

Abstract
Despite the central role of the liver in the regulation of glucose and lipid metabolism there are currently no methods to directly assess hepatic oxidative metabolism in humans in vivo. By utilizing a novel 13C-labeling strategy in combination with 13C magnetic resonance spectroscopy we show that rates of mitochondrial oxidation and anaplerosis in human liver can be directly determined noninvasively. Using this approach we found the mean rates of hepatic TCA cycle flux (VTCA) and anaplerotic flux (VANA) to be 0.43 ± 0.04 μmol (g-liver-min)−1 and 0.60 ± 0.11 μmol (g-liver-min)−1, respectively, in fourteen healthy, lean, individuals. We also found the ratio VANA/VTCA to be 1.39 ± 0.22, which is several fold lower than recently published estimates using an indirect approach. This method will be useful for understanding the pathogenesis of non-alcoholic fatty liver disease and type 2 diabetes as well as assessing the effectiveness of new therapies targeting these pathways in man.
Keywords: liver, metabolism, TCA cycle, in vivo13C magnetic resonance spectroscopy
Introduction
The liver plays a central role in the regulation of normal glucose and lipid homeostasis and in the pathogenesis of type 2 diabetes. Both hepatic mitochondrial tricarboxylic acid flux (VTCA) and anaplerotic flux (VANA) are critical functions in this regard given their major roles in hepatic fatty acid oxidation and gluconeogenesis. However, despite their importance, there are currently no methods to directly assess these fluxes in human liver. Indirect estimates of hepatic fluxes in humans have been obtained recently by examining the isotopomer labeling patterns of plasma glucose using a multiple tracer method in combination with ex vivo analysis by 13C and 2H nuclear magnetic resonance (NMR)1–3, a method derived from techniques originally developed using radioactive tracers4,5. The relative rate of hepatic anaplerosis to TCA cycle flux was determined from the 13C isotopomers of the C2 carbon of plasma glucose following the ingestion of [U-13C] propionate; using this technique the ratio VANA/VTCA was found to be approximately five. Absolute anaplerotic and TCA cycle fluxes were then calculated from measurements of whole body glucose turnover combined with rates of gluconeogenesis determined from deuterium labeling of C2 and C5 glucose from ingested 2H water. However, this approach assesses rates of anaplerosis and TCA cycle flux indirectly and the fluxes are necessarily expressed in terms of body-weight or lean body-mass, rather than per amount of liver tissue, which complicates interpretation of the data when comparing results in subjects with differing body compositions and body weights.
In contrast, in vivo magnetic resonance spectroscopy (MRS) offers the major advantage of being able to directly assess rates of intracellular metabolism in an organ specific manner6–11. Hepatic energetics have been investigated in patients with type 2 diabetes using 31P-MRS techniques. Szendroedi et al. observed that the concentrations of adenosine tri-phosphate (ATP) and inorganic phosphate (Pi) were decreased in the liver of overweight individuals with type 2 diabetes and correlated inversely with hepatic insulin sensitivity12. This group has also observed that the rate of hepatic ATP turnover (unidirectional Pi → ATP flux, assessed by 31P-saturation-transfer-MRS) was decreased in a similar cohort of overweight, type 2 diabetics13, suggesting that hepatic mitochondrial metabolism may be compromised in type 2 diabetes. While novel, the 31P-saturation-transfer-MRS approach does not directly assess hepatic mitochondrial oxidative function14. Studies utilizing 13C MRS have employed [2-13C] acetate infusions to directly assess mitochondrial oxidative function in skeletal muscle7,8,11,15 and in glial cells in brain16,17 by monitoring the rate of 13C label incorporation into C4 glutamate. However, this 13C labeling scheme is unsuitable for liver where the presence of significant quantities of intracellular lipid obscures the detection of [4-13C] glutamate. To circumvent this problem we implemented a novel 13C labeling strategy and infused [1-13C] acetate and monitored 13C label incorporation into liver C5- and C1-glutamate, which, given their chemical shifts, we reasoned would be clear of interfering lipid resonances in vivo18–20.
Using this approach we show that 13C enrichment in C5- and C1-glutamate can be directly detected in human liver without interference from intrahepatic lipid peaks and demonstrate that these data, in turn, can be used to directly assess the absolute fluxes of hepatic VTCA and VANA. Assumptions underlying the MRS methodology and modeling analysis were also validated using analogous experiments in rat liver, ex vivo. The successful development of this 13C MRS technique has yielded the first direct measurements of hepatic mitochondrial oxidative and anaplerotic rates in man and will enhance the investigation of hepatic metabolism in normal and diabetic humans as well as the etiology and treatment of non-alcoholic fatty liver disease (NAFLD).
Results
We recruited twelve young, lean, healthy, participants from the local New Haven, CT community for these studies (see Online Methods). Subjects were screened to eliminate factors that would alter normal hepatic metabolism; all had a normal birth weight, no history of chronic metabolic diseases, were non-smokers and were not taking any medications. Body mass index for this cohort (22.8 ± 2.0 kg m−2) was within the normal range. All subjects had normal (<5.5%) levels of liver triglyceride content assessed by 1H MRS (Supplementary Table 1). Written, informed consent was obtained from each subject after the purpose, nature and potential complications of the studies were explained. All procedures were approved by the Yale University Human Investigation Committee.
13C-MRS assessment of hepatic TCA cycle flux
During the infusion study, the plasma acetate concentration and 13C enrichment were elevated within 5 minutes after commencing the infusion of [1-13C] acetate and plateaued by 20 minutes. There were no significant differences in plasma acetate kinetics with or without a priming dose of [1-13C] acetate and results were combined (Fig. 1). Localized 13C-MRS of the liver (Fig. 2a) was performed throughout the infusion protocol, and 13C labeling of the hepatic glutamate and bicarbonate pools was observed (Fig. 2b) due to the oxidation of [1-13C] acetate via the TCA cycle. Enrichment of these metabolites could be detected without interference from the lipid carboxyl group peak at ~172.5 ppm. Using this novel labeling strategy, initial entry of 13C-acetate into the TCA cycle results in enrichment at the C5 carbon of glutamate, followed by labeling at C1-glutamate as the 13C label traverses a 2nd turn of the cycle. The bicarbonate pool also becomes enriched due to the loss of 13C labeled carbon dioxide (CO2) during the decarboxylation reactions of several TCA cycle intermediates as well as exchange with the plasma CO2/bicarbonate pool. A more detailed description of the 13C labeling scheme can be found in the Online Methods.
Figure 1.
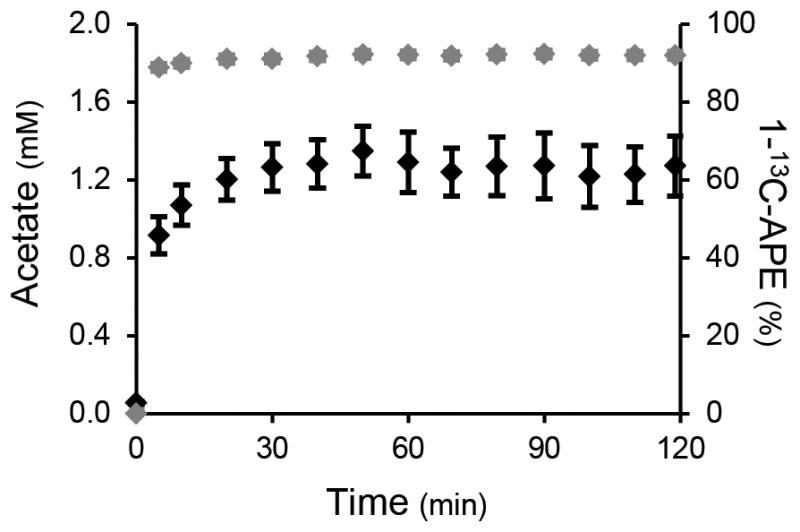
Plasma acetate concentration (black diamonds) and 1-13C enrichment (grey diamonds) during the [1-13C] acetate infusion protocol (n = 12).
Figure 2.
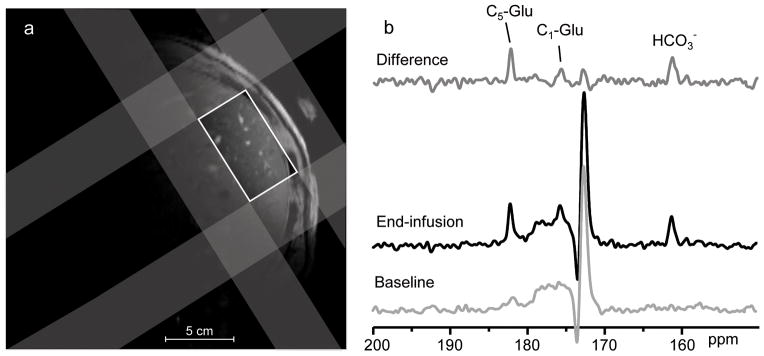
(a) Axial gradient-echo image of the torso acquired during a single breath-hold. 1H-decoupled 13C MR spectra were acquired from a localized region within the liver. Voxel selection was accomplished using outer volume suppression (shaded regions) applied in 3 dimensions to suppress signals arising from outside the liver and define the volume of interest (white box). (b) Localized 13C spectra of the human liver acquired prior to and at the end of a 120min infusion of 1-13C-acetate. 13C-labeling at the C5 position of glutamate (C5-Glu) was observed due to the oxidation of [1-13C] acetate via the TCA cycle. Labeling at C1-glutamate (C1-Glu) and of the bicarbonate (HCO3−) pool was also observed as the 13C label traversed a 2nd turn of the TCA cycle.
13C enrichment of the C5-glutamate pool was detected during the initial 10 min of the [1-13C] acetate infusion and rapidly attained steady state (Fig. 3). 13C labeling at C1-glutamate occurred more slowly but also plateaued by the end of the 120 min infusion protocol. Hepatic glutamate concentration (5.79 ± 1.31 μmol g−1, n = 12) was measured by natural abundance localized 13C-MRS on a separate occasion.
Figure 3.
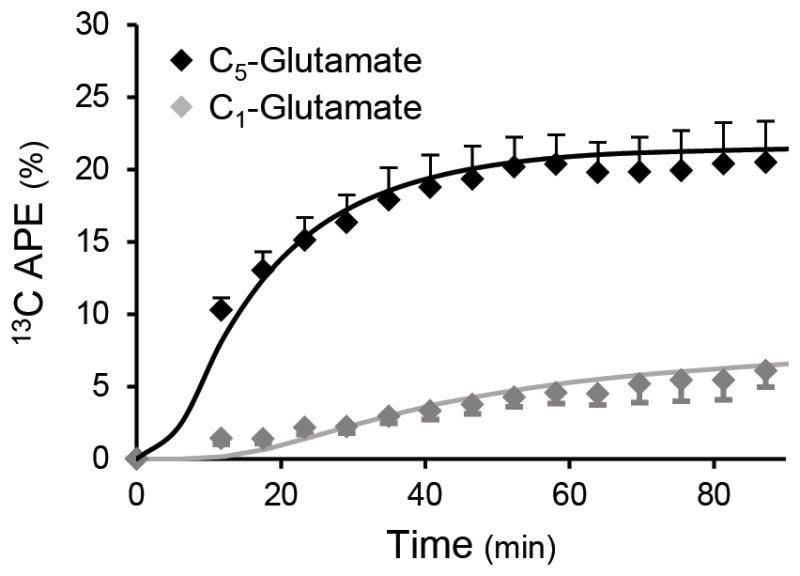
Time-courses of hepatic C5- and C1-glutamate enrichment determined by localized in vivo 13C-MRS during an infusion of [1-13C] acetate (n = 12). The kinetics of liver glutamate enrichment for each subject was determined by the metabolic model of hepatic metabolism. The average fit of the model to the glutamate enrichment data is superimposed.
Metabolic Modeling
C5- and C1- glutamate 13C enrichment profiles for each subject were fitted to a model of liver oxidative metabolism (Fig. 4). This model was adapted from previous schemes used to simulate acetate metabolism in muscle and brain8,17,21 and incorporated modifications to reflect the specific biochemical pathways of hepatic metabolism. These included: 1) anaplerotic fluxes to account for the entry of metabolites into the TCA cycle; and 2) gluconeogenesis from TCA cycle intermediates via phosphoenolpyruvate (PEP). The modeling scheme is described in detail in the Online Methods.
Figure 4.

Metabolic model of liver acetate oxidative metabolism used to estimate hepatic TCA cycle flux (VTCA) and anaplerosis (VANA). Carbon positional enrichment denoted in red occurs during the initial incorporation of label from 1-13C-acetate to glutamate on the first turn of the TCA cycle. Positional enrichment denoted in blue occurs during the 2nd turn of the TCA cycle, with label originating from internal scrambling at succinate or from bicarbonate (HCO3−)/13CO2 via anaplerosis. A full description of the metabolic model can be found in the Online Methods section.
For each subject the precision in the rates of hepatic metabolism estimated by the metabolic model was determined by performing 500 Monte-Carlo simulations of the dataset followed by probability distribution analysis. Representative data for the estimates of TCA cycle flux and anaplerosis are shown for one of the subjects in the Supplementary Information (Fig. S1). For this group of lean, healthy subjects the mean rates of VTCA and VANA were 0.43 ± 0.04 and 0.60 ± 0.11 μmol (g-liver-min)−1, respectively; the ratio VANA/VTCA was 1.39 ± 0.22 (Table 1, Supplementary Fig. S2).
Table 1.
Average hepatic mitochondrial metabolic fluxes estimated by metabolic modeling of the time-courses of liver glutamate enrichment for each subject during an infusion of [1-13C] acetate. VTCA = hepatic TCA cycle flux. VANA = rate of anaplerosis. VAC = rate of acetate utilization.
| n | Mean | SEM | |
|---|---|---|---|
| VTCA (μmol g-min−1) | 12 | 0.43 ± 0.04 | |
| VANA (μmol g-min−1) | 12 | 0.60 ± 0.11 | |
| VAC (μmol g-min−1) | 12 | 0.10 ± 0.01 | |
| VANA/VTCA | 12 | 1.39 ± 0.22 | |
Discussion
In this study, we have developed and applied a novel 13C-MRS method to directly assess rates of hepatic oxidative and anaplerotic flux in human liver in vivo. This was accomplished by utilizing a novel 13C labeling scheme, which enriches the carboxyl groups of liver glutamate and avoids the interfering resonances of intrahepatic lipid. Time-courses of 13C enrichment of hepatic glutamate were detected with sufficient sensitivity and time resolution to permit modeling of the data to yield rates of hepatic VTCA and VANA in humans. To ensure accurate simulation of the kinetics of 13C-label turnover, a sophisticated metabolic model of liver oxidative metabolism was developed for these analyses. Using this approach we found that the mean rates of hepatic VTCA and VANA were 0.43 ± 0.04 and 0.60 ± 0.11 μmol (g-liver-min)−1, respectively, yielding a VANA/VTCA ratio of 1.39 ± 0.22.
To validate this methodology, we performed analogous infusion experiments in adult male rats (see Supplementary Information). Positional enrichment of liver glutamate at multiple time points was determined ex vivo by 13C-NMR and LC/MS/MS of the extracted tissue. Profiles of 13C glutamate enrichment were constructed from the individual time points, and this composite dataset was fitted using our model of hepatic acetate metabolism. Using this approach we found the liver VTCA flux was estimated to be approximately 1.2 μmol (g-liver-min)−1, which is comparable to VTCA flux rates determined by Beylot et al. in rats in vivo and in perfused livers22. We also estimated hepatic VTCA flux rates in these animals indirectly by an independent method wherein we measured the ratio of VANA/VTCA flux through steady state labeling of C2, C3 and C4 glutamate during an infusion of [3-13C] lactate (Supplementary Table S2) and multiplied this fraction by rates of hepatic gluconeogenesis. This was calculated from endogenous glucose production, assessed by [3-3H] glucose turnover assuming a 90% contribution from the liver, and from the contribution of pyruvate to gluconeogenesis, which was determined during the infusion of [3-13C] lactate (see Supplementary Information). TCA cycle flux estimated using this independent method was found to be 1.4 ± 0.4 μmol (g-liver-min)−1 (Supplementary Table S2), in excellent agreement with VTCA derived using dynamic metabolic modeling of the liver C5 and C1 glutamate enrichment time course data.
We found the ratio of VANA/VTCA in humans to be 1.39 ± 0.22, which is over 300% lower than has been recently been published by Sunny et al.3. By assessing the isotopomer splitting pattern in C2 plasma glucose from ingested U-13C propionate they found rates of VANA to be approximately five times greater than VTCA flux in normal subjects3. Such high rates of anaplerosis are incompatible with the extent of 13C labeling at C1-glutamate that we have observed in the present study in human liver following an infusion of [1-13C] acetate. We believe that this discrepancy in VANA/VTCA may be due to important limitations regarding the use of U-13C propionate as a tracer for assessing hepatic metabolism in vivo. Specifically, propionate enters the TCA cycle as an anaplerotic substrate via succinyl CoA, which requires obligate utilization via gluconeogenesis and/or pyruvate recycling. Furthermore, in contrast to acetate or lactate, Puchowicz et al. have demonstrated very large periportal-perivenous gradients for propionate uptake across the liver23. To examine these issues we measured glucose turnover in a group of rats infused intravenously with low (333 μmol kg−1) and high (666 μmol kg−1) doses of [U-13C] proprionate that are within the range used by previous studies2,24,25. We found that plasma glucose concentrations increased by ~25 mg dl−1 during the high dose propionate infusions compared to the low dose propionate infusions. Furthermore, this increase in plasma glucose concentration during the high dose propionate infusion was associated with an ~30% increase in rates of hepatic glucose production (Supplementary Fig. S3), compared to the low dose infusion. This stimulation of hepatic glucose production by propionate is similar to the ~40% increase in rates of endogenous glucose production [from 7.3 to 10.0 mg (kg-min)−1], observed when the dose of [U-13C] propionate given to rats was increased from 222 to 825 μmol24,25. In contrast, doubling the infusion rates of [1-13C] acetate or [3-13C] lactate had no effects on glucose production, the ratio of pyruvate carboxylase flux (VPC) to VTCA or the ratio of pyruvate dehydrogenase flux (VPDH) to VTCA. (Supplementary Fig. S3). Taken together, these data demonstrate that [U-13C] propionate at doses in the range used to assess rates of mitochondrial metabolism can alter rates of anaplerosis making it unsuitable as a tracer for hepatic metabolism. These substrate driven effects of [U-13C] propionate on VANA will likely be further exaggerated when the isotope is given orally since first pass portal vein delivery will probably result in much higher concentrations of propionate being presented to the periportal hepatocytes than when administered systemically. Furthermore, in contrast to the in vivo MRS method described here, the [U-13C] propionate isotope method assumes negligible flux through hepatic pyruvate dehydrogenase, which is not likely to be the case in normal liver (Supplementary Table S2)26 and does not account for the effects of 13C label recycling via CO2 which modulates the observed labeling patterns27.
The methodology described in this article uses a dynamic approach to monitor metabolite kinetics and estimate metabolic rates. While the MRS techniques involved in data acquisition are technically demanding, and the metabolic modeling analyses require high quality data collected over an extended duration (90–120 min of infusion are required to obtain sufficient time points to establish the kinetics of glutamate enrichment), the technique provides reliable, direct estimates of hepatic oxidative metabolism. Relatively modest increments in plasma acetate concentrations are observed (up to ~1 mM) minimizing the likelihood that the provision of exogenous substrate will perturb metabolism in the liver, or other organs. Metabolite enrichment in the target tissue is directly assessed avoiding assumptions about the contributions of other tissues such as the kidney, which may impact other (whole-body) techniques. Additionally, the dynamic approach does not inherently require isotopic steady-state conditions for either the substrate precursor or the target metabolite(s); however, since both the peripheral circulation and the liver were repeatedly sampled, isotopic steady-state was confirmed for these studies (Fig. 1 and 3).
We also performed a Monte-Carlo simulation for each dataset, combined with probability distribution analysis to determine the precision with which the metabolic model fitted the in vivo data (Supplementary Fig. S1). This analysis, which determined uncertainty based upon spectral noise and deviation of the data from the best fit, showed that reliable estimates of hepatic metabolism were obtained on each of the subjects in this study. It further demonstrates that this method can be used to track hepatic metabolism in individual subjects opening up the possibility of monitoring the impact of interventions which may perturb the liver. The estimated fluxes are sensitive to assumptions underlying the model of hepatic metabolism; therefore, actual error may exceed the estimates reported.
In summary, the use of traditional 13C-MRS techniques to measure tissue specific rates of mitochondrial VTCA flux is not possible in liver due to the presence of dominant intrahepatic lipid peaks which obscure the detection of conventional 13C-labeling schemes. By using an innovative labeling strategy, in combination with localized in vivo 13C-MRS, we have directly observed rates of mitochondrial oxidation and anaplerosis in human liver. A sophisticated metabolic model of hepatic metabolism was developed to simulate the kinetics of 13C-label turnover, which generated estimates of liver oxidative fluxes. This method will be useful for understanding the potential role of altered hepatic mitochondrial fat oxidation in the pathogenesis of NAFLD and type 2 diabetes as well as for assessing the effectiveness of novel therapies aimed at increasing rates of hepatic fatty oxidation in man to reverse NAFLD.
Online Methods
Human Subjects
All procedures were approved by the Yale University Human Investigation Committee; written, informed consent was obtained from each participant after explanation of the purpose, nature and potential complications of the study. Twelve young, lean, healthy volunteers (10 male, 2 female) were recruited from the local community and were prescreened to be in good health, lean, non-smokers and taking no medications. Subjects were also selected to have normal levels of intrahepatic lipid content determined using 1H MRS.
In vivo 13C MRS: general procedure
Studies were performed on a 4.0 Tesla Bruker Medspec system using a custom-built 13C/1H probe consisting of a 9 cm diameter 13C surface coil with a pair of elliptical, 14.5 × 11 cm, 1H coils arrayed in quadrature for imaging, shimming and decoupling. Subjects were positioned supine with the probe attached to a rigid support to minimize extraneous movement. The 13C/1H probe was curved in design to site the radiofrequency coils in an optimal position over the lateral aspect of the liver. Scout images of the torso were acquired during an end-expiration breath-hold to confirm correct positioning of the probe and to define a volume for localized 13C spectroscopy. Field homogeneity was optimized using a respiratory-gated FASTMAP routine28. 1H linewidths from an 8 cm3 volume within the region of interest, acquired using a respiratory-gated STEAM sequence, were typically ~25 Hz. Each participant underwent two MRS sessions typically separated by less than 8 days: a natural-abundance 13C scan to determine the hepatic concentration of glutamate, and an infusion study to assess hepatic TCA cycle flux. The natural abundance measurement was performed prior to the infusion study for eight of the 12 participants; for the remaining individuals there was a sufficient interval after the infusion study to ensure full washout of 13C-label before the natural abundance scan.
13C-MRS assessment of hepatic glutamate concentrations
Localized 13C spectra were acquired using a custom-written adiabatic pulse-acquire sequence with WALTZ16 decoupling and 3 dimensional outer volume suppression (OVS) to minimize signal arising from the chest wall and select a region of interest within the liver (Figure 2). The size of the localized volume varied from subject to subject depending on the morphology of the liver but was typically ~225 cm3. Due to the relatively long T1 relaxation times of the carboxyl groups, spectra were acquired with a repetition time (TR) of 10 s in blocks of 32 transients and repeated until sufficient signal had been acquired to detect the natural abundance glutamate signal (typically 50–60 minutes). The hepatic concentration of glutamate was determined from the in vivo C5-glutamate signal relative to the fully-relaxed (TR = 30 s) signal obtained from a 25 mM glutamate phantom, using a voxel of identical size and location, after correction for coil loading effects.
13C-MRS assessment of hepatic TCA cycle flux
Following an overnight fast, intravenous catheters were inserted into an antecubital vein in each arm. Localized 13C spectra were obtained over a 20 min baseline period and throughout a 120 min infusion of [1-13C]acetate (99% enriched, 350 mmolL−1 sodium salt). Subjects underwent either a continuous acetate infusion protocol (n = 6) at a rate of 3.0 mg(kg-min)−1, or a primed-continuous infusion protocol (n = 6), with a 5 min priming dose at 150% of the continuous acetate infusion rate. Spectra were acquired as described above but with nuclear Overhauser enhancement (nOe) applied during the TR (1 ms hard-pulse every 50 ms) to increase 13C sensitivity. Blood samples were obtained at regular intervals throughout the infusion protocol for the measurement of plasma acetate concentration and 13C1 fractional enrichment by high-field 1H NMR at 500 MHz or by gas-chromatography/mass-spectrometry (GC/MS)21. To enhance signal/noise, data acquired during the infusion were summed using a four spectrum rolling average; difference spectra were created for each time point by subtracting the baseline spectrum. Glutamate enrichment at the C5 and C1 positions was determined from each difference spectrum relative to the baseline bicarbonate signal, which was assumed to be 1.1% enriched (i.e. natural abundance for 13C). Slight differences in nOe for bicarbonate and C5 glutamate in liver and the detection efficiency of C1 versus C5 glutamate using this pulse-sequence were accounted for by means of correction factors determined in vivo and from a glutamate phantom experiment, respectively.
Metabolic Modeling
The time courses of hepatic glutamate enrichment for each subject were fitted to a metabolic model of the TCA cycle (Fig 4) using CWave software (developed by Dr. Graeme Mason, Yale University). This model consists of a series of isotopic and mass balance differential equations that describe the hepatic metabolism of acetate (see Supplementary Information). This model was developed from previous schemes used to examine acetate metabolism in muscle21 and brain8,17,29 modified to account for liver-specific phenomena including gluconeogenesis from TCA cycle intermediates via phosphoenolpyruvate (PEP), the entry of metabolites into the TCA cycle via anaplerosis, and 13CO2 recycling.
Acetate uptake occurs via the monocarboxylate transporter; under the systemic acetate concentrations in these studies (~1 mM) acetate transport is assumed to be saturated and acetate is converted to acetyl-CoA within the hepatocyte at the rate, VAC. AcetylCoA enters the TCA cycle labeling citrate and α-ketoglutarate (α-KG) at the C5 position. Entry of unlabeled substrates derived from fatty acids or pyruvate is accounted for by the flux VDIL, which dilutes the enrichment of acetyl CoA. Glutamate and α-KG are in rapid exchange (VX) via asparate-aminotransferase, and equilibration results in the enrichment of 5-13C-glutamate, which can be detected by 13C-MRS. As the TCA cycle progresses, the C1 and C4 positions of the TCA cycle intermediates succinate and malate are equally labeled due to the stereo-chemical symmetry of the succinate molecule. To preserve mass balance of the TCA cycle intermediates, carbon-entry from unlabeled α-KG occurs at the level of succinate.
Key branch points in this model of hepatic metabolism occur at oxaloacetate where the 13C-label may undergo several fates. From OAA, the 13C label may enter a second turn of the TCA cycle; [4-13C] OAA forms [1-13C]citrate and eventually enriches [1-13C] glutamate, which is also detectable by 13C-MRS. [1-13C] OAA labels the carboxyl side-chain (C6) of citrate; 13C at this position is lost as CO2 during the decarboxylation of citrate. Oxaloacetate may also exchange with the aspartate pool, which delays enrichment at [1-13C] citrate and is modeled at a rate equivalent to VX. The action of PEP-carboxykinase (VPEPCK) decarboxylates OAA forming 13CO2 and 1-13C-phosphoenol-pyruvate (PEP) from 13C4 and 13C1-OAA, respectively. PEP is a precursor for gluconeogenesis or may be converted to pyruvate via pyruvate kinase (PK). Pyruvate can also potentially be directly formed from malate through the action of malic enzyme (ME). Our experimental data demonstrates that there is substantial dilution of [1-13C] with respect to [5-13C] glutamate, indicating that unlabeled carbons must enter the TCA cycle post-succinate via anaplerosis. We assume that any 1-13C-pyruvate formed via PK or ME is unavailable for recycling into OAA through pyruvate-carboxylase (VPC). This approach is supported by experiments in rodents (see Supplementary Information) where we observe negligible labeling of [1-13C] lactate or alanine (surrogates for [1-13C] pyruvate) during a 1-13C-acetate infusion and in published literature which suggests compartmentation of the gluconeogenic and anaplerotic pools30 and/or that the PK+ME flux is relatively low31–33. Therefore, only unlabeled pyruvate and 13CO2 may enter the TCA cycle via VPC. Anaplerosis (VANA) is treated as a parameter that is equivalent to VPC and VPEPCK. Backwards scrambling of 13C-label between C1 and C4 OAA due to the action of fumarase is treated as a simple, rapid inter-conversion between the two species (VFUM).
Target functions of C5- and C1-glutamate enrichment during the [1-13C] acetate infusion were created for each subject as described above. The hepatic concentration of glutamate was estimated from a natural abundance 13C-MRS measurement. Due to the high activity of carbonic anhydrase, liver bicarbonate enrichment estimated from the in vivo 13C spectra was used as a surrogate for the 13CO2 input function. The liver bicarbonate concentration was calculated to be 12 mM, based on an assumed plasma bicarbonate concentration of 24 mM and a pH difference of 0.3 between liver and blood. Concentrations of the other hepatic metabolic intermediates were estimated from the literature, using average values scaled to the estimated concentration of glutamate for each subject. Due to the low concentrations of these intermediates relative to glutamate, labeling of these metabolites has a negligible impact on the kinetics of glutamate enrichment or the modeled rates of hepatic metabolism. The parameters VAC, VDIL, VTCA and VANA were estimated by numerical simulation of the differential equations that contribute to the metabolic model. Based on previous studies, VX was assumed to be significantly faster than VTCA and non-limiting. Each simulated parameter was constrained to be greater than or equal to zero.
1H MRS assessment of intrahepatic lipid content
To assess liver lipid content, localized 1H spectra were obtained from a (15 × 15 × 15) mm3 voxel placed in 3 different locations of the liver. Spectra were acquired using a respiratory-gated stimulated echo-acquisition mode (STEAM) sequence, with chemical shift selective peak suppression (CHESS). Total intrahepatic lipid (IHL) content was estimated from comparison of a water-suppressed lipid spectrum and a lipid-suppressed water spectrum, with the appropriate peak for each spectrum on-resonance.
Statistical Analysis
Data are expressed as mean ± standard error of the mean (SEM). The precision in the fit of the metabolic model to the raw data was determined by probability distribution analysis of 500 Monte-Carlo simulations of the model with random Gaussian noise added to the fitted time courses. Probability distribution analysis was performed for each rate parameter on the resultant Monte-Carlo dataset using the distribution fitting function (dfittool) in Matlab.
Supplementary Material
Acknowledgments
The authors would like to thank A. Impellizeri, Y. Kosover, I. Smolgovsky, M. Smolgovsky, G. Solomon, C. Parmelee and the staff of the Yale Center for Clinical Investigation Hospital Research Unit for their technical support, and the volunteers for their participation in these studies. We also acknowledge the contributions of Dr. G. Mason who assisted in the implementation and interpretation of the metabolic modeling of the rat [1-13C] acetate infusion data.
This publication was supported by grants from the United States Public Health Service: R24 DK-085638, R01 AG-23686, R01 DK-49230, P30 DK-45735, UL1 RR-024139, a Distinguished Clinical Investigator Award from the American Diabetes Association (KFP) and an investigator initiated grant from Pfizer, Inc. (KFP). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the United States National Center for Research Resources or National Institutes of Health.
Footnotes
Author Contributions
D.E.B, R.J.P, J.T, J.B, K.F.P, D.L.R and G.I.S designed the experimental protocols. D.E.B, R.J.P, S.D, G.W.C and K.F.P performed the studies. D.E.B, N.J, R.J.P, S.D, G.W.C and D.L.R analyzed the data. D.E.B, R.J.P, K.F.P, D.L.R and G.I.S contributed to the writing of the manuscript.
References
- 1.Jones JG, Solomon MA, Cole SM, Sherry AD, Malloy CR. An integrated (2)H and (13)C NMR study of gluconeogenesis and TCA cycle flux in humans. Am J Physiol Endocrinol Metab. 2001;281:E848–856. doi: 10.1152/ajpendo.2001.281.4.E848. [DOI] [PubMed] [Google Scholar]
- 2.Jones JG, Solomon MA, Sherry AD, Jeffrey FM, Malloy CR. 13C NMR measurements of human gluconeogenic fluxes after ingestion of [U-13C]propionate, phenylacetate, and acetaminophen. Am J Physiol. 1998;275:E843–852. doi: 10.1152/ajpendo.1998.275.5.E843. [DOI] [PubMed] [Google Scholar]
- 3.Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011;14:804–810. doi: 10.1016/j.cmet.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Landau BR, et al. 14C-labeled propionate metabolism in vivo and estimates of hepatic gluconeogenesis relative to Krebs cycle flux. Am J Physiol. 1993;265:E636–647. doi: 10.1152/ajpendo.1993.265.4.E636. [DOI] [PubMed] [Google Scholar]
- 5.Landau BR, et al. Use of 2H2O for estimating rates of gluconeogenesis. Application to the fasted state. J Clin Invest. 1995;95:172–178. doi: 10.1172/JCI117635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shulman GI, et al. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N Engl J Med. 1990;322:223–228. doi: 10.1056/NEJM199001253220403. [DOI] [PubMed] [Google Scholar]
- 7.Befroy DE, et al. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes. 2007;56:1376–1381. doi: 10.2337/db06-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lebon V, et al. Effect of triiodothyronine on mitochondrial energy coupling in human skeletal muscle. J Clin Invest. 2001;108:733–737. doi: 10.1172/JCI11775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mason GF, et al. Simultaneous determination of the rates of the TCA cycle, glucose utilization, alpha-ketoglutarate/glutamate exchange, and glutamine synthesis in human brain by NMR. J Cereb Blood Flow Metab. 1995;15:12–25. doi: 10.1038/jcbfm.1995.2. [DOI] [PubMed] [Google Scholar]
- 10.Mason GF, et al. Measurement of the tricarboxylic acid cycle rate in human grey and white matter in vivo by 1H-[13C] magnetic resonance spectroscopy at 4.1T. J Cereb Blood Flow Metab. 1999;19:1179–1188. doi: 10.1097/00004647-199911000-00002. [DOI] [PubMed] [Google Scholar]
- 11.Petersen KF, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szendroedi J, et al. Abnormal hepatic energy homeostasis in type 2 diabetes. Hepatology. 2009;50:1079–1086. doi: 10.1002/hep.23093. [DOI] [PubMed] [Google Scholar]
- 13.Schmid AI, et al. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care. 2011;34:448–453. doi: 10.2337/dc10-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Befroy DE, Rothman DL, Petersen KF, Shulman GI. (3)(1)P-magnetization transfer magnetic resonance spectroscopy measurements of in vivo metabolism. Diabetes. 2012;61:2669–2678. doi: 10.2337/db12-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitchell CS, et al. Resistance to thyroid hormone is associated with raised energy expenditure, muscle mitochondrial uncoupling, and hyperphagia. J Clin Invest. 2010;120:1345–1354. doi: 10.1172/JCI38793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boumezbeur F, et al. Altered brain mitochondrial metabolism in healthy aging as assessed by in vivo magnetic resonance spectroscopy. J Cereb Blood Flow Metab. 2010;30:211–221. doi: 10.1038/jcbfm.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lebon V, et al. Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J Neurosci. 2002;22:1523–1531. doi: 10.1523/JNEUROSCI.22-05-01523.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li S, Yang J, Shen J. Novel strategy for cerebral 13C MRS using very low RF power for proton decoupling. Magn Reson Med. 2007;57:265–271. doi: 10.1002/mrm.21148. [DOI] [PubMed] [Google Scholar]
- 19.Li S, et al. In vivo 13C magnetic resonance spectroscopy of human brain on a clinical 3 T scanner using [2-13C]glucose infusion and low-power stochastic decoupling. Magn Reson Med. 2009;62:565–573. doi: 10.1002/mrm.22044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sailasuta N, et al. Clinical NOE 13C MRS for neuropsychiatric disorders of the frontal lobe. J Magn Reson. 2008;195:219–225. doi: 10.1016/j.jmr.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Befroy DE, et al. Increased substrate oxidation and mitochondrial uncoupling in skeletal muscle of endurance-trained individuals. Proc Natl Acad Sci U S A. 2008;105:16701–16706. doi: 10.1073/pnas.0808889105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beylot M, Soloviev MV, David F, Landau BR, Brunengraber H. Tracing hepatic gluconeogenesis relative to citric acid cycle activity in vitro and in vivo. Comparisons in the use of [3-13C]lactate, [2-13C]acetate, and alpha-keto[3-13C]isocaproate. J Biol Chem. 1995;270:1509–1514. doi: 10.1074/jbc.270.4.1509. [DOI] [PubMed] [Google Scholar]
- 23.Puchowicz MA, et al. Zonation of acetate labeling across the liver: implications for studies of lipogenesis by MIDA. Am J Physiol. 1999;277:E1022–1027. doi: 10.1152/ajpendo.1999.277.6.E1022. [DOI] [PubMed] [Google Scholar]
- 24.Jin ES, et al. Glucose production, gluconeogenesis, and hepatic tricarboxylic acid cycle fluxes measured by nuclear magnetic resonance analysis of a single glucose derivative. Anal Biochem. 2004;327:149–155. doi: 10.1016/j.ab.2003.12.036. [DOI] [PubMed] [Google Scholar]
- 25.Jones JG, Carvalho RA, Franco B, Sherry AD, Malloy CR. Measurement of hepatic glucose output, krebs cycle, and gluconeogenic fluxes by NMR analysis of a single plasma glucose sample. Anal Biochem. 1998;263:39–45. doi: 10.1006/abio.1998.2796. [DOI] [PubMed] [Google Scholar]
- 26.Alves TC, et al. Regulation of hepatic fat and glucose oxidation in rats with lipid-induced hepatic insulin resistance. Hepatology. 2011;53:1175–1181. doi: 10.1002/hep.24170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones JG, et al. Measurement of gluconeogenesis and pyruvate recycling in the rat liver: a simple analysis of glucose and glutamate isotopomers during metabolism of [1,2,3-(13)C3]propionate. FEBS Lett. 1997;412:131–137. doi: 10.1016/s0014-5793(97)00764-3. [DOI] [PubMed] [Google Scholar]
- 28.Shen J, Rycyna RE, Rothman DL. Improvements on an in vivo automatic shimming method [FASTERMAP] Magn Reson Med. 1997;38:834–839. doi: 10.1002/mrm.1910380521. [DOI] [PubMed] [Google Scholar]
- 29.Patel AB, de Graaf RA, Rothman DL, Behar KL, Mason GF. Evaluation of cerebral acetate transport and metabolic rates in the rat brain in vivo using 1H-[13C]-NMR. J Cereb Blood Flow Metab. 2010;30:1200–1213. doi: 10.1038/jcbfm.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katz J, Wals P, Lee WN. Isotopomer studies of gluconeogenesis and the Krebs cycle with 13C-labeled lactate. J Biol Chem. 1993;268:25509–25521. [PubMed] [Google Scholar]
- 31.Heath DF, Rose JG. [14C]bicarbonate fixation into glucose and other metabolites in the liver of the starved rat under halothane anaesthesia. Metabolic channelling of mitochondrial oxaloacetate. Biochem J. 1985;227:851–865. doi: 10.1042/bj2270851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petersen KF, Blair JB, Shulman GI. Triiodothyronine treatment increases substrate cycling between pyruvate carboxylase and malic enzyme in perfused rat liver. Metabolism. 1995;44:1380–1383. doi: 10.1016/0026-0495(95)90133-7. [DOI] [PubMed] [Google Scholar]
- 33.Fernandez CA, Des Rosiers C. Modeling of liver citric acid cycle and gluconeogenesis based on 13C mass isotopomer distribution analysis of intermediates. J Biol Chem. 1995;270:10037–10042. doi: 10.1074/jbc.270.17.10037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.