenged with acute disuse or 3 days of cyclic loading [61]. These findings raise the possibility that β1 integrins in osteocytes may only play a minor role in mechanotransduction during normal ambulatory loading in both the growing and adult skeletons. To date, no conclusive in vivo data have been presented to indicate a major role for osteocytic β1 integrin in mechanical sensing and transduction.
Other investigators have hypothesized that the osteocyte cell body may have another method of sensing mechanical strain in vivo. Nicollela and colleagues argued that the complex microarchitecture of the osteocytic perilacunar space can give rise to very high local strains under physiological loading [62]. However, in vivo experimental data support of this hypothesis is limited. Other investigators have suggested primary cilia as another candidate for mechanosensation and mechanotransduction. Primary cilia are microtubule-based organelles that protrude beyond the surface of many eukaryotic cells but have no role in cell motility. Rather, they sense and transduce chemical and fluid-flow mechanical signals [63]. A recent study by Malone et al. demonstrated the presence of primary cilia on MC3T3-E1 osteoblasts and MLO-Y4 osteocytes in vitro [64], and led the authors to posit that primary cilia in osteocytes and osteoblasts may be required to mediate both osteogenic and pro-osteoclastogenic responses (COX2 upregulation and lowered OPG/RANKL ratio, respectively) to fluid flow. However, some questions remain to be addressed concerning primary cilia as mediators of skeletal mechanotransduction, particularly in osteocytes. First, primary cilia lengths range from 2 μm in chondrocytes [65] to 30 μm in kidney epithelial cells [66]. This length is necessary for the primary cilia to deflect under fluid flow (i.e. short cilia are too rigid to deflect). The perilacunar space between osteocyte cell bodies and lacunar wall is less than 1 μm [67], which is simply not enough space for primary cilia to function as effective sensors of fluid flow within the confined spaces of lacunae as they do elsewhere. Second, bones from mice in which primary cilia were deleted from osteoblasts and osteocytes by knocking out Kif3a, (a subunit of the kinesin II intraflagellar transport protein responsible for primary cilia formation and function) exhibited largely normal skeletal phenotypes. Some attenuation of responsiveness to experimental high force mechanical loading was observed, but the extent to which this reflects changes in osteoblasts or osteocytes is unknown. Finally, patients with polycystic kidney disease (PKD) have dysfunctional primary cilia due to genetic mutations of polycystin 1 (PKD1) and polycystin 2 (PKD2) [68], but also show no abnormalities in skeletal phenotype [69]. This lack of change in skeletal phenotype is also demonstrated in the PKD1 and PKD2 knockout mouse models [70, 71].
In summary, growing evidence has clearly established the osteocyte and its network as the principal mechanosensory element in bone. In the scenario developed by Weinbaum and colleagues and more recently Kamioka et al., the sensitivity of osteocytes to mechanical stimuli appears to result from a distinctive anatomy, in particular its rigid dendritic processes relative to its cell body, and from structural features of its pericellular environment. However, speculation about the possible role of primary cilia and focally elevated lacunar strains has also been proposed. In all of these instances, fluid flow through the LCS is the primary mechanical stimulus. Still, numerous questions remain about osteocyte mechanotransduction, in particular the nature of intracellular signaling pathways activated by the sensing mechanisms, and the way osteocyte networks act to integrate these signals leading to appropriate modeling or remodeling responses.
4. Osteocyte Communication
Although osteocytes live in highly restricted circumstances, they do not exist in isolation. Rather, osteocytes exist in networks that allow them to communicate with each other and with other cells on bone surfaces. Each network is a functional syncytium – not a true syncytium, which refers explicitly to a “multinucleated mass of cytoplasm that results from the fusion of cells” [72], like muscle cells, osteoclasts and slime molds. The term “osteocytic syncytium”, while growing in popularity, is incorrect. Communication within the osteocyte network is important for effective integration of both local and global stimuli (e.g. mechanical load), while communication to surface cells is essential for osteocytes to relay sensory inputs from within the bone to bone surface sites where bone resorption and formation are initiated. Osteocytes use several mechanisms, both direct (gap junctional intercellular communication) and indirect communication (paracrine signaling) to accomplish these signaling tasks.
Direct cell-cell communication
Where osteocytes connect to their neighbors, at the ends of their dendritic processes, they form gap junctional connections that render their cytoplasms continuous, allowing the movement of molecules < 1 kDa and the transmission of electrical potential within the osteocytic network (i.e. the functional syncytium). Among the molecules that pass through gap junctions are monovalent and divalent mineral ions (Na+, K+, Ca2+, Mg2+), nucleotides, and other low molecular weight signaling molecules like inositol phosphate, and cyclic nucleotides. Doty et al. were the first to observe that osteocytes are connected to each other via gap junctions in vivo [12], while Marotti et al. demonstrated that this form of communication exists between osteocytes and osteoblasts, bone lining cells and even beyond to the endothelial cells of the bone capillaries [73].
Gap junctions are transmembrane channels formed by the linkage of two membrane protein complexes (termed connexons or hemichannels) on adjacent cells. Each connexon consists of a hexamer of transmembrane proteins (connexins) that form a pore. The most abundant of these proteins in bone is connexin 43 (Cx43), although others such as connexins 45 and 46 are also present [74]. The association between two opposing connexin hemichannels to form a functional gap junction is non-covalent and reversible.
Much of our knowledge about gap junctions in bone cells, particularly at the mechanistic level, originates from in vitro studies, since cells in culture express connexins, form hemichannels and establish gap junctions when in contact. Yellowley et al. were the first to identify the existence of functional gap junctions between MLO-Y4 osteocyte-like cells, and their abundant expression of Cx43 [75]. In response to fluid shear stress in vitro, MLO-Y4 osteocyte-like cells redistribute Cx43 from the cytoplasm to the cell surface [76], and enhance osteocytic intercellular communication by increasing overall Cx43 expression [77].
Functional analysis of gap junctions, both in vivo and in vitro, frequently relies on the use of antagonistic drugs such as carbenoxolone or mefloquine [78]. There are certain caveats to the use of channel blocking drugs, since they may act upon more than one type of channel and their selectivity may vary depending on the concentration used. More recently, efforts to understand the role of Cx43 in osteocyte communication and mechanotransduction in vivo have utilized different strategies based on the development of transgenic mice with cell-selective deletions in Cx43. Civitelli et al. and Donahue et al. created transgenic mice where Cx43 was ablated at different points within the osteoblastic and osteocytic lineage [79, 80]. Both strains generally exhibited normal skeletal growth and development, but each differed from wild-type mice in its response to mechanical challenge, [81–83]. These alterations in mechanoresponsiveness suggested a role for Cx43 gap junctional communication in regulating communications among osteocytes, osteoblasts and osteoclasts during bone modeling and remodeling (discussed in later sections)
The fact that osteoblast and osteocyte-specific deletion of Cx43 proteins did not lead to major deficiencies in bone formation and resorption does not necessarily rule out an important role for gap junctions in preserving osteocyte connectivity and communication. In astrocytes, for example, loss of Cx43 function results in compensation by other connexins [84]. Civitelli and colleagues also reported an increase in Cx45 protein expression in calvarial Cx43-null osteoblasts, suggesting that Cx43 function in osteocytes may also be compensated by other connexin proteins [85]. Currently, a detailed analysis of osteocyte connectivity in Cx43 deficient mice has not been reported.
Paracrine Signaling by Small (Short Range) Metabolites
Aside from direct cell-to-cell contact, osteocytes have been shown in vitro to communicate with other osteocytes and bone cells via delivery of small signaling molecules (i.e. metabolites like prostaglandins, nitric oxide and ATP) over a short distance through the extracellular fluid. In this context, two types of membrane channels permitting movement of such signaling molecules between the cytoplasm and the extracellular space have received particular attention: hemichannels and pannexin channels. In vitro studies on osteocytes indicate that hemichannels, which are non-junctional connexons (essentially half of a gap junction) can exist on cell surfaces and function in molecular transport. In low-density (no cell contact) cell culture systems that have few gap junctions, osteocytes have been shown to increase prostaglandin E2 (PGE2) expression, ATP release and cell surface expression of Cx43 after fluid flow treatment [41, 44]. Furthermore, application of fluid shear stress on osteocytes also promotes PI3K signaling which leads to the binding of integrin α5 to open Cx43 hemichannels [86]. These studies demonstrate a possible mechanism for hemichannels to aid in osteocytic communication via autocrine/paracrine signaling in response to mechanical stimuli. However, it should be noted that integrin α5 partners exclusively with β1 [87], and so its actions would likely be restricted to osteocyte cell bodies in vivo. Moreover, the importance of osteocytic hemichannel function has been questioned since all studies to date have been performed in vitro, while in vivo hemichannel function in other systems such as brain [88], heart [89], and astrocytic neurons [90] remains controversial. Thus the role of hemichannels in osteocyte-osteocyte communication in situ has yet to be established.
Pannexin channels are non-junctional membrane channels in vertebrates that have a sequence homology to the proteins that form gap junctions in invertebrates (the “innexins”) [91]. Critically, vertebrate pannexin channels exist only in an uncoupled form – unlike connexin–based channels, they do not form cell-cell junctions. Pannexins exist in a wide range of cells in situ [92], and have been demonstrated to regulate the movement of paracrine signaling molecules between intracellular and extracellular spaces [93], arguing that they are a more likely mode of autocrine/paracrine signaling among osteocytes than Cx43 hemichannels [92]. Recent data demonstrate that pannexin 1 (Panx1) channels are mechanically sensitive [94], and that association of Panx1 channels with purinergic P2X7 receptors (P2X7R) aids in ATP transport from the cell to the extracellular space [92]. While pannexin channels have not yet been reported to function in osteocytes, Panx1 channels are known to play a role in ATP release by osteoblasts.
Paracrine Signaling by Macromolecules
Osteocytes both secrete and respond to a variety of macromolecular signaling molecules. Among the macromolecules produced by osteocytes are sclerostin (24 kDa), FGF23 (32 kDa), and RANKL (20–30 kDa), while those for which osteocytes express receptors include PTH/PTHrP (ca 9 kDa). These signals and others will be discussed in later sections in the context of their physiological functions. However, we note here that the use of large signaling molecules by osteocytes has often been viewed as surprising, given the spatial constraints of solute movement through the LCS. Few if any in situ demonstrations of macromolecular signaling to and from osteocytes in vivo have been provided. Macromolecular signaling by osteocytes has usually been inferred from histochemical demonstrations of the signaling molecules or their receptors in osteocytes at the gene or protein level, or more recently by functional analysis of osteocyte-specific conditional knockouts produced by Cre-lox technology [95, 96]. In any event, the size range of most macromolecular signals makes it likely that they will require mechanical loading-induced convective fluid flow for effective transport through the LCS.
In summary, osteocytes are known to transfer small signaling molecules to each other and other bone cells directly via gap junctions, or indirectly via hemichannels or pannexin channels. Although in vitro data suggest that hemichannels and pannexins aid in osteocyte autocrine/paracrine signaling, in vivo validation is required to elucidate their physiological significance. Osteocytes can also communicate via secretion of macromolecules, which require mechanical loading induced-interstitial fluid flow to assist in their transport to their target cells.
5. Osteocyte-osteoblast communication
Osteocytes communicate with osteoblasts and lining cells at bone surfaces by direct (gap junctions) [12, 97], and by indirect (extracellular paracrine) signaling pathways. In each case, signal transmission is subject to the same constraints discussed previously: size of the signaling molecule, gap junctional selectivity, interstitial fluid flow patterns and resistance of the pericellular matrix. In addition, the spectrum of signaling molecules utilized by osteoblasts and osteocytes is similar, undoubtedly reflecting their close developmental relationship. Among the signals common to osteocytes and osteoblasts are small molecules often considered “ubiquitous” in cell physiology, like nitric oxide (NO), nucleotides/nucleosides, prostanoids, as well as larger peptides such as insulin-like growth factors (IGFs). Osteocytes also express the PTH/PTHrP receptor, indicating that these entombed cells can respond not only to a local paracrine factors, but also to the endocrine regulator PTH [95].
Investigations of paracrine signaling from osteocytes have focused on anabolic signals produced by osteocytes in vitro and in vivo and found to be enhanced by mechanical stimuli. These investigations were consistent with the concept of bone adaptation in response to mechanical demand [32]. Cyclic AMP (cAMP) [98] and prostaglandin E2 (PGE2) [99] were early signaling molecules shown to be upregulated by mechanical loading of bone cells. Prostaglandin production in bone tissue is also stimulated by mechanical loading in conjunction with increased expression of cyclooxygenase-2 (COX-2) [100, 101], the inducible form of the enzyme that converts fatty acids into prostanoids [102]. Similarly, osteocytic cells in vitro were shown to increase COX-2 expression and prostaglandin production in response to diverse mechanical stimuli including fluid flow [103, 104], substrate deformation [105] and hydrostatic pressure [34]. Of the prostaglandins, PGE2 is the most extensively studied with respect to bone anabolic effects, although prostacyclin (PGI2) has also been reported to have similar actions [106, 107]. Sorting out pathways of prostaglandin signaling between osteocytic and osteoblastic cell populations presents problems, particularly in situ, since both osteoblasts and osteocytes produce prostaglandins. Moreover, osteocytes and osteoblasts appear to express different subsets of prostaglandin receptors. Both osteoblasts and osteocytes express EP4 receptors, but a second class, EP2 receptors are only found on osteocytes [108]. The functional significance of this difference is not clear.
Nitric oxide (NO) is also another mechanical loading-induced anabolic molecule produced by osteocytes [109]. NO is synthesized by a family of nitric oxide synthases (NOSs), including the endothelial, inducible and neuronal isoenzymes (eNOS, and iNOS and nNOS) respectively [110, 111]. NO has a biphasic effect on osteoblastic activity: low concentrations promote osteoblast proliferation and differentiation, while high concentrations have been reported to promote osteoblast apoptosis in vitro [112]. Osteoblasts derived from singly defective eNOS, iNOS or nNOS mice demonstrate enhanced nitric oxide production in response to mechanical stimuli [113], Mice with defective eNOS [106] or nNOS [114] also display altered skeletal phenotypes compared to wild type mice, however whether they result from changes in osteoclasts, osteoblasts or osteocytes in situ remains to be established. Both prostaglandins and NO have half-lives on the order of seconds, and thus their actions are limited in range [115]. Their rapid secretion after mechanical loading and their anabolic effects on osteoblasts are consistent with this short-range action. However, this behavior suggests that if osteocytes utilize these molecules to regulate osteoblasts, then only the osteocytes closest to bone surfaces are involved in signaling, or some mechanism must be available to propagate the signals throughout the osteocyte network. To date, such a propagation mechanism has not been established.
IGF-1 is a well-established anabolic regulator of bone growth in vivo [116]. A principal mediator of growth hormone (GH) actions on skeletal growth, IGF-1 and its binding proteins are produced by a wide range of connective tissue cells that act as a local regulator independent of GH. Osteocytes express IGF-1, and a conditional knockout of IGF-1 from mouse osteocytes leads to severe impairments to longitudinal and periosteal bone growth and development of thinner calvarial bone [117]. In addition, osteocytic expression of IGF-1 is mechanically sensitive, as mechanical loading on rat tail vertebrae leads to an increase in trabecular and cortical osteocytic IGF-1 mRNA as early as 6 hours after mechanical loading [118]. Four-point bending of rat tibiae also increases osteocytic IGF-1 mRNA expression, which corresponds to an increase in endosteal bone formation [119].
While osteocytes can stimulate osteoblastic activity by the mechanisms described above, some of their most dramatic actions on osteoblasts are inhibitory. Osteocytes are the major producers of sclerostin (the protein product of the SOST gene) [120], although osteoclasts and chondrocytes have also been reported to secrete sclerostin in aged mice [121, 122]. Sclerostin antagonizes the canonical Wnt signaling pathway, which is the major driver of osteoblast bone formation. In brief, sclerostin binds to members of a Wnt co-receptor family, the lipoprotein receptor-related proteins (LRP 4, LRP 5/6), thereby limiting their ability to interact with Wnts and their co-receptor Frizzled (Frz) [123, 124]. Osteocytes also produce dickkopf-1 (DKK-1), another inhibitor of the Wnt signaling pathway that binds Lrp4 [124] and Lrp5/6 co-receptors to inhibit canonical Wnt signaling and downstream bone formation [125]. Although mechanisms of interaction between sclerostin and DKK-1 are not fully understood, they can act synergistically to inhibit osteoblast activity. Sclerostin and DKK-1 do not bind simultaneously to LRP5/6 co-receptors, and DKK-1 can displace sclerostin from previously formed sclerostin-Lrp5 complexes [126].
Mechanical loading decreases expression of both sclerostin and DKK-1 expression in association with increased bone formation [127, 128], whereas unloading increases osteocytic secretion of these proteins and inhibits bone formation [129–131]. Of particular note, sclerostin is the only paracrine regulator of osteoblastic activity whose expression in osteocytes was found to be directly proportional to mechanical strain in bone [128]. Taken together, these results support the concept that osteocytes, largely through sclerostin, play a major role in determining the size and shape of bone by limiting its expression of paracrine factors based on their ability to sense and respond to mechanical demands.
Recognition that sclerostin acts as a negative regulator of bone formation but has no effects on resorption has made it an attractive therapeutic target, particularly as a bone anabolic agent. Specifically, monoclonal anti-sclerostin antibodies have been shown to activate bone formation in both animals [132, 133] and humans [134]. Initial human clinical trials of the monoclonal sclerostin antibody (AMG-785) demonstrate that AMG-785 treatment significantly increases bone mineral density (BMD) up to 5.3% in the lumbar spine, and 2.8% in the hip [134]. These increases in BMD are comparable to those achieved by parathyroid hormone and bisphosphonate therapies that are currently used to treat osteoporosis [135]. The success of monoclonal antibodies directed against an osteocyte product is particularly intriguing since antibodies, even monovalent Fab antibody fragments, are too large to enter the LCS even with convective flow [19]. While antibodies might gain limited access to young osteocytes and their secretory products at sites of bone formation where the matrix has not yet mineralized, this suggests that AMG-785 must be interacting with sclerostin at, or near the surface of bone.
In summary, osteocytes communicate with each other and cells within the osteoblastic lineage (i.e. osteoblasts and lining cells) by direct cellular contact via gap junctional signaling and by paracrine signaling. Osteocytes produce macromolecules (e.g. IGF-1, sclerostin) as well as small molecules (e.g. PGE2, NO, nucleotides) that act as regulators of osteoblast activity. In addition, production of many of these paracrine signals is subject to regulation by mechanical stimuli, often in a manner consistent with demonstrated anabolic and catabolic actions of mechanical load. Thus, mechanical loading upregulates expression of factors with demonstrated anabolic actions on osteoblasts (PGE2, PGI2, NO, and IGF-1), while reduction or absence of mechanical load downregulates those factors but upregulates osteoblast inhibitors like sclerostin (Figure 2). The relationship between sclerostin expression by osteocytes and osteoblast activity under varying conditions of mechanical load has been especially well established, supporting the concept that this osteocyte product is a major mediator of mechanically regulated bone formation. These findings have led to early clinical successes in the development of bone anabolic agents.
Figure 2.
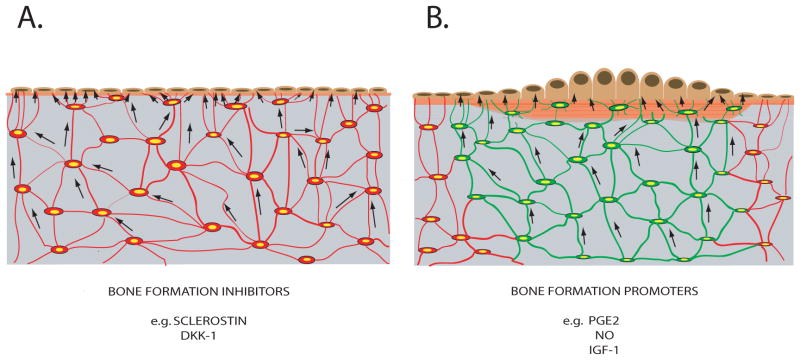
Schematic summary of osteocytes in bone and their signaling to regulate bone formation. A) Osteocytes underlying resting surface of bone lining cells. These osteocytes constitutively produce inhibitors of bone formation (i.e. sclerostin and DKK-1; shown in red). When osteocyte production of the bone formation inhibitors ceases, bone formation is activated. In such circumstances, (B), osteocytes underlying regions of active bone formation can produce signals that promote osteoblast activity (i.e. PGE2, NO, and IGF-1; shown in green)
6. Osteocyte-Osteoclast Communication
Osteocytes regulate bone resorption as well as formation. However, elucidating the communication pathways between osteocytes and osteoclasts (and their progenitors) has proved less straightforward than for osteoblasts, and uncovered some initially unexpected regulatory relationships. The idea that cells buried within bone can control the recruitment and subsequent differentiation of hematopoietic osteoclast progenitors at bone surfaces was first proposed by Frost in the early 1960s [136, 137]. As we describe below, subsequent experimentation validated this concept and established several key elements of the signaling pathway, including a consistent, obligate link between osteocyte death (specifically apoptotic, or regulated death) and the initiation of bone resorption. While this was a surprising finding, it is not a unique circumstance within the skeletal system. Cell death is also coupled to resorption in the growth plate, albeit the dying cell in that case is the hypertrophic chondrocyte [138].
Noble and colleagues were some of the first investigators to associate the presence of apoptotic osteocytes with areas of high bone turnover in normal and pathological bone [139]. Verborgt et al. made similar observations, as apoptotic osteocytes were found to co-localize with areas of osteoclastic bone resorption in fatigue damaged bone, suggesting that apoptotic osteocytes may serve as beacons for targeted osteoclast remodeling [140]. Indeed, osteoclasts have been shown to engulf apoptotic osteocytes [141, 142], thus establishing the fact that osteoclasts are capable of removing damaged osteocytes from a remodeling site. However, these studies did not establish a causal relationship between osteocyte cell death and osteoclastic resorption, nor did they discern whether osteocyte cell death preceded osteoclastic activity or resorption by osteoclasts led to osteocyte cell death.
Bentolila et al. developed a fatigue-loaded rat ulnae model based on the in vivo ulnar loading model by Lanyon and Torrance [143]. In the ulnar fatigue model, intracortical resorption is activated 10–14 days after microdamage induction [144]. Verborgt et al. used this model to study the relationship between osteocyte apoptosis and osteoclast resorption, and found that osteocyte apoptosis occurs as early as one day after fatigue loading [140], thus supporting the hypothesis that osteocyte apoptosis precedes the onset of osteoclastic bone resorption. Definitive demonstration of a causal relationship between osteocyte apoptosis and initiation of microdamage-induced bone resorption was provided by Cardoso et al, who showed that preventing osteocyte apoptosis with a pan-caspase inhibitor led to complete abrogation of the osteoclastic remodeling response [145]. Subsequent studies demonstrated that bone resorption induced by estrogen deficiency (ovariectomy) [146] and disuse (hindlimb unloading) [147] were also preceded by localized increases in osteocyte apoptosis, which was completely prevented by blocking apoptosis with the pan-caspase inhibitor. Tatsumi and colleagues also observed increases in osteocyte death and resorption in an osteocyte ablation mouse model [148], but did not examine the nature of osteocyte cell death.
Taken as a whole, the data from the above studies point to osteocyte apoptosis as a principal trigger for osteoclast resorption and generated many hypotheses to explain how the loss of osteocytes causes osteoclast resorption. Many investigators speculated that loss of osteocytes results in a loss of inhibitory signals towards bone resorption [149–152]. For instance, Maejima-Ikeda et al. found that osteocyte homogenates isolated from chick embryo calvariae markedly inhibited osteoclast pit resorption [152]. The authors also found a novel “bone-resorption-inhibitory protein” within the osteocyte homogenate with a molecular mass of 18.5 kDa, which was hypothesized to be responsible for the osteoclast inhibitory properties of the osteocyte homogenate. However, no subsequent studies identified this inhibitory protein. A later in vitro study by Heino et al. suggested that osteocytes constitutively secrete inhibitory molecules such as TGF-β (25 kDa) that prevent osteoclastogenesis and osteoclast resorption [151]. Gu et al. also reported that the presence of live osteocytes in rat organ cultures inhibited osteoclast pit resorption, while devitalized bone promoted osteoclast activity in vitro [150].
While these studies are consistent with the hypothesis that osteocytes provide inhibitory signals to functioning osteoclasts, they do not address the issue of how osteocyte loss can trigger the initial stages of bone resorption. Bone does not contain a pool of differentiated osteoclasts for osteocytes to repress in a constitutive manner. Rather, osteoclasts must be produced on demand from monocyte precursors recruited from the circulatory system [153, 154]. The recruitment process involves several steps including the binding of precursor cells to endothelium, and transmigration across both the endothelium and the layer of lining cells to the bone surface [155]. At that point the precursor cells differentiate into osteoclasts and begin to resorb bone in a directed fashion to reach the target region. The mechanisms that trigger localized endothelial activation and precursor cell transmigration are likely similar to those involved in leukocyte chemotaxis toward sites of inflammation [156–158]. Specific mechanisms by which osteocytes could activate endothelial cells and direct osteoclasts through bone are not well understood, but they have been speculated to involve production of factors that promote angiogenesis, endothelial activation and osteoclastogenesis (e.g. VEGF, IL-6 and/or RANKL) [159, 160]. Further compelling evidence against the idea that osteocytes release bone from a resorption-repressed state is that osteocyte-deficient bone such as allograft does not resorb in vivo. Indeed, Shimizu et al. showed experimentally that dying bone was a more effective substrate for osteoclastic resorption in vitro than bone in which cells had been killed by successive freezing and thawing [161]. Ito et al. showed that RANKL and VEGF impregnated into devitalized mouse allografts triggered extensive bone resorption, suggesting that bone itself (likely osteocytes) might be the source for pro-osteoclastic signals in vivo [162]. Thus, these studies argue against the concept that the loss of osteocytes creates a permissive environment for resorption. Rather, pro-resorptive signals emanate from within bone itself, providing positive regulatory signals to initiate osteoclast activity in bone.
The “essential” pro-resorptive signal needed to trigger the differentiation of osteoclast progenitors is RANKL. Several papers recently demonstrated that osteocytes are the dominant source for RANKL production in vivo [163, 164]. Nakashima et al. demonstrated that transgenic deletion of osteocyte RANKL using a DMP promoter results in a severe osteopetrotic phenotype [163]. Xiong et al. observed similarly and further demonstrated that the BMDs of osteocyte-specific RANKL-deficient mouse bones were increasingly higher than wild type controls up to 6 months of age [164]. These observations indicate the importance of RANKL production by young osteocytes in the modeling and remodeling processes of growing bone. MLO-Y4 osteocyte-like cell cultures also constitutively express RANKL [165], suggesting that these cells may model the behavior of young osteocytes in growing bone. In contrast, Kartsogiannis et al. and Kennedy et al. showed with both gene expression and immunohistochemistry that adult osteocytes produce little if any RANKL constitutively [166, 167].
A key feature underlying the causal link between osteocyte apoptosis and the initiation of bone resorption is that not all of the osteocytes die following a remodeling stimulus. Rather, in fatigue damage (the most extensively studied model), surviving osteocytes that surround microcracks express an active anti-apoptotic response that includes upregulation of Bcl-2 [168]. This “penumbra” formation around apoptotic cells in a region of localized tissue damage is typically observed in cases of ischemia, e.g. in the heart [169] and brain [170]. Moreover, it is these immediately adjacent surviving non-apoptotic osteocytes, rather than the dying cells, that upregulate RANKL and downregulate constitutively expressed OPG [166]. The spatial extent of RANKL upregulation has been shown to be within a few hundred microns of the apoptosis zone [166], which is far enough for RANKL signals to travel to the nearest endocortical/periosteal bone surface or vascular surface to which osteoclast precursors are recruited (Figure 3).
Figure 3.

Schematic summary of the role of osteocytes in triggering bone resorption. A) Microcracks in bone caused by fatigue loading lead to highly localized osteocyte apoptosis (shown in white) surrounding the microcrack. B) Recent studies show that surviving osteocytes immediately neighboring the region of apoptosis upregulate production of pro-osteoclastogenic signals (i.e. RANKL, and others). This increase in RANKL signaling is caused by the osteocyte apoptosis, not the bone microdamage itself. Healthy osteocytes farther from the microdamage site do not produce osteoclastogenic signals. Osteoclasts are then recruited to resorb damaged and apoptotic osteocytes during the microdamage repairing process.
What remains poorly understood are the signals that apoptotic osteocytes use to alert their surviving neighbors, and those that the surviving cells use to propagate RANKL upregulation through the network of osteocytes from regions of apoptosis to bone surfaces. As previously discussed, signaling throughout the network of living (surviving) osteocytes can operate through both direct (gap junctional) and extracellular paracrine mechanisms, subject to the constraints of molecular size, presence or absence of junctions/pores, fluid flow and the properties of pericellular matrix within the LCS. Apoptotic debris, and/or the release of inflammatory signaling molecules such as the alarmins (i.e., high mobility group box protein 1, HMGB-1) from apoptotic cells have been posited to activate nearby survivors. However, apoptotic debris is too large to travel through the LCS, and is an unlikely candidate for effective communication within the osteocyte compartment. Kogianni et al. demonstrated that apoptotic debris could stimulate resorption by existing osteoclasts in vitro [171], but there are no in vivo data that apoptotic debris cause activation of RANKL. Bidwell and colleagues have shown production of HMGB-1 by apoptotic MLO-Y4 osteocyte-like cells [172], and speculated that this factor might stimulate osteoclast formation, but these in vitro findings remain to be corroborated in vivo. Lastly, HMGB-1 release from apoptotic cells is a comparatively late phenomenon, associated with a terminal degradation of cells (also known as secondary necrosis) that occurs 4–5 days after challenge [173]. In contrast, in vivo activation of RANKL signaling from osteocytes near apoptotic cells occurs within 24 hours in the bone fatigue model [166].
In addition to molecules capable of triggering RANKL upregulation, other paracrine signals have been identified in other tissues where local injury leads to the recruitment of phagocytic cells. These are divided into “find-me” signals and “eat-me” signals. Candidate “find-me” signal molecules include ATP [174], membrane-derived lipids sphingosine-1-phosphate [175] and lysophosphatidylcholine (LPC) [176], and CX3CL1/fractalkine [177]. Understanding of this apoptosis-driven production of these signals is currently quite limited. “Eat-me” signals consist of markers that apoptotic cells express on their external membrane surfaces de novo (e.g. phosphatidylserine and calreticulin). These signals trigger the engulfment of apoptotic cells by phagocytic cells, which only become relevant once osteoclasts reach the area of apoptosis (i.e. once the response is already well established).
In summary, current studies indicate that osteoclastogenesis and activity is regulated by osteocytes mainly via paracrine signaling. It is now widely accepted that apoptotic osteocytes mediate targeted osteoclast resorption via RANKL. However, data suggest that dying osteocytes themselves do not produce the critical pro-osteoclastic cytokine RANKL, but rather induce expression and/or secretion of inflammatory cytokines by neighboring healthy osteocytes. The mechanisms of communication between dying osteocytes and neighboring osteocytes that lead to osteoclast recruitment are currently unknown.
7. Osteocytes as regulators of mineral homeostasis
Osteocytes have been considered as possible regulators of systemic mineral metabolism essentially since the contribution of the skeleton to mineral homeostasis was first appreciated. This consideration was based largely on the potential for mineral exchange throughout the LCS, which directly opposes the osteocytes and their processes and accounts for a tremendous amount of bone surface potentially available for rapid exchange. The surface area adjacent to osteocytes and their processes is estimated to be approximately 400 times greater than that of Haversian and Volkmann’s canals [97], and 100 times greater than trabecular bone surfaces [178]. Moreover, enlarged osteocytic lacunae and disrupted pericellular mineralized matrix that occurs during osteocytic perilacunar resorption (a phenomenon termed “osteocytic osteolysis” by Bélanger in 1969 [179]) had been previously described by von Recklinghausen in rickets and osteomalacia patients in 1910 [180]. Baud’s early seminal electron microscopy studies on parathyroid hormone treated mice also observed this phenomenon [181]. Local osteocyte-mediated exchange of mineral ions between bone and tissue fluid is also rapid compared with osteoclastic bone resorption. Recruitment of osteoclasts takes approximately one week, and it has been estimated that the amount of mineral released from osteoclastic resorption only accounts for approximately 0.1% of the total serum mineral content [182]. The concept that osteocytes could act as effector cells controlling plasma calcium levels fell out of favor with the recognition that osteoclastic bone remodeling was a major target for regulation by calcitropic hormones (PTH, calcitonin and 1,25(OH)2D3) and was disrupted in a variety of metabolic bone diseases. The concept of osteocyte osteolysis became particularly contentious over the possibility of artifacts introduced by histological sectioning and histomorphometric methods. However, in recent years, interest in osteocytes as potential regulators of mineral metabolism has re-intensified, particularly in circumstances of severe calcium demand such as lactation. For example, Bonewald and colleagues demonstrated osteocytic osteolysis using back-scattered electron microscopy, which reduced artifacts that had previously impeded accurate measurements of perilacunar bone loss [183]. Other evidence that osteocytes can modify their bony walls was recently reported by Sharma et al. following rat ovariectomy [184]. These observations provided strong evidence that some osteocyte perilacunar remodeling can also occur in stress states that are not calcium-driven.
Local Regulation of Mineral Homeostasis
While osteocytes lack the raw synthetic and degradative power of osteoblasts and osteoclasts, they possess several mechanisms to regulate the exchange of mineral ions between the matrix and interstitial fluid. These mechanisms were mentioned previously in the context of forming the LCS and maintaining its patency. Osteocytes have a modest capacity both to produce and degrade the organic constituents of bone matrix. They require MMPs (in particular MT1-MMP) to establish canalicular fluid space around their processes [7]. They produce other proteases as well (to be discussed below) and also been reported to express enzymes found in osteoclasts and functionally implicated in bone resorption (e.g. cathepsin K, tartrate-resistant acid phosphatase (TRAP) [183]). From a synthetic standpoint, osteocytes do not produce appreciable amounts of type I collagen. However, they synthesize several noncollagenous bone matrix proteins capable of interacting with mineral ions modulating the formation and growth of bone mineral crystals. Among these matrix proteins are three small integrin-binding ligand, N-linked glycoproteins (SIBLING proteins): osteopontin (OPN), dentin matrix protein-1 (DMP1) and MEPE, which have been shown to inhibit mineralization. McKee and colleagues demonstrated that the lamina limitans, a thin, non-mineralized matrix layer covering bone surfaces throughout the LCS and adjacent to osteocytes, contains more OPN than the surrounding mineralized matrix [185]. The expression of OPN in the osteocytic pericellular matrix is further enhanced in pathological conditions such as hypophosphatemic rickets [186]. The abundance of calcium-binding proteins like OPN in the lamina limitans suggests that osteocytes may maintain a loosely packed pericellular mineral compartment that can provide easily accessible mineral to keep up with the body’s calcium and phosphate demands.
Global Mineral Homeostasis
While a local role for osteocytes in mineral metabolism via osteocytic osteolysis has a long history, much more recent findings have established a novel systemic role as well. The key finding was the identification of a molecule specifically localized to osteocytes by immunohistochemistry as PHEX (PHosphate-regulating Endopeptidase homolog, X-linked), the product of the hyp gene that is mutated in X-linked hypophosphatemic rickets (HYP) [187]. HYP is characterized by reduced tubular phosphate reabsorption, phosphate wasting and skeletal deficiencies that include under mineralized osteoid, “blurred” microtrabecular architecture and pseudofractures, resulting in impaired mechanical properties and bone growth abnormalities [188]. Subsequently it has become clear that PHEX, and other proteins synthesized by osteocytes (e.g. DMP-1, MEPE and FGF-23) act in concert not only to regulate skeletal mineral homeostasis locally, but also to control serum phosphate levels globally by actions on the kidney. The most direct link between osteocytes and the kidney is provided by FGF-23, a phosphaturic hormone produced by osteoblasts and osteocytes that acts on kidney tubule cells to suppress tubular phosphate reabsorption. Circulating FGF-23 levels are elevated in HYP as well as in autosomal dominant hypophosphatemic rickets (ARHR), a similar condition that results from a deficiency of DMP1 [189, 190].
How deficiencies in PHEX and DMP1 contribute to elevated serum FGF23 levels and the other consequences of hypophosphatemia are not fully understood. However, studies using DMP1 deficient and DMP1/PHEX double deficient mice indicate that PHEX and DMP1 appear to regulate FGF23 via a common pathway [191]. PHEX is a protease that binds to SIBLING proteins like MEPE, DMP1 and OPN via the acidic serine aspartate-rich MEPE-associated (ASARM) motif [192, 193]. In HYP, defective PHEX exposes MEPE for proteolytic cleavage of ASARM by cathepsin B proteases [194]. The resulting free ASARM peptides can act locally on bone to inhibit mineralization [193, 195]. Another consequence of PHEX absence in HYP patients and in mutant mice is diminished turnover of OPN in bone. The resulting accumulation of OPN in osteoid can also inhibit normal mineralization [186].
8. Conclusion
Osteocytes are formidably multi-functional cells, with demonstrated capability to orchestrate virtually every aspect of skeletal metabolism from wholesale tissue modeling/remodeling to the exchange of mineral from bone surfaces. Osteocytes are able to sense and integrate both mechanical and chemical signals from their environment, and trigger appropriate responses from effector cells (i.e. osteoblasts and osteoclasts). The regulatory sphere of influence of osteocytes is broad, encompassing local (osteocytic osteolysis), regional (modeling and remodeling) and systemic (regulation of kidney function via endocrine signaling) processes. Many of the distinctive properties of osteocytes are a direct function of the unique environment they inhabit (i.e. buried within the mineralized matrix of bone). Living in a network of caverns has forced them to be dependent on fluid transport through the LCS for critical exchanges of nutrients, metabolites, wastes and information. As a result, osteocytes are reliant on mechanical loading of bone to move fluids efficiently, and have also developed exquisitely sensitive means to detect changes in tissue-level load based on differences in fluid flow through the LCS. Moreover, interacting with their neighbors only through a few contact points at the end of dendritic processes or by paracrine signaling through the LCS has constrained the ways in which communication throughout the osteocyte network may occur. The constraints of cave-dwelling have not prevented osteocytes from having long lives, nor has it impaired their ability to communicate with neighbors or to influence homeostatic events both inside and outside of bone. The recognition that osteocytes possess such diverse capabilities has come from research whose range is equally diverse – from histology and imaging, to cellular/molecular biology to experimental biomechanics, to engineering-based modeling approaches. The challenges now are to define the mechanisms that underlie these capabilities and apply that knowledge to the prevention and treatment of skeletal disorders. Undoubtedly continuing use of all these approaches will be required.
Acknowledgments
This work was supported by grants AR41210, AR57139, and AR60445 from the National Institute of Arthritis, Musculoskeletal and Skin Diseases.
Footnotes
Disclosures – None
The authors have stated that they have no conflict of interest.
Contributor Information
Dr. Mitchell B. Schaffler, Email: mschaffler@ccny.cuny.edu, University: City College of New York, Department: Biomedical Engineering, Phone: 212-650-5070, Fax: 212-650-6727
Dr. Wing-Yee Cheung, Email: wcheung@ccny.cuny.edu, University: City College of New York, Department: Biomedical Engineering
Dr. Robert Majeska, Email: rmajeska@ccny.cuny.edu, University: City College of New York, Department: Biomedical Engineering
Dr. Oran Kennedy, Email: Oran.Kennedy@nyumc.org, University: New York University, Department: Orthopaedic Surgery
References
- 1.Franz-Odendaal TA, Hall BK, Witten PE. Buried alive: how osteoblasts become osteocytes. Developmental dynamics : an official publication of the American Association of Anatomists. 2006;235:176–190. doi: 10.1002/dvdy.20603. [DOI] [PubMed] [Google Scholar]
- 2.Palumbo C. A three-dimensional ultrastructural study of osteoid-osteocytes in the tibia of chick embryos. Cell and tissue research. 1986;246:125–131. doi: 10.1007/BF00219008. [DOI] [PubMed] [Google Scholar]
- 3.Palumbo C, Palazzini S, Zaffe D, Marotti G. Osteocyte differentiation in the tibia of newborn rabbit: an ultrastructural study of the formation of cytoplasmic processes. Acta anatomica. 1990;137:350–358. doi: 10.1159/000146907. [DOI] [PubMed] [Google Scholar]
- 4.Doty SB, Morey-Holton ER, Durnova GN, Kaplansky AS. Cosmos 1887: morphology, histochemistry, and vasculature of the growing rat tibia. FASEB J. 1990;4:16–23. doi: 10.1096/fasebj.4.1.2153083. [DOI] [PubMed] [Google Scholar]
- 5.Dallas SL, Veno PA. Live imaging of bone cell and organ cultures. Methods Mol Biol. 2012;816:425–457. doi: 10.1007/978-1-61779-415-5_26. [DOI] [PubMed] [Google Scholar]
- 6.Zhang K, Barragan-Adjemian C, Ye L, Kotha S, Dallas M, Lu Y, Zhao S, Harris M, Harris SE, Feng JQ, Bonewald LF. E11/gp38 selective expression in osteocytes: regulation by mechanical strain and role in dendrite elongation. Mol Cell Biol. 2006;26:4539–4552. doi: 10.1128/MCB.02120-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holmbeck K, Bianco P, Pidoux I, Inoue S, Billinghurst RC, Wu W, Chrysovergis K, Yamada S, Birkedal-Hansen H, Poole AR. The metalloproteinase MT1-MMP is required for normal development and maintenance of osteocyte processes in bone. Journal of cell science. 2005;118:147–156. doi: 10.1242/jcs.01581. [DOI] [PubMed] [Google Scholar]
- 8.Inoue K, Mikuni-Takagaki Y, Oikawa K, Itoh T, Inada M, Noguchi T, Park JS, Onodera T, Krane SM, Noda M, Itohara S. A crucial role for matrix metalloproteinase 2 in osteocytic canalicular formation and bone metabolism. Journal of Biological Chemistry. 2006;281:33814–33824. doi: 10.1074/jbc.M607290200. [DOI] [PubMed] [Google Scholar]
- 9.Bloch SL, Kristensen SL, Sorensen MS. The Viability of Perilabyrinthine Osteocytes: A Quantitative Study Using Bulk-Stained Undecalcified Human Temporal Bones. Anatomical Record-Advances in Integrative Anatomy and Evolutionary Biology. 2012;295:1101–1108. doi: 10.1002/ar.22492. [DOI] [PubMed] [Google Scholar]
- 10.McNamara LM, Majeska RJ, Weinbaum S, Friedrich V, Schaffler MB. Attachment of osteocyte cell processes to the bone matrix. Anat Rec (Hoboken) 2009;292:355–363. doi: 10.1002/ar.20869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamioka H, Kameo Y, Imai Y, Bakker AD, Bacabac RG, Yamada N, Takaoka A, Yamashiro T, Adachi T, Klein-Nulend J. Microscale fluid flow analysis in a human osteocyte canaliculus using a realistic high-resolution image-based three-dimensional model. Integr Biol (Camb) 2012;4:1198–1206. doi: 10.1039/c2ib20092a. [DOI] [PubMed] [Google Scholar]
- 12.Doty SB. Morphological evidence of gap junctions between bone cells. Calcified tissue international. 1981;33:509–512. doi: 10.1007/BF02409482. [DOI] [PubMed] [Google Scholar]
- 13.You LD, Weinbaum S, Cowin SC, Schaffler MB. Ultrastructure of the osteocyte process and its pericellular matrix. Anat Rec A Discov Mol Cell Evol Biol. 2004;278:505–513. doi: 10.1002/ar.a.20050. [DOI] [PubMed] [Google Scholar]
- 14.Thompson WR, Modla S, Grindel BJ, Czymmek KJ, Kirn-Safran CB, Wang L, Duncan RL, Farach-Carson MC. Perlecan/Hspg2 deficiency alters the pericellular space of the lacunocanalicular system surrounding osteocytic processes in cortical bone. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2011;26:618–629. doi: 10.1002/jbmr.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noonan KJ, Stevens JW, Tammi R, Tammi M, Hernandez JA, Midura RJ. Spatial distribution of CD44 and hyaluronan in the proximal tibia of the growing rat. J Orthop Res. 1996;14:573–581. doi: 10.1002/jor.1100140411. [DOI] [PubMed] [Google Scholar]
- 16.Busse B, Djonic D, Milovanovic P, Hahn M, Puschel K, Ritchie RO, Djuric M, Amling M. Decrease in the osteocyte lacunar density accompanied by hypermineralized lacunar occlusion reveals failure and delay of remodeling in aged human bone. Aging Cell. 2010;9:1065–1075. doi: 10.1111/j.1474-9726.2010.00633.x. [DOI] [PubMed] [Google Scholar]
- 17.Addison WN, Masica DL, Gray JJ, McKee MD. Phosphorylation-dependent inhibition of mineralization by osteopontin ASARM peptides is regulated by PHEX cleavage. J Bone Miner Res. 2010;25:695–705. doi: 10.1359/jbmr.090832. [DOI] [PubMed] [Google Scholar]
- 18.Gericke A, Qin C, Sun Y, Redfern R, Redfern D, Fujimoto Y, Taleb H, Butler WT, Boskey AL. Different forms of DMP1 play distinct roles in mineralization. J Dent Res. 2010;89:355–359. doi: 10.1177/0022034510363250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tami AE, Schaffler MB, Knothe Tate ML. Probing the tissue to subcellular level structure underlying bone’s molecular sieving function. Biorheology. 2003;40:577–590. [PubMed] [Google Scholar]
- 20.Thompson WR, Majid AS, Czymmek KJ, Ruff AL, Garcia J, Duncan RL, Farach-Carson MC. Association of the alpha(2)delta(1) subunit with Ca(v)3.2 enhances membrane expression and regulates mechanically induced ATP release in MLO-Y4 osteocytes. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2011;26:2125–2139. doi: 10.1002/jbmr.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L, Wang Y, Han Y, Henderson SC, Majeska RJ, Weinbaum S, Schaffler MB. In situ measurement of solute transport in the bone lacunar-canalicular system. Proc Natl Acad Sci U S A. 2005;102:11911–11916. doi: 10.1073/pnas.0505193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piekarski K, Munro M. Transport mechanism operating between blood supply and osteocytes in long bones. Nature. 1977;269:80–82. doi: 10.1038/269080a0. [DOI] [PubMed] [Google Scholar]
- 23.Weinbaum S, Cowin SC, Zeng Y. A model for the excitation of osteocytes by mechanical loading-induced bone fluid shear stresses. Journal of biomechanics. 1994;27:339–360. doi: 10.1016/0021-9290(94)90010-8. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Ciani C, Doty SB, Fritton SP. Delineating bone’s interstitial fluid pathway in vivo. Bone. 2004;34:499–509. doi: 10.1016/j.bone.2003.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ciani C, Doty SB, Fritton SP. An effective histological staining process to visualize bone interstitial fluid space using confocal microscopy. Bone. 2009;44:1015–1017. doi: 10.1016/j.bone.2009.01.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knothe Tate ML, Niederer P, Knothe U. In vivo tracer transport through the lacunocanalicular system of rat bone in an environment devoid of mechanical loading. Bone. 1998;22:107–117. doi: 10.1016/s8756-3282(97)00234-2. [DOI] [PubMed] [Google Scholar]
- 27.Li W, You L, Schaffler MB, Wang L. The dependency of solute diffusion on molecular weight and shape in intact bone. Bone. 2009;45:1017–1023. doi: 10.1016/j.bone.2009.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Price C, Zhou X, Li W, Wang L. Real-time measurement of solute transport within the lacunar-canalicular system of mechanically loaded bone: direct evidence for load-induced fluid flow. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2011;26:277–285. doi: 10.1002/jbmr.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang B, Zhou X, Price C, Li W, Pan J, Wang L. Quantifying load-induced solute transport and solute-matrix interactions within the osteocyte lacunar-canalicular system. J Bone Miner Res. 2012 doi: 10.1002/jbmr.1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weinstein RS, O’Brien CA, Almeida M, Zhao H, Roberson PK, Jilka RL, Manolagas SC. Osteoprotegerin prevents glucocorticoid-induced osteocyte apoptosis in mice. Endocrinology. 2011;152:3323–3331. doi: 10.1210/en.2011-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knothe Tate ML. “Whither flows the fluid in bone?” An osteocyte’s perspective. Journal of biomechanics. 2003;36:1409–1424. doi: 10.1016/s0021-9290(03)00123-4. [DOI] [PubMed] [Google Scholar]
- 32.Wolff J. The law of bone remodelling. Springer-Verlag; Berlin ; New York: 1986. [Google Scholar]
- 33.Takai E, Mauck RL, Hung CT, Guo XE. Osteocyte viability and regulation of osteoblast function in a 3D trabecular bone explant under dynamic hydrostatic pressure. J Bone Miner Res. 2004;19:1403–1410. doi: 10.1359/JBMR.040516. [DOI] [PubMed] [Google Scholar]
- 34.Liu C, Zhao Y, Cheung WY, Gandhi R, Wang L, You L. Effects of cyclic hydraulic pressure on osteocytes. Bone. 2010;46:1449–1456. doi: 10.1016/j.bone.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adachi T, Aonuma Y, Ito S, Tanaka M, Hojo M, Takano-Yamamoto T, Kamioka H. Osteocyte calcium signaling response to bone matrix deformation. Journal of biomechanics. 2009;42:2507–2512. doi: 10.1016/j.jbiomech.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 36.Rubin CT, Lanyon LE. Regulation of bone formation by applied dynamic loads. J Bone Joint Surg Am. 1984;66:397–402. [PubMed] [Google Scholar]
- 37.Fritton SP, McLeod KJ, Rubin CT. Quantifying the strain history of bone: spatial uniformity and self-similarity of low-magnitude strains. Journal of biomechanics. 2000;33:317–325. doi: 10.1016/s0021-9290(99)00210-9. [DOI] [PubMed] [Google Scholar]
- 38.You J, Yellowley CE, Donahue HJ, Zhang Y, Chen Q, Jacobs CR. Substrate deformation levels associated with routine physical activity are less stimulatory to bone cells relative to loading-induced oscillatory fluid flow. J Biomech Eng. 2000;122:387–393. doi: 10.1115/1.1287161. [DOI] [PubMed] [Google Scholar]
- 39.Burger E, Veldhuijzen JP. Influence of mechanical factors on bone formation, resorption and growth in vitro. In: Hall BK, editor. Bone. 1993. [Google Scholar]
- 40.Martin RB, Burr DB, Sharkey NA. Mechanical Properties of Bone. In: Martin RB, Burr DB, Sharkey NA, editors. Skeletal Tissue Mechanics. Springer-Verlag; New York: 1998. [Google Scholar]
- 41.Genetos DC, Kephart CJ, Zhang Y, Yellowley CE, Donahue HJ. Oscillating fluid flow activation of gap junction hemichannels induces ATP release from MLO-Y4 osteocytes. J Cell Physiol. 2007;212:207–214. doi: 10.1002/jcp.21021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klein-Nulend J, Semeins CM, Ajubi NE, Nijweide PJ, Burger EH. Pulsating fluid flow increases nitric oxide (NO) synthesis by osteocytes but not periosteal fibroblasts--correlation with prostaglandin upregulation. Biochemical and biophysical research communications. 1995;217:640–648. doi: 10.1006/bbrc.1995.2822. [DOI] [PubMed] [Google Scholar]
- 43.Lu XL, Huo B, Park M, Guo XE. Calcium response in osteocytic networks under steady and oscillatory fluid flow. Bone. 2012;51:466–473. doi: 10.1016/j.bone.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cherian PP, Siller-Jackson AJ, Gu S, Wang X, Bonewald LF, Sprague E, Jiang JX. Mechanical strain opens connexin 43 hemichannels in osteocytes: a novel mechanism for the release of prostaglandin. Mol Biol Cell. 2005;16:3100–3106. doi: 10.1091/mbc.E04-10-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anderson EJ, Kaliyamoorthy S, Iwan J, Alexander D, Knothe Tate ML. Nano-microscale models of periosteocytic flow show differences in stresses imparted to cell body and processes. Annals of biomedical engineering. 2005;33:52–62. doi: 10.1007/s10439-005-8962-y. [DOI] [PubMed] [Google Scholar]
- 46.Han Y, Cowin SC, Schaffler MB, Weinbaum S. Mechanotransduction and strain amplification in osteocyte cell processes. Proc Natl Acad Sci U S A. 2004;101:16689–16694. doi: 10.1073/pnas.0407429101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.You L, Cowin SC, Schaffler MB, Weinbaum S. A model for strain amplification in the actin cytoskeleton of osteocytes due to fluid drag on pericellular matrix. J Biomech. 2001;34:1375–1386. doi: 10.1016/s0021-9290(01)00107-5. [DOI] [PubMed] [Google Scholar]
- 48.Nakamura H, Kenmotsu S, Sakai H, Ozawa H. Localization of CD44, the hyaluronate receptor, on the plasma membrane of osteocytes and osteoclasts in rat tibiae. Cell and tissue research. 1995;280:225–233. doi: 10.1007/BF00307793. [DOI] [PubMed] [Google Scholar]
- 49.Florian JA, Kosky JR, Ainslie K, Pang Z, Dull RO, Tarbell JM. Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ Res. 2003;93:e136–142. doi: 10.1161/01.RES.0000101744.47866.D5. [DOI] [PubMed] [Google Scholar]
- 50.Bellin RM, Kubicek JD, Frigault MJ, Kamien AJ, Steward RL, Jr, Barnes HM, Digiacomo MB, Duncan LJ, Edgerly CK, Morse EM, Park CY, Fredberg JJ, Cheng CM, LeDuc PR. Defining the role of syndecan-4 in mechanotransduction using surface-modification approaches. Proc Natl Acad Sci U S A. 2009;106:22102–22107. doi: 10.1073/pnas.0902639106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reilly GC, Haut TR, Yellowley CE, Donahue HJ, Jacobs CR. Fluid flow induced PGE2 release by bone cells is reduced by glycocalyx degradation whereas calcium signals are not. Biorheology. 2003;40:591–603. [PubMed] [Google Scholar]
- 52.Wang Y, McNamara LM, Schaffler MB, Weinbaum S. Strain amplification and integrin based signaling in osteocytes. J Musculoskelet Neuronal Interact. 2008;8:332–334. [PMC free article] [PubMed] [Google Scholar]
- 53.McKee MD, Nanci A. Osteopontin and the bone remodeling sequence. Colloidal-gold immunocytochemistry of an interfacial extracellular matrix protein. Ann N Y Acad Sci. 1995;760:177–189. doi: 10.1111/j.1749-6632.1995.tb44629.x. [DOI] [PubMed] [Google Scholar]
- 54.Nakamura I, Pilkington MF, Lakkakorpi PT, Lipfert L, Sims SM, Dixon SJ, Rodan GA, Duong LT. Role of alpha(v)beta(3) integrin in osteoclast migration and formation of the sealing zone. J Cell Sci. 1999;112 ( Pt 22):3985–3993. doi: 10.1242/jcs.112.22.3985. [DOI] [PubMed] [Google Scholar]
- 55.Roca-Cusachs P, Gauthier NC, Del Rio A, Sheetz MP. Clustering of alpha(5)beta(1) integrins determines adhesion strength whereas alpha(v)beta(3) and talin enable mechanotransduction. Proc Natl Acad Sci U S A. 2009;106:16245–16250. doi: 10.1073/pnas.0902818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tzima E, del Pozo MA, Shattil SJ, Chien S, Schwartz MA. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO J. 2001;20:4639–4647. doi: 10.1093/emboj/20.17.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miyauchi A, Gotoh M, Kamioka H, Notoya K, Sekiya H, Takagi Y, Yoshimoto Y, Ishikawa H, Chihara K, Takano-Yamamoto T, Fujita T, Mikuni-Takagaki Y. AlphaVbeta3 integrin ligands enhance volume-sensitive calcium influx in mechanically stretched osteocytes. Journal of bone and mineral metabolism. 2006;24:498–504. doi: 10.1007/s00774-006-0716-x. [DOI] [PubMed] [Google Scholar]
- 58.Wu D, Ganatos P, Spray DC, Weinbaum S. On the electrophysiological response of bone cells using a Stokesian fluid stimulus probe for delivery of quantifiable localized picoNewton level forces. Journal of biomechanics. 2011;44:1702–1708. doi: 10.1016/j.jbiomech.2011.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Radel C, Carlile-Klusacek M, Rizzo V. Participation of caveolae in beta1 integrin-mediated mechanotransduction. Biochemical and biophysical research communications. 2007;358:626–631. doi: 10.1016/j.bbrc.2007.04.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Friedland JC, Lee MH, Boettiger D. Mechanically activated integrin switch controls alpha5beta1 function. Science. 2009;323:642–644. doi: 10.1126/science.1168441. [DOI] [PubMed] [Google Scholar]
- 61.Phillips JA, Almeida EA, Hill EL, Aguirre JI, Rivera MF, Nachbandi I, Wronski TJ, van der Meulen MC, Globus RK. Role for beta1 integrins in cortical osteocytes during acute musculoskeletal disuse. Matrix Biol. 2008;27:609–618. doi: 10.1016/j.matbio.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 62.Nicolella DP, Moravits DE, Gale AM, Bonewald LF, Lankford J. Osteocyte lacunae tissue strain in cortical bone. Journal of biomechanics. 2006;39:1735–1743. doi: 10.1016/j.jbiomech.2005.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lancaster MA, Gleeson JG. The primary cilium as a cellular signaling center: lessons from disease. Current opinion in genetics & development. 2009;19:220–229. doi: 10.1016/j.gde.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Malone AM, Anderson CT, Tummala P, Kwon RY, Johnston TR, Stearns T, Jacobs CR. Primary cilia mediate mechanosensing in bone cells by a calcium-independent mechanism. Proc Natl Acad Sci U S A. 2007;104:13325–13330. doi: 10.1073/pnas.0700636104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wann AK, Knight MM. Primary cilia elongation in response to interleukin-1 mediates the inflammatory response. Cell Mol Life Sci. 2012;69:2967–2977. doi: 10.1007/s00018-012-0980-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wheatley DN. Landmarks in the first hundred years of primary (9+0) cilium research. Cell Biol Int. 2005;29:333–339. doi: 10.1016/j.cellbi.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 67.Wassermann FYJ. Fine Structure of the Osteocyte Capsule and of the Wall of the Lacunae in Bone. Cell and tissue research. 1965;67:636–652. [Google Scholar]
- 68.Mochizuki T, Tsuchiya K, Nitta K. Autosomal dominant polycystic kidney disease: recent advances in pathogenesis and potential therapies. Clin Exp Nephrol. 2012 doi: 10.1007/s10157-012-0741-0. [DOI] [PubMed] [Google Scholar]
- 69.Harris PC, Torres VE. Polycystic Kidney Disease, Autosomal Dominant. GeneReviews Access Date Access 2002 [Google Scholar]
- 70.Torres VE, Harris PC. Polycystic kidney disease: genes, proteins, animal models, disease mechanisms and therapeutic opportunities. J Intern Med. 2007;261:17–31. doi: 10.1111/j.1365-2796.2006.01743.x. [DOI] [PubMed] [Google Scholar]
- 71.Xiao Z, Dallas M, Qiu N, Nicolella D, Cao L, Johnson M, Bonewald L, Quarles LD. Conditional deletion of Pkd1 in osteocytes disrupts skeletal mechanosensing in mice. FASEB J. 2011;25:2418–2432. doi: 10.1096/fj.10-180299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Synctium. The Merriam-Webster’s Collegiate Dictionary Merriam Webster Mass Market; 2004. [Google Scholar]
- 73.Palazzini S, Palumbo C, Ferretti M, Marotti G. Stromal cell structure and relationships in perimedullary spaces of chick embryo shaft bones. Anat Embryol (Berl) 1998;197:349–357. doi: 10.1007/s004290050145. [DOI] [PubMed] [Google Scholar]
- 74.Stains JP, Civitelli R. Cell-to-cell interactions in bone. Biochemical and biophysical research communications. 2005;328:721–727. doi: 10.1016/j.bbrc.2004.11.078. [DOI] [PubMed] [Google Scholar]
- 75.Yellowley CE, Li Z, Zhou Z, Jacobs CR, Donahue HJ. Functional gap junctions between osteocytic and osteoblastic cells. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2000;15:209–217. doi: 10.1359/jbmr.2000.15.2.209. [DOI] [PubMed] [Google Scholar]
- 76.Cheng B, Zhao S, Luo J, Sprague E, Bonewald LF, Jiang JX. Expression of functional gap junctions and regulation by fluid flow in osteocyte-like MLO-Y4 cells. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2001;16:249–259. doi: 10.1359/jbmr.2001.16.2.249. [DOI] [PubMed] [Google Scholar]
- 77.Cheng B, Kato Y, Zhao S, Luo J, Sprague E, Bonewald LF, Jiang JX. PGE(2) is essential for gap junction-mediated intercellular communication between osteocyte-like MLO-Y4 cells in response to mechanical strain. Endocrinology. 2001;142:3464–3473. doi: 10.1210/endo.142.8.8338. [DOI] [PubMed] [Google Scholar]
- 78.D’Hondt C, Ponsaerts R, De Smedt H, Bultynck G, Himpens B. Pannexins, distant relatives of the connexin family with specific cellular functions? BioEssays : news and reviews in molecular, cellular and developmental biology. 2009;31:953–974. doi: 10.1002/bies.200800236. [DOI] [PubMed] [Google Scholar]
- 79.Castro CH, Stains JP, Sheikh S, Szejnfeld VL, Willecke K, Theis M, Civitelli R. Development of mice with osteoblast-specific connexin43 gene deletion. Cell communication & adhesion. 2003;10:445–450. doi: 10.1080/cac.10.4-6.445.450. [DOI] [PubMed] [Google Scholar]
- 80.Zhang Y, Paul EM, Sathyendra V, Davison A, Sharkey N, Bronson S, Srinivasan S, Gross TS, Donahue HJ. Enhanced osteoclastic resorption and responsiveness to mechanical load in gap junction deficient bone. PloS one. 2011;6:e23516. doi: 10.1371/journal.pone.0023516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Grimston SK, Goldberg DB, Watkins M, Brodt MD, Silva MJ, Civitelli R. Connexin43 deficiency reduces the sensitivity of cortical bone to the effects of muscle paralysis. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2011;26:2151–2160. doi: 10.1002/jbmr.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grimston SK, Brodt MD, Silva MJ, Civitelli R. Attenuated response to in vivo mechanical loading in mice with conditional osteoblast ablation of the connexin43 gene (Gja1) Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2008;23:879–886. doi: 10.1359/JBMR.080222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lloyd SA, Lewis GS, Zhang Y, Paul EM, Donahue HJ. Connexin 43 deficiency attenuates loss of trabecular bone and prevents suppression of cortical bone formation during unloading. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2012;27:2359–2372. doi: 10.1002/jbmr.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scemes E, Dermietzel R, Spray DC. Calcium waves between astrocytes from Cx43 knockout mice. Glia. 1998;24:65–73. doi: 10.1002/(sici)1098-1136(199809)24:1<65::aid-glia7>3.0.co;2-#. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lecanda F, Warlow PM, Sheikh S, Furlan F, Steinberg TH, Civitelli R. Connexin43 deficiency causes delayed ossification, craniofacial abnormalities, and osteoblast dysfunction. J Cell Biol. 2000;151:931–944. doi: 10.1083/jcb.151.4.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Batra N, Burra S, Siller-Jackson AJ, Gu S, Xia X, Weber GF, DeSimone D, Bonewald LF, Lafer EM, Sprague E, Schwartz MA, Jiang JX. Mechanical stress-activated integrin alpha5beta1 induces opening of connexin 43 hemichannels. Proc Natl Acad Sci U S A. 2012;109:3359–3364. doi: 10.1073/pnas.1115967109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Reddy KV, Mangale SS. Integrin receptors: the dynamic modulators of endometrial function. Tissue Cell. 2003;35:260–273. doi: 10.1016/s0040-8166(03)00039-9. [DOI] [PubMed] [Google Scholar]
- 88.Spray DC, Ye ZC, Ransom BR. Functional connexin “hemichannels”: a critical appraisal. Glia. 2006;54:758–773. doi: 10.1002/glia.20429. [DOI] [PubMed] [Google Scholar]
- 89.Suadicani SO, Vink MJ, Spray DC. Slow intercellular Ca(2+) signaling in wild-type and Cx43-null neonatal mouse cardiac myocytes. Am J Physiol Heart Circ Physiol. 2000;279:H3076–3088. doi: 10.1152/ajpheart.2000.279.6.H3076. [DOI] [PubMed] [Google Scholar]
- 90.Iglesias R, Dahl G, Qiu F, Spray DC, Scemes E. Pannexin 1: the molecular substrate of astrocyte “hemichannels”. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:7092–7097. doi: 10.1523/JNEUROSCI.6062-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Baranova A, Ivanov D, Petrash N, Pestova A, Skoblov M, Kelmanson I, Shagin D, Nazarenko S, Geraymovych E, Litvin O, Tiunova A, Born TL, Usman N, Staroverov D, Lukyanov S, Panchin Y. The mammalian pannexin family is homologous to the invertebrate innexin gap junction proteins. Genomics. 2004;83:706–716. doi: 10.1016/j.ygeno.2003.09.025. [DOI] [PubMed] [Google Scholar]
- 92.Thi MM, Islam S, Suadicani SO, Spray DC. Connexin43 and pannexin1 channels in osteoblasts: who is the “hemichannel”? The Journal of membrane biology. 2012;245:401–409. doi: 10.1007/s00232-012-9462-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sosinsky GE, Boassa D, Dermietzel R, Duffy HS, Laird DW, MacVicar B, Naus CC, Penuela S, Scemes E, Spray DC, Thompson RJ, Zhao HB, Dahl G. Pannexin channels are not gap junction hemichannels. Channels (Austin) 2011;5:193–197. doi: 10.4161/chan.5.3.15765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bao L, Locovei S, Dahl G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 2004;572:65–68. doi: 10.1016/j.febslet.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 95.Powell WF, Jr, Barry KJ, Tulum I, Kobayashi T, Harris SE, Bringhurst FR, Pajevic PD. Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J Endocrinol. 2011;209:21–32. doi: 10.1530/JOE-10-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lau KH, Baylink DJ, Zhou XD, Rodriguez D, Bonewald LF, Li Z, Ruffoni D, Muller R, Kesavan C, Sheng MH. Osteocyte-derived insulin-like growth factor I is essential for determining bone mechanosensitivity. Am J Physiol Endocrinol Metab. 2013;305:E271–281. doi: 10.1152/ajpendo.00092.2013. [DOI] [PubMed] [Google Scholar]
- 97.Marotti G, Ferretti M, Muglia MA, Palumbo C, Palazzini S. A quantitative evaluation of osteoblast-osteocyte relationships on growing endosteal surface of rabbit tibiae. Bone. 1992;13:363–368. doi: 10.1016/8756-3282(92)90452-3. [DOI] [PubMed] [Google Scholar]
- 98.Rodan GA, Bourret LA, Harvey A, Mensi T. Cyclic AMP and cyclic GMP: mediators of the mechanical effects on bone remodeling. Science. 1975;189:467–469. doi: 10.1126/science.168639. [DOI] [PubMed] [Google Scholar]
- 99.Somjen D, Binderman I, Berger E, Harell A. Bone remodelling induced by physical stress is prostaglandin E2 mediated. Biochimica et biophysica acta. 1980;627:91–100. doi: 10.1016/0304-4165(80)90126-9. [DOI] [PubMed] [Google Scholar]
- 100.Forwood MR, Kelly WL, Worth NF. Localisation of prostaglandin endoperoxide H synthase (PGHS)-1 and PGHS-2 in bone following mechanical loading in vivo. The Anatomical record. 1998;252:580–586. doi: 10.1002/(SICI)1097-0185(199812)252:4<580::AID-AR8>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 101.Forwood MR. Inducible cyclo-oxygenase (COX-2) mediates the induction of bone formation by mechanical loading in vivo. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1996;11:1688–1693. doi: 10.1002/jbmr.5650111112. [DOI] [PubMed] [Google Scholar]
- 102.Giuliano F, Warner TD. Origins of prostaglandin E2: involvements of cyclooxygenase (COX)-1 and COX-2 in human and rat systems. J Pharmacol Exp Ther. 2002;303:1001–1006. doi: 10.1124/jpet.102.041244. [DOI] [PubMed] [Google Scholar]
- 103.Li J, Rose E, Frances D, Sun Y, You L. Effect of oscillating fluid flow stimulation on osteocyte mRNA expression. J Biomech. 2012;45:247–251. doi: 10.1016/j.jbiomech.2011.10.037. [DOI] [PubMed] [Google Scholar]
- 104.Jiang JX, Cheng B. Mechanical stimulation of gap junctions in bone osteocytes is mediated by prostaglandin E2. Cell Commun Adhes. 2001;8:283–288. doi: 10.3109/15419060109080738. [DOI] [PubMed] [Google Scholar]
- 105.Mikuni-Takagaki Y. Mechanical responses and signal transduction pathways in stretched osteocytes. Journal of bone and mineral metabolism. 1999;17:57–60. doi: 10.1007/s007740050065. [DOI] [PubMed] [Google Scholar]
- 106.Fortier I, Patry C, Lora M, Samadfan R, de Brum-Fernandes AJ. Immunohistochemical localization of the prostacyclin receptor (IP) human bone. Prostaglandins Leukot Essent Fatty Acids. 2001;65:79–83. doi: 10.1054/plef.2001.0292. [DOI] [PubMed] [Google Scholar]
- 107.Nakalekha C, Yokoyama C, Miura H, Alles N, Aoki K, Ohya K, Morita I. Increased bone mass in adult prostacyclin-deficient mice. J Endocrinol. 2010;204:125–133. doi: 10.1677/JOE-09-0376. [DOI] [PubMed] [Google Scholar]
- 108.Fortier I, Gallant MA, Hackett JA, Patry C, de Brum-Fernandes AJ. Immunolocalization of the prostaglandin E2 receptor subtypes in human bone tissue: differences in foetal, adult normal, osteoporotic and pagetic bone. Prostaglandins, leukotrienes, and essential fatty acids. 2004;70:431–439. doi: 10.1016/j.plefa.2003.08.024. [DOI] [PubMed] [Google Scholar]
- 109.Zaman G, Pitsillides AA, Rawlinson SC, Suswillo RF, Mosley JR, Cheng MZ, Platts LA, Hukkanen M, Polak JM, Lanyon LE. Mechanical strain stimulates nitric oxide production by rapid activation of endothelial nitric oxide synthase in osteocytes. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1999;14:1123–1131. doi: 10.1359/jbmr.1999.14.7.1123. [DOI] [PubMed] [Google Scholar]
- 110.Knowles RG, Moncada S. Nitric-Oxide Synthases in Mammals. Biochemical Journal. 1994;298:249–258. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Caballero-Alias AM, Loveridge N, Lyon A, Das-Gupta V, Pitsillides A, Reeve J. NOS isoforms in adult human osteocytes: Multiple pathways of NO regulation? Calcified Tissue Int. 2004;75:78–84. doi: 10.1007/s00223-003-0161-y. [DOI] [PubMed] [Google Scholar]
- 112.Mancini L, Moradi-Bidhendi N, Becherini L, Martineti V, MacIntyre I. The biphasic effects of nitric oxide in primary rat osteoblasts are cGMP dependent. Biochemical and biophysical research communications. 2000;274:477–481. doi: 10.1006/bbrc.2000.3164. [DOI] [PubMed] [Google Scholar]
- 113.Bakker AD, Huesa C, Hughes A, Aspden RM, van’t Hof RJ, Klein-Nulend J, Helfrich MH. Endothelial nitric oxide synthase is not essential for nitric oxide production by osteoblasts subjected to fluid shear stress in vitro. Calcif Tissue Int. 2013;92:228–239. doi: 10.1007/s00223-012-9670-x. [DOI] [PubMed] [Google Scholar]
- 114.van’t Hof RJ, MacPhee J, Libouban H, Helfrich MH, Ralston SH. Regulation of bone mass and bone turnover by neuronal nitric oxide synthase. Endocrinology. 2004;145:5068–5074. doi: 10.1210/en.2004-0205. [DOI] [PubMed] [Google Scholar]
- 115.Hakim TS, Sugimori K, Camporesi EM, Anderson G. Half-life of nitric oxide in aqueous solutions with and without haemoglobin. Physiological measurement. 1996;17:267–277. doi: 10.1088/0967-3334/17/4/004. [DOI] [PubMed] [Google Scholar]
- 116.Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CL, Wu Y, Liu JL, Ooi GT, Setser J, Frystyk J, Boisclair YR, LeRoith D. Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest. 2002;110:771–781. doi: 10.1172/JCI15463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sheng MH, Zhou XD, Bonewald LF, Baylink DJ, Lau KH. Disruption of the insulin-like growth factor-1 gene in osteocytes impairs developmental bone growth in mice. Bone. 2013;52:133–144. doi: 10.1016/j.bone.2012.09.027. [DOI] [PubMed] [Google Scholar]
- 118.Lean JM, Jagger CJ, Chambers TJ, Chow JW. Increased insulin-like growth factor I mRNA expression in rat osteocytes in response to mechanical stimulation. The American journal of physiology. 1995;268:E318–327. doi: 10.1152/ajpendo.1995.268.2.E318. [DOI] [PubMed] [Google Scholar]
- 119.Reijnders CM, Bravenboer N, Tromp AM, Blankenstein MA, Lips P. Effect of mechanical loading on insulin-like growth factor-I gene expression in rat tibia. J Endocrinol. 2007;192:131–140. doi: 10.1677/joe.1.06880. [DOI] [PubMed] [Google Scholar]
- 120.Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Lowik CW, Reeve J. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19:1842–1844. doi: 10.1096/fj.05-4221fje. [DOI] [PubMed] [Google Scholar]
- 121.Ota K, Quint P, Ruan M, Pederson L, Westendorf JJ, Khosla S, Oursler MJ. Sclerostin is expressed in osteoclasts from aged mice and reduces osteoclast-mediated stimulation of mineralization. Journal of cellular biochemistry. 2013;114:1901–1907. doi: 10.1002/jcb.24537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Roudier M, Li X, Niu QT, Pacheco E, Pretorius JK, Graham K, Yoon BR, Gong J, Warmington K, Ke HZ, Black RA, Hulme J, Babij P. Sclerostin is expressed in articular cartilage but loss or inhibition does not affect cartilage remodeling during aging or following mechanical injury. Arthritis and rheumatism. 2013;65:721–731. doi: 10.1002/art.37802. [DOI] [PubMed] [Google Scholar]
- 123.Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883–19887. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 124.Leupin O, Piters E, Halleux C, Hu S, Kramer I, Morvan F, Bouwmeester T, Schirle M, Bueno-Lozano M, Fuentes FJ, Itin PH, Boudin E, de Freitas F, Jennes K, Brannetti B, Charara N, Ebersbach H, Geisse S, Lu CX, Bauer A, Van Hul W, Kneissel M. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. The Journal of biological chemistry. 2011;286:19489–19500. doi: 10.1074/jbc.M110.190330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li J, Sarosi I, Cattley RC, Pretorius J, Asuncion F, Grisanti M, Morony S, Adamu S, Geng Z, Qiu W, Kostenuik P, Lacey DL, Simonet WS, Bolon B, Qian X, Shalhoub V, Ominsky MS, Zhu Ke H, Li X, Richards WG. Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone. 2006;39:754–766. doi: 10.1016/j.bone.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 126.Balemans W, Piters E, Cleiren E, Ai M, Van Wesenbeeck L, Warman ML, Van Hul W. The binding between sclerostin and LRP5 is altered by DKK1 and by high-bone mass LRP5 mutations. Calcified tissue international. 2008;82:445–453. doi: 10.1007/s00223-008-9130-9. [DOI] [PubMed] [Google Scholar]
- 127.Robling AG, Bellido T, Turner CH. Mechanical stimulation in vivo reduces osteocyte expression of sclerostin. Journal of musculoskeletal & neuronal interactions. 2006;6:354. [PubMed] [Google Scholar]
- 128.Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, Mantila SM, Gluhak-Heinrich J, Bellido TM, Harris SE, Turner CH. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283:5866–5875. doi: 10.1074/jbc.M705092200. [DOI] [PubMed] [Google Scholar]
- 129.Gaudio A, Pennisi P, Bratengeier C, Torrisi V, Lindner B, Mangiafico RA, Pulvirenti I, Hawa G, Tringali G, Fiore CE. Increased sclerostin serum levels associated with bone formation and resorption markers in patients with immobilization-induced bone loss. J Clin Endocrinol Metab. 2010;95:2248–2253. doi: 10.1210/jc.2010-0067. [DOI] [PubMed] [Google Scholar]
- 130.Moriishi T, Fukuyama R, Ito M, Miyazaki T, Maeno T, Kawai Y, Komori H, Komori T. Osteocyte network; a negative regulatory system for bone mass augmented by the induction of Rankl in osteoblasts and Sost in osteocytes at unloading. PloS one. 2012;7:e40143. doi: 10.1371/journal.pone.0040143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Moustafa A, Sugiyama T, Prasad J, Zaman G, Gross TS, Lanyon LE, Price JS. Mechanical loading-related changes in osteocyte sclerostin expression in mice are more closely associated with the subsequent osteogenic response than the peak strains engendered. Osteoporos Int. 2012;23:1225–1234. doi: 10.1007/s00198-011-1656-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Li X, Ominsky MS, Warmington KS, Morony S, Gong J, Cao J, Gao Y, Shalhoub V, Tipton B, Haldankar R, Chen Q, Winters A, Boone T, Geng Z, Niu QT, Ke HZ, Kostenuik PJ, Simonet WS, Lacey DL, Paszty C. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2009;24:578–588. doi: 10.1359/jbmr.081206. [DOI] [PubMed] [Google Scholar]
- 133.Ominsky MS, Vlasseros F, Jolette J, Smith SY, Stouch B, Doellgast G, Gong J, Gao Y, Cao J, Graham K, Tipton B, Cai J, Deshpande R, Zhou L, Hale MD, Lightwood DJ, Henry AJ, Popplewell AG, Moore AR, Robinson MK, Lacey DL, Simonet WS, Paszty C. Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2010;25:948–959. doi: 10.1002/jbmr.14. [DOI] [PubMed] [Google Scholar]
- 134.Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26:19–26. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- 135.Shen L, Xie X, Su Y, Luo C, Zhang C, Zeng B. Parathyroid hormone versus bisphosphonate treatment on bone mineral density in osteoporosis therapy: a meta-analysis of randomized controlled trials. PLoS One. 2011;6:e26267. doi: 10.1371/journal.pone.0026267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hattner R, Epker BN, Frost HM. Suggested sequential mode of control of changes in cell behaviour in adult bone remodelling. Nature. 1965;206:489–490. doi: 10.1038/206489a0. [DOI] [PubMed] [Google Scholar]
- 137.Frost HM. Introduction to Biomechanics. Charles C Thomas Pub Ltd; 1967. [Google Scholar]
- 138.Hatori M, Klatte KJ, Teixeira CC, Shapiro IM. End labeling studies of fragmented DNA in the avian growth plate: evidence of apoptosis in terminally differentiated chondrocytes. J Bone Miner Res. 1995;10:1960–1968. doi: 10.1002/jbmr.5650101216. [DOI] [PubMed] [Google Scholar]
- 139.Noble BS, Stevens H, Loveridge N, Reeve J. Identification of apoptotic changes in osteocytes in normal and pathological human bone. Bone. 1997;20:273–282. doi: 10.1016/s8756-3282(96)00365-1. [DOI] [PubMed] [Google Scholar]
- 140.Verborgt O, Gibson GJ, Schaffler MB. Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2000;15:60–67. doi: 10.1359/jbmr.2000.15.1.60. [DOI] [PubMed] [Google Scholar]
- 141.Elmardi AS, Katchburian MV, Katchburian E. Electron microscopy of developing calvaria reveals images that suggest that osteoclasts engulf and destroy osteocytes during bone resorption. Calcif Tissue Int. 1990;46:239–245. doi: 10.1007/BF02555002. [DOI] [PubMed] [Google Scholar]
- 142.Cerri PS, Boabaid F, Katchburian E. Combined TUNEL and TRAP methods suggest that apoptotic bone cells are inside vacuoles of alveolar bone osteoclasts in young rats. J Periodontal Res. 2003;38:223–226. doi: 10.1034/j.1600-0765.2003.02006.x. [DOI] [PubMed] [Google Scholar]
- 143.Torrance AG, Mosley JR, Suswillo RF, Lanyon LE. Noninvasive loading of the rat ulna in vivo induces a strain-related modeling response uncomplicated by trauma or periostal pressure. Calcified tissue international. 1994;54:241–247. doi: 10.1007/BF00301686. [DOI] [PubMed] [Google Scholar]
- 144.Bentolila V, Boyce TM, Fyhrie DP, Drumb R, Skerry TM, Schaffler MB. Intracortical remodeling in adult rat long bones after fatigue loading. Bone. 1998;23:275–281. doi: 10.1016/s8756-3282(98)00104-5. [DOI] [PubMed] [Google Scholar]
- 145.Cardoso L, Herman BC, Verborgt O, Laudier D, Majeska RJ, Schaffler MB. Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2009;24:597–605. doi: 10.1359/JBMR.081210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Emerton KB, Hu B, Woo AA, Sinofsky A, Hernandez C, Majeska RJ, Jepsen KJ, Schaffler MB. Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone. 2010;46:577–583. doi: 10.1016/j.bone.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cabahug PCLD, Kennedy O, Majeska RJ, Tuthill A, Judex S, Schaffler MB. Inhibition of osteocyte apoptosis prevents trabecular bone loss after unloading of mouse long bone. Orthopaedic Research Society; San Antonio, Texas, Univted States: 2013. [Google Scholar]
- 148.Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, Ito M, Takeshita S, Ikeda K. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell metabolism. 2007;5:464–475. doi: 10.1016/j.cmet.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 149.Martin RB. Toward a unifying theory of bone remodeling. Bone. 2000;26:1–6. doi: 10.1016/s8756-3282(99)00241-0. [DOI] [PubMed] [Google Scholar]
- 150.Gu G, Mulari M, Peng Z, Hentunen TA, Vaananen HK. Death of osteocytes turns off the inhibition of osteoclasts and triggers local bone resorption. Biochemical and biophysical research communications. 2005;335:1095–1101. doi: 10.1016/j.bbrc.2005.06.211. [DOI] [PubMed] [Google Scholar]
- 151.Heino TJ, Hentunen TA, Vaananen HK. Osteocytes inhibit osteoclastic bone resorption through transforming growth factor-beta: enhancement by estrogen. J Cell Biochem. 2002;85:185–197. doi: 10.1002/jcb.10109. [DOI] [PubMed] [Google Scholar]
- 152.Maejima-Ikeda A, Aoki M, Tsuritani K, Kamioka K, Hiura K, Miyoshi T, Hara H, Takano-Yamamoto T, Kumegawa M. Chick osteocyte-derived protein inhibits osteoclastic bone resorption. Biochem J. 1997;322 ( Pt 1):245–250. doi: 10.1042/bj3220245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Collin-Osdoby P, Rothe L, Bekker S, Anderson F, Huang Y, Osdoby P. Basic fibroblast growth factor stimulates osteoclast recruitment, development, and bone pit resorption in association with angiogenesis in vivo on the chick chorioallantoic membrane and activates isolated avian osteoclast resorption in vitro. J Bone Miner Res. 2002;17:1859–1871. doi: 10.1359/jbmr.2002.17.10.1859. [DOI] [PubMed] [Google Scholar]
- 154.Webber DM, Menton D, Osdoby P. An in vivo model system for the study of avian osteoclast recruitment and activity. Bone Miner. 1990;11:127–140. doi: 10.1016/0169-6009(90)90053-i. [DOI] [PubMed] [Google Scholar]
- 155.Parfitt AM. Osteoclast precursors as leukocytes: importance of the area code. Bone. 1998;23:491–494. doi: 10.1016/s8756-3282(98)00140-9. [DOI] [PubMed] [Google Scholar]
- 156.Kindle L, Rothe L, Kriss M, Osdoby P, Collin-Osdoby P. Human microvascular endothelial cell activation by IL-1 and TNF-alpha stimulates the adhesion and transendothelial migration of circulating human CD14+ monocytes that develop with RANKL into functional osteoclasts. J Bone Miner Res. 2006;21:193–206. doi: 10.1359/JBMR.051027. [DOI] [PubMed] [Google Scholar]
- 157.McGowan NW, Walker EJ, Macpherson H, Ralston SH, Helfrich MH. Cytokine-activated endothelium recruits osteoclast precursors. Endocrinology. 2001;142:1678–1681. doi: 10.1210/endo.142.4.8204. [DOI] [PubMed] [Google Scholar]
- 158.Formigli L, Fiorelli G, Benvenuti S, Tani A, Orlandini GE, Brandi ML, Zecchi-Orlandini S. Insulin-like growth factor-I stimulates in vitro migration of preosteoclasts across bone endothelial cells. Cell Tissue Res. 1997;288:101–110. doi: 10.1007/s004410050797. [DOI] [PubMed] [Google Scholar]
- 159.Al-Dujaili SA, Lau E, Al-Dujaili H, Tsang K, Guenther A, You L. Apoptotic osteocytes regulate osteoclast precursor recruitment and differentiation in vitro. Journal of cellular biochemistry. 2011;112:2412–2423. doi: 10.1002/jcb.23164. [DOI] [PubMed] [Google Scholar]
- 160.Cheung WY, Liu C, Tonelli-Zasarsky RM, Simmons CA, You L. Osteocyte apoptosis is mechanically regulated and induces angiogenesis in vitro. Journal of orthopaedic research : official publication of the Orthopaedic Research Society. 2011;29:523–530. doi: 10.1002/jor.21283. [DOI] [PubMed] [Google Scholar]
- 161.Shimizu H, Sakamoto M, Sakamoto S. Bone resorption by isolated osteoclasts in living versus devitalized bone: differences in mode and extent and the effects of human recombinant tissue inhibitor of metalloproteinases. J Bone Miner Res. 1990;5:411–418. doi: 10.1002/jbmr.5650050415. [DOI] [PubMed] [Google Scholar]
- 162.Ito H, Koefoed M, Tiyapatanaputi P, Gromov K, Goater JJ, Carmouche J, Zhang X, Rubery PT, Rabinowitz J, Samulski RJ, Nakamura T, Soballe K, O’Keefe RJ, Boyce BF, Schwarz EM. Remodeling of cortical bone allografts mediated by adherent rAAV-RANKL and VEGF gene therapy. Nat Med. 2005;11:291–297. doi: 10.1038/nm1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF, Penninger JM, Takayanagi H. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17:1231–1234. doi: 10.1038/nm.2452. [DOI] [PubMed] [Google Scholar]
- 164.Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17:1235–1241. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zhao S, Zhang YK, Harris S, Ahuja SS, Bonewald LF. MLO-Y4 osteocyte-like cells support osteoclast formation and activation. J Bone Miner Res. 2002;17:2068–2079. doi: 10.1359/jbmr.2002.17.11.2068. [DOI] [PubMed] [Google Scholar]
- 166.Kennedy OD, Herman BC, Laudier DM, Majeska RJ, Sun HB, Schaffler MB. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone. 2012;50:1115–1122. doi: 10.1016/j.bone.2012.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kartsogiannis V, Zhou H, Horwood NJ, Thomas RJ, Hards DK, Quinn JM, Niforas P, Ng KW, Martin TJ, Gillespie MT. Localization of RANKL (receptor activator of NF kappa B ligand) mRNA and protein in skeletal and extraskeletal tissues. Bone. 1999;25:525–534. doi: 10.1016/s8756-3282(99)00214-8. [DOI] [PubMed] [Google Scholar]
- 168.Verborgt O, Tatton NA, Majeska RJ, Schaffler MB. Spatial distribution of Bax and Bcl-2 in osteocytes after bone fatigue: complementary roles in bone remodeling regulation? Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2002;17:907–914. doi: 10.1359/jbmr.2002.17.5.907. [DOI] [PubMed] [Google Scholar]
- 169.Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M, Voipio-Pulkki LM. Apoptosis in human acute myocardial infarction. Circulation. 1997;95:320–323. doi: 10.1161/01.cir.95.2.320. [DOI] [PubMed] [Google Scholar]
- 170.Lipton P. Ischemic cell death in brain neurons. Physiological reviews. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- 171.Kogianni G, Mann V, Noble BS. Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J Bone Miner Res. 2008;23:915–927. doi: 10.1359/jbmr.080207. [DOI] [PubMed] [Google Scholar]
- 172.Bidwell JP, Yang J, Robling AG. Is HMGB1 an osteocyte alarmin? Journal of cellular biochemistry. 2008;103:1671–1680. doi: 10.1002/jcb.21572. [DOI] [PubMed] [Google Scholar]
- 173.Borde C, Barnay-Verdier S, Gaillard C, Hocini H, Marechal V, Gozlan J. Stepwise release of biologically active HMGB1 during HSV-2 infection. PLoS One. 2011;6:e16145. doi: 10.1371/journal.pone.0016145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, Barbour SE, Milstien S, Spiegel S. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 2008;22:2629–2638. doi: 10.1096/fj.08-107169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Peter C, Waibel M, Keppeler H, Lehmann R, Xu G, Halama A, Adamski J, Schulze-Osthoff K, Wesselborg S, Lauber K. Release of lysophospholipid ‘find-me’ signals during apoptosis requires the ATP-binding cassette transporter A1. Autoimmunity. 2012;45:568–573. doi: 10.3109/08916934.2012.719947. [DOI] [PubMed] [Google Scholar]
- 177.Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, Melville L, Melrose LA, Ogden CA, Nibbs R, Graham G, Combadiere C, Gregory CD. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood. 2008;112:5026–5036. doi: 10.1182/blood-2008-06-162404. [DOI] [PubMed] [Google Scholar]
- 178.Mullender MG, van der Meer DD, Huiskes R, Lips P. Osteocyte density changes in aging and osteoporosis. Bone. 1996;18:109–113. doi: 10.1016/8756-3282(95)00444-0. [DOI] [PubMed] [Google Scholar]
- 179.Belanger LF. Osteocytic osteolysis. Calcified tissue research. 1969;4:1–12. doi: 10.1007/BF02279101. [DOI] [PubMed] [Google Scholar]
- 180.Recklinghausen F. Untersuchungen über Rachitis und Osteomalazie. Jena 1910 [Google Scholar]
- 181.Baud CA. Submicroscopic structure and functional aspects of the osteocyte. Clin Orthop Relat Res. 1968;56:227–236. [PubMed] [Google Scholar]
- 182.Teti A, Zallone A. Do osteocytes contribute to bone mineral homeostasis? Osteocytic osteolysis revisited. Bone. 2009;44:11–16. doi: 10.1016/j.bone.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 183.Qing H, Ardeshirpour L, Pajevic PD, Dusevich V, Jahn K, Kato S, Wysolmerski J, Bonewald LF. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2012;27:1018–1029. doi: 10.1002/jbmr.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sharma D, Ciani C, Marin PA, Levy JD, Doty SB, Fritton SP. Alterations in the osteocyte lacunar-canalicular microenvironment due to estrogen deficiency. Bone. 2012;51:488–497. doi: 10.1016/j.bone.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.McKee MD, Farach-Carson MC, Butler WT, Hauschka PV, Nanci A. Ultrastructural immunolocalization of noncollagenous (osteopontin and osteocalcin) and plasma (albumin and alpha 2HS-glycoprotein) proteins in rat bone. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1993;8:485–496. doi: 10.1002/jbmr.5650080413. [DOI] [PubMed] [Google Scholar]
- 186.Barros NM, Hoac B, Neves RL, Addison WN, Assis DM, Murshed M, Carmona AK, McKee MD. Proteolytic processing of osteopontin by PHEX and accumulation of osteopontin fragments in Hyp mouse bone, the murine model of X-linked hypophosphatemia. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2013;28:688–699. doi: 10.1002/jbmr.1766. [DOI] [PubMed] [Google Scholar]
- 187.Thompson DL, Sabbagh Y, Tenenhouse HS, Roche PC, Drezner MK, Salisbury JL, Grande JP, Poeschla EM, Kumar R. Ontogeny of Phex/PHEX protein expression in mouse embryo and subcellular localization in osteoblasts. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2002;17:311–320. doi: 10.1359/jbmr.2002.17.2.311. [DOI] [PubMed] [Google Scholar]
- 188.Bergwitz C, Juppner H. FGF23 and Syndromes of Abnormal Renal Phosphate Handling. In: Kuro-o M, editor. Endocrine FGFs and Klothos. 2012. pp. 41–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Liu S, Zhou J, Tang W, Menard R, Feng JQ, Quarles LD. Pathogenic role of Fgf23 in Dmp1-null mice. Am J Physiol Endocrinol Metab. 2008;295:E254–261. doi: 10.1152/ajpendo.90201.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab. 2006;291:E38–49. doi: 10.1152/ajpendo.00008.2006. [DOI] [PubMed] [Google Scholar]
- 191.Martin A, Liu S, David V, Li H, Karydis A, Feng JQ, Quarles LD. Bone proteins PHEX and DMP1 regulate fibroblastic growth factor Fgf23 expression in osteocytes through a common pathway involving FGF receptor (FGFR) signaling. FASEB J. 2011;25:2551–2562. doi: 10.1096/fj.10-177816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Martin A, David V, Laurence JS, Schwarz PM, Lafer EM, Hedge AM, Rowe PS. Degradation of MEPE, DMP1, and release of SIBLING ASARM-peptides (minhibins): ASARM-peptide(s) are directly responsible for defective mineralization in HYP. Endocrinology. 2008;149:1757–1772. doi: 10.1210/en.2007-1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.David V, Martin A, Hedge AM, Drezner MK, Rowe PS. ASARM peptides: PHEX-dependent and -independent regulation of serum phosphate. Am J Physiol Renal Physiol. 2011;300:F783–791. doi: 10.1152/ajprenal.00304.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Guo R, Rowe PS, Liu S, Simpson LG, Xiao ZS, Quarles LD. Inhibition of MEPE cleavage by Phex. Biochemical and biophysical research communications. 2002;297:38–45. doi: 10.1016/s0006-291x(02)02125-3. [DOI] [PubMed] [Google Scholar]
- 195.Addison WN, Nakano Y, Loisel T, Crine P, McKee MD. MEPE-ASARM peptides control extracellular matrix mineralization by binding to hydroxyapatite: an inhibition regulated by PHEX cleavage of ASARM. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2008;23:1638–1649. doi: 10.1359/jbmr.080601. [DOI] [PubMed] [Google Scholar]