

1 Introduction
Classical atomistic simulations, also known as Molecular Mechanics simulations, use simple potential-energy functions to model molecular systems at the atomic level. In this representation, atoms or groups of atoms are represented as spherical particles that interact through relatively simple potential functions such as Hooke’s law, Lennard-Jones and Coulomb potentials. These representations are then used to sample the conformational phase space of the molecules via simulation techniques such as Monte Carlo, ligand-docking and molecular dynamics (MD). In MD simulations, the particles obey classical equations of motion, generally Newton’s laws or Langevin dynamics, which allow for the characterization of the time evolution of the molecular structures, their fluctuations and interactions, and therefore the investigation of the system’s kinetic and thermodynamical properties.
Since their introduction to the Physics community during the 1950s,1 MD methods have grown in complexity with refinements both of the accuracy of the energy functionals and of the sophistication of the methods used for the sampling of the relevant phase space. The force fields used in biomolecular simulations include a set of potentials based on physical models, along with a set of associated parameters which are obtained by fitting to experimental and/or quantum simulations. The potentials are mathematical functions of the nuclear coordinates only, since the Born-Oppenheimer approximation2 justifies the omission of the electronic degrees of freedom. Bonded atoms are represented by two-body, three-body and four-body terms, based on bond distances and bond and dihedral angles. Non-bonded interactions, commonly modeled by Lennard-Jones and Coulomb potentials, are generally described by pairwise interactions.
Long-range electrostatic interactions are crucial for the stability of proteins, nucleic acids, glycomolecules, lipids, and other macromolecules, and their interactions with solvent, ions, and other molecules. Electrostatic interactions in biomolecular simulations have typically been modeled using the atom-centered ‘partial-charge’ approximation in which the full charge density of the system is replaced by point, fractional charges assigned to every atom. For instance, the simplest models of water assign one partial charge to each atom of the molecule. If higher accuracy is required to reproduce a specific property of water, then extra charges (representing, for instance, the lone pairs) and/or multipoles and/or polarization are added to the water model. Even in the “simple”, purely partial-charge model of biomolecules, the long-range Coulomb interactions quickly become the computational bottleneck. Their treatment demands carefully constructed algorithms in order to avoid artifacts3 and to take advantage of the existing and emerging computer architectures.
Historically, simple models of biological molecules have been used mainly out of necessity due to restraints imposed by absence of experimental data (or data at low resolution) and algorithmic and computational limitations. In many cases, very simple models have proved quite successful, such as the coarse-grained and lattice models used to identify the main features of protein folding. However, biological systems in general are very complex and experimental data is often at too low resolution to infer details of, for instance, molecular recognition processes. Since one of the main reasons to use molecular modeling is to predict molecular properties that are difficult to observe experimentally, molecular modeling often finds itself in a paradoxical situation where its predictions cannot be validated experimentally. What has become clear, though, is that the monopole electrostatic approximation is inadequate, and that the importance of an accurate representation of electrostatics cannot be overemphasized for challenging situations such as molecular recognition and computer-aided drug design.4
In this review, we primarily treat electrostatic methods for classical biomolecular simulations in explicit solvent, with special emphasis on the accuracy of the electrostatic representation. The first part of the article is dedicated to reviewing computational methods for dealing with the long-range nature of the electrostatic interactions, including traditional methods as well as recent extensions and developments. The second part of the article is dedicated to reviewing current methods for representing the molecular electronic charge cloud, that go well beyond the point-charge representation. In addition, we discuss various multiscale approaches at the Quantum Mechanics/Molecular Mechanics level and at the Molecular Mechanics/Coarse-Grain level. The general trends in both cases are towards more accuracy and proper treatment of electrostatics. We finish the article by describing other challenging developments and giving our perspective on the current state of the field.
1.1 The electrostatic problem
A classical electrostatic description first requires a choice for the representation of the continuous quantum electronic density. While the positive nuclear charge can be considered to be discrete on the atomic scale, the representation of the continuous negative electronic charge distribution is considerably more challenging. First, one has to decide how much of the molecular electronic density should be assigned to each atom. This “charge assignment” problem is nontrivial, as it is desirable that the charge distribution assigned to a particular atom be transferable, i.e., relatively independent of the molecular or condensed phase environment in which the atom finds itself. Second, one has to decide how the assigned atomic density should be represented mathematically.
The partitioning of the extended molecular charge distribution is a key issue for improving the accuracy of current force fields for large-scale biomolecular simulations. Traditionally, electrostatic interactions have been modeled using a set of fixed atom-centered point charges or “partial charges”. In fact, popular classical MD codes for biomolecular simulations assign partial charges to virtually every atom in the system. The most popular methods for extracting charges from molecular wavefunctions are based on a fitting of the atomic charges to the molecular electrostatic potential (MEP) computed with ab initio or semiempirical methods5 outside the van der Waals surface. These nonbonded potentials are then expressed as a sum of spherically symmetric atom-atom potentials. Such a description is known to represent an important source of errors for current force fields,6 primarily because the monopoles or partial charges can vary enormously with conformational changes.7 In fact, a realistic physical molecular representation generally requires dipole moments (e.g., to model the lone pairs), and quadrupole moments (e.g., to model the π-bonds). Alternatively, more charge sites can be added to the molecule.8 In principle, such a “distributed multipole” description can exactly describe the potential due to the true charge density, at points distant from the expansion centers where “penetration” effects are negligible.6 However, even with these improvements, the fit to the MEP remains poor in regions near the atomic nuclei where the charge densities overlap. As a consequence, the electrostatic interaction energy must be corrected for the “penetration” effects at close range.9 Finally, even if a distributed multipole representation gives excellent agreement with the MEP outside the van der W aals surface, and even if continuous functions (such as Gaussians) are used to represent the electrostatic potential inside the van der Waals surface, a totally reliable and stable representation of the electronic density requires the inclusion of polarization to account for the variation of the electronic density caused by intra-and inter-molecular conformational changes, as well as general changes in the atomic environment. Additional complications arise when there are changes in ionization states, as discussed in Section 5.
To begin, consider the simplest representation of electrostatics given by the monopolar approximation. The long-range effects are most pronounced for this potential, which decays with distance r as 1/r. The charge-assignment scheme based on fitting to the MEP is mathematically an under-determined problem since, in principle, innumerable distributions of different charges can all produce the same electrostatic potential outside a surface that encloses all the charges. This process is particularly poor for the case of “buried charges” for atoms in the interior of the molecule. However, once a set of charges has been assigned, the calculation of the Coulomb electrostatic potential and energy due to the given set of charges is a well-posed mathematical problem: knowing all the partial charges qi and their positions ri, one can compute their interactions, no matter how complicated the configurations of charges may be. In practice such computations are not trivial, due to two main considerations: the finite system size and the size of the molecules. Since all simulations have finite-size constraints, the long-range nature of the interactions, as well as other quantities that characterize the properties of the system in the thermodynamic limit, are dealt with through boundary conditions. With respect to molecular size, for a system with N charges, the electrostatic energy is computed via a sum over N(N −1)/2 pairs, and is therefore an
 (N2) calculation. Thus, a direct computation of all the pair interactions is often too costly for typical biomolecular sizes.
(N2) calculation. Thus, a direct computation of all the pair interactions is often too costly for typical biomolecular sizes.
An additional problem related to size is that biomolecules need to be solvated. Since molecular biology takes place, at least partially, in an aqueous environment, and since the physiological environment of a biomolecule cannot often be adequately represented with present computer resources (not to mention the fact that many of the details of this environment are often unknown), most simulations of proteins, nucleic acids and membranes assume a water environment. As each water molecule is represented by at least three charges, a large amount of the simulation time is consumed in the calculation of water-water interactions. Such an explicit solvent environment is often desirable because it captures the delicate interactions between the solutes, water and ions, and the formation of hydrogen bonds. All of these interactions can sometimes be crucial determinants for the molecular structure or function. However, the high cost of computing the water-water interactions and of fully sampling the phase space often makes the use of implicit solvent methods desirable. These methods represent the solvent as a dielectric continuum where the solute-solvent interactions are described in the spirit of a mean-field approach.
Computationally, the electrostatic potential for a system with explicit solvent is computed by either solving the Poisson’s equation or explicitly adding the individual charge potentials. For the monopolar case, these equations are given by
| (1) |
and
| (2) |
where qj, j = 1, … N represent all the partial and ionic charges, including those of water. Instead, in the implicit solvent representation, the solvent is replaced by a dielectric continuum function ε(r) that varies in space. The charge density in the resulting Poisson’s equation contains the charges due to the solute, plus another term that accounts for the counterions and co-ions in the solvent. If the mean-field, Debye-Hückel approximation is used for the salt, then the resulting Poisson-Boltzmann equation (PBE) is
| (3) |
where Nsolute is the number of charges in the solute, cs is the bulk number concentration of the s-type ions of charge qs, and Σs csqs = 0. This PBE already contains an approximation, as the exact potential of mean force that would enter the exponential has been approximated by qsφ0(r). The PBE is a non-linear differential equation, and an expansion of the exponential leads to a second approximation, in which the second term of Eq. (3) is replaced by the linear term . The linear approximation is only really valid in the limit of extremely small ion concentrations. There has been considerable debate in the literature about these approximations, and we will not cover these issues here. The major difficulty with the PBE derives from the sharp and fast variations of the dielectric function, whose values for a protein in solution are not clearly determined, and whose representation is essentially arbitrary. Its description must involve a function capable of fast variations in space, but with numerically stable gradients. At present, solving the PBE numerically is still a relatively slow process. Hence, other simpler implicit solvent models, such as the Generalized Born approximation, have been developed.10–15 In addition to the polar (dielectric) screening effects, implicit models have to reproduce the apolar components of the water interactions,16,17 mainly the van der Waals contacts and the entropic cost of creating a cavity within the solvent. Often, implicit models are used for quick estimates of free energies, especially solvation free energies.18 In this article, we will not review implicit solvent methods, and will simply refer the reader to relevant review articles.12–15,19–21
Let us now turn to the problem of the finite size of the system, which necessitates a proper implementation of boundary conditions. Boundary conditions may be conveniently divided into periodic boundary conditions (PBC) and non-periodic boundary conditions (NPBC). Generally vacuum and implicit solvent simulations are coupled to NPBC, while explicit solvent simulations generally are carried out with PBC (NPBC in explicit solvent can also be implemented with real-space methods). Intermediate approaches, where the solute is surrounded by a thin layer of explicit waters, have also been treated with both NPBC and PBC. In these models, special care is needed for the boundary between the explicit and continuum descriptions, as this may become a source of artifacts. Although both NPBC and PBC have been criticized as introducing artifacts in the simulations, there is growing consensus on the soundness of PBC in the treatment of long-range forces. A brief review of these issues is presented in Section 2.8.
In this work, we review important algorithmic developments used in biomolecular simulations with explicit solvent. We will not consider the treatment of systems that have a finite size, such as quasi two-dimensional systems in slab geometry. Methods developed for this geometry include several adaptations of the Ewald summation,22–26 Lekner summation,27–33 and the MMM2D method.34,35 Real-space methods, such as multigrid or fast-multipoles methods, can naturally deal with these types of geometries.
2 Methods for computing the long-range electrostatic interactions
In this section, we discuss algorithms currently used to compute the long-range electrostatic interactions in an efficient manner. We start the discussion with Ewald-based methods, which are more specifically described in Section 2.1 to Section 2.3. The Ewald summation was the first method to give an exact calculation of the electrostatic potential. Its importance cannot be understated, as the method has become one of the corner stones upon which many other methods for the numerical computation of electrostatic interactions are based.
Ewald introduced the method in 1921 to compute the electrostatic energy of ionic crystals,36 whose mathematical description is given by a Coulomb series that is only conditionally convergent. The 1921 paper is a follow-up of Ewald’s earlier work on the theory of X-ray crystallography and the development of the concept of reciprocal lattice (by Ewald and Laue).37 Ewald’s aim was to calculate the electrostatic potential and field due to dipoles, but he faced the problem that these quantities diverge at each dipole’s position. The calculation can be easily done using Fourier transforms, but Fourier methods were not well known at the time and without them, the problem is mathematically challenging. As described in Ref.,38 a vital contribution came from a discussion with Peter Debye in 1911 during one of the Arnold Sommerfeld research group retreats, where Debye suggested the use of theta functions. Ewald later applied a similar approach to solve the electrostatic lattice potential – this method is now known as the Ewald summation.36 Ewald’s contributions turned out to be seminal: he is one of the most important contributors to the theory of X-ray crystallography, and the side project on lattice potentials36 that led to the discovery of the Ewald summation has become one of the most important techniques in computer simulations of charge-containing systems.
In concrete terms, by means of a Jacobi Theta transform, the slowly, conditionally convergent Coulomb sum was converted into two sums that converge rapidly and absolutely, with the conditionally convergent nature of the series hidden in a surface term.39 This last term depends on the dipole moment of the unit cell U, the dielectric constant of the surrounding medium, and the order of summation –that is, on the asymptotic shape of the array of copies of U– and vanishes when the surrounding medium has an infinite dielectric constant (“tin-foil” boundary conditions). Although the surface term is only rarely implemented, its use has been recommended when calculating the dielectric constant of the sample.40
Ewald-based methods use the error function erf and the complementary error function erfc to re-write the Coulomb interaction as 1/r = erfc(βr)/r + erf(βr)/r, with . The first term is short-ranged and gives rise to the so-called direct sum, which is an explicit sum over all pairs of charges within a distance cutoff. The second term is long-ranged, varies slowly, and is typically handled in reciprocal space via Fourier transforms. Such a splitting of the Coulombic interactions involves a smooth function of r and therefore avoids any discontinuities in the forces, or higher-order derivatives of 1/r. Ewald-based methods include the original Ewald summation,36 the particle-particle particle-mesh method first introduced by Hockney and Eastwood,41,42 the particle-mesh Ewald algorithm developed by Darden and co-workers,43,44 and the Fast Fourier-Poisson method, introduced by York and Yang.45 Although the multigrid method solves the “reciprocal” part of the Ewald summation in real space instead of employing Fourier Transforms,46 we still consider this to be an Ewald-based method, since it uses a similar formalism to deal with the electrostatic interactions.
2.1 Ewald summations
The molecular solute and its solvent are placed in a unit simulation cell U, defined by the unit lattice vectors a1, a2 and a3 (not necessarily orthogonal), and whose volume is given by V = a1 · a2 × a3. Any other lattice vector is defined as n = n1a1 + n2a2 + n3a3 for all integer triplets (n1, n2, n3), with n1, n2, n3 not all zero. Under PBC these lattice vectors generate successive copies of the unit cell that fill space. A standard way39,47,48 of treating Coulombic interactions under PBC is to model the system as a large but finite collection of copies of the unit cell U, which is then immersed in a dielectric medium. Each particle at position ri within the cell interacts with all the other particles in U, with their and its own periodic images, as well as with the reaction field induced in the surrounding dielectric medium. Infinite lattice sums are obtained in the limit of an infinite collection. The traditional Coulomb (monopolar) potential φ0(ri) acting on charge i is the same as in Eq. (2), except that one must take into account all the periodic images, i.e.,
| (4) |
where the prime indicates that terms with i = j and n = 0 are omitted and Σn ≡ Σn1 Σ n2 Σ n3.
The infinite Coulomb series conditionally converges slowly to a finite limit only if U is electrically neutral. When it is not, the standard practice is to add a uniform background charge density (the “neutralizing plasma”), that fixes the problem with the monopole term, without disturbing the higher-order terms. The Ewald method has also been extended to higher-order multipoles.49–54 To motivate the formalism, first note that the electrostatic potential φ(r1) at position r1 due to a set of point multipoles at position r2 is given by the Taylor’s expansion53
| (5) |
where the subscript 1 or 2 on ∇ denotes differentiation at the point r1 or r2, and the symbols p, Q, O, and H denote dipole, quadrupole, octupole and hexadecapole respectively. The different “dot” products stand for the usual tensor contraction, i.e., H::∇∇∇∇= Σi, j,k,l Hi jkl (d/dxi)(d/dxj)(d/dxk)(d/dxl). For simplicity of notation, we introduce the multipolar operator L̂i by:
| (6) |
Of course, since ∇j = −∇i when applied to any function that depends on |ri − rj|, the corresponding operator L̂j becomes L̂j = (qj − pj · ∇i + Qj∶∇i∇i − Oj⋮ ∇i∇i∇i + Hj∷ ∇i∇i∇i∇i).
Here we illustrate an extension of the Ewald method to a system of N point charges and multipoles at positions r1, r2,…, rN within the unit cell, with q1 + q2 + ··· + qN = 0. As mentioned above, the edges of the unit cell are denoted by vectors aα, α = 1,2,3, which need not be orthogonal. The conjugate reciprocal vectors are defined by the relations (the Kronecker delta), for α, β = 1,2,3. The point charge and multipoles at position ri have fractional coordinates sαi, α = 1,2,3 defined by . The charges and multipoles interact with each other, and with their periodic images. Thus each and every component of a multipole set {qi, pi, Qi, Oi, Hi} at position ri interacts with each and every component of another multipole set {qj, pj, Qj, Oj, Hj} at positions rj, j ≠ i, as well as with their periodic images at positions rj + n1a1 + n2a2 + n3a3 for all integer triplets (n1, n2, n3). It also interacts with its own periodic images at ri + n1a1 + n2a2 + n3a3 for all such triplets with n1, n2, n3 not all zero. The electrostatic energy of the unit cell is then written as:
| (7) |
where the outer sum is over the vectors n = n1a1 + n2a2 + n3a3, the prime indicating that terms with i = j and n = 0 are omitted.
As in the usual Ewald or particle-mesh treatments of Coulombic interactions under PBC, this sum is split into a short-range term which is handled in the direct sum, plus a long-range, smoothly varying term, handled in the reciprocal sum by means of Fourier methods. In molecular systems, corrections are introduced to account for the “masked pairs”, which are atom pairs (i, j) ∈
 , where
is the masked list, whose nonbonded interactions should not be calculated, since they are accounted for by other terms in the potential. These masked pairs are easily omitted from the direct sum. However, since all pair interactions are unavoidably included in the Fourier treatment of the reciprocal sum, the reciprocal part of the masked pair contributions must be separately subtracted. Similarly the self-energy, which is the reciprocal part of the interactions of multipole components with themselves, must be removed. The electrostatic energy can then be written as a sum of four terms, the direct, the reciprocal, the adjusted and the self energy terms: U = Udir + Urec + Uadj + Uself. The reciprocal energy Urec requires a generalization of the structure factor S(m) to include the multipolar interactions. The generalized structure factor S(m) can be written as a function of the reciprocal lattice vectors
, with m1, m2, m3 integers not all zero, as:
, where
is the masked list, whose nonbonded interactions should not be calculated, since they are accounted for by other terms in the potential. These masked pairs are easily omitted from the direct sum. However, since all pair interactions are unavoidably included in the Fourier treatment of the reciprocal sum, the reciprocal part of the masked pair contributions must be separately subtracted. Similarly the self-energy, which is the reciprocal part of the interactions of multipole components with themselves, must be removed. The electrostatic energy can then be written as a sum of four terms, the direct, the reciprocal, the adjusted and the self energy terms: U = Udir + Urec + Uadj + Uself. The reciprocal energy Urec requires a generalization of the structure factor S(m) to include the multipolar interactions. The generalized structure factor S(m) can be written as a function of the reciprocal lattice vectors
, with m1, m2, m3 integers not all zero, as:
| (8) |
where exp(2πim ·rj) = exp(2πi(m1s1j + m2s2j + m3s3j)), sαj, α = 1,2,3 are the fractional coordinates of site j, defined above, and L̃j(m) is the Fourier transform of the multipolar operator L̂j, given by L̃j(m) = qj + 2πipj · m −(2π)2Qj ∶m m − (2π)3iOj⋮ m m m + (2π)4Hj∷ m m m m. The reciprocal energy simply becomes
| (9) |
The direct sum energy is given by
| (10) |
where the asterisk over the sum denotes that the terms with n = 0 and either j = i or (i, j) ∈
are omitted. The adjusted part contributions are given by
| (11) |
Finally, one has to compute the self-energy term. For instance, up to quadrupoles, this term is given by:52
| (12) |
The self-energy is not physical; it is a consequence of the Ewald summation and goes to zero as β → 0. The electrostatic field and force on atom i at position ri are computed as the negative gradient of the electrostatic potential φ(ri) and electrostatic energy U(ri), respectively:
| (13) |
| (14) |
An important difference with the monopolar case is that, for non-zero multipoles, the force is no longer parallel to the field, and a torque τ(ri) needs to be calculated. With the use of local reference frames,50,53 these point torques can be converted into atomic forces, necessary to carry out atomistic MD. Other quantities one needs to compute are the electrostatic “self field” and the new contributions to the stress tensor Π, necessary for constant pressure simulations.55–57 While the direct and adjusted parts are computed in the standard way, , where (Fjin)β is the β component of the direct force between particles i and j, whose relative position is given by rjin = rj − ri + n (a similar expression, with (i, j) ∈ M and n = 0 is obtained for the adjusted part). The computation of the reciprocal tensor, on the other hand, gives new terms compared to the monopolar contribution. The energy of the unit cell can be written as E = E(s11, …, s3N ; a), where sαi, α = 1,2,3, denote the fractional coordinates of atom i,i = 1, …,N, and a is the 3 × 3 matrix having the lattice vectors aα, α = 1,2,3 as columns (the volume V of the unit cell is given by the determinant of a, and a−1 is the 3 × 3 matrix having the reciprocal lattice vectors as rows). Following Nosé and Klein,57 the 3 × 3 stress tensor π satisfies
| (15) |
for α = 1,2,3. The application of this equation to the direct and adjusted components of the energy recovers the previous expression. The reciprocal contribution includes a term already present in the monopolar case, that originates on the a-dependence of the volume V and of the kernel exp(−π2m2/β2)/m2. In the monopolar case the structure factor S(m) is independent of the vectors aα, α = 1,2,3. The multipolar interactions, as shown by Eq. (8) and Eq. (9), introduce a dependence of S(m) on a and these additional contributions need to be explicitly computed.53
Finally, these equations are valid for total multipoles, i.e., for the general case where each multipolar degree of freedom may have a permanent and/or an induced component. If there are induced multipoles, the polarization energy must also be added to the total electrostatic energy. Such polarization effects are further discussed in Section 3.4.
All these equations recover the traditional Ewald expression when the multipoles are set to zero. The erfc function in the Ewald direct sum originates in Gaussian screening functions centered at each point particle. The reciprocal sum is given by the Fourier series representation of the solution to Poisson’s equation in PBC, using the sum of the compensating Gaussians, again centered at each point particle, as a source distribution. The relative contributions and computational cost of the direct and reciprocal sum can be tuned by varying the width (the β parameter in the previous expressions) of the Gaussian distributions. Narrow Gaussians (large βs) make the erfc in the direct sum decay faster, and so a shorter cutoff is needed for the direct sum while more terms are needed in the reciprocal space. Wide Gaussians have the opposite effect: the exponential in the reciprocal sum decays fast and fewer terms are needed, while the direct sum needs more terms. In particular, if the Gaussians are chosen to vanish (within a prescribed tolerance) at a standard cutoff distance independent of system size N, conventionally taken to coincide with the cutoff of the Lennard-Jones interactions, the direct sum is
(N) but the reciprocal sum becomes
(N2). This convention has become standard in the particle mesh methods described below, where the computation of the reciprocal sum is made to scale as
(N log(N)) with the use of Fast Fourier Transforms. For higher order multipoles, it is far more convenient to use a short cutoff for the direct sum, and to carry most of the calculation in reciprocal space.53
2.2 Particle-Mesh methods that use Fourier Transforms
The particle-mesh (PM) methods are based on the Ewald approach of splitting the electrostatic potential and energy into a direct and a reciprocal part, but compute the reciprocal part on a mesh. The direct part is made to scale as
(N) via the use of a fixed cutoff (generally, 8 or 9 Å), so that the overall scaling of the method is determined by the reciprocal part. PM methods compute the reciprocal potential by solving Poisson’s equation of a smooth charge distribution on a grid. This can be done via Fast Fourier Transform (FFT) under PBC or via a discrete Poisson solver in real space under a wide variety of boundary conditions. FFT PM methods include the particle-particle particle-mesh method41,42 (P3M), the particle-mesh Ewald algorithm43,44 (PME), and the Fast Fourier-Poisson45 (FFP) method.
All the FFT PM methods transform the reciprocal sum into a sum over coefficients of the discrete Fourier transform of a gridded charge distribution, whose calculation is accelerated to
(N log N) via three dimensional FFTs. Although the methods can be used with different switching functions41 (that give the relative weight of direct and reciprocal parts), the use of Gaussian screening functions as in the original Ewald method and an optimal Green’s function58 greatly improves the accuracy of PM methods. The methods differ in how they transform the continuous charge density due to the sum of compensating Gaussians onto a regular three-dimensional grid, and how they compensate for the loss of accuracy introduced in this process. These methods have been carefully studied and compared in the literature.3,42,59–65 Of these, the PME algorithm of Darden at al.43,44 is perhaps the most broadly used. The method approximates the complex exponentials in the reciprocal sum by an Euler spline interpolation, based on cardinal B-splines in the smooth PME.44 B-splines are factored into three one-dimensional B-splines which can be differentiated analytically via recursion relations. The forces are obtained analytically from the gradient of the energy in the smooth PME method at very low cost, requiring only two FFTs (as opposed to four FFTs for equivalent accuracy in the P3M method). The actual steps in the algorithms for either P3M or PME are well-known and described in the literature, and so are the differences and similarities between the two methods.3,42,60,61,63–65 The PME method for molecular simulations with explicit solvent has been implemented in most of the major biomolecular codes, such as AMBER,66 CHARMM,67,68 NAMD,69 GROMACS,70 DESMOND,71 DL_POLY,72,73 and others.
Another variant of the Particle Mesh approach is the Fast Fourier Poisson (FFP) method.45 This method directly samples the compensating Gaussians onto the grid, and avoids the loss of accuracy from interpolation by the use of a clever identity, later used in multigrid methods. Due to the cost of sampling the Gaussians, this method is not competitive for the modest accuracies appropriate for present force fields. It appears, however, to be more efficient than the above methods for simulations with high accuracy requirements.
Recent methodological advances include the use of non-uniform FFTs,74 and the Staggered Mesh Ewald (StME) algorithm by Cerutti et al.,75 that uses an old technique named “interlacing”41,76 to improve the accuracy of the smooth PME method by averaging the results of two separate reciprocal space calculations. Here, the original mesh in the PME method is replaced by two separate coarser meshes, each less than one-third of the original size. The resulting StME improves the accuracy of the computed forces, and exceeds the efficiency of the smooth PME with similarly optimized parameters. In addition, the method also offers advantages for parallel implementations because it permits the use of coarser meshes without requiring higher orders of charge interpolation and because the two reciprocal space calculations can be run independently. The interlacing technique has also been recently applied to the P3M algorithm77 with analytical and ik-differentiation. The resulting algorithms compare favorably against the non-interlaced versions and against the interlaced smooth particle-mesh Ewald algorithm. The success of interlacing is due to the fact that it suppresses the aliasing effects introduced by the FFTs when calculating the forces.
A promising combination of the PME and multigrid ideas has been recently introduced in the form of the “Multi-Level Ewald” (MLE) method,78 that splits the convolution required by standard P3M algorithms into a series of convolutions over slab-shaped subregions of the original simulation cell. A convolution over the entire cell is performed using a much coarser mesh. The MLE approximation does not seem to introduce significant errors compared to the PME method. It is anticipated that the division of the single convolution into many independent subproblems will be useful for extending the parallel scaling of molecular simulations.
Goedecker et al.79–81 developed the particle-particle particle-scaling (P3S) algorithm for Coulombic interactions in free boundary conditions. This is also an
(N log(N)) algorithm that uses interpolating scaling functions for the representation of the charge density and FFT for the Poisson’s equation. The algorithm is ideally suited for computing the potential of a localized charge distribution.
Although parallel implementations are not within the scope of this review, we add a comment here. The parallel scaling of the PME methods is usually limited by the scaling of the three-dimensional FFT that requires an “all-to-all” communication. For typical meshes on commodity networks, the number of messages to be sent and their associated latency is the primary determinant of FFT communication time.71 To improve the overall parallel performance the latency-sensitive FFT computations are typically interleaved with the latency-tolerant (and dominant) cutoff limited calculations. The division of work can be either explicit,70,82 in which case a number of processors can be dedicated to the reciprocal PME computations, or adaptive. The latter approach is used in the NAMD program69 taking advantage of its message-driven architecture. It allows for great scalability of the resulting MD code.83
2.3 Particle-Mesh methods in real space
The multigrid method as first applied to biomolecular simulations46 is a PM method where the equivalent of the reciprocal sum is computed on real space grids via multigrid techniques.84–86 Multigrid methods scale as
(N) with the number of particles, and has good prospects for parallelization. Parallelization, however, is extremely dependent on the computer architectures and therefore a constantly moving target. Recent breakthroughs in the treatment of FFT allow the PME methods in codes such as NAMD and DESMOND to be highly parallelizable and extremely competitive. At present, it is not possible to predict how the FFT PM and the multigrid methods (or any other method, for that matter) will compare in the next parallelization breakthrough. First introduced in the 1970s by Brandt,84,87,88 multigrid methods can solve linear and non-linear elliptic partial differential equations and integro-differential equations.86 Their appeal derives from the ease of handling any sort of boundary conditions; great functionality in implicit solvent methods, like Poisson-Boltzmann approaches;89–94 possible advantages in the implementation of highly accurate, continuous electrostatics; possible adaptability in efficient multiple-time step integrators that separate fast and slow interactions;95 etc. In particular, multigrid or the related multilevel methods90–94 have been very successful in the static calculation of electrostatic potentials for large, complex molecules in implicit solvent, specially with the use of finite elements. In this article, we are interested in classical molecular dynamics (MD) with explicit solvent and therefore review multigrid methods as applied to this particular case.
Multigrid algorithms introduce successive auxiliary grids with larger mesh spacings in order to eliminate all different frequency components of the error in Fourier space.84,87,88 The resulting accelerated convergence on all length scales allows for the solution of elliptical partial differential equations on grids with K points in
(K) operations.84 For condensed-matter systems, this translates into a truly
(N) linearly scaling method. By comparison, other well-known relaxation methods such as the Gauss-Seidel, the alternating direction implicit method and the iterative Successive Over Relaxation (SOR) methods exhibit a convergence rate that is lower than that of multigrid methods. For instance, it has been shown that an optimal SOR method scales as
(N4/3), because the cost per iteration also increases as the size of the system increases.96
A first attempt to use multigrid in the context of biomolecular simulations consisted of an adaptive multigrid technique implemented into the original FMM.97 This technique is competitive with the FMM only at low accuracy (comparable to FMM with 4th-6th order multipoles). Molecular dynamics was not implemented, but given what is known about lack of energy conservation for FMM with lower-order multipoles, this method was not deemed to be an acceptable solution for large-scale MD simulations. Later, Sagui and Darden46 introduced a multigrid method appropriate for biomolecular MD simulations with explicit solvent. Along general lines, the method divides the pair interactions into the short-range or direct sum and the long-range sum using the partition scheme proposed in the FFP45 method, except that the solution of the long-range sum is carried out via real-space multigrid. The latter consists of: (i) a charge assignment scheme, which both interpolates and smoothly assigns the charges onto a grid; (ii) the solution of Poisson’s equation on the grid via multigrid methods; (iii) the back-interpolation of the “reciprocal” forces from the grid to the particle space, to be added to the direct and adjusted forces. For classical calculations, the charge assignment scheme (i.e., the spreading of the charges on the mesh) was found to be crucial for the performance of the method. To obtain reasonable accuracies, the charge density on the grid must be as smooth as possible. This means that the charges must be spread over a fairly large number of grid points, that can quickly become an important time consuming part of the algorithm. The use of the FFP partition scheme, a deferred defect correction scheme84,85,98 for the discretization on the grid, and Gaussians as interpolating functions gives relative force errors of ~10−4, and relative energy errors of approximately 10−5 with excellent energy conservation (this was denoted as the LGM method).
Since then, several other variations of multigrid have attempted to speed up the method.99–102 The challenge has been to obtain a method that is efficient, while preserving accuracy, since relative force errors for reliable biomolecular MD trajectories need to be of the order of 10−4. Goedecker and Chauvin100 proposed a modified multigrid V-cycle scheme that employs wavelets and is more efficient than the standard V-cycle. Instead of transferring the residue to the coarser grid levels, this scheme works with copies of the charge density on the different grid levels that were obtained from the underlying charge density on the finest grid by wavelet transformations. The authors show that halfway V-cycles with the restriction and prolongation steps based on wavelet theory are the most efficient approach for the solution of the three-dimensional Poisson’s equation on the grid. Currently, it is not clear whether wavelets will significantly improve efficiency when applied to charge and force interpolation.
Two later studies103,104 have applied convolutions to accelerate the charge assignment and force interpolation steps in the LGM method (which simply used separability of three-dimensional Gaussians to pre-tabulate one-dimensional Gaussians). Both schemes involve computing an on-mesh charge distribution ρS(rm) by convolving the point charge distribution ρ(r) with a charge spreading function S: ρS(rm) = ∫ S(rm − r)ρ(r)d3r. The set of mesh points at which S(r) is nonzero (the support of S(r)) partially determines the computational expense of the charge spreading step, with a large support considerably increasing the cost of the calculation. These schemes effectively reduce the number of mesh points at which the charge spreading function is nonzero by proceeding in two stages. In the first stage, charges are spread onto the mesh, and in the next stage a convolution with a Gaussian is performed to further spread the charges. Shan et al.104 used another Gaussian (with a smaller support) for the first stage of charge spreading, and this same Gaussian was used for the final interpolation to calculate the forces. Banerjee and Board,103 on the other hand, used a set of weights on the grid, which then were further convolved with the second Gaussian. These same weights are used for the back interpolation to compute the forces, but in a different manner. On a single-processor, both convolution-based methods reduce the computational burden by a factor of about two when compared to the earlier LGM implementation. Parallelization studies were not reported in Ref.104 The convolutions implemented in Ref.103 perform well in parallel environment, although the multigrid implementation itself performed poorly in terms of communication in constrained parallel environments. A parallel multigrid implementation that appears more successful and is available as an open source code has been released by Izaguirre et al.102
Among other novel applications of the multigrid method we find the Multi-Level Ewald (MLE) method78 that combines PME and multigrid ideas and was briefly described before; the implementation of multigrid in dissipative particle dynamics (DPD) by Groot;105 and its application to higher-order multipoles by Sagui et al.53 Groot incorporated electrostatics into DPD by solving for the electrostatic field locally on a grid with a multigrid method. The method captures the essential features of electrostatic interactions and satisfies screening of the potential near a charged surface and the Stillinger-Lovett moment conditions.106 The method is specially applicable to (coarse-grained) simulations of mixed surfactants near oil-water interfaces, or to simulations of Coulombic polymer-surfactant interactions. We discuss the inclusion of electrostatic interactions in DPD in more detail in Section 4.3. Some recent applications of the real-space multigrid method to quantum MD can be found in Ref.107
2.4 Fast Multipole Methods
Fast Multipole Methods (FMMs) are real-space methods that divide the simulation cell in a hierarchy of subcells. Coulombic interactions are treated exactly for particles within the same or neighboring subcells (unlike in the lattice sums), and the potential for the more distant particles is evaluated through multipolar expansions. This splitting of the Coulombic interactions is not a smooth function of r and thus, these methods need high multipolar order for good energy conservation.108 Since historically the first hierarchical N-body algorithms were developed for calculating gravitational forces in astrophysical simulations,109 the original FMM110,111 also used NPBC. The method was soon extended to PBC in three dimensions by Schmidt and Lee112 and Lambert et al.,113 who realized that the periodic images have the same multipole expansion about their centers as the main box and they can easily be shifted to the center of the main box. The PBC actually simplify the algorithm and help to the load balancing in parallel implementations.114
Here, we review the original Fast Multipole Method of Greengard and Rokhlin110,115–119 only very briefly, as the method has been reviewed several times.3,120 The main elements of the method are: (i) multipole expansions in terms of spherical coordinates; (ii) octal tree constructions, which consist of successive subdivisions of the initial cell into self-similar subcells in order to calculate the expansion of the far field created by the particles inside the cell; (iii) translation operators for transformations of multipole expansions. The key of the tree methods is to compute interactions between well separated clusters through multipole expansions with rigorous bounds on the error. For a given precision ε, it is necessary to use terms in the expansion,117 where p is the multipolar order. The major hurdle in the original FMM scheme is the conversion of multi-pole to local translations in the “downward pass”, a transformation which requires approximately 189N p4 operations. Later developments include the combination of multipole expansions with exponential or plane wave expansions, which diagonalize the translation operators.117 A still later version of the adaptive FMM for the Laplace equation uses compression techniques and diagonal forms for the translation operators to accelerate the convergence of the method.121 For relatively uniform charge distributions, the method scales as , where s is the number of charges per box at the finest level.
The relative efficiency of FMM versus PM based approaches for biomolecular simulations in explicit solvent is still an open question. In an older study by Pollock & Glosli,42 it was concluded that the P3M approach is significantly more efficient than FMM for any conceivable system size, despite the superior asymptotic scaling of the latter (O(N) versus O(N log N)). They also discussed other advantages of the P3M method, the most obvious one being the ease of coding. Also, non-cubic periodic cells can be more easily implemented with the particle mesh methods. Although parallel implementations of FMM are supposed to scale better than P3M or PME with the number of processors, we are not aware of any recent comparisons involving state-of-the-art codes. Skeel et al.99 carried out a comparison between multigrid and FMM methods. Their results suggest that the multigrid methods are superior to FMMs. The FMM approach, on the other hand, has found a niche in solid state physics and scattering problems. The method, that has both non-adaptive and adaptive versions, may be better suited to problems involving highly non-uniform particle densities, since the tree approach lends itself well to an adaptive approach,122 whereas the grid size in non-uniform distributions in the PM methods will grow faster than linearly with the number of particles. However, local finer subgrids, especially in multigrid methods, can circumvent this problem, as can the use of non-uniform FFTs.74 The truncation of the multipole expansion in FMM results in a potential energy that is discontinuous as a function of the positions of the charged particles. Specifically, there is a small leap in energy when a particle moves from one FMM cell to another. Thus, FMM suffers from a lack of energy conservation unless very high accuracy is employed,46,108,123 whereas the PM methods have very good energy conservation properties. Efficient parallel implementations are discussed in Refs.114,124 A comprehensive and insightful review of the method is given by Kurzak and Pettitt.120 One of the most recent enhancements of the FMM method is given by the automatic generation of highly optimized FFTs (that incorporate knowledge of symmetries in the input arrays) for translations of multipole expansions in spherical harmonics.125 Also, a very interesting recent development is the extension of an adaptive tree code and the FMM to charge densities with high angular momentum.126 This is an
(
 ) method aimed at classical molecular simulations with stochastic boundaries that may include polarizable force fields and/or QM/MM potentials, and shows a lot of promise for the more sophisticated force fields under present development. We also note that the FMM is the method of choice in some electronic structure calculations, which are not reviewed here.
) method aimed at classical molecular simulations with stochastic boundaries that may include polarizable force fields and/or QM/MM potentials, and shows a lot of promise for the more sophisticated force fields under present development. We also note that the FMM is the method of choice in some electronic structure calculations, which are not reviewed here.
2.5 Local Methods
A different approach was followed by Maggs and Rossetto,127 who showed that the Coulomb interaction is recovered in the thermodynamic limit of Maxwell electrodynamics, where the magnetic field B and the current J produced by moving charges have vanished. Since the electric field E obeys Gauss’ law, div E = 4πρ, in the static limit the electric field satisfies E = −∇ φ and the electrostatic potential is the solution of Poisson’s equation ∇2φ = −4πρ. This elliptic partial differential equation demands a global solution in space, which is the origin of the high computational cost of electrostatics. In full electrodynamics, however, electromagnetic waves propagate with finite speed of light c as Ḃ = −c curl E and Ė = c curl B −4πJ. In contrast to the Poisson equation, solutions to these hyperbolic differential equations require only local operations. Since Mawxell’s equations conserve Gauss’ law if it is obeyed as an initial condition, Coulomb interactions can be generated by coupling charges directly to the time varying electromagnetic fields, and then solving for their dynamics.
In the original Monte Carlo (MC) algorithm, the “electromagnetic field” is propagated via diffusive dynamics through the lattice. Rather than quenching the curl E degrees of freedom in the electric field to zero, one can perform an integration over them and obtain the Coulomb interaction even though the electrostatic energy is greater than its minimum at equilibrium. Performing this integration while maintaining Gauss’ law in each MC step requires only local operations and obviates the need for computing long-range interactions at every timestep.
This insight can be applied to MD by propagating Maxwell equations with a tunable speed of light c and by coupling the resulting fields to the motions of the particles,128 with Langevin thermostats on both particle and magnetic field degrees of freedom. It is sufficient to make c small enough for the quasi-static approximation to be valid (the static properties of the system were shown to be independent of the value of c). The resulting equations are integrated on a grid,128,129 and the integration verifies the presence of an effective Coulombic 1/r interaction. The principal technical challenge lies in the efficient coupling of charges and fields. As in all mesh based methods, the charges need to be interpolated from the continuum onto the electrostatic grid to generate the current J, and the electric field must be extrapolated to compute the electrostatic force. Similarly to the multigrid method, high accuracy requires smooth interpolation over a large volume, which becomes the dominant computational part of the algorithm. The current implementation130 uses truncated Gaussians for the interpolation of the point charges to the electric grid with relative force errors greater than 1 × 10−3 which are considered too large for atomistic biomolecular simulations. The method is particularly suited to Poisson-Boltzmann models of solutions131 and has similar advantages to multigrid methods:
(N) scaling, ease of parallelization, flexibility for all types of boundary conditions, and the ability to handle inhomogeneous dielectric environments.
2.6 Truncation Methods
Initially, long-range forces in macromolecular simulations were often dealt with by means of truncation schemes in which the electrostatic interactions are computed up to some cutoff distance.132,133 Such approaches were mandated by the lack of sufficient computer power to carry out the simulations using an “infinite” cutoff or an Ewald method – at the time, the difference between
(N) vs
(N2) or
(N3/2) scaling was considered prohibitive. The idea of truncating the electrostatic interactions is based on the facts that the Coulomb potential decreases monotonically with atomic distance and that water provides high dielectric screening. Truncation is also appealing because of its intrinsic simplicity, its ability to handle all sort of boundary conditions, and the belief that the calculation of the interactions has lower computational costs.
Unfortunately, simulations with electrostatics cutoffs soon revealed that they can lead to incorrect results that depend on the truncation scheme and give rise to severe artifacts in the structure, dynamics or thermodynamics of the system.133–136 Such artifacts have been reported in simulations of water,137–143 peptides,144,145 DNA,134,135,146,147 proteins,148–154 and lipids.136,140,155–160 Truncation works better in homogeneous bulk systems, but the presence of interfaces, especially when the dielectric constant changes rapidly across an interface, can cause severe problems. For instance, truncation schemes in lipid membrane simulations induce artificial structure at the scale of the truncation distance, and this results in a change of large-scale properties such as the elasticity and phase behavior of the membrane, which may lose its fluid characteristics and acquire an artificial gel-like ordering.155,156,160 With an exact treatment of electrostatics. the correct structure and phase behavior of the membrane are recovered. In bulk systems, the electrostatic screening length has also been used in structural coarse-graining based on the potential of mean force to justify the use of a system-specific cutoff.161
To alleviate many of the artifacts, various switching and shifting functions have been introduced. Even then, artifacts may still be expected, unless the cutoff is large. Indeed, most cutoffs methods seem to perform satisfactorily in the 20–30 Å range, but such large cutoffs can put these schemes at a disadvantage with respect to state-of-the-art full electrostatics implementations in major biomolecular simulation packages, where focused efforts have been invested in parallelizing the FFTs subroutines. Independently of the cutoff method (including most of the switched and shifted schemes currently in use), simulations with cutoffs smaller than 15 Å are generally not recommended.
As more artifacts of the cutoff schemes became known, newer, more sophisticated schemes were introduced; along with other insights, such as the artificial layering of water induced by group-based cutoffs.162 Most of the new truncation schemes are based on the work of Wolf et al.163 Since in the straightforward truncation and shifting schemes, the truncation sphere inside the cutoff radius was found to be not electro-neutral, charge neutrality through a neutralizing term has been imposed in the new schemes. Although scaling remains
(N), the new functions contain an erfc-function and a damping parameter that has to be adjusted together with the cutoff radius. Methods based on the Wolf et al. functions include corrections for energy conservation by Zahn et al.,164 and various versions of the damped shifted force scheme165,166 and charge-neutral condition.167
The performance of truncation schemes has been compared to Ewald summation and Ewald summation based methods in several works.166,168–171 In the case of bulk systems, the new truncation methods showed encouraging behavior. However, when the systems were made more inhomogeneous, the simulation behavior deviated more from the correct solution.166,171 In addition, the performance depends strongly on the choice of parameters.171 One study that addressed a system with interfaces168 concluded that these new truncation methods are suitable for simulations of lipid bilayers. We would like to point out, however, that this particular study remains inconclusive: the bilayer simulations were performed for only a single nanosecond. This is such a short time that the lipids do not have time to move around and mix. At the very minimum, simulations of tens of nanoseconds155,156,160 are required for that, since typical diffusion coefficients for lipids are ~ 10−8 cm2/sec.
Other studies have looked at protein folding.170,172 For a 23-residue protein, a reaction-field method with a cutoff of 14 Å gave similar results to PME for the “committer probability” that a protein conformation will commit to the native state before it commits to the unfolded state.172 Another study looked at the folding of the villin headpiece.170 The authors found that force-shifted truncation schemes tend to subtly shift the balance between hydrophobic and hydrophilic interactions, favoring the more compact structures. The effects do not seem too dramatic in the folded state but increase for the unfolded state, where the force-shifted truncation scheme increases the helicity and compactness of the structure. Unfortunately the effects of truncating both the Lennard-Jones and the electrostatic interactions at the same time are rather entangled, since truncating the Lennard-Jones potential is a well-known source of additional errors. Also, not all biomolecules are equally sensitive to the treatment of electrostatics. In particular, the first fully solvated simulations of the folding of the villin headpiece were carried out with a crude 8 Å residue-based cutoff applied to the long-range nonbonded protein-water and water-water interactions (both electrostatic and van der Waals).173
Among the most recent truncation schemes are the linear-combination-based isotropic periodic sums (LIPS)174 and the zero-dipole summation method.175,176 The LIPS method is a variation of the isotropic periodic sum (IPS) of Wu and Brooks,177,178 where the effects of the approximated long-range interactions are included in the potential function through “imaginary” images.177,178 The IPS method comes in two flavors: the IPSn method for point charges, and the IPSp method for polar molecules. The IPSp method was created to correct failures of the IPSn method when applied to polar molecules. However, unless extremely long cutoff radii are used, the IPSn method does not properly describe water-vapor in interfacial systems and its convergence is poor. The LIPS method in principle addresses some of these difficulties by estimating both homogeneous and heterogeneous systems for nonpolar and polar molecules.174 The method is much more accurate but it still needs 22–30 Å cutoff radii for convergence. The zero-dipole (ZD) summation method by Fukuda et al.175,176 represents another truncation scheme that seems to perform relatively well. Building on previous shift truncation schemes, the authors obtain an energy formula that employs a pairwise function inspired by the Ewald direct sum, such that the neutrality of the charges and the dipoles is enforced in a subdomain inside the cutoff sphere. The method shows good convergence in the energy and the dielectric constant of bulk TIP3P waters for several values of the damping parameter. The radial distribution function also shows good agreement, with the typical oscillations at the cutoff (which seem of considerably smaller magnitude than in other schemes). The distance-dependent Kirkwood factor, that was previously known to show a hole at the cutoff radius, increases properly with radial distance as expected. This force, however, is exactly the same direct force as given by the Ewald method, which clearly would require relatively large cutoffs, since the reciprocal component is missing. It is not clear how the method will apply to cases where charge or dipole neutrality cannot be achieved (such as one single ion in a water bath).
At present it looks as if many of the artifacts associated with the more elemental cutoff methods of the past have been corrected, but it is clear that there are several important issues that need to be addressed. Here, we list the main ones:
Damping parameter: As discussed, the better performing truncation methods163–167 use a damping parameter (the potential is a variation of the Ewald direct sum, and the force is the Ewald direct force plus a constant), and the results do depend strongly on it. In many cases, the ideal value of the damping parameter appears to be system-dependent.166,171 In addition, the accuracy obtained for a given parameter could vary as the conformation of the system varies, which is not a desirable feature.
Cutoff radii. To avoid serious artifacts, large cutoff radii are needed; most methods seem to perform well with cutoffs in the 20–30 Å range, but results with cutoffs shorter than 15 Å should be closely scrutinized. Obviously, the size of the cutoff has to increase with the size of the solute, as no one wants the solute to experience the artifacts at the boundary, even if they are small. Also, there may be “internal awareness” of fixed charges across the solute, especially since biomolecular solutes tend to have rather low dielectric constants. (The cutoff radius will increase with the size of the solute up to a certain value beyond which it will not make a difference).
Definition of “quantitative”. Another issue which we identified in several papers that claim quantitative results, is the lack of a proper “exact” model against which to measure such results. Ideally, one would like for example an Ewald summation with an enormous number of terms, or an infinite cutoff. Since this is often not practical, most researchers fall back into some Ewald-based lattice method, such as the PME. However, these lattice methods are not exact; their accuracy depends on the parameters chosen and this has to be carefully considered. For instance, it is not unusual to confuse the tolerance in implementations such as AMBER or TINKER, as the accuracy of the PME calculation. It is not: given a tolerance ε and a cutoff radius Rc, the algorithm will determine a damping parameter β such that erfc(βRc)/Rc < ε. The total error will be determined by this, by the order of the spline interpolation for the reciprocal space, and by the size of the mesh of reciprocal space, which are not always reported, resulting in pseudo-exact reference calculations whose accuracies can vary greatly from one paper to another.
Cost of the calculations. Meaningful comparisons between truncation methods and a full treatment of electrostatics have become inextricably linked with issues of computer performance and parallelization. At present, advancements in computer architectures coupled with algorithmic developments have made possible the inclusion in most MD packages of full long-range interactions, which can now be computed at a cost that is smaller than that of a typical cutoff calculation. This situation, however, may change as parallelization paradigms shift along new technologies. In particular, Graphic Processing Units (GPUs) have the ability to switch the balance, such that even very large cutoffs are feasible (of course, increasing the cutoff also increases the communication costs, though not as badly as those for FFTs).
System properties, presence of interfaces and inhomogeneities. Most of the tests have been performed using liquid systems that are homogeneous or close to being homogeneous. Inhomogeneities and interfaces can (and often do) render cutoff methods a questionable choice. A common example of such systems in the biomolecular context is the lipid bilayer:155,156 the water phase has a dielectric constant close to 80, but inside the bilayer interior the value is only about 5. Carbon nanotubes and the interiors of membrane proteins are examples of other such systems.179,180
Domain of validity. The domain of validity of the truncation methods needs to be clearly determined, as it is human nature to use a tool developed for a particular application for a completely different set of problems. Here we give three examples, where truncation methods may need some extra work, or the newer versions may need to be evaluated very carefully. First, consider the case where the interactions are not spherically symmetric, such as when dipoles and higher order multipoles are included. This case is further discussed in Section 3.1. Second, in QM/MM methods. For instance, it has been shown that while the association of oppositely charged ions is well-described by an Ewald-based method under PBC or by full electrostatics evaluation under spherical boundary conditions, an electrostatic cutoff with either boundary condition causes an artificial and sharp decrease in the potential of mean force at large ion separations (where it should be zero).181 This would result in artificial overstabilization of oppositely charged ions in QM/MM calculations. Similar problems are seen in the association of ions.181 Third, in non-equilibrium phenomena, such as heat transfer in water. For instance, the thermal conductivities of common water models were recently compared using equilibrium Green-Kubo (GK) measurements and non-equilibrium MD (NEMD).182 Before this study, there were conflicting reports on the thermal conductivity of various water models, between groups that used NEMD and a group that used the GK formula. This recent study showed that the source of disagreement was the use of a cutoff for the electrostatic interactions in the GK formalism.182 The authors also showed that various correlation functions, such as the heat flux potential energy self-correlation, show strong dependence on the cutoff radius (with 20 Å still not converging to the infinite radius limit182). Indeed, the need to include the full long-range electrostatics contributions to the heat flux has been discussed in several studies of liquids.183–185
To finish this section, we will review reaction field methods, as they represent a popular option offered in most biomolecular simulation packages. Reaction field methods can be considered as a special case of truncations schemes: the pair-wise interactions are computed only up to a cutoff, but the effect of the neglected electrostatic interactions are included in a mean-field manner.
2.6.1 Reaction Field Methods
Even with the speed-up provided by the state-of-the-art, full electrostatics methods, more efficient algorithms for the computation of electrostatics are always in great demand. One particular popular choice that represents a compromise between the full evaluation of pair interactions and implicit methods is the reaction field method.
There are several variants of the reaction field method,186–190 but the basis for all of them is Onsager’s original idea from 1936191 based on a molecule in a cavity. Molecules are polarizable and cause the surrounding dielectric to become polarized. This polarization then creates a reaction field that acts on the molecules in the cavity. The Onsager form is not used directly in simulations, but most implementations are based on the formulation of Barker and Watts from 1973.186 In brief, the cavity has a size given by a cutoff radius rcut for each atom. Outside this, the medium is described by a dielectric constant εRF. The electrostatic potential within the cavity is then given by
| (16) |
Here, the first term is the direct electrostatic interaction between the atoms that are inside the cavity (rcut), the second term is the actual reaction field term, and the third term shifts the potential to zero at rcut. The last term is usually added to remedy problems that arise from particles entering and leaving the cavity which leads to problems with the conservation of energy. The reaction field method, in the above form, has been implemented in virtually all of the classical MD packages, and generally performs well enough for many properties, but not always. For example, the structural properties of membranes are reasonably well preserved but the dynamical aspects may present artifacts.156 We note that there are several variants of the reaction field approach.187–189,192
A recent assessment of simulation protocols based on membrane proteins has shown that the reaction field method has serious problems in non-homogeneous systems and at worst can lead to artifacts such a strong continuous flow of water through nanoscale protein channels.180 Only using an Ewald-based method together with an appropriate combination of other parameters and thermostat restores the physically correct situation. The failure of the reaction field method, as implemented in most software, is easy to understand in qualitative terms. Beyond a cutoff (typically 1.4–2.0 nm), the electrostatic interactions are computed through a mean-field approximation with the dielectric constant as the main input. When a system is very inhomogeneous such as in the case of membrane proteins or carbon nanotubes, such an approximation is not valid. In systems with carbon nanotubes or membrane proteins connecting two water reservoirs, the result is typically an unphysical, continuing flow of water.179,180 Another assessment of reaction-field electrostatics showed that group based cutoffs for reaction-field electrostatics in simulations of DNA performed badly whereas atom-based cutoffs provided reliable results.193 These results show that more studies on these issues are needed.
The latest development in reaction field methods is the so-called “image-based reaction field” method of Lin et al.190 We are not aware of independent tests of this method but the idea is very interesting: the use of FMM together with the multiple image charge-method for direct electrostatic interactions. Their approach uses Gauss-Radau quadratures and the Neumann image charges and hence differs from the Barker-Watts method.
2.7 Charge-group methods
In addition to the summation method for the electrostatic problem, many software packages contain additional methods to speed up the simulation. One such method is the so-called charge-group or group-based cutoff method. The idea of charge-groups is simple: Neighboring atoms belonging to a molecule are assigned to a group such that the net charge of the group is zero. Net zero charge is typically needed to avoid artificial multipole interactions. Then, instead of computing distances between all charges separately, the geometric centers of the groups are used for distance calculation. If this distance is less than the cutoff, electrostatic interactions between all the charges belonging to the groups are computed; if it is larger, no interactions are computed. It is easy to see that this procedure can give a large speedup since the number of pair calculations is significantly reduced: If the charges of a TIP4P water molecule are grouped together, there is no need to compute 16 pair distances – only one is sufficient. Since the partial charges within each TIP4P molecule are close to each other, grouping is well justified. In addition, since virtually all atoms have partial charges, charge-groups are a very appealing method to increase efficiency. A possible problem is that when the groups are too large, unphysical artifacts may arise. Hence, the spatial size of charge-groups is of primary concern and a bad choice of charge-groups may lead to unphysical behavior.
Charge-groups can be used with cutoff, reaction field and Ewald-based methods. The net charge of the groups can have quite an impact in lipid bilayers when using the cutoff methods (for which neutral groups are recommended), but not when using the PME method.159 If the groups are not carefully chosen, problems still arise even when PME and reaction field electrostatics are used.179,180 Identification of possible artifacts is difficult because a bad charge-group choice does not render the simulation algorithm unstable: One must use basic physical properties and reference simulations to validate the choice, and in general it is not enough to use only one physical property. There are three major determinants for the validity of the simulations when using the charge-group approach: the spatial size of the group, the net charge of the group and the update frequency for the reciprocal sum.
In addition to the speedup, one of the possible advantages of the group-based cutoffs over atom-based cutoffs was that it might be possible to avoid artifacts arising from unaccounted multipolar interactions. While the idea is attractive, potential problems were pointed out already by Steinbach and Brooks133 almost two decades ago. In particular, based on their NVE simulations, they pointed out that a charge-group based cutoff leads to significant unphysical heating. Such heating is masked by thermostats in NVT simulations.
The GROMACS70 software package together with the GROMOS force-field194 is able to exploit charge-groups to speed up the simulations. Other software and force-fields, such as CHARMM68 and AMBER66 also use charge-groups. In the following, we discuss GROMACS/GROMOS since we have recently tested it in detail.179,180 Similar issues may occur with other software and force-field combinations but we are not aware of detailed tests. GROMACS implements charge-groups in an elegant way and can give a speed-up of about an order of magnitude.70 It also has the option of choosing the frequency for updating the reciprocal sum in the PME method. This can also provide an additional significant speed when combined with charge-groups. Updating the long-range part every 10 time steps is generally a safe choice in simulations of membrane bilayers, but the situation can be drastically different for studies of transport of molecules through nanotubes and proteins.180 The only way to avoid artifacts is to examine the physical validity of the results and, preferably, to compare them with simulations using conservative or no updates. In addition to these issues, it has also been shown that the reaction field is not stable for DNA simulations when group-based cutoffs are used.193
2.8 Treatments and artifacts of boundary conditions
The effect of the boundary conditions on the simulation results has been a contentious subject for about four decades. Even with current computer capabilities, one can only simulate an extremely small sample of a given bulk system and, in order to reproduce bulk behavior, a careful treatment of the boundary conditions needs to be implemented. If a liquid-vacuum or liquid-continuous solvent interface is used (for instance, a solute immersed in a spherical water cluster that is surrounded by vacuum or implicit solvent), then many issues arise195–202 with respect to the proper potentials or reaction fields needed to prevent solvent evaporation of the cluster, artificial ordering of the solvent molecules at the interface, and spurious surface tension effects and pressure increase. These fields must restore the long-range nature of the electrostatic interactions and avoid the electrostatics discontinuities at the interface. Ewald-based methods under PBC, on the other hand, avoid all these issues at the cost of introducing an artificial periodicity in the system. Although other methods have been proposed to diminish the artificial long-range periodicity of the system, such as hyperspherical boundary conditions,203–206 these methods have not been used in mainstream applications.
Since eliminating surface effects in solvent clusters has proven rather elusive, a considerable amount of work has been dedicated to the nature and the magnitude of the “periodic artifacts” in biomolecular simulations using Ewald-based methods and PBC. In order to minimize these problems, it is crucial to account for finite-size effects and self-interactions.207 These days,such analytical corrections are standard in most codes, but in the past, reported periodic “artifacts” were later traced down to the lack of these correction terms. In addition, the conditionally convergent nature of the Ewald series results in a surface term39 that depends on the dipole moment of the unit cell and the dielectric constant of the surrounding medium, and that vanishes when the surrounding medium has an infinite dielectric constant (“tin-foil” boundary conditions). The use of the surface term has been recommended for calculating the dielectric permittivity, dipole time correlation functions and the Kirkwood g factor.40 Once all these correction terms are in place, the remaining possible artifacts are expected to diminish with increasing solvent around the solute, as the interaction of the solute with its periodic images vanishes. Systems with a small amount of solvent and/or a solvent with low dielectric constant are more susceptible to periodicity-induced artifacts, as the interactions of the solute with its images will not be screened enough. It is hard to come up with a prescription on how much water to add around a solute, since this will be rather system-dependent. Thus a globular protein that is not charged (and stays folded) will generally require less water than an equally sized protein that has net charge and/or unfolds/changes shape. Accordingly, the width of the layer of water around the solute can vary from 8 Å to a safe 12–15Å thickness. Unless there is plenty of solvent, the shape of the cell is also important. For example, a tetragonal lattice with a high aspect ratio (unequal side over one of the equal sides) might initially accommodate well a long segment of DNA, but it might hinder its rotation along axes perpendicular to the long side. In cases like this, it is better to choose a more “spherically symmetric” cell such as a truncated octahedron.
Under the previous conditions, the use of Ewald techniques provides accurate, mainly finite-size independent calculations of thermodynamics solution properties. For instance, many investigators believed that the long-range periodic artifacts act to stabilize the system, i.e., that the Ewald summation suppresses conformational fluctuations. However, crystal simulations comparing the use of PME versus truncation schemes with short (9 Å) and long (18 Å) cutoffs showed that even though the Root-Mean-Square (RMS) from the crystal structure is slightly smaller, the RMS fluctuations obtained with the PME method are larger and in better agreement with experimental B-factors than those obtained with the long cutoff (the short cutoff is unstable).148 The Ewald potential has clear anisotropies; for instance a single ideal dipole will behave very differently when it is totally isolated from a dipole that is placed in a unit cell and then repeated periodically. Smith and Pettitt208 studied the rotational potential energy surface for a linear dipole, a quadrupole and the BPTI protein. They showed that the transition between hindered rotation (due to periodic anisotropy) and free rotation involves an energy change below kBT for solvents with a reasonably high relative permittivity.
In addition to the entirely electrostatic corrections, other thermodynamic corrections that depend on the temperature, salt concentration, dielectric constant, and other thermodynamic properties of the system may be required,209,210 as their absence may result in apparent “periodic” artifacts. For instance, probably a few of the reported artifacts in the literature can be attributed to the absence of complete sampling in systems with explicit solvent under PBC (which are far more expensive to sample exhaustively than implicit solvent systems). A recent example involves an octo-alanine peptide with charged termini in explicit solvent, for which severe effects due to the use of Ewald sums were reported in the past.211 Later, more extensive simulations of the system212 have shown that there are no periodicity-induced artifacts. The study indicates that incomplete sampling is likely to affect the results to a much larger extent than any artifacts induced by the use of Ewald sums. These conclusions seem to be supported by other long-time simulations.213–215 Another example is provided by the free energy maps of an extremely simple system such as zwitterionic tri-alanine, which illustrates how careful, extensive sampling of the system with explicit solvent is needed in order to reproduce the correct free energy map within a given force-field model.216
Free energy maps have been used as a test for the different electrostatic treatments. Free energy studies are rather tricky and challenging for all but the simplest systems. However, the effect of boundary conditions on calculated free energies has also been studied for various complex systems.217 In particular, ion hydration free energies provide simple systems to study these effects, and have been shown to exhibit remarkably system size consistency207 in Ewald calculations in high dielectric solvents. For instance, with electrostatics and thermodynamic corrections included, free energies of solvation of small molecular ions have been computed accurately even when only relatively few waters surround the ion,207 For low dielectric solvents, analytical, finite-size corrections, that also include the size of the ion, have been derived209,218–220 for ionic charging free energies, (however, these corrections have not been extended to more complex systems). The general conclusion of these studies is that Ewald-based treatments combined with PBC can get correct ion hydration free energies from molecular calculations that lead to well-characterized results appropriate to the thermodynamic limit.209 Notice that size-consistency is a necessary but not a sufficient condition for correct ionic charging free energies. For instance, these energies were computed with both Ewald in PBC and spherical boundary conditions.221,222 Size consistent results were obtained in both cases, but the results differed substantially. The difference between the two boundary conditions was traced to the electric potential drop across the vacuum-water interface in the spherical cluster. Using Gauss’ law together with the spherical symmetry, a rigorous treatment of the charging process in cluster interiors is possible. The results for a variety of simple ions using this treatment are in quantitative agreement with those using Ewald summation, even when low dielectric solvents (with system size corrections) were used.
On the other hand, for truly non-periodic systems with a strictly localized charge distribution such as isolated molecules or polymer chains, or partially periodic systems such as crystal or liquid slabs, periodic boundary conditions are no longer applicable and the application of plane wave methods for the solution of Poisson’s equation under free boundary conditions is not accurate. One of the best approaches to deal with these systems is given by the particle-particle particle-scaling (P3S) algorithm of Goedecker et al.79–81 The algorithm uses interpolating scaling functions based on wavelet theory for the representation of the charge density and FFT for the Poisson’s equation, resulting in an
(N log(N)) scaling. The forces of the P3S method are analytical derivatives of the energy which guarantees energy conservation. The method competes well with the FMM for systems with up to 105 particles; for larger systems, the FMM method may become more efficient.
3 Accurate representation of the electronic charge
3.1 Charge assignment schemes
Accurate representations of the interactions of the molecular electronic clouds can be achieved through the inclusion of higher-order multipoles, polarizability, and continuous electrostatic functions. Of course, the gain in accuracy has to be balanced with the increased simulation cost, and new algorithms are needed in order to propagate the more physical representations of the electronic density in a classical simulation in a cost-effective fashion.
Electrostatic interactions have traditionally been modeled using an atom-centered point charge (‘partial charge’) representation of the molecular charge density. The most popular methods for extracting charges from molecular wavefunctions are based on fitting atomic charges to the molecular electrostatic potential (MEP), computed with ab initio, density functional theory or semiempirical wavefunctions. The charge fitting procedure consists of minimizing the squared deviation between the Coulombic potential produced by the atomic charges and the MEP.7 These non-bond potentials are then expressed as a sum of spherically isotropic atom-atom potentials. Such representations are believed to be an important source of error in current force fields:6 mathematically this charge assignment is an under-determined problem since innumerable charge distributions can reproduce the MEP outside a surface that encloses all the charges, which is particularly problematic for the “buried charges” in atoms well inside the molecule.
Improvements in the MEP fitting procedure include applying restraints to the MEP.223 Another possibility is the enhancement of the numerical stability via a weighting function based on atomic densities to filter points that are too close or too far to the nuclei.224 This function is employed to avoid the discontinuity encountered in earlier methods. Other methods that have been developed or employed to calculate partial point charges include the wavefunction mapping Class IV and Class V models,225–229 natural bond orbital analysis,230 topological analysis based on quantum theory of atoms in molecules,231,232 the electrostatic potential expansion and analysis,233–235 maximally localized Wannier functions,236–240 and the full density screening (FDS) method.241
The fit to the MEP can be improved either by adding more charge sites8 or by including higher order multipoles at the atomic sites or bonds. The use of off-center charges and/or higher order atomic point multipoles can significantly improve the treatment of electrostatics6,231,232,236,242–246 and numerous schemes have been developed to enable the partitioning of the electronic cloud into multipoles.5,231,236,242,247–253 Figure 1 illustrates the improvement in the classical MEP for a ethylene molecule using classical multipoles computed through maximally localized Wannier functions254 (WFs). WFs are obtained from a localization procedure and provide a chemically intuitive way of partitioning the electronic charge. They are distributed in space, which allows for a good physical representation of the charge density in terms of the different multipole moments.236 In addition, the Wannier Centers are intimately related to the polarization255–257 of a molecule. The different multipole moments are efficiently calculated within the WF formalism. In particular, the ethylene molecule shown in this figure is characterized by six WFs: two associated with the C–C bond, and four associated with the C–H bonds. Figure 1 shows the MEP as obtained from a quantum based calculation (left); from a representation that only has charges (middle), and from a representation that has charges and quadrupoles (right). Notice that the dipoles for a Wannier function representation are zero, because the Wannier centers sit on the center of charge. Recently, it has been shown that the fit to the MEP can be further improved when a comprehensive set of local reference axis systems that exploit all the molecular symmetries is introduced, and when the atomic multipole moments are fitted to the quantum mechanically derived electrostatic potential.258
Figure 1.

MEP of the ethylene molecule C2H4 outside the van der Waals surface. Three planes of the molecule are shown. Left colummn: DFT calculation with standard norm-conserving pseudopotentials with a plane wave cutoff of at least 70 Ry and the BLYP density functional. Middle colummn: classical MEP as computed from the Wannier charges (monopoles) alone. Right column: classical MEP as computed from the Wannier charges and quadrupoles.236
Even with these improvements the fit to the MEP remains poor in regions near the atomic nuclei. These include regions where the charge densities of interacting fragments overlap, e.g. hydrogen bonding regions. As a consequence, the electrostatic interaction energy must be corrected for “penetration” effects at close range9 The penetration error can be ameliorated to some extent by the use of damping functions that model the “de-screening” effect resulting from the charge density overlap, and several empirical or Gaussian–based models have been proposed.259–264
In addition to fixed distributed multipoles, an enhanced accuracy requires the inclusion of polarization and, when possible, of the effect of charge density overlap. Polarization is the result of the change in the molecular charge density due to the presence of other molecules or to changes in the geometry of the molecule. These effects are known as inter- and intra-molecular polarization respectively,6 and may be modeled by the implementation of an explicit polarization energy contribution. Several methods have been developed including the Drude oscillator,265,266 fluctuating charge267,268 and induced dipole model.269–271 This has given rise to several polarizable force fields272–276 that use “point” objects (point charges, point dipoles) to represent polarization effects. A review of polarization is given in Section 3.4.
3.2 Point Multipoles
The distributed multipole analysis first introduced by Stone6 assigns distributed multipole moments to several sites in the molecule (i.e., atoms and bond midpoints), and gives a more accurate representation of the electrostatic potential than one-center, molecular multipole expansion. The generalization of Ewald summation to atomic multipoles up to quadrupoles was given by Smith.49 Since then, a few groups50–53,270 have extended the Ewald method or reaction field methods277 to take into account multipoles at the atomic and other point sites. However, the multipoles greatly increase the cost of calculations within the Ewald framework. For instance, the Ewald formalism generalized to charges, dipoles and quadrupoles costs approximately 100 times more than a representation with only charges, thus rendering multipolar representations in biomolecular simulations prohibitively expensive. In order to surmount this difficulty, PME-based methods have been introduced. A first approach for large-scale biomolecular simulations was introduced in 2000 by Toukmaji et al.,50 who developed –in addition to the classical Ewald treatment – a PME based treatment of fixed and induced point dipoles. Both methods have been implemented in AMBER vs. 6–12. The PME based implementation is quite efficient, being less than a factor of two more expensive than a calculation including only charges. Since then, a polarizable empirical force field based on the classical Drude oscillator model278 was implemented in CHARMM in 2005, and further dipolar implementations have been carried out for the PME279 and the P3M280 methods. Even these very simple representations of polarizability make a big difference for improving the modeling of various biomolecular systems.266,281–295
A systematic and efficient implementation of higher order multipoles in a Cartesian tensor formalism was introduced by Sagui et al.53,54 There, the long-range electrostatic interactions are divided in two sums according to the usual Ewald scheme: the direct sum, which evaluates the fast-varying, particle-particle interactions, considered up to a given cutoff in real space; and the “reciprocal” sum, which evaluates the smoothly varying, long-range part of the interaction. When implementing multipoles, one has to take care of additional physics that is not present in the usual treatment of charges. First, the higher-order multipoles produce additional contributions to the reciprocal virial, which arise from the dependence of the structure factor on the reciprocal lattice vector. Second, all the multipolar components that appear in the expressions of energy, forces, etc. are given in a global coordinate system. It is therefore necessary to transform the local multipole moments –defined in reference to a molecular framework– to a global framework before the calculation of the electrostatic potential starts. This is achieved by defining “frames” (local orthogonal coordinate systems). Third, to carry out MD, the torques produced by every multipole need to be converted into atomic forces.53 In the PME-based Cartesian multipole method, the direct part was greatly accelerated using special techniques,296 while the reciprocal part was implemented in three different ways: using an Ewald scheme, a PME-based approach and a multigrid-based approach. The standard matrix implementation of multipole interactions up to hexadecapole-hexadecapole costs three orders of magnitude more than charge-charge interactions. Instead, due to the use of the PME and the factorizability of B-splines, the multipolar PME is very efficient. By transferring more of the computation of the interactions to reciprocal space and using a small cutoff for the direct sum, it is possible to preserve the accuracy of the calculations at a moderate cost. In fact, a calculation of interactions up to hexadecapole-hexadecapole costs less than a factor of 9 than a regular AMBER implementation with only charge-charge interactions for standard accuracies. This code has been adapted to achieve a fast implementation of the AMOEBA (Atomic Multipole Optimized Energetics for Biomolecular Applications) force field of Ren and Ponder.270,297–299 The force field includes fixed atomic multipoles up to quadrupole level as well as inducible atomic dipoles using a Thole damping model. A PME-based implementation of the multipolar code for AMOEBA has been released in AMBER vs. 9–12.
It is generally believed that because higher-order multipolar interactions decay faster than the monopolar case, it is equally accurate and more efficient to use a cutoff for these interactions. In our experience, this is rarely the case. First of all, the use of cutoffs may be accurate for highly screened systems and definitely faster than the use of Ewald summations, but it is not faster than PME-based implementations. For instance, a comparison53 between the two approaches was performed for a water bath of over 4,000 water molecules whose electrostatic interactions included all terms up to hexadecapole-hexadecapole. A PME-based calculation for which most of the interaction is computed in reciprocal space was carried out to obtain a relative RMS force error of ε = 5 ×10−4. Then, the calculation was repeated using a standard cutoff of 8 Å for the Coulomb summation, which turned out to be 6 times more expensive and with a force error two orders of magnitude larger than the complete PME-based calculation. It is fair to argue that the cutoff should only be used for quadrupoles and higher order multipoles. A similar calculation where the charges and dipoles of the model were set to zero (so that all the interactions were quadrupole-quadrupole or higher order and converged absolutely) resulted in a relative RMS force error greater than 0.01 at a 7 Å cutoff and about 8 ×10−3 at an 8 Å cutoff. The run with the 7 Å cutoff costs approximately three times more than the optimized PME which uses a 3.6 Å cutoff for the direct sum and moves most of the calculation to reciprocal space.
Artifacts associated with the use of cutoffs for dipolar interactions are well-documented in the literature. Just as an example, MD simulations of polarizable water show that an ordinary spherical cutoff overestimates the induced polarization, resulting in an overestimation of the interaction energy, a sharper liquid structure and slower dynamics, as compared to the Ewald summation and reaction field methods277 (with the latter showing more size effects than the Ewald summation). The arguments presented in our discussion, however, do not preclude situations in which a cutoff approach may find a proper niche. It is well-known that neighboring molecules in the condensed phase interact cooperatively thereby enhancing the long-ranged nature of the dipolar effects and their correlation functions. A different situation is presented by molecules in the gas phase. A recent MC study of various dipolar and hydrogen-bonding small molecules in the saturated, superheated and supersaturated vapor phases compared the Ewald summation (the method of choice for MC simulations) with a spherical truncation scheme based on neutral groups.300 It was found that for densities not too close to the critical density, the results using the group-based spherical cutoff were statistically identical to those obtained with the Ewald summation, albeit at a considerably lower cost. A review of simple dipolar fluids bearing an electric or magnetic dipole moment under various boundary conditions and with different rheological properties is given in Ref.301
3.3 Continuous representations of molecular charge density
The use of distributed point charges or point multipoles for the description of the molecular charge density fails at medium to short range due to the so-called penetration error.6,302 The penetration error can be avoided by an explicit representation of the charge density by means of continuous functions. Continuous functions provide a natural description of the charge density overlap at close distances, as well as increased accuracy for the calculation of electrostatic properties and interactions.
Several methods have been proposed to improve the calculations required by a continuous description of the molecular charge. One of these methods involves Gaussian multipole functions,303 which models the charge density of each fragment by means of distributed atom-centered spherical Gaussian multipoles. These multipoles are obtained by expanding the ab initio density of a fragment using the Distributed Multipole Analysis6 (DMA), modified so that the expansion basis corresponds to Gaussian multipoles. Due to the correspondence of the Gaussian multipoles to distributed point multipoles obtained with DMA these two approaches may be combined to increase the computational efficiency.9 For example, the electrostatic interaction of a formamide-formaldehyde hydrogen bonded dimer was computed with varying multipole orders and compared against the Hayes-Stone intermolecular perturbation theory304 (IMPT). A calculation with full Gaussian multipoles of 4th order (i.e., up to hexadecapoles) for each fragment gave errors of 0.28 kJ/mol and of 0.3 to 0.36 kJ/mol for hybrid Gaussian/point multipoles, depending on the switching cutoff. When the multipole order was reduced to two (quadrupoles) for each fragment the error increased to 1.59 kJ/mol for the full Gaussian multipole interaction.9
Another example is provided by the “pixel by pixel” Semi-Classical Density Sum (SCDS) method.305 This method relies on the use of molecular densities obtained from ab initio gas phase molecular orbital calculations of isolated fragments. These densities are gridded on a rectangular mesh to yield “e-pixels” of density. These pixels are post-processed to screen out points near the cores and condense pixels that are further away from the cores into “super pixels”. The resulting gridded densities, along with point nuclear charges are used to calculate the intermolecular electrostatic and polarization interactions by means of numerical integration. The use of the gridded densities yield electrostatic energies which are in good agreement with intermolecular decomposition results. For example, for the di-formaldehyde hydrogen-bonded dimer the error in electrostatic energy interaction was found to be between 2.2 and 2.6 kJ/mol.305 The SCDS method has been applied to the simulation of a series of crystalline energetic organic materials.306 The calculated lattice energies are in good agreement with experimental sublimation enthalpies. Pulitzer and Ma have used a modification of this procedure to investigate non-covalent interactions in RDX (hexahydro-1,3,5-trinitro-s-triazine) and compared this to a point charge representation.307 Their results show that point charges are inadequate to model electrostatic interactions in these molecular crystals.
Volkov and Coppens308–310 have introduced a method based on the Hansen-Coppens formalism,311 which describes the static density of a molecular fragment by superposing aspherical pseudo-atoms composed of nucleus-centered density units. A database of these pseudo-atoms has been developed using theoretically calculated densities.308 The deformation density bond peaks obtained for molecules formed from pseudo-atoms from the database are reproduced to within 0.1–0.2 e/Å. These densities were then used to calculate intermolecular electrostatic interactions for a range of dimers and compared to reference data obtained from the Morokuma-Ziegler decomposition.312,313 The results obtained with the initial version of the database were on average smaller by about 25 kJ/mol. This was attributed to the limitations of the method used for the evaluation of the interactions. In this case the monomer densities obtained from the pseudo-atoms were used to obtain point multipoles via a Buckingham type expansion, which resulted in the loss of accuracy.308,309 This loss of accuracy is due to the approximation of the electrostatic interaction by distributed multipoles, which resulted in significant penetration errors.
Subsequently, the exact potential and multipole method (EP/MM) was developed.314 This method calculates the intermolecular electrostatic interaction from the pseudoatom fragment by combining the evaluation of the exact potential at short range and the use of multipoles at long range. For the short-range calculation the 6-D integral is separated into two terms that comprise the multiplication of the density on fragment A with the electrostatic potential of B and vice versa. The resulting 3-D integrals are then evaluated by numerical quadrature. Intermolecular electrostatic energies obtained by the hybrid method give average errors of 10 kJ/mol with respect to the reference Morokuma-Ziegler intermolecular electrostatics. The use of a continuous representation for the short-range interactions recovered the charge density overlap effect resulting in an increase in accuracy.
Sundholm has developed the “direct approach to gravitation and electrostatics” (DAGE) method.315 In this procedure, the electrostatic interaction is obtained by numerical integration after expanding the potential using finite-element based functions. The electrostatic interaction between molecules has also been calculated by using promolecular charge densities.316 This method has been recently implemented into the AMOEBA force field for poly-aromatic hydrocarbons PAHs.317 This AMOEBA modification has been fitted to reproduce SAPT reference calculations. This method also allows for the radial change of atomic densities.
A classical polarizable force field parametrized from ab initio data using force matching has been developed and applied to study lithium iodide (Li–I).318 In this model, each fragment is represented by a core charge density and an electronic density distribution. The total electronic density of the system is thus defined as the sum of a reference density δ0 and a density response δρ. The formulation is based on a second-order expansion of the energy density, and δρis modeled by means of Gaussian basis functions up to l = 1 (p-orbitals) obtained from density functional linear response theory. Results for the Li–I test system with this force field have comparable accuracy to results obtained from ab initio Car-Parrinello MD calculations (CPMD).318,319
This method has been extended by combining it with a frozen-density model described by the Kim-Gordon theory.320,321 The density change is determined by density functional linear response theory. This extension does not rely on empirical pair potentials. Cartesian Gaussian functions are used to model both the frozen and response densities. In this case, the Thomas-Fermi kinetic energy functional and the local-density approximation exchange functionals are used. This model has been shown to reproduce experimental structural and thermodynamic properties of a series of alkali halides in different phases.321
The Gaussian charge polarizable model (GCPM) has been developed for simulations of water.322 This method relies on the use of three spherical Gaussian functions to describe the charge density; an induced point dipole at the center of mass of the molecule; and a Buckingham-type exponential term for dispersion. Results with this model yield accurate intermolecular dimer interactions as well as condensed phase properties such as vapor pressures, densities, dielectric constants and self-diffusion coefficients.322 GPCM simulations on liquid water compare well to CPMD ab initio simulations.323 This model has also been employed to simulate water in extreme environments (773 ≤ T ≤ 1373).324
More recently, the Gaussian electrostatic model (GEM) has been introduced.253,325,326 In this method, the molecular charge density obtained from quantum mechanical (QM) calculations is fitted to a linear combination of Gaussian functions centered at specific sites. Auxiliary basis sets (ABS) for Coulomb fitting are generally employed.327 The fitted densities of the fragments are employed to calculate each of the components of the intermolecular interaction separately, The GEM fragments are fitted to reproduce gas phase ab initio QM intermolecular interaction results from the constrained space orbital variation (CSOV) energy decomposition scheme.328,329 The use of continuous functions also provides a more accurate description of molecular properties compared to conventional point charges as shown in Figure 2.
Figure 2.

Electrostatic potential (ESP) isosurfaces for a water molecule in two orientations. The ESPs were calculated from Merz-Kollman (MK) fitted charges, GEM using the P1 auxiliary basis set, and from B3LYP/6–31G(d) calculated density (ab initio). MK and P1 were both fitted from the same B3LYP/6–31G(d) calculated density.
The initial implementation of the full force field involved the analytical fitting of QM densities to s-type functions (GEM-0).326 This allowed the calculation of intermolecular interactions with average errors below 0.84 kJ/mol for each component as well as for the total intermolecular energy. Subsequent extension to higher angular momentum functions provided the added advantage of distributed multipoles and a decrease in the overall number of Gaussian primitives. These distributed multipoles have recently been shown to be applicable in the AMOEBA force field.330 Results for Coulomb and Exchange intermolecular interactions for homo-dimers of water, formamide and benzene, as well as a water-benzene heterodimer showed errors below 0.84 kJ/mol for the larger auxiliary basis sets.253 Electrostatic interactions are obtained by directly calculating the Coulomb integrals between the interacting densities. Exchange interactions are calculated by the Wheatley-Price overlap model.331,332
The calculation of intermolecular interactions with GEM requires the computation of a large number of two-center electron integrals. The analytical calculation of these integrals is computationally costly, and thus a number of methods have been explored in order to speed up the computations. Significant improvements were achieved by using reciprocal space methods based on Ewald sums.253,333 In this way the integrals are calculated in direct and reciprocal space, depending on the exponent of the Gaussian. The use of PME and FFP allowed the calculation of the interaction energy and force for several water boxes in PBC with a five site model with 110 Hermite primitive functions. These calculations could be further sped up by reducing the size of the mesh for the sampling of the Gaussians by using the Gaussian split Ewald approach.104
Alternative fitting methods have been also explored. This has enabled the reduction of numerical instabilities in the fitting procedure and the number of fitting sites.334 Overall, the use of numerical fits produces results similar to the analytical fits with errors around 0.84 kJ/mol. The reduction of fitting sites allowed the energy/force calculation of a 4096 water box with a three-site model with 46 Hermite primitive functions using PME was performed in 2.29 s. By contrast, the same system using TIP3P charges was done in 0.2 s using SANDER on the same computer.334 More recently the numerical fitting has been improved by introducing spherical grids coupled to a Gaussian type weighting function for discarding points near the core.335
Continuous functions have also been employed for the calculation of second-order interactions. Among these, damping functions based on spherical Gaussian distributions (among others) have been proposed.336,337 Induced Gaussian dipoles have been implemented for an induced polarization model.271 Giese and York have used atom-centered dipolar Gaussian functions to simulate the polarizability change for semiempirical quantum models in combined QM/MM simulations.338 This method was extended to describe exchange and dispersion interactions where the exchange interactions were modeled by the overlap method using simple Slater overlap monopole functions.339
3.4 Polarization
Polarization refers to the redistribution of the electron density of a molecule in the presence of an external electric field. In biomolecules, this arises naturally from the charge distribution of other molecules or atoms. Polarization generates non-additive, attractive inter- or intra- molecular interactions, which can result in significant many-body polarization energy. In principle, polarization may be described entirely in terms of classical electrostatics and, as such, it should be distinguished from the dispersion interaction that arises from instantaneous fluctuations of molecular charge distributions, which is entirely quantum in origin. In classical MD simulations, polarization effects are included either implicitly or explicitly. The traditional, implicit representation completely avoids the many-body calculation by including a mean polarization effect in the functional form of the interaction potential. As a consequence, the magnitude of the (permanent) dipole moment of dipolar molecules is overestimated with respect to its gas phase value, and the dynamic dipolar fluctuations that take place during the evolution of the system are ignored. Classical electrostatic models that take into account the polarization explicitly appeared as early as 1950s. For instance, Barker discussed the electrostatic energy of molecules with “permanent and induced dipoles” and presented an iterative procedure to solve for the total electric field.340 Two years later, Buckingham and Pople reported studies on imperfect gases, using a similar induced-dipole approach.341,342 Since then, there has been a number of studies that consider polarization many-body effects343–350 Nonetheless, it is mostly fixed atomic charges that are found in the classical force fields commonly in use today. Only in the past decade or so, there has been a systematic effort to develop general polarizable force fields for molecular modeling.
To illustrate quantum effects of polarization that are not captured by a point-charge description, Figure 3 shows the MEP around a water molecule with and without a positive point charge (+1) placed 2 Å away from the molecule on the molecular plane, along one of the OH bonds. The MEP is calculated for both classical unpolarizable point charges (TIP3P) and full QM (B3LYP/6-31G*). The differences previously pointed out for the isolated water molecule (Figure 2) increase even further when the water molecule interacts with the point positive charge, whose presence distorts the MEP in the classical calculation far more than it does in the QM calculation. The response of the water molecule in the QM calculation is mediated by the polarization of the electronic charge, a phenomenon that is not captured in the point-charge water model.
Figure 3.
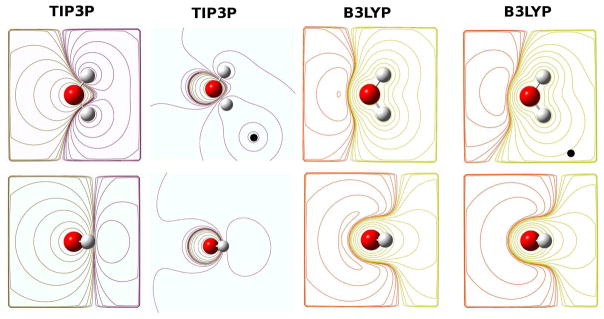
Electrostatic potential (ESP) isosurfaces for a water molecule in two orientations without (1st and 3rd columns) and with (2nd and 4th columns) a positive unit point charge. The black dot in the top panel of 2nd and 4th columns shows the position of the point charge. The ESPs were calculated from TIP3P point charges (1st and 2nd columns), and from B3LYP/6–31G(d) calculated density (3rd and 4th columns).
The development of polarizable electrostatic models has been largely driven by the interest in modeling solvent effects, mainly those of water. According to Guillot,351 more than half of the fifty or so classical water potentials published over the three decades by the early 2000s are “polarizable”, led by the polarizable and dissociable water model of Stillinger and David344 and the polarizable electropole model by Barnes and coworkers.345 Both models employed molecule-based, point induced dipoles to account for the polarization many-body effect in condensed-phase water. An iterative procedure was used to obtain the self-consistent induced dipole
| (17) |
and the polarization energy
| (18) |
where is induced dipole i, Tij the interaction matrix between and , αi the polarizability of site i, and the electric field due to the permanent charge distribution. Stillinger and David introduced a polynomial “damping” term to modify the Coulombic electric field at distances comparable to the oxygen electron radius, which effectively accounts for the spatial effect that is missing in the point dipole representation. The damping effect takes place at short distances, less than or comparable to the atomic radius, and vanishes at longer distances. It turns out that this short-range damping has become a critical feature in many of the recent polarizable force fields.
The SPCP model,352 published in 1989 in an MD study of water, continued with the induced-dipole approach using a predictive rather than an iterative solution. A few subsequent polarizable water models, such as PTIP4P,353 CKL,354 NEMO,355 NCC,356 RPOL,357 Dang-Chang,358 have all adopted the induced dipole framework to treat polarization, mostly with a single isotropic polarizable site on each molecule.
Several other models approached the polarization effect with variable point charges instead of inducible dipoles. For example, in the SPC-FQ and TIP4P-FQ267 water models, the partial charges would flow intramolecularly from one atom to another according to the electronegativity equalization theory, similarly to the charge equilibration method reported earlier.359 Note that the electronegativity equalization method (EEM) was previously introduced and applied to molecular systems including alanine dipeptide and deoxyribose.360 The PPC model employed an empirical formula for charge variation as a function of the local field.361 Stern et al. combined the fluctuating charge with the induced point dipole representations in the POL5 water model.362 Another family of models has utilized the Drude Oscillator to account for polarization effects.363,364 The Drude model makes use of two point charges joined by a spring that provides the dipole induction, and historically has been used to model the dispersion interactions365 as well as ion polarization.366 Yu et al. developed a Charge-On-Spring (COS) model for liquid water.265 In an improved COS model, a virtual site was added to better reproduce the molecular quadrupole moments and host the molecular polarizability.367 In the SWM4 models, an either negatively or positively charged Drude particle is attached to the oxygen atom.266,294 While the former seems to be a more physical representation of mobile electrons, in practice both COS and SWM4 perform similarly well in liquid water simulations.
Some recent water models have become even more sophisticated in treating polarization effects. The polarizable SPC model by Bernardo et al.,368 ab initio TTM series models369–373 and AMOEBA water model270,299,374 all utilize an interactive, distributed atomic polarizability with a Thole’s damping scheme.375 The most serious drawback of the point dipole models is the so-called “polarization catastrophe”. This phenomenon occurs when two mutually interacting induced dipoles with atomic polarizabilities αi and αj diverge at a finite distance R = (4αiαj)1/6. In an MD simulation, this situation leads to non-physical forces and velocities. The advantages of the Thole model are that anisotropic molecular polarizabilities can be achieved via isotropic and transferable atomic polarizabilities, and the polarization catastrophe is effectively avoided by smearing out the point representation of charge distribution. The TTM2-F model also incorporates geometry-dependent atomic charges via lookup tables, which allows capturing the dipole derivative with respect to the O-H bond stretch.372 The AMOEBA model uses permanent atomic charge, dipole and quadrupole moments for the accurate representation of permanent charge distributions.270,297 The electrostatics in QMPFF is represented by a combination of positive atomic point charges and diffuse electron clouds that change shapes (locations in earlier versions) when polarized.376,377 The use of a continuous charge distribution instead of point charges or multipoles is more physical and faces fewer problems associated with penetration and the polarization catastrophe. Elking and coworkers271 compared a Gaussian charge density approach with the Thole model, and suggested that the Gaussian model performed slightly better in reproducing molecular polarizability tensors. Recently, the charge penetration and transfer effects have been incorporated into the DPP2 water model.378,379 Voth and coworkers took a very different approach to model polarization in water. Their POLARFLEX model is based on the multistate empirical valence bond (MS-EVB) method, which describes the water electronic structure with three basis states.380 In a later improvement of POLARFLEX, Gaussian charge distributions replaced point charges, and the bond angle was modified381
The individual contribution of atoms in a molecule to the polarizability is not a physical observable; it is only the molecular polarizability that can be measured. The atomic dipole induction, Drude oscillator, and fluctuating charge models mentioned above are different empirical schemes that describe molecular responses. Detailed comparison among these models has been provided by Rick and Stuart,382 along with the underlying mathematics, and a discussion of the advantages and disadvantages of each approach. The induced-dipole and Drude oscillator approaches both have a great deal in common, with subtle differences in the detailed physics and model implementation. Both models can benefit from a Thole-like damping in order to avoid the polarization catastrophe and to produce an anisotropic molecular polarization response with scalar atomic polarizabilities. Note that the original Applequist interactive atomic polarizability model,383 where the induced atomic dipoles polarized other atoms in the same molecule, used no such damping and resulted in a molecular anisotropy that is too low. As a result, the atom polarizabilities are all smaller than those used by the Thole model. The distributed polarizability model by Dykstra and coworkers is additive,384,385 without intramolecular mutual induction. Atomic polarizability tensors are thus required to produce the anisotropic molecular response. The fluctuating charge model is based on a different physical principle. The intramolecular charge-flow is limited by the chemical connectivity, and thus it cannot capture certain out-of-plane polarizations or treat single ions. It has, however, the potential to naturally handle geometry-dependent charge flow missing in point induced dipole or Drude models. Olson and Sundberg386 and subsequently Applequist387 proposed a model consisting of both polarizable atomic charges and dipoles, which would allow intramolecular charge-flow upon polarization. Stern et al. developed a force field that made use of both the fluctuating charge and the induced dipoles.388 Alternatively, formulae for ab initio atomic polarizabilities for charge, dipole and high order moments have been given by Stone250 and Karlström.389 Even though the overall molecular polarizabilities are recovered exactly in the ab initio approach, determining such atomic polarizabilities from quantum mechanics is not trivial.
3.5 Charge transfer
Charge transfer (CT) refers to the interaction energy arising when electrons of one molecule delocalize into the orbitals of other molecules. Stone suggested that CT is part of the induction energy, and should not be confused with basis set superposition error (BSSE) when separating out the CT contribution.6 The BSSE is an artifact caused by using dimer basis sets on the individual molecules in a supramolecular calculation, effectively mixing orbitals of different molecules. In the limit of an infinitely large basis set, the BSSE disappears, and the CT becomes part of induction. In practice, when finite basis sets are used, the separation of CT from polarization is basis-set dependent and somewhat arbitrary.390 In an ab initio calculation of a water dimer, Stone showed that the CT contribution was about −4 kJ/mol,390 about 20% of the total interaction energy. Mo et al. developed an energy decomposition analysis (EDA) based on a block-localized wavefunction (BLW-ED) approach, which allows separating out the polarization and BSSE-free CT energy.391 The CT here refers to the energy related to relaxing the occupied molecular orbitals of one molecule into the virtual molecular orbitals of the other, after correcting for the BSSE. They suggest that the CT between two water molecules in a trimer is −4.1 to −4.7 kJ/mol,392 consistent with Stone’s findings. Moreover, while the CT contribution in benzene-ion (e.g. Li+, Na+, K+ and ) complexes is a fraction of the polarization energy, it becomes much more significant in the complexes of carbon monoxide and transition metals (e.g. Ni, Pd, Pt and their monovalent ions). A grand canonical treatment was incorporated into the X-pol method393 to describe charge transfer. Application of the GC-X-Pol to ten molecular complexes showed that the CT energy from this approach follows the trend of the BLW results.394 Khaliullin et al. reported a study on water dimer and other complexes using the absolutely localized molecular orbitals energy decomposition (ALMO-ED) scheme.395 It was found that, unlike the polarization energy, the CT energy was sensitive to the water dimer orientation (flap angle). In some orientations, the CT was comparable to the polarization contribution (−6 kJ/mol), while in others the CT contribution was slightly less (−4 kJ/mol). The CT and polarization energy became noticeable only at short-range (near the minimum-energy separation of a dimer), and exhibited similar distance dependence, which indicates that an “over-polarized” classical model might be able to capture both effects. Recently, Wu et al. employed the constrained space orbital variations (CSOV)396 and reduced variational space (RVS)397 decomposition methods to examine the Zn2+-water complexes.398 According to RVS, the CT energy was ~20% of the polarization energy at the HF/CEP-41G(2d) level, which reduced to 5% when a large basis set, aug-cc-pVTZ (water)/6-31G* (Zn), was used. Furthermore, as the Zn2+-water cluster size increases, the percentage of CT in the total induction energy (CT and polarization) decreases. The anti-cooperativity suggests that in bulk water the CT between Zn2+ and solvent may become much less significant relative to the polarization many-body effect. Thus, in the parameterization of AMOEBA force field for Zn2+, the CT contribution was effectively absorbed into the other interaction by matching AMOEBA with the QM polarization as well as the total QM binding energy. By contrast, cooperative many-body CT effect was reported for Cl− anion solvation in water.399
There has been very limited effort in modeling the CT with classical mechanics. A pairwise empirical exponential function was proposed by Clavaguera-Sarrio et al. for modeling uranyl ( ) in water.400 The parameters were derived empirically by fitting the distance-dependent QM CT energy profile. Hagberg et al. followed up with a similar study using the same functional form and obtained rather reasonable bulk properties from MD simulations.401 In the absence of an explicit CT term, the modeled uranyl-water dimer interaction energy in the medium separation (2.5–4 Å) was not “attractive” enough, similar to what was observed for Zn2+-water.398 In the recent DPP2 water model, the CT was accounted for also by using this pairwise function.379 It is however unclear if this pairwise approach would be applicable in general, given the cooperative and anti-cooperative many-body effects discussed above.
Korchowiec and Uchimaru402 devised a self-consistent charge and configuration method based on electronegativity equalization. The system energy is expanded with respect to the charge reorganization and truncation after the second order led to the so-called CT energy , where dq is the change in partial charge, and μ and η are the chemical potential and hardness parameters. However, it has been pointed out that, at the dissociation limit, the exact energy should be linear in charges, and thus a quadratic model gives rise to incorrect charge transfer.403,404 Lee and Rick recently integrated this method into the fixed-charge based TIP4P,405 TIP4P/2005406 and polarizable TIP4P-FQ267 water models to treat the charge transfer. While this formalism is in principle equivalent to that underlying the original fluctuating charge model, the specific treatment offers a way to avoid arbitrary charge flow among molecules. In their study, the CT effect was shown to be anti-cooperative going from the water dimer (0.020 e) to liquid water (0.008 e).407
3.6 Polarizable Force fields
Most current force fields for biological or synthetic molecules represent the charge distributions with fixed point charges at the atomic centers.408–411 A major hurdle preventing the widespread use of polarizable force fields is the concern over computational expense. Many-body polarization requires a computationally expensive procedure such as self-consistent iterations in order to compute the induced dipoles. Even though polarization is recognized as an important physical phenomenon, the “need” for explicit consideration of polarization is perhaps not yet fully established. Many of the polarizable water models discussed above provide notable improvement in properties such as the liquid dielectric constants and self-diffusion coefficients over simple models such as TIP3P, SPC or SPC/E. However, more sophisticated and carefully parameterized fixed-charge models such as TIP4P-EW,412 TIP4P/2005,406 and TIP5P413 also perform rather well, at least for pure water. An important lesson drawn from the development of water models is that improving the representation of the permanent charge distribution (for the atomic charge model) is perhaps as important as the inclusion of polarization effects.
Nonetheless, an increasing number of studies shows that the lack of polarization can be a serious limitation, particularly for ionic systems and chemical processes that involve a change of environment. Ions have long been investigated with polarizable force fields in solution and water-vapor interface.281,285,414–423 Perera and Berkowitz414–419 were the first to predict that halide ions (Cl−, Br− and I−, but not F−) in water clusters favor surface solvation, as opposed to bulk solvation. They correctly attributed this to polarization effects. These predictions were confirmed by photelectron spectroscopy424,425 and quantum studies.420,426 After a few years of debate in which the polarization explanation was cast into doubt, it is understood that surface solvation of an ion in a spherical water cluster is due mainly to both ion and water polarization, coupled to the charge and size of the ion.285 As an example, Figure 4 shows the total free energy and total potential energy at 300K for a Cl− ion in a spherical water cluster as a function of the distance r of the ion from the center of the cluster.285 The top panel shows results for the SPC/E model427 that assigns fixed point charges to the O and H atoms, while the bottom panel shows results for the RPOL357 water model, that assigns both (smaller) fixed point charges and atomic polarizabilities to the O and H atoms. Clearly, the fixed-charge model predicts bulk solvation, while the polarizable model predicts surface solvation with a minimum in the free energy at the surface of the cluster. The polarization effect is coupled to the size of the ion: F− remains inside the cluster but the remaining three halide ions are on the surface. Since RPOL has a larger moment than SPC/E it has been argued that it is not polarization but a large dipole moment the one responsible for surface solvation. But that is not necessarily the case: by (artificially) reducing the fixed charges of RPOL such that its total dipole moment is smaller than that of SPC/E, one can still see surface solvation for I− for RPOL (but not for SPC/E). Finally, the charge is also important, with large positive ions still able to show surface solvation in the polarizable water model.285
Figure 4.

Total free energy
 (r) (smooth dark line) and total potential energy Utot (r) (noisy dark line) for a Cl− ion in a spherical cluster of 238 waters as a function of the distance r of the ion from the center of the cluster. Top panel: SPC/E water model. Bottom panel: RPOL water model. The entropic T S(r) contribution (obtained simply as the difference between Utot (r) and
(r)) is plotted just to show that this contribution is not negligible. Reprinted with permission from Ref. 285 Copyright 2005 American Institute of Physics.
(r) (smooth dark line) and total potential energy Utot (r) (noisy dark line) for a Cl− ion in a spherical cluster of 238 waters as a function of the distance r of the ion from the center of the cluster. Top panel: SPC/E water model. Bottom panel: RPOL water model. The entropic T S(r) contribution (obtained simply as the difference between Utot (r) and
(r)) is plotted just to show that this contribution is not negligible. Reprinted with permission from Ref. 285 Copyright 2005 American Institute of Physics.
Unfortunately, due to the use of the extra-thermodynamic assumption and the surface potential, interpreting single-ion hydration free energies from experimental data on electrolytes can be rather convoluted. The hydration free energy of some monovalent ions such as Na+ and K+ from different sources can differ by as much as 10 kcal/mol or 10%. For fixed charge models, the parameterization of ion parameters (van der Waals radius and minimum-energy well depth) depends on the choice of “experimental” targets. On the other hand, the polarizable models, which can be parameterized from gas-phase ab initio data, have shown encouraging predictive power for hydration properties, as demonstrated on a series of halide, alkali and alkaline earth metal ions.294,428–430 Previous studies have pointed out that a fixed-charge force field will underestimate the Mg2+-guanine interaction energy by as much as 30% due to the absence of the many-body polarization energy.431 Attempts have been made to model transition metal Zn2+,398,432 lanthanides (Gd3+,433) and actinides (Th2+,434) with polarizable force fields. Accurate modeling of such high-valence ions with classical mechanics will likely need additional physics beyond polarization, such as charge transfer and ligand-field effects.
Molecular association such as protein-ligand binding requires the ligand to leave the high dielectric water environment before forming a complex with the protein target. Free energy simulations of ligand binding to trypsin and matrix metalloproteinase suggested that neglecting polarization can lead to an overestimation of the protein-ligand attraction, relative to the solvation strength of the ligands.432,435 An earlier study of benzamidinium chloride inhibitors binding to trypsin using fixed charge force fields showed that the binding affinities from the thermodynamic integration calculations were indeed mostly overestimated. The authors attributed this to the underestimation of ligand hydration free energy.436 However, in other work on hydrophobic ligands binding to lysozyme, simulations using fixed-charged force fields underestimate the binding free energy substantially, perhaps due to sampling issues.437 As expected, the lack of polarizability causes the imbalance between the bulk solvation and protein environment, and it seems the errors will unlikely “cancel” systematically even when comparing affinities between ligands. The recent development of fixed-charge GROMACS biomolecular force field resorted to parameterizing different charges for proteins to account for the differences in water and cyclohexane environments.438
A number of reviews on polarizable force fields has been published over the last decade.291,298,382,439–441 Nonetheless, it is fair to say that the development of general polarizable force fields remains in its early stages. The use of polarizable electrostatics in protein simulations dates back to 1976.442 The SIBFA (Sum of Interactions Between Fragments Ab initio computed) force field by Gresh and coworkers is one of the most sophisticated ab initio force fields with rigorous electrostatic terms including polarization, charge transfer and ligand field contribution for open shell metals.276,443–445 Its primary focus has been on biological systems including complex metalloproteins.446–453 Improving SIBFA’s computational efficiency and implementing SIBFA in MD simulations are goals being pursued by the researchers. Karlström and coworkers have been developing the Non-Empirical Molecular Orbital (NEMO) force field, based on atomic charges, dipoles and dipole polarizabilities, for various organic compounds.355,401,454–457 The AMBER community has long been interested in polarizable force fields,50,354,458 and a new generation of polarizable force fields is actively being developed.459–463 The electrostatic component of the force field consists of permanent point charges and inducible atomic dipoles. Kaminski et al. reported an ab initio polarizable protein force field (PFF) based on inducible dipoles and point charges.464 The dispersion parameters were later refined through condensed-phase simulations.465 To cut the computational cost, the mutual induction was subsequently modified to a limited iteration scheme (POSSIM).466,467 One challenge is that the analytical derivative is unavailable for such a potential energy surface, which prevents the use of MD. Ren and Ponder have been developing an atomic multipole-based polarizable force field, Atomic Multipole Optimized Energetics for Biomolecular Applications (AMOEBA). After the initial the water model,298,299,374 this force field has been extended to ions,398,428–430,434 organic molecules,297,468–471 and proteins.272 AMOEBA has been applied in a few studies on the thermodynamics of protein-ligands bindings.432,435,472–475 Several independent studies using AMOEBA have also been reported.476–485 The AMOEBA force field is available through the TINKER,486 AMBER273 and OpenMM487 software packages. Particle-mesh Ewald for multipole interactions,53 parallelization and multi-time step MD have all been implemented. CHARMM now also incorporates a classical Drude oscillator polarizable force field.294,488 This force field now covers alcohols,489 aromatics,440 amides,287 ions,295,488 and polypeptides.490 To overcome the deficiency in the Lennard-Jones mixing rule for unlike atom pairs, it was found necessary to use pair-specific van der Waals parameters in order to obtain accurate hydration free energies.491 Instead of using a self-consistent iteration scheme, an extended Lagrangian approach was used to propagate the induction, which led to improved computational efficiency and scalability.492 The Drude force field was shown to give an excellent dipole potential for a lipid monolayer.289 Patel and Brooks have also developed a CHARMM based polarizable force field, but with the fluctuating charge scheme.493,494 With this force field, Patel and coworkers have investigated ions solvation, water-vapor interface, ion channels and membranes.495–498 This force field also employs the extended Lagrangian method for charge fluctuations. Gao et al. developed the polarizable intermolecular potential for fluids (PIPF), which has been applied to alkanes, alcohols and amides.499,500 Later Gao and coworkers presented the X-Pol framework combing fragment-based electronic structure theory with molecular mechanical force fields.393,501,502 This model was successful in a MD simulation of a solvated protein.393,501,503 For non-biological systems, polarizable force fields are also emerging. Smith and coworkers applied a many-body polarizable force field in the simulations of ion-conducting polyethylene oxide (PEO).504–506 Chen et al. developed polarizable force fields to examine the phase equilibrium.507,508 Cummings and coworkers also formulated a novel Gaussian charge polarizable force field to model water, ions and and PEO.322,509,510 A polarizable force field for ionic liquids was reported to provide accurate thermodynamics and transport properties for a wide range of molecules.511
Most fixed-charge force fields share common potential energy functions and simulation algorithms, and differ mostly in the parameterization procedures. Most polarizable force fields rely on both gas-phase ab initio data and experimental properties for parameterization, although to a differing extent. While fixed charge force field simulation technology has become rather mature over the years, one expects to see the growth of polarizable force fields along with their increasing application to unconventional molecular systems, where traditional force fields have been challenged.
Finally, we briefly refer to models that deal with polarization within a continuum solvent environment. Intermolecular polarization is greatly affected by the presence of a solvent. For implicit treatments of solvent as a continuum dielectric, Maple et al.512 proposed a self-consistent method to combine classical polarizable models with the Poisson-Boltzmann (PB) based solvation reaction field. In their approach, after the solute charge distribution in gas-phase is determined, the PB equation is solved to obtain the reaction field, which is then fed back to the polarizable solute. The interactions are iterated until self-consistency is reached. This scheme is in principle applicable to any polarizable model, induced dipole or charged based. Note that in this classical treatment of “polarizable continuum”, the polarization includes both solvent and intra-solute contributions. The former is also treated in typical self-consistent reaction-field methods. Schnieders and coworkers reported a similar PB based approach for solute with polarizable atomic multipoles.513 The authors further developed a Generalized-Kirkwood (GK) method to describe the reaction field of a polarizable solute.514 The GK method is analogous to the well-known Generalized Born (GB) approach515 for solute with point charges, but extended to treat point dipole and quadrupole moments. The GB and GK aim to approximate the PB solution in a computationally efficient way. Truchon et al. also reported a PB based implicit solvent approach to polarizable solutes.516,517 In their model, however, the solute polarization is not described by classical models of induced dipoles or charges. The molecular polarization is treated via an effective “inner” dielectric constant and the charge distortion is solved numerically from the Poisson equation. While this is an interesting approach to polarization, it is unclear how to mix different “inner” dielectrics together in an inhomogeneous biochemical environment.
4 Electrostatics in multiscale modeling
4.1 Long-range electrostatics in QM/MM
Classical simulations have been extremely useful to study many biosystems. However, a key drawback of most classical methods is the inability to properly describe chemical reactions. Warshel and Levitt therefore introduced the QM/MM concept442 in 1976. This hybrid approach uses quantum methods in a small region (the QM subsystem) where a high level of electronic description is needed, and it simulates the remaining atoms (including solvent) via molecular mechanics. QM/MM methods have been applied to study a myriad of systems, including reactions in solution, reactions in zeolites and enzymatic reactions, along with an enormous number of solid state systems that describe electronic processes. The QM/MM approximation has often been the method of choice for modeling reactions in biomolecular systems.518,519
The treatment of long-range electrostatics in QM/MM simulations presents special challenges due to the presence of the quantum mechanical subsystem. A crucial issue that needs to be considered is the representation of the continuous QM charge density, which affects the calculation of the long-range interactions as well as the treatment of the boundary between the QM and MM subsystems. Several methods have been developed to account for long-range electrostatics in QM/MM calculations. These can be broadly separated into two approaches based on the use of boundary potentials and periodic boundary conditions.
In boundary potential models, a number of molecules are represented explicitly. These include all QM atoms and part of the MM subsystem. The remaining infinite bulk, which may include part of the protein, is represented by an effective boundary potential. One of the first examples of these type of methods is the local reaction field method for empirical valence bond/molecular mechanics (EVB/MM).520,521 This method is based on the local reaction field, which separates the system into groups of atoms and calculates the short- and long-range contributions of each group. The short-range part is evaluated explicitly. The long-range part is approximated by a multipole expansion to fourth order.187 This method has also been extended for ab initio QM/MM and employed for simulations of cations in solution.522
The generalized solvent boundary potential (GSBP)202 has also been extended for QM/MM.523 The GSBP method enables the hybrid explicit/implicit representation of irregularly shaped solute/solvent systems. In this method, an inner region including the QM subsystem, a portion of the protein, and maybe solvent, are explicitly represented. The remaining atoms of the protein are explicitly represented with fixed positions and the rest of the solvent is replaced by a dielectric continuum. The contributions from all the atoms in the outer region are included in the QM/MM calculation. A crucial issue with this (and other) methods is the representation of the continuous QM charge density. In this particular implementation, the density is approximated by Mulliken point charges.523
The initial extension of GSBP for QM/MM was implemented for a semi-empirical quantum method and tested by simulating the reaction of human carbonic anhydrase II. The results from the QM/MM-GSBP method were compared to stochastic boundary condition simulations with various treatments for the electrostatics. Structural and energetic results from GSBP were shown to be in better agreement with experimental data than the cutoff or extended electrostatic (stochastic boundary) methods.523
The QM/MM-GSBP method involves solving the Poison-Boltzmann equation many times over. This is not efficient for some QM/MM applications, such as geometry optimizations. To this end, the solvated macromolecule boundary potential (SMBP) has been developed. SMBP is largely based on GSBP, with the main difference that a different ansatz is used and the PB equation to update the individual contributions to the PMF is solved when needed.524 SMBP has also been developed to enable efficient sampling of phase-space MD simulations. Another difference with GSBP is that the interaction of the QM charge density with the boundary potential is modeled by projecting the boundary potential onto a set of virtual surface charges. This allows the introduction of these virtual charges in the calculation of the Fock matrix.524
SMBP has been validated by simulating the reactions of two different enzymes: chorismate mutase and p-hydrozybenzoate hydroxylase.525 The use of SMBP allows the separation of the electrostatic effects due to the solvent and the outer macromolecule. The results show that the inclusion of long-range electrostatic effects is especially important for reactions where significant charge is transferred.
A similar method based on the spherical solvent boundary potential (SSBP) has recently been proposed.526 This method is similar to GSBP, however in its current implementation it is only valid for an arbitrary charge distribution inside a spherical cavity. Therefore, this implementation requires the use of a large spherical cutoff to include the solute and only the solvent outside the sphere is represented by the boundary potential.
The second family of methods relies on periodic boundary and lattice summation methods for the inclusion of long-range electrostatic effects in QM/MM simulations. The initial implementation of Ewald sums for QM/MM was developed for semiempirical Hamiltonians.527 Here, only the QM/MM long-range interactions are calculated while the QM/QM long-range contribution is omitted. Thus, in the direct sum the QM/QM interactions are calculated at the QM level. For the QM/MM interactions the QM charge density is approximated by point charges. This method was tested by determination of solvation free energy of a chloride ion in water by FEP calculation with MC simulations. Subsequently, studies of acetic acid in water via MC simulations gave results in good agreement with OPLS simulations.528
Nam et al. have developed the QM/MM-Ewald method where all long–range contributions are explicitly taken into account.181 This method takes advantage of a re-writing of the energy expression for a QM/MM system under periodic boundary conditions. Briefly, the total energy for a system of this type can be decomposed into three terms (QM, QM/MM and MM) each subject to periodic boundary conditions. The QM and QM/MM periodic boundary terms can be re-expressed as a sum of the non-periodic (real space) contribution and a periodic boundary correction term. In this implementation the terms involving the periodic boundary correction with QM charge densities are calculated with conventional Ewald sums and the QM charge for reciprocal space calculations is approximated by Mulliken charges. The MM reciprocal term is calculated with PME.181
The QM/MM-Ewald method was tested with simulations of ion association and dissociative phosphoryl transfer reactions. The results were compared with periodic boundary and spherical boundary MD simulations both with medium (11.5 Å) and without electrostatic cutoffs. The results show long-range electrostatic effects influence the reactions and significant artifacts are observed when cutoffs are used.181
This method was subsequently modified to perform not only the MM reciprocal space part with PME but also the QM/MM reciprocal space contribution and implemented in the AMBER suite of programs.529 The use of PME for the QM/MM reciprocal space calculations results (depending on the QM subsystem size) in a time reduction of slightly more than twice with respect to the initial implementation. Recently, this method has been applied to include long–range electrostatic effects in the explicit polarization (X–pol) potential.530
A similar approach has been developed using Ladd sums531 in the framework of semiempirical QM models combined with classical methods. In this method, the lattice sum is done in direct space only. Similar to the boundary potential methods, the lattice methods require the representation of the QM charge density for the long-range interactions. Here, two representation were studied, Mulliken charges and the exact charge distribution. It was shown that both schemes give similar results. This method was tested by a simulation of chloride and lithium ions in SPC/E water.531
A linear-scaling method based on a multigrid approach has been implemented for DFT based QM/MM simulations.532 This method is based on the Gaussian expansion of the electrostatic potential (GEEP)533 and a modified Ewald lattice summation for the long-range QM/MM potential. GEEP allows the decomposition of the MM electrostatic potential in terms of Gaussian functions which are efficiently mapped on different grids. Results for an analytically solvable system show errors in energy below 0.03 % with respect to the reference. Tests on two α-quartz crystals and solvated Gly-Ala dipeptide show that proper treatment of the long-range interactions are required for the simulation of these systems.
4.2 Continuous functions in QM/MM
As mentioned in Section 3.3, the use of continuous functions can improve significantly the description of electrostatic interactions. The accurate treatment of electrostatics is especially important when quantum and classical methods are combined. When conventional point charges or multi-poles are employed, care must be taken to avoid over–polarization of the continuous charge density of the QM subsystem, which is an undesirable effect that can be rather dramatic when the QM and MM subsystems are in close proximity. The most extreme example involves the cutting of covalent bonds across the QM/MM boundary. For example large dipole moments arise along the bond that was cut in partitioning schemes where point charges close to the QM subsystem are explicitly included. This results in large errors in calculated barriers, proton affinities and deprotonation errors.534,535 A common approximation in these cases involves the neglect of the interaction between all QM atoms and the MM charges of the MM group where the bond was cut. Another possibility involves the re–distribution of the charges (and in some cases bond dipoles) of the MM boundary atom (or atoms).529,536
This redistribution procedure has been extended in the tuned and balanced redistributed charge algorithm (TBRC).535,537 Here, in addition to the charge re-distribution, the QM boundary atom is a tuned fluorine link atom that reproduces the partial charge of the uncapped region of the QM subsystem. The re-distributed charges (or dipoles) can be further improved by including the polarization effects of the QM subsystem on these charges.538 The TBRC method has also been extended to blur the charges near the QM/MM boundary537 via a Slater function (expressed by six Gaussian functions, STO-6G). Results for deprotonation energies for a range of reactions give mean unsigned errors of 1.6 kcal/mol.537
Das et al. developed the delocalized Gaussian MM charge (DGMM) method where a spherical Gaussian function is used to blur the MM charges that are close to the QM subsystem.534 The use of the Gaussian function ensures that the Coulomb interaction between any QM atom and the blurred charge will always be finite. This method was tested on a series of systems including rotational barriers of butane, proton affinities and deprotonation energies. Their results show that the use of DGMM gave results in agreement with full QM calculations compared to conventional QM/MM calculations where the charges of MM groups with a dangling bond are neglected.534
A similar method has been recently proposed to include charge penetration effects in molecular modeling.263 In this case the MM charge is represented by a point charge that represents the nuclear charge, core and inner valence electrons. The outer valence electrons are modeled by a smeared charge via a Slater-type orbital. Results for a test set of complexes showed a reduction in the error of electrostatic and induction interactions between the QM and MM subsystems from 8.1 to 2.8 kcal/mol and 1.9 to 1.4 kcal/mol, respectively.
An alternative algorithm employs an angular momentum-based expansion scheme of the point charges into partial wave orbitals to blur and polarize the MM charges.539 Here, a combination of Laguerre and spherical harmonics with maximum angular momentum of 1 (p-orbitals) are used for the blurring. Results for geometry optimizations of the canonical water dimer and a hydrogen sulfide dimer show good agreement with full QM reference calculations. In addition the interaction energy of BH4 with the heme group of inducible nitric oxide synthase show that the inclusion of polarization on the blurred charges has a significant effect on the electronic energy and the QM/MM interaction energy.539
The use of frozen densities as described by GEM has been shown to provide a more accurate environment in hybrid QM/MM calculations.540 The QM/GEM implementation was used to investigate the polarization response of the QM wavefunction on ten water dimers and two dimers involving a cation and a water molecule. The results showed that the use of frozen densities gives the correct polarization environment for the QM wavefunction as opposed to point charges when neutral fragments are considered. In the case of a charged fragment in the MM subsystem, it is possible to use damping functions to recover the penetration effects.540
The density fragment interaction method (DFI) employs multiple QM fragments field approximation.541 In this case the total one-electron density and Hamiltonian matrices are block-diagonal and the interaction between the different QM fragments is approximated by a mean field method via a self consistent calculation. This provides not only a continuous description of the charge density for multiple fragments but also includes explicit polarization of the QM fragments. In practice only a small subset of the full system is separated into the various QM subsystems. DFI was tested by calculating excitation energies for a magnesium sensitive dye. The total system was separated into two fragments, one comprises the dye, Mg2+ and four water molecules for the first fragment and the solvent around a sphere of 3 or 8 Å around the dye for the second. The remaining atoms were treated by a classical force field. The results show that DFI can accurately predict the spectral shift of the dye caused by the binding of the cation.541 DFI has recently been reformulated and applied to the simulation of liquid water.542
Wesolowski and Warshel have developed the frozen density embedding theory (FDET).543–546 In this method the QM subsystem is embedded in a microscopic environment represented by fragments with frozen densities, which are expressed as a function of two density dependent variables. This method has been recently extended to consider multi-level simulations.547,548
4.3 Electrostatics in coarse-grained models
In the past ten years, multiscale modeling and systematic coarse-graining have emerged as real alternatives to reach longer simulation times and larger systems. According to the Web of Science, in 2000 there were 481 articles published with multiscale simulation as the topic. In 2005 that number had jumped to 2,112, and in 2011 to 10,175. This rapid increase is a consequence of the facts that there has been a clear need for such methods and that there have been significant leaps in developing more rigorous methods and algorithms. Several recent reviews about multiscale modeling are available.549–554
The aim in multiscale modeling is to construct mappings that can be used to obtain physically important information and parameters from smaller scales and to use such information to construct larger scale, or coarse-grained, models. The opposite, when information from a large scale model is used in a more microscopic model, is called fine-graining. The main research emphasis has been on coarse-graining, but lately, some mapping schemes for fine-graining have emerged as well.552,555–560 While fine-graining has several appealing aspects, e.g., equilibration and better sampling of configurational space, the re-mapping of the system back to a more microscopic one is non-trivial and may need to involve structural relaxation procedures.557 In addition, free energy properties between the models may differ due to reduction of degrees of freedom in the coarse-grained models. Despite such difficulties, multiscale modeling has emerged as a promising approach. In more microscopic scales, QM/MM modeling represents a multiscale approach when quantum accuracy is needed.
Electrostatics poses several challenges to multiscale modeling: First, the 1/r potential is long-ranged and cannot be simply integrated away. This is also well-known from the standard virial expansion (or Mayer expansion): if Coulombic interactions are included, the expansion does not converge. These issues have led to different approaches, including the implicit inclusion of charges via iterative coarse-graining procedures, and the explicit inclusion of charges and attempts to try to include some aspects of Coulomb interactions. We discuss some of the issues next, together with some of the most common coarse-grained methods.
Perhaps the most intuitive way of coarse-graining is based on structural information. In particular, the potential of mean force (PMF) and the pair correlation function are related through
| (19) |
Since g(r) is a standard measurement in any particle system, and since it is an equilibrium quantity, the relation above can be inverted to determine first the PMF and also the underlying pairwise interactions. This is sometimes called the Henderson theorem561–563 and it is the molecular level analog to the Hohenberg-Kohn theorem for an electron system.564 See, for example, Ref.553 for a detailed discussion, a prescription to apply it in practice and its relation to other coarse-graining methods. How does Eq. (19) help to the coarse-graining procedure? The basic idea is simple: Assume that we have a given g(r) for an atomistic system, then any coarse-grained representation of the same system must produce the same g(r). Using an atomistic system as a reference, we can define new coarse-grained units, see for instance Figure 5. When new coarse-grained particles are defined, one then tries to find a coarse-grained potential that reproduces the pair correlation function.553,565,566 Since g(r) represents a free energy, direct inversion does not immediately yield the interaction potential. Instead, Eq. (19) is typically used as a starting point to determine the pair interactions through an iterative process. The most common inversion methods are the Boltzmann inversion and Inverse Monte Carlo.
Figure 5.
Example of coarse-graining. Instead of computing the interactions between all hydrogens and oxygens of water molecules, one can construct a higher level representation as shown by the yellow circles. For the coarse-grained representation to be a meaningful one, both systems must produce the same g(r) as described in the text. In the coarse-grained representation, the electrostatic interactions are included in the new effective coarse-grained potential (see text) that is obtained first by inverting Eq. (19) and then iterating it.161,553,565,566
Such an iterative inversion procedure can, at best, be a flexible, consistent and quick method to provide interaction potentials for a particular system, since the level of coarse-graining can be chosen at will. The resulting potentials do not, in general, have a functional form but need to be tabularized. The reason why a simple functional form, such as the Lennard-Jones, does not emerge is that the iterative procedure includes higher order correlations and long-range interactions, that are implicitly present in the pair correlation function. It is difficult to separate the electrostatic contribution out of the pair correlation function – Coulomb interactions are implicitly included in the correlations. One possible way to study the importance of Coulomb interactions is through the decay of ion-ion correlations. For example, Lyubartsev et al.161 studied the decay of the effective potential of aqueous NaCl solutions between 0.55 M and 4.1 M and found that in those cases the potential was screened after about 1 nm. Interestingly, they found that the coarse-grained potentials obtained at 0.87 M were transferable to other concentrations. It is not clear, however, how general that result is, and if it holds at different temperatures and in the presence of more complex molecules or different ions. It might also be useful to evaluate the goodness of this approach under the Stillinger-Lovett second-moment condition.
Inversion methods are not limited to potentials. Izvekov and Voth567 have developed a multiscale approach based on force matching. In their approach, instead of deriving the interaction potentials from structural data, they use the atomistic potentials for obtaining coarse-grained interactions. In particular, they investigated the effect of Coulomb interactions in a system containing methanol and water. They considered the case where long-range electrostatic interactions were explicitly included and compared these results to the case in which only the short-ranged coarse-grained interactions were present. The differences between the two cases were found to be relatively minor. They did not, however, test a case in which ions were present. Izvekov et al.568 have also developed a separate method to replace long-range Coulomb interactions by short-ranged potentials through force matching. Like Lyubartsev et al.,161 they found very good agreement with the underlying systems of pure water and NaCl in aqueous solution. The advantages and issues concerning this method are similar to those of the inversion methods.
The recent MARTINI model of Marrink et al.569 represents a different approach. Rather than using structural data from microscopic simulations, it is based on re-parameterization of the interactions. The model uses a four-to-one mapping for particles and has been systematically parameterized using partitioned free energies. The model takes the same approach as traditional force-fields and, unlike the PMF based methods, its force-field includes explicit charges (for the new coarse-grained particles) with the exception of water. The original MARTINI water model does not have charges and, hence, a relative dielectric constant is included in the Coulomb interactions. This creates problems, whose solution has been attempted with more recent water models, as discussed below. The model has been successfully used in a variety of problems including properties of membranes and membrane proteins,570 charged monolayers571 and lipoprotein complexes.572
Dissipative Particle Dynamics (DPD)573,574 also represents a very popular coarse-grained approach. In its traditional formulation, DPD uses soft non-bonded conservative interactions (linear forces) amended with a momentum conserving Langevin thermostat that can be used together with any conservative potential.553 The conservative force due to j on particle i is given in DPD as
| (20) |
where αi j defines the strength of the interaction and the cutoff rc is typically chosen to be unity in dimensionless units. This force has a finite value at zero particle separation. The DPD particles may be interpreted as a kind of density. It was shown by Forrest and Suter575 that averaging Lennard-Jones interactions over a long time leads to this form. DPD particles are described by their size – or range of interactions – and amplitude of interactions. For the case of polymers, the latter can be derived from solubilities. This soft form of the potential means that particles can overlap and slide through each other, which creates a problem for the inclusion of electrostatic interactions, since the 1/r form can lead to oppositely charged particles substantially overlapping and collapsing onto each other.
There have been several attempts to include electrostatics in the DPD approach. The first one is due to Groot105 whose approach follows the same idea as in self-gravitating systems: the potential has to be somehow regularized at short distances. This can be done by smearing out the charges. When the separation of oppositely charged particles is large, the potential has the usual 1/r form but there must be a penalty at distances shorter than some cutoff Rele. There are several possible ways to choose that cutoff function, and Groot’s choice was a linear function
| (21) |
Here, the parameter Rele defines the radius over which the charge is smeared out. It has to be chosen in such a way that the potential of ion pair formation is of order kBT. Then, the electrostatic field is solved on a grid as in the PME o multigrid methods. Importantly, this approach fulfills the Stillinger-Lovett second moment condition.
Another method is that of González-Melchor et al.,576 who proposed a closely related method that utilizes the standard Ewald summation instead of a grid solution. Instead of Eq. (21), they used
| (22) |
for smearing out the charge. As in Eq. (21), the above equation has only one length scale (λ) that defines the screening length. The two methods are very similar, and the use of Ewald sums makes the implementation of González-Melchor et al. particularly easy.
Praprotnik et al.552 have developed a very different multiscale method from the above. In their approach, termed Adaptive Resolution, the spatial resolution of the system is tuned adaptively. Unlike the previously described methods that apply to the whole system at once, the Adaptive Resolution method allows for a more microscopic treatment of the system in the regions of interest, while other regions, such as bulk water, may be treated at the continuum level, thereby speeding up the simulation times. This approach is very appealing since it represents a true multiscale method. However, it also poses several challenges. First, momentum conservation (as well as other conservation laws) must hold independently of the scales throughout the system, including the transition zones or interfaces between different scales. Evaluating the free energy is also challenging due to changes in the entropic contribution at different scales. What perhaps is even more demanding – but has been accounted for in simpler systems – is electrostatics: When water and ions move between continuum and particle descriptions, charge must be conserved as must be the range of interactions. The current treatment is based on reaction field electrostatics.552 It will be interesting to see how this method applies to more complex systems.
Finally, let us briefly discuss the issue of water – and the importance of charges – in coarse-grained models. As already mentioned, the MARTINI water carries no charges and is a re-parameterization of Lennard-Jones interactions to match the compressibility and density of water. The lack of charges creates artifacts, such as water freezing. These problems have not been reported with the inversion methods, potential or force, as the self-consistency guarantees the reproduction of the original radial distribution functions. Thus, other coarse-grained water models have been introduced. These include the polarizable MARTINI water,577 and the so-called Mercedes Benz578 and BMW (Big Multipole Water)579 models. The polarizable MARTINI water has two equal and opposite charges to capture the most important features of dielectric response. Electrostatic interactions are then computed either through reaction field or PME. The Mercedes Benz model does not include explicit electrostatic interactions, but uses a Tersoff type penalty function to enforce tetrahedral coordination to model hydrogen bonding. However, due to the lack of charges, it is totally transparent to external and local electric fields. The model is conceptually simple, although computationally somewhat demanding, and reproduces many of water’s anomalies and the qualitative phase diagram correctly. The BMW model contains three explicit charges and its surface tension and dielectric properties are in good agreement with experiments, thus making it a good candidate for simulations of interfacial systems. Reproduction of interfacial properties, in particular dipole orientation (or total lack of it), is one of the challenges of coarse-grained models. In general, interfaces at different scales pose a challenge to multiscale modeling: For example, close to a protein, water and ions may have preferential orientation and locations and switching between representations does not necessarily preserve the dielectric properties even if restraints are applied. This, together with response to external fields, are some of the areas where more work is expected to emerge in the future.
5 Other developments
In this section we review some interesting developments covering other aspects of electrostatics in the context of classical biomolecular simulations. One of the recent developments has been the extension of the charge equilibration model to obtain the split charge formalism of Nistor et al.580 The charge equilibration model is based on the concept of electronegativity equalization, where the polarization is obtained by the migration of charge density (condensed to partial charges) among sites within a given molecule.267,581–587 The idea of the split charge method of Nistor et al. is that charge is allowed to move along covalent bonds: the bonds are parameterized in terms of charge subject to conservation of charge and the neutrality condition for isolated molecules. The term split charge refers to the charge that flows between neighboring atoms along the bond that joins them in a molecule. It is important to notice that it is not the same as the polarizable bond approach of, e.g., Chelli et al.588 The split charge model of Nistor et al. introduces physically well-motivated bond-hardness and atomic hardness terms. It also allows extension for bond breaking, thus allowing simulations of chemical reactions. One should notice that this model is parameterized in terms of bonds only. It does not include parameterization of atoms which can provide yet another improvement. It is also easily treated by the fast multipole and Ewald based methods. Such a model is potentially very interesting as the split charge would take changes in the (dielectric) environment of a molecule properly into account in a natural way. This includes surfaces, changes in pKas and so on. Verstraelen et al.589 have performed independent testing of the split charge model.
An area where exciting developments are expected to take place is in the calculation of protein ionization constants or pKas 590–594 (which generally is carried out with implicit solvent models, although occasionally explicit solvents are also employed). In the traditional approaches that we consider in this review, it is assumed that the ionic charges on the amino acid residues are invariant to conformational changes. For a given pH, the residues are either charged or neutral at the beginning of a simulation, and this charge distribution is kept constant throughout the entire simulation (except, possibly, for polarization effects). However, 30% of the residues are ionizable and their pKas depend on the pH of the surrounding environment (and thus on salt concentration, temperature, and interactions with neighboring residues). The pKa of a residue at a given pH determines the protonation state of the residue, and the protonation equilibria of a polypeptide are tightly coupled to its conformation and folding pathways. Any change in the protonation states of a polypeptide considerably alters the charge distribution, at least around the ionizable residue, and results in an important change in the protein electrostatics. Unfortunately, the dynamic protonation equilibria are not considered in regular MD due to the high cost associated with these calculations: if there are i ionizable groups in a protein of R residues, then there are 2i ~ 2R/3 possible ionization states to consider, whose relative free energies have to be evaluated in order to determine the protonation equilibria. Obviously, this can quickly become prohibitively expensive in all but the smallest proteins, and the electrostatic problem becomes intrinsically coupled to the sampling problem. Thus, many approximations are needed. In turn, this results in loss of accuracy and dependency on the parameters of the model. A more accurate method to compute the total solvation free energies of proteins as function of pH was introduced by Vorobjev et al.593 The method uses a fast adaptive multigrid boundary element (FAMBE) to solve the Poisson equation. The electrostatic free energies of the ionizable sites are calculated in atomic detail for neutral and charged states and a precise dielectric surface interface is defined, which is then adaptively tsellated. The ionization partition function is computed directly for proteins with less than 20–25 ionizable groups, or via the Tanford-Schellman integral for larger number of ionizable groups. The FAMBE-pH method was tested on three proteins to compute the pH-dependent solvation free energies and the results indicate accurate predictions. Clearly, this is an exciting area of active research,21,591 where further important developments are expected.
Other interesting development has been the application of accurate electrostatics to the refinement of X-ray structure factors. X-ray crystal structures with subatomic resolution have been rapidly increasing in the Protein Data Bank, and they represent an ideal opportunity for studying valence electron distributions and the ensuing electrostatic properties of macromolecules. Recently, Schnieders, Fenn et al. used polarizable, Cartesian Gaussian atomic multipoles for X-ray crystal structure refinement.595–598 Usual calculations of structure factors are based on the isolated atom model (IAM) that assumes a spherical symmetric electron density around each atom, thus neglecting the anisotropies that result from bonding and the chemical environment. Another problem of the use of traditional force fields for restraining X-ray refinement is the absence of a proper treatment of electrostatics. The Cartesian Gaussian multipoles offer an efficient alternative to the Hansen-Coppens formulation based on Slater-type orbitals.311 The Gaussian multipoles allow for the calculation of structures factors via direct summation in reciprocal space using FFTs, which can lead to a speed-up relative to direct summation of over an order of magnitude for aspherical refinement of ultra-high resolution sets.595 The authors originally used four peptide crystals to compare this approach and the AMOEBA force field, and found that an atomic multipole expansion up to hexadecapoles (or addition of interatomic scattering sites) occasionally may be necessary to explain bond electron density.595 The authors also applied the AMOEBA force field to “re-refine” high-resolution lysozyme and trypsin data sets, which yielded an improvement in the crystallographic refinement statistics and overall potential energy, as well as additional information about pKa values at enzymatic active sites, hydrogen-bonding structure, and molecular stabilization.596 Re-refinement of a DNA duplex resulted in a complete hydrogen-bond network that agrees precisely with the Dickerson hydration model.599
6 Summary and Perspective
Electrostatic interactions are crucial for biomolecular simulations, as their calculation is the most time consuming when computing the total classical forces, and their representation has profound consequences for the accuracy of classical force fields. At present, classical codes for biomolecular simulations, where electrostatics is represented by point charges, can carry out full evaluation of electrostatics at comparable cost than cutoff methods. They represent mature software whose evolution is largely driven by parallelization issues springing from the successive generations of computer architectures. At the cutting edge of the technology, very large systems and millisecond time scales are now feasible.83,600 As simulations on such and longer scales grow more widespread with improvements in supercomputer power, commodity networks, and GPU hardware,601 the validity of the point-charge representation will become under scrutiny. We expect a sizeable amount of work devoted to characterize the effect of point-charge electrostatics in extremely long MD simulations, since undesirable artifacts have already been spotted.
Hints to these problems have been gleaned by Raval et al.602 This group has performed a comprehensive study involving at least 100μs MD simulations for 24 proteins used in recent CASP competitions. These long-time simulations were applied to both the experimental structures and homology models. For most systems, the structures drift away from the native state, even when starting from the experimental structure. Even though the authors only used two force fields, they concluded that this is most likely a limitation of the available point-charge force fields.
As computing power increases, it becomes clear that other components of molecular mechanics representations, including permanent electrostatics, penetration effects, charge-transfer, exchange-repulsion, dispersion, and polarization also need to be addressed or improved in order to truly achieve chemical accuracy. Present models explore the ability to perform simulations using more sophisticated methods, such as those that employ multipoles or continuous functions. The use of continuous functions for simulations enables the accurate representation of the simulated systems by avoiding problems such as penetration errors. In addition, continuous functions provide very accurate results for intermolecular Coulomb interactions. In spite of the presence of various models for treating polarization effects, the progress toward the development and applications of general-purpose polarizable force fields is still limited. The concern with computational speed and the lack of understanding of the importance of polarization effects are likely the major obstacles. In terms of hybrid QM/MM methods, many issues remain partially or completely unaddressed. Further developments in this area include the more wide-spread use of methods to include long-range electrostatic interactions, general ways to address the critical QM/MM boundary and QM/MM implementations where the MM environment is able to respond to changes in the QM subsystem (polarizable MM). Systematic investigations and the consequent understanding of the various intermolecular forces in a wide range of biological systems and different nano-bio applications would help us elucidate the gains in accuracy brought about by these elaborate force fields, especially with respect to the non-bonded interactions. This, in turn, will require the development of new parallel implementations to allow for the efficient evaluation of long-range electrostatic interactions with more accurate representations of the molecular charge density.
Finally, we would like to note that advancements in MD in general require efficient algorithms not only for computing the different components of the molecular interaction potentials but also for sampling the molecular configurations. Two different approaches tackle the issue of sampling. Coarse-grained modeling allows for both larger systems and longer time scales. The challenge here concerns issue of transferability and accuracy, especially in a multiscale framework linking it to atomistic molecular mechanics simulations, and common trends are moving away from “phenomenological” models to models that have a rigorous statistical mechanics foundation. This is particularly challenging when coupling continuum and particle representations as, for example, in adaptive resolution schemes:552 charge neutrality must be conserved and interfaces between different representations should not generate artificial polarization or multipole moments. In the purely atomistic realm, conformational space sampling relies on enhanced sampling techniques that allow the system to visit different regions of phase space, even if these regions are separated by high free energy barriers. This also represents an active area of research and numerous new methods have been introduced in the last decade, such as non-equilibrium methods,216,603–610 combined with various versions of Replica Exchange Molecular Dynamics,609,611–615 adiabatic molecular dynamics,616,617 and others.
Acknowledgments
We would like to thank Robert E. Duke for sharing with us his experience with truncation methods. We would like to thank the support we have received for this work. G.A.C. thanks Wayne State University, M.K. thanks the Natural Sciences and Engineering Research Council of Canada, P.R. thanks the Robert A. Welch Foundation (F-1691), and the National Institutes of Health (R01GM106137), and C.S thanks NSF grants 1021883 and 1148144.
Biographies

Pengyu Ren:
Pengyu Ren received his Ph.D. in Chemical Engineering from the University of Cincinnati in 1999. After a postdoctoral training at Washington University Medical School, he joined The University of Texas at Austin as Faculty in 2005. He is now an Associate Professor of Biomedical Engineering at UT Austin, working on biomolecular modeling and simulations with emphasis on protein-ligand binding and force field development.

G. Andrés Cisneros:
G. Andres Cisneros received a B.Sc. in Chemistry from the National Autonomous University of Mexico (UNAM) and a Ph.D. in Chemistry from Duke University. Afterward, he worked as a postdoctoral fellow in the National Institute of Environmental Health Sciences/NIH. He is currently an Assistant Professor in the Department of Chemistry at Wayne State University. His research interests include development and application of accurate methods for chemical/biochemical simulations with emphasis on force field and QM/MM development, and the study of DNA replication/repair systems and ionic liquids.

Celeste Sagui:
C. Sagui received her PhD in Physics from the University of Toronto in 1995 and performed postdoctoral work at McGill University and National Institutes of Environmental and Health Sciences. In 2000, she joined North Carolina State University, where she has been Professor since 2009. Her research interests include statistical mechanics, condensed matter theory and complex systems, phase separation and nucleation processes, computational biology and biomolecular simulations. Her work has also focused on methodological developments for the accurate and efficient treatment of electrostatics, and the development of free-energy methods for large-scale biomolecular simulations.

Mikko Karttunen:
Mikko Karttunen completed his PhD in condensed matter physics at the Department of Physics at McGill University in Montreal in 1999. He then moved to the Polymer Theory Group of Prof. Kurt Kremer at the Max Planck Institute for Polymer Research in Mainz for postdoctoral fellowship. From Mainz he moved to Helsinki University of Technology in Finland where he worked as a senior researcher and later as an Academy of Finland Fellow and started his own computational biological and soft matter physics group. In 2006 he moved for a faculty position at the Dept. of Applied Mathematics at the University of Western Ontario in Canada. He is currently a Professor of Chemistry at University of Waterloo in Canada.
References
- 1.Alder BJ, Wainwright TE. J Chem Phys. 1957;27:1208. [Google Scholar]
- 2.Born M, Oppenheimer R. Annalen der Physik. 1927;84:0457. [Google Scholar]
- 3.Sagui C, Darden TA. Annu Rev Biophys Biomol Struct. 1999;28:155. doi: 10.1146/annurev.biophys.28.1.155. [DOI] [PubMed] [Google Scholar]
- 4.Marshall GR. J Comput Aided Mol Des. 2013;27:107. doi: 10.1007/s10822-013-9634-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Proft FD, Alsenov CV, Peeters A, Langenaeker W, Geerlings P. J Comp Chem. 2002;23:1198. doi: 10.1002/jcc.10067. [DOI] [PubMed] [Google Scholar]
- 6.Stone AJ. International Series of Monographs on Chemistry. Oxford University Press; Oxford: 1996. The Theory of Intermolecular Forces. [Google Scholar]
- 7.Francl MM, Chirlian LA. In: Rev Comp Ch. Lipkowitz K, Boyd DB, editors. Vol. 14 VCH Publishers; New York, NY: 1999. [Google Scholar]
- 8.Dixon R, Kollman P. J Comp Chem. 1997;18:1632. [Google Scholar]
- 9.Wheatley R, Mitchell J. J Comp Chem. 1994;15:1187. [Google Scholar]
- 10.Cramer CJ, Truhlar DG. In: Rev Comp Ch. Lipkowitz K, Boyd DB, editors. Vol. 6 VCH Publishers; New York, NY: 1995. [Google Scholar]
- 11.Cramer CJ, Truhlar DG. Chem Rev. 1999;99:2161. doi: 10.1021/cr960149m. [DOI] [PubMed] [Google Scholar]
- 12.Bashford D, Case D. Ann Rev Phys Chem. 2000;51:129. doi: 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- 13.Simonson T. Curr Opin Struct Bio. 2001;11:243. doi: 10.1016/s0959-440x(00)00197-4. [DOI] [PubMed] [Google Scholar]
- 14.Feig M, Brooks CL. Curr Opin Struct Bio. 2004;14:217. doi: 10.1016/j.sbi.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 15.Baker NA. Curr Opin Struct Bio. 2005;15:137. doi: 10.1016/j.sbi.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 16.Gallicchio E, Levy RM. J Comp Chem. 2004;25:479. doi: 10.1002/jcc.10400. [DOI] [PubMed] [Google Scholar]
- 17.Wagoner J, Baker NA. J Comp Chem. 2004;25:1623. doi: 10.1002/jcc.20089. [DOI] [PubMed] [Google Scholar]
- 18.Roux B, Simonson T. Biophys Chem. 1999;78:1. doi: 10.1016/s0301-4622(98)00226-9. [DOI] [PubMed] [Google Scholar]; Ctr Calcul Atom & Molec; CECAM Workshop on Implicit Solvent Models for Biomolecular Simulations; Lyon, France. May, 1998. [Google Scholar]
- 19.Tomasi J, Persico M. Chem Rev. 1994;94:2027. [Google Scholar]
- 20.Tomasi J, Mennucci B, Cammi R. Chem Rev. 2005;105:2999. doi: 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- 21.Chen J, Brooks CL, III, Khandogin J. Curr Opin Struct Bio. 2008;18:140. doi: 10.1016/j.sbi.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heyes DM, Barber M, Clarke JHR. J Chem Soc, Faraday Trans. 1977;2:1485. [Google Scholar]
- 23.Hautman J, Klein ML. Mol Phys. 1992;75:379. [Google Scholar]
- 24.de Leeuw SW, Perram JW. Mol Sim. 1979;37:1313. [Google Scholar]
- 25.Nijboer BRA, de Wette FW. Physica. 1957;23:309. [Google Scholar]
- 26.Yeh IC, Berkowitz ML. J Chem Phys. 1999;111:3155. [Google Scholar]
- 27.Lekner J. Physica A. 1989;157:826. [Google Scholar]
- 28.Lekner J. Physica A. 1991;176:485. [Google Scholar]
- 29.Lekner J. Mol Sim. 1998;20:357. [Google Scholar]
- 30.Sperb R. Mol Sim. 1994;13:189. [Google Scholar]
- 31.Sperb R. Mol Sim. 1998;20:179. [Google Scholar]
- 32.Sperb R. Mol Sim. 1999;22:199. [Google Scholar]
- 33.Strebel R, Sperb R. Mol Sim. 2001;27:61. [Google Scholar]
- 34.Arnold A, Holm C. Chem Phys Lett. 2002;354:324. [Google Scholar]
- 35.Arnold A, Holm C. Comp Phys Comm. 2002;148:327. [Google Scholar]
- 36.Ewald P. Ann Phys. 1921;64:253. [Google Scholar]
- 37.Ewald P. Fifty Years of X-ray Diffraction. N.V.A. Oosthoek’s Uitgeversmaatschappij; Utrecht, the Netherlands: 1962. [Google Scholar]
- 38.Juretschke H, Wagenfeld H. Current Contents/Engineering Technology & Applied Sciences. 49. 1985. p. 20. [Google Scholar]
- 39.DeLeeuw SW, Perram JW, Smith ER. Proc R Soc Lond. 1980;A373:27. [Google Scholar]
- 40.Boresch S, Steinhauser O. Ber Bunseges Phys Chem. 1997;101:1019. [Google Scholar]
- 41.Hockney RW, Eastwood JW. Computer Simulation using Particles. McGraw-Hill; New York: 1981. [Google Scholar]
- 42.Pollock E, Glosli J. Comp Phys Comm. 1996;95:93. [Google Scholar]
- 43.Darden TA, York DM, Pedersen LG. J Chem Phys. 1993;98:10089. [Google Scholar]
- 44.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. J Chem Phys. 1995;103:8577. [Google Scholar]
- 45.York D, Yang W. J Chem Phys. 1994;101:3298. [Google Scholar]
- 46.Sagui C, Darden TA. J Chem Phys. 2001;114:6578. [Google Scholar]
- 47.Smith ER. J Stat Phys. 1994;77:449. [Google Scholar]
- 48.Caillol JP. J Chem Phys. 1994;101:6080. [Google Scholar]
- 49.Smith W. CCP5 Information Quarterly. 1982;4:13. [Google Scholar]
- 50.Toukmaji A, Sagui C, Board JA, Darden T. J Chem Phys. 2000;113:10913. [Google Scholar]
- 51.Nymand TM, Linse P. J Chem Phys. 2000;112:6152. [Google Scholar]
- 52.Aguado A, Madden P. J Chem Phys. 2003;119:7471. [Google Scholar]
- 53.Sagui C, Pedersen LG, Darden TA. J Chem Phys. 2004;120:73. doi: 10.1063/1.1630791. [DOI] [PubMed] [Google Scholar]
- 54.Sagui C, Roland C, Pedersen LG, Darden TA. In: New Algorithms for Macromolecular Simulations. Leimkuhler B, Chipot C, Elber R, Laaksonen A, Mark A, Schlick T, Schuette C, Skeel R, editors. Vol. 49. Springer Verlag; Berlin: 2005. p. 297. [Google Scholar]
- 55.Smith W. CCP5 Information Quarterly. 1987;26:43. [Google Scholar]
- 56.Alejandre J, Tildesley DJ, Chapela GA. J Chem Phys. 1995;102:4574. [Google Scholar]
- 57.Nose S, Klein M. Mol Phys. 1983;50:1055. [Google Scholar]
- 58.Ferrell R, Bertschinger E. J Mod Phys. 1994;5:933. [Google Scholar]
- 59.Toukmaji A, Board JA. Comp Phys Comm. 1996;95:78. [Google Scholar]
- 60.Darden T, Toukmaji A, Pedersen L. J Chim Phys. 1997;94:1346. [Google Scholar]
- 61.Deserno M, Holm C. J Chem Phys. 1998;109:7678. [Google Scholar]
- 62.Deserno M, Holm C. J Chem Phys. 1998;109:7694. [Google Scholar]
- 63.Sagui C, Darden TA. In: Simulation and Theory of Electrostatic Interactions in Solution. Pratt LR, Hummer G, editors. Vol. 492. AIP; Melville, NY: 1999. p. 104. [Google Scholar]
- 64.Ballenegger V, Cerda JJ, Lenz O, Holm C. J Chem Phys. 2008;128:034109. doi: 10.1063/1.2816570. [DOI] [PubMed] [Google Scholar]
- 65.Wang H, Dommert F, Holm C. J Chem Phys. 2010;133:034117. doi: 10.1063/1.3446812. [DOI] [PubMed] [Google Scholar]
- 66.Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods R. J Comput Chem. 2005;26:1668. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. J Comp Chem. 1983;4:187. [Google Scholar]
- 68.Brooks BR, et al. J Comp Chem. 2009;30:1545. [Google Scholar]
- 69.Phillips J, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel R, Kale L, Schulten K. J Comp Chem. 2005;26:1781. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hess B, Kutzner C, van der Spoel D, Lindahl E. J Chem Theo Comp. 2008;4:435. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 71.Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossvary I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE. SC’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing; New York: ACM Press; 2006. [Google Scholar]
- 72.Bush IJ, Todorov IT, Smith W. Comp Phys Comm. 2006;175:323. [Google Scholar]
- 73.Smith W. Mol Sim. 2006;32:933. [Google Scholar]
- 74.Hedman F, Laaksonen A. Chem Phys Lett. 2006;425:142. [Google Scholar]
- 75.Cerutti DS, Duke RE, Darden TA, Lybrand TP. J Chem Theory Comput. 2009;5:2322. doi: 10.1021/ct9001015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen L, Langdon AB, Birdsall CK. J Comp Phys. 1974;14:200. [Google Scholar]
- 77.Neelov A, Holm C. J Chem Phys. 2010;132:234103. doi: 10.1063/1.3430521. [DOI] [PubMed] [Google Scholar]
- 78.Cerutti DS, Case DA. J Chem Theory Comput. 2010;6:443. doi: 10.1021/ct900522g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Genovese L, Deutsch T, Neelov A, Goedecker S, Beylkin G. J Chem Phys. 2006;125:074105. doi: 10.1063/1.2335442. [DOI] [PubMed] [Google Scholar]
- 80.Neelov A, Ghasemi SA, Goedecker S. J Chem Phys. 2007;127:024109. doi: 10.1063/1.2746328. [DOI] [PubMed] [Google Scholar]
- 81.Genovese L, Deutsch T, Goedecker S. J Chem Phys. 2007;127:054704. doi: 10.1063/1.2754685. [DOI] [PubMed] [Google Scholar]
- 82.Duke RE. Amber 9 manual: amber.scripps.edu/doc9/amber9.pdf. In: Case DA, et al., editors. AMBER 9. University of California; San Francisco: 2006. p. 225. [Google Scholar]
- 83.Schulten K, Phillips JC, Kale LV, Bhatele A. In: Petascale Computing: Algorithms and Applications. Bader D, editor. Chapman & Hall/CRC Press; 2008. p. 165. [Google Scholar]
- 84.Brandt A. Math Comput. 1977;31:333. [Google Scholar]
- 85.Briggs EL, Sullivan DJ, Bernholc J. Phys Rev B. 1996;54:14362. doi: 10.1103/physrevb.54.14362. [DOI] [PubMed] [Google Scholar]
- 86.Beck TL. Rev Mod Phys. 2000;72:1041. [Google Scholar]
- 87.Brandt A. SIAM J Num Anal. 1994;31:1695. [Google Scholar]
- 88.Brandt A. In: Multiscale and Multiresolution Methods: Theory and Applications. Barth TJ, Chan TF, Haimes R, editors. Springer Verlag; Heidelberg: 2001. [Google Scholar]
- 89.Honig B, Nicholls A. Science. 1995;268:1144. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 90.Holst M, Saied F. J Comp Chem. 1993;14:105. [Google Scholar]
- 91.Holst M, Kozack RE, Saied F, Subramaniam S. Proteins Struct Fun Gen. 1994;18:231. doi: 10.1002/prot.340180304. [DOI] [PubMed] [Google Scholar]
- 92.Holst M, Saied F. J Comp Chem. 1995;16:337. [Google Scholar]
- 93.Ripoll DR, Vorobjev YN, Liwo A, Vila JA, Scheraga HA. J Mol Biol. 1996;264:770. doi: 10.1006/jmbi.1996.0676. [DOI] [PubMed] [Google Scholar]
- 94.Vorobjev YN, Scheraga HA. J Comp Chem. 1997;18:569. [Google Scholar]
- 95.Qian X, Schlick T. J Chem Phys. 2002;116:5971. [Google Scholar]
- 96.Press W, Teukolsky SA, Vettering WT, Flannery BP. Numerical Recipes in FOR-TRAN: The Art of Scientific Computing. 2 Cambridge University Press; Cambridge: 1992. [Google Scholar]
- 97.Zaslavsky L, Schlick T. Appl Math Comput. 1998;97:237. [Google Scholar]
- 98.Hackbush W. Multigrid Methods and Applications. Springer-Verlag; Berlin: 1985. [Google Scholar]
- 99.Skeel RD, Tezcan I, Hardy DJ. J Comp Chem. 2002;23:673. doi: 10.1002/jcc.10072. [DOI] [PubMed] [Google Scholar]
- 100.Goedecker S, Chauvin C. J Theo Comp Chem. 2003;2:483. [Google Scholar]
- 101.Tsukerman I. IEEE Trans Magn; 9th Joint Magnetism and Magnetic Materials Conference/International Magnetics Conference; Anaheim, CA. Jan 05–09, 2004.2004. p. 2158. [Google Scholar]
- 102.Izaguirre JA, Hampton SS, Matthey T. J Parall Distrib Comp. 2005;65:949. [Google Scholar]
- 103.Banerjee S, Board JAJ. J Comp Chem. 2005;26:957. doi: 10.1002/jcc.20220. [DOI] [PubMed] [Google Scholar]
- 104.Shan Y, Klepeis JL, Eastwood MP, Dror RO, Shaw DE. J Chem Phys. 2005;122:054101. doi: 10.1063/1.1839571. [DOI] [PubMed] [Google Scholar]
- 105.Groot RD. J Chem Phys. 2003;118:11265. [Google Scholar]
- 106.Lovett R, Stillinger FH. J Chem Phys. 1991;94:7353. [Google Scholar]
- 107.Bernholc J, Hodak M, Lu W. J Phys: Condens Matter; CECAM Workshop on Linear-Scaling Ab Initio Calculation – Applications and Future Directions; Lyon, France. Sep 03–06, 2007.2008. p. 294205. [Google Scholar]
- 108.Bishop T, Skeel R, Schulten K. J Comp Chem. 1997;18:1785. [Google Scholar]
- 109.Barnes J, Hut P. Nature. 1986;324:446. [Google Scholar]
- 110.Greengard L, Rokhlin V. J Comp Phys. 1987;73:325. [Google Scholar]
- 111.Board JA, Causey JW, Leathrum JF, Windemuth A, Schulten K. Chem Phys Lett. 1992;198:89. [Google Scholar]
- 112.Schmidt KE, Lee MA. J Stat Phys. 1991;63:1223. [Google Scholar]
- 113.Lambert CG, Darden TA, Board JA. J Comp Phys. 1996;126:274. [Google Scholar]
- 114.Kurzak J, Pettitt BM. J Parall Distrib Comp. 2005;65:870. [Google Scholar]
- 115.Greengard LF. The Rapid Evaluation of Potential Fields in Particle Systems. The MIT Press; Cambridge MA: 1988. [Google Scholar]
- 116.Greengard LF. Science. 1994;265:909. doi: 10.1126/science.265.5174.909. [DOI] [PubMed] [Google Scholar]
- 117.Greengard L, Rokhlin V. Acta Numerica. 1997;6:229. [Google Scholar]
- 118.Greengard L, Rokhlin V. J Comput Phys. 1997;135:280. [Google Scholar]
- 119.White CA, Headgordon M. J Chem Phys. 1994;101:6593. [Google Scholar]
- 120.Kurzak J, Pettitt BM. Mol Sim. 2006;32:775. doi: 10.1080/08927020600991161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cheng H, Greengard L, Rokhlin V. J Chem Phys. 1999;155:468. [Google Scholar]
- 122.Fenley M, Olson W, Chua K, Boschitsch A. J Comp Chem. 1994;17:976. [Google Scholar]
- 123.Zhou R, Berne B. J Chem Phys. 1995;103:9444. [Google Scholar]
- 124.Ogata S, Campbell T, Kalia R, Nakano A, Vashishta P, Vemparala S. Comp Phys Comm. 2003;153:445. [Google Scholar]
- 125.Kurzak J, Mirkovic D, Pettitt BM, Johnsson SL. Int J High Performance Computing Applications. 2008;22:219. doi: 10.1177/1094342008090915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Giese TJ, York DM. J Comp Chem. 2008;29:1895. doi: 10.1002/jcc.20946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Maggs AC, Rossetto V. Phys Rev Lett. 2002;88:196402. doi: 10.1103/PhysRevLett.88.196402. [DOI] [PubMed] [Google Scholar]
- 128.Rottler J, Maggs AC. Phys Rev Lett. 2004;93:170201. doi: 10.1103/PhysRevLett.93.170201. [DOI] [PubMed] [Google Scholar]
- 129.Pasichnyk I, Duenweg B. J Phys: Cond Mat. 2004;16:S3999. [Google Scholar]
- 130.Rottler J. J Chem Phys. 2007;127:134104. doi: 10.1063/1.2769358. [DOI] [PubMed] [Google Scholar]
- 131.Rottler J, Maggs AC. Soft Matter. 2011;7:3260. [Google Scholar]
- 132.Brooks CL, Pettitt BM, Karplus M. J Chem Phys. 1985;83:5897. [Google Scholar]
- 133.Steinbach PJ, Brooks BR. J Comp Chem. 1994;15:667. [Google Scholar]
- 134.Auffinger P, Westhof E. Encyclopedia of Computational Chemistry. John Wiley and Sons; New York: 1998. [Google Scholar]
- 135.Cheatham TE, III, Kollman PA. J Am Chem Soc. 1997;119:4805. [Google Scholar]
- 136.Karttunen M, Rottler J, Vattulainen I, Sagui C. Curr Top Membr. 2008;60:49. [Google Scholar]
- 137.Alper HE, Levy RM. J Chem Phys. 1989;91:1242. [Google Scholar]
- 138.Alper HE, Bassolino D, Stouch TR. J Chem Phys. 1993;98:9798. [Google Scholar]
- 139.Alper HE, Bassolino-Klimas D, Stouch TR. J Chem Phys. 1993;99:5547. doi: 10.1021/bi00210a010. [DOI] [PubMed] [Google Scholar]
- 140.Feller SE, Pastor RW, Rojnuckarin A, Bogusz S, Brooks BR. J Phys Chem. 1996;100:17011. [Google Scholar]
- 141.Brunsteiner M, Boresch S. J Chem Phys. 2000;112:6953. [Google Scholar]
- 142.Mark P, Nilsson L. J Comp Chem. 2002;23:1211. doi: 10.1002/jcc.10117. [DOI] [PubMed] [Google Scholar]
- 143.Hess B. J Chem Phys. 2002;116:209. [Google Scholar]
- 144.Smith PE, Pettitt BM. J Chem Phys. 1991;95:8430. [Google Scholar]
- 145.Schreiber H, Steinhauser O. Biochemistry. 1992;31:5856. doi: 10.1021/bi00140a022. [DOI] [PubMed] [Google Scholar]
- 146.York DM, Yang WT, Lee H, Darden T, Pedersen LG. J Am Chem Soc. 1995;117:5001. [Google Scholar]
- 147.Norberg J, Nilsson L. Biophys J. 2000;79:1537. doi: 10.1016/S0006-3495(00)76405-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.York DM, Darden TA, Pedersen LG. J Chem Phys. 1993;99:8345. [Google Scholar]
- 149.Saito M. J Chem Phys. 1994;101:4055. [Google Scholar]
- 150.Ibragimova GT, Wade RC. Biophys J. 1998;74:2906. doi: 10.1016/S0006-3495(98)77997-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zuegg J, Greedy JE. Biochemistry. 1999;38:13862. doi: 10.1021/bi991469d. [DOI] [PubMed] [Google Scholar]
- 152.Faraldo-Gómez JD, Smith GR, Sansom MSP. Eur Biophys J. 2002;31:217. doi: 10.1007/s00249-002-0207-5. [DOI] [PubMed] [Google Scholar]
- 153.Tieleman DP, Hess B, Sansom MSP. Biophys J. 2002;83:2393. doi: 10.1016/s0006-3495(02)75253-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kessel A, Tieleman DP, Ben-Tal N. Eur Biophys J. 2004;33:16. doi: 10.1007/s00249-003-0345-4. [DOI] [PubMed] [Google Scholar]
- 155.Patra M, Karttunen M, Hyvönen MT, Falck E, Lindqvist P, Vattulainen I. Biophys J. 2003;84:3636. doi: 10.1016/S0006-3495(03)75094-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Patra M, Karttunen M, Hyvönen MT, Falck E, Vattulainen I. J Phys Chem B. 2004;108:4485. doi: 10.1063/1.1824033. [DOI] [PubMed] [Google Scholar]
- 157.Anézo C, de Vries AH, Höltje HD, Tieleman DP. J Phys Chem B. 2003;107:9424. [Google Scholar]
- 158.Khelashvili GA, Scott HL. J Chem Phys. 2004;120:9841. doi: 10.1063/1.1724814. [DOI] [PubMed] [Google Scholar]
- 159.Wohlert J, Edholm O. Biophys J. 2004;87:2433. doi: 10.1529/biophysj.104.044222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Patra M, Hyvönen MT, Falck E, Sabouri-Ghomi M, Vattulainen I, Karttunen M. Comp Phys Comm. 2007;176:14. [Google Scholar]
- 161.Lyubartsev AP, Karttunen M, Vattulainen I, Laaksonen A. Soft Mater. 2002;1:121. [Google Scholar]
- 162.van der Spoel D, van Maaren PJ. J Chem Theory Comput. 2006;2:1. doi: 10.1021/ct0502256. [DOI] [PubMed] [Google Scholar]
- 163.Wolf D, Keblinski P, Phillpot S, Eggebrecht J. J Chem Phys. 1999;110:8254. [Google Scholar]
- 164.Zahn D, Schilling B, Kast S. J Phys Chem B. 2002;106:10725. [Google Scholar]
- 165.Fennell CJ, Gezelter JD. J Chem Phys. 2006;124:234104. doi: 10.1063/1.2206581. [DOI] [PubMed] [Google Scholar]
- 166.McCann B, Acevedo O. J Chem Theory Comput. 2013;9:944. doi: 10.1021/ct300961e. [DOI] [PubMed] [Google Scholar]
- 167.Yonezawa Y. J Chem Phys. 2012;136:244103. doi: 10.1063/1.4729748. [DOI] [PubMed] [Google Scholar]
- 168.Kikugawa G, Apostolov R, Kamiya N, Taiji M, Himeno R, Nakamura H, Yonezawa Y. J Comput Chem. 2009;30:110. doi: 10.1002/jcc.21035. [DOI] [PubMed] [Google Scholar]
- 169.Hansen J, Schroeder T, Dyre J. J Phys Chem B. 2012;116:5738. doi: 10.1021/jp300750g. [DOI] [PubMed] [Google Scholar]
- 170.Piana S, Lindorff-Larsen K, Dirks RM, Salmon JK, Dror RO, Shaw DE. Plos One. 2012;7:e39918. doi: 10.1371/journal.pone.0039918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Nguyen T, Carrillo J, Dobrynin A, Brown W. J Chem Theory Comput. 2013;9:273. doi: 10.1021/ct300718x. [DOI] [PubMed] [Google Scholar]
- 172.Robertson A, Luttmann E, Pande VS. J Comp Chem. 2008;29:694. doi: 10.1002/jcc.20828. [DOI] [PubMed] [Google Scholar]
- 173.Duan Y, Kollman P. Science. 1998;282:740. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 174.Takahashi KZ, Narumi T, Suh D, Yasuoka K. J Chem Theory Comput. 2012;8:4503. doi: 10.1021/ct3003805. [DOI] [PubMed] [Google Scholar]
- 175.Fukuda I, Yonezawa Y, Nakamura H. J Chem Phys. 2011;134:164107. doi: 10.1063/1.3582791. [DOI] [PubMed] [Google Scholar]
- 176.Fukuda I, Kamiya N, Yonezawa Y, Nakamura H. J Chem Phys. 2012;137:054314. doi: 10.1063/1.4739789. [DOI] [PubMed] [Google Scholar]
- 177.Wu X, Brooks B. J Chem Phys. 2005;122:044107. doi: 10.1063/1.1836733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wu X, Brooks BR. J Chem Phys. 2009;131:024107. doi: 10.1063/1.3160730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wong-ekkabut J, Miettinen MS, Dias C, Karttunen M. Nature Nanotech. 2010;5:555. doi: 10.1038/nnano.2010.152. [DOI] [PubMed] [Google Scholar]
- 180.Wong-ekkabut J, Karttunen M. J Chem Theory Comput. 2012;8:2905. doi: 10.1021/ct3001359. [DOI] [PubMed] [Google Scholar]
- 181.Nam K, Gao J, York DM. J Chem Theory and Comput. 2005;1:2. doi: 10.1021/ct049941i. [DOI] [PubMed] [Google Scholar]
- 182.Sirk TW, Moore S, Brown EF. J Chem Phys. 2013;138:064505. doi: 10.1063/1.4789961. [DOI] [PubMed] [Google Scholar]
- 183.Roemer F, Lervik A, Bresme F. J Chem Phys. 2012;137:074503. doi: 10.1063/1.4739855. [DOI] [PubMed] [Google Scholar]
- 184.Zhang M, Lussetti E, de Souza L, Muller-Plathe F. J Phys Chem B. 2005;109:15060. doi: 10.1021/jp0512255. [DOI] [PubMed] [Google Scholar]
- 185.Muscatello J, Bresme F. J Chem Phys. 2011;135:234111. doi: 10.1063/1.3670965. [DOI] [PubMed] [Google Scholar]
- 186.Barker JA, Watts RO. Mol Phys. 1973;26:789. [Google Scholar]
- 187.Lee FS, Warshel A. J Chem Phys. 1992;97:3100. [Google Scholar]
- 188.Alper H, Levy RM. J Chem Phys. 1993;99:9847. [Google Scholar]
- 189.Tironi IG, Sperb R, Smith PE, van Gunsteren WF. J Chem Phys. 1995;102:5451. [Google Scholar]
- 190.Lin Y, Baumketner A, Deng S, Xu Z, Jacobs D, Cai W. J Chem Phys. 2009;131:154103. doi: 10.1063/1.3245232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Onsager L. J Am Chem Soc. 1936;58:1486. [Google Scholar]
- 192.Wang L, Hermans J. J Phys Chem. 1995;99:12001. [Google Scholar]
- 193.Ni B, Baumketner A. J Mol Model. 2011;17:2883. doi: 10.1007/s00894-011-0975-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Oostenbrink C, Soares TA, van der Vegt NFA, van Gunsteren WF. Eur Biophys J. 2005;34:273. doi: 10.1007/s00249-004-0448-6. [DOI] [PubMed] [Google Scholar]
- 195.Berkowitz ML, McCammon JA. Chem Phys Lett. 1982;90:215. [Google Scholar]
- 196.Brooks BR, Karplus M. J Chem Phys. 1983;79:6312. [Google Scholar]
- 197.Belch AC, Berkowitz ML. Chem Phys Lett. 1985;113:278. [Google Scholar]
- 198.King G, Warshel A. J Chem Phys. 1989;91:3647. [Google Scholar]
- 199.Juffer AH, Berendsen HJC. Mol Phys. 1993;79:623. [Google Scholar]
- 200.Beglov D, Roux B. J Chem Phys. 1994;100:9050. [Google Scholar]
- 201.Essex JW, Jorgensen WL. J Comp Chem. 1995;16:951. [Google Scholar]
- 202.Im W, Bernèche S, Roux B. J Chem Phys. 2001;114:2924. [Google Scholar]
- 203.Caillol JM. J Chem Phys. 1992;96:1455. [Google Scholar]
- 204.Caillol JM. J Chem Phys. 1999;111:6528. [Google Scholar]
- 205.Caillol JM, Gilles D. J Stat Phys. 2000;100:905. [Google Scholar]
- 206.Hanassab S, Vandernoot T. Mol Sim. 2003;29:527. [Google Scholar]
- 207.Hummer G, Pratt LR, Garcia AE. J Phys Chem. 1996;100:1206. [Google Scholar]
- 208.Smith PE, Pettitt BM. J Chem Phys. 1996;105:4289. [Google Scholar]
- 209.Hummer G, Pratt L, Garcia A. J Phys Chem A. 1998;102:7885. [Google Scholar]
- 210.Hummer G, Pratt LR, Garcia AE, Neumann M. Treatment of electrostatic interactions in computer simulations and calculation of thermodynamic properties such as free energies and pressures. Workshop on Treatment of Electrostatic Interactions in Computer Simulations of Condensed Media; SANTA FE, NM. JUN 23–25, 1999.1999. [Google Scholar]
- 211.Weber W, Hunenberger P, McCammon J. J Phys Chem B. 2000;104:3668. [Google Scholar]
- 212.Villarreal MA, Montich GG. J Biomol Struct Dyn. 2005;23:135. doi: 10.1080/07391102.2005.10507054. [DOI] [PubMed] [Google Scholar]
- 213.Monticelli L, Colombo G. Theo Chem Acc. 2004;112:145. [Google Scholar]
- 214.Monticelli L, Simões C, Belvisi L, Colombo G. J Phys: Condens Matter. 2006;18:S329. [Google Scholar]
- 215.Reif MM, Kraeutler V, Kastenholz MA, Daura X, Hünenberger PH. J Phys Chem B. 2009;113:3112. doi: 10.1021/jp807421a. [DOI] [PubMed] [Google Scholar]
- 216.Babin V, Roland C, Darden TA, Sagui C. J Chem Phys. 2006;125:204909. doi: 10.1063/1.2393236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Levy RM, Gallicchio E. Ann Rev Phys Chem. 1998;49:531. doi: 10.1146/annurev.physchem.49.1.531. [DOI] [PubMed] [Google Scholar]
- 218.Figueirido F, Buono GD, Levy RM. J Phys Chem B. 1997;101:5622. [Google Scholar]
- 219.Hummer G, Pratt LR, Garcia AE. J Chem Phys. 1997;107:9275. [Google Scholar]
- 220.Sakane S, Ashbaugh HS, Wood RH. J Phys Chem B. 1998;102:5673. [Google Scholar]
- 221.Darden T, Pearlman D, Pedersen L. J Chem Phys. 1998;109:10921. [Google Scholar]
- 222.Herce D, Darden T, Sagui C. J Chem Phys. 2003;119:7621. doi: 10.1063/1.1829635. [DOI] [PubMed] [Google Scholar]
- 223.Bayly C, Cieplak P, Cornell W, Kollman P. J Phys Chem. 1993;97:10269. [Google Scholar]
- 224.Hu H, Lu Z, Yang W. J Chem Theo Comp. 2007;3:1004. doi: 10.1021/ct600295n. [DOI] [PubMed] [Google Scholar]
- 225.Storer JW, Giesen DJ, Cramer CJ, Truhlar DG. J of Comp-Aided Mol Design. 1995;9:87. doi: 10.1007/BF00117280. [DOI] [PubMed] [Google Scholar]
- 226.Li J, Zhu T, Cramer CJ, Truhlar DG. J Phys Chem A. 1998;102:1820. [Google Scholar]
- 227.Li J, Williams B, Cramer CJ, Truhlar DG. J Chem Phys. 1999;110:724. [Google Scholar]
- 228.Winget P, Thompson JD, Xidos JD, Cramer CJ, Truhlar DG. J Phys Chem A. 2002;106:10707. [Google Scholar]
- 229.Marenich AV, Jerome SV, Cramer CJ, Truhlar DG. J Chem Theo Comp. 2012;8:527. doi: 10.1021/ct200866d. [DOI] [PubMed] [Google Scholar]
- 230.Reed AE, Curtiss LA, Weinhold F. Chem Rev. 1988;88:899. [Google Scholar]
- 231.Popelier P. Atoms in Molecules: An Introduction. Prentice Hall; Harlow, England: 2000. [Google Scholar]
- 232.Popelier PLA, Joubert L, Kosov DS. J Phys Chem A. 2001;105:8254. [Google Scholar]
- 233.Simmonett AC, Gilbert ATB, Gill PMW. Mol Phys. 2005;103:2789. [Google Scholar]
- 234.Sigfridsson E, Ryde U. J Comp Chem. 1998;19:377. [Google Scholar]
- 235.Lee TS, York DM, Yang W. J Chem Phys. 1995;102:7549. [Google Scholar]
- 236.Sagui C, Pomorski P, Darden TA, Roland C. J Chem Phys. 2004;120:4530. doi: 10.1063/1.1644800. [DOI] [PubMed] [Google Scholar]
- 237.Kirchner B, Hutter J. J Chem Phys. 2004;121:5133. doi: 10.1063/1.1785780. [DOI] [PubMed] [Google Scholar]
- 238.Stengel M, Spaldin N. Phys Rev B. 2006;73:075121. [Google Scholar]
- 239.Rotenberg B, Salanne M, Simon C, Vuilleumier R. Phys Rev Lett. 2010;104:138301. doi: 10.1103/PhysRevLett.104.138301. [DOI] [PubMed] [Google Scholar]
- 240.Molina JJ, Lectez S, Tazi S, Salanne M, Dufreche JF, Roques J, Simoni E, Madden PA, Turq P. J Chem Phys. 2011;134:014511. doi: 10.1063/1.3518101. [DOI] [PubMed] [Google Scholar]
- 241.Wang B, Truhlar DG. J Chem Theo Comp. 2012;8:1989. doi: 10.1021/ct2009285. [DOI] [PubMed] [Google Scholar]
- 242.Coombes D, Price S, Willock D, Leslie M. J Phys Chem. 1996;100:7352. [Google Scholar]
- 243.Price S. In: Rev Comp Ch. Lipkowitz K, Boyd DB, editors. Vol. 14. VCH Publishers; New York, NY: 1999. p. 225. [Google Scholar]
- 244.Kosov DS, Popelier PLA. J Phys Chem A. 2000;104:7339. [Google Scholar]
- 245.Popelier PLA, Kosov DS. J Chem Phys. 2001;114:6539. [Google Scholar]
- 246.Price SL, Leslie M, Welch GWA, Habgood M, Price LS, Karamertzanis PG, Day GM. Phys Chem Chem Phys. 2010;12:8478. doi: 10.1039/c004164e. [DOI] [PubMed] [Google Scholar]
- 247.Claverie P. Int J Quantum Chem. 1971;5:273. [Google Scholar]
- 248.Sokalski W, Poirier R. Chem Phys Lett. 1983;98:86. [Google Scholar]
- 249.Wheatley RJ. Chem Phys Lett. 1993;208:159. [Google Scholar]
- 250.Stone A, Alderton M. Mol Phys. 1985;56:1047. [Google Scholar]
- 251.Bader R. Atoms in Molecules: A Quantum Theory. Clarendon Press; Oxford: 1990. [Google Scholar]
- 252.Stone AJ. J Chem Theo Comp. 2005;1:1128. doi: 10.1021/ct050190+. [DOI] [PubMed] [Google Scholar]
- 253.Cisneros GA, Piquemal JP, Darden TA. J Chem Phys. 2006;125:184101. doi: 10.1063/1.2363374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Wannier G. Phys Rev. 1937;52:191. [Google Scholar]
- 255.King-Smith R, Vanderbilt D. Phys Rev B. 1993;47:1651. doi: 10.1103/physrevb.47.1651. [DOI] [PubMed] [Google Scholar]
- 256.Vanderbilt D, King-Smith R. Phys Rev B. 1993;48:1993. doi: 10.1103/physrevb.48.4442. [DOI] [PubMed] [Google Scholar]
- 257.Marzari N, Vanderbilt D. Phys Rev B. 1997;56:12847. [Google Scholar]
- 258.Kramer C, Gedeck P, Meuwly M. J Comp Chem. 2012;33:1673. doi: 10.1002/jcc.22996. [DOI] [PubMed] [Google Scholar]
- 259.Kairys V, Jensen JH. Chem Phys Lett. 1999;315:140. [Google Scholar]
- 260.Freitag MA, Gordon MS, Jensen JH, Stevens WJ. J Chem Phys. 2000;112:7300. [Google Scholar]
- 261.Piquemal JP, Gresh N, Giessner-Prettre C. J Phys Chem A. 2003;107:10353. doi: 10.1021/jp035748t. [DOI] [PubMed] [Google Scholar]
- 262.Cisneros GA, Tholander SNI, Parisel O, Darden TA, Elking D, Perera L, Piquemal JP. Int J Quantum Chem. 2008;108:1905. doi: 10.1002/qua.21675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Wang B, Truhlar DG. J Chem Theo Comp. 2010;6:3330. doi: 10.1021/ct1003862. [DOI] [PubMed] [Google Scholar]
- 264.Stone AJ. J Phys Chem A. 2011;115:7017. doi: 10.1021/jp112251z. [DOI] [PubMed] [Google Scholar]
- 265.Yu H, Hansson T, van Gunsteren WF. J Chem Phys. 2003;118:221. [Google Scholar]
- 266.Lamoureux G, Alexander D, MacKerell J, Roux B. J Chem Phys. 2003;119:5185. [Google Scholar]
- 267.Rick SW, Stuart SJ, Berne BJ. J Chem Phys. 1994;101:6141. [Google Scholar]
- 268.Ribeiro MCC, Almeida LCJ. J Chem Phys. 1999;110:11445. [Google Scholar]
- 269.Caldwell J, Dang LX, Kollman PA. J Am Chem Soc. 1990;112:9144. [Google Scholar]
- 270.Ren P, Ponder JW. J Phys Chem B. 2003;107:5933. [Google Scholar]
- 271.Elking D, Darden TA, Woods RJ. J Comp Chem. 2007;28:1261. doi: 10.1002/jcc.20574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Ponder JW, Wu C, Ren P, Pande VS, Chodera JD, Schnieders MJ, Haque I, Mobley DL, Lambrecht DS, Robert A, DiStasio, Head-Gordon M, Clark GNI, Johnson ME, Head-Gordon T. J Phys Chem B. 2010;114:2549. doi: 10.1021/jp910674d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Case DA, Cheatham TE, III, Darden TA, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. J Comp Chem. 2005;26:1668. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Day PN, Jensen JH, Gordon MS, Webb SP, Stevens WJ, Krauss M, Garmer D, Basch H, Cohen D. J Chem Phys. 1996;105:1968. [Google Scholar]
- 275.Gagliardi L, Lindh R, Karlström G. J Chem Phys. 2004;121:4494. doi: 10.1063/1.1778131. [DOI] [PubMed] [Google Scholar]
- 276.Gresh N, Claverie P, Pullman A. Int J Quantum Chem. 1979;253 Symp 11. [Google Scholar]
- 277.Nymand TM, Linse P. J Chem Phys. 2000;112:6386. [Google Scholar]
- 278.Anisimov VM, Lamoureux G, Vorobyov IV, Huang N, Roux B, MacKerell AD. J Chem Theory Comput. 2005;1:153. doi: 10.1021/ct049930p. [DOI] [PubMed] [Google Scholar]
- 279.Wang W, Skeel R. J Chem Phys. 2005;123:164107. doi: 10.1063/1.2056544. [DOI] [PubMed] [Google Scholar]
- 280.Cerda JJ, Ballenegger V, Lenz O, Holm C. J Chem Phys. 2008;129:234104. doi: 10.1063/1.3000389. [DOI] [PubMed] [Google Scholar]
- 281.Jungwirth P, Tobias D. J Phys Chem B. 2002;106:6361. [Google Scholar]
- 282.Baucom J, Transue T, Fuentes-Cabrera M, Krahn J, Darden T, Sagui C. J Chem Phys. 2004;121:6998. doi: 10.1063/1.1788631. [DOI] [PubMed] [Google Scholar]
- 283.Vrbka L, Mucha M, Minofar B, Jungwirth P, Brown E, Tobias D. Curr Opin Coll Interface Sci. 2004;9:67. [Google Scholar]
- 284.Harder E, Eaves J, Tokmakoff A, Berne B. Proc Natl Acad Sci USA. 2005;102:11611. doi: 10.1073/pnas.0505206102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Herce D, Perera L, Darden T, Sagui C. J Chem Phys. 2005;122:024513. doi: 10.1063/1.1829635. [DOI] [PubMed] [Google Scholar]
- 286.Babin V, Baucom J, Darden TA, Sagui C. J Phys Chem B. 2006;110:11571. doi: 10.1021/jp061421r. [DOI] [PubMed] [Google Scholar]
- 287.Harder E, Anisimov VM, Whitfield TW, MacKerell AD, Jr, Roux B. J Phys Chem B. 2008;112:3509. doi: 10.1021/jp709729d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Vladimirov E, Ivanova A, Roesch N. J Chem Phys. 2008;129:194515. doi: 10.1063/1.3013456. [DOI] [PubMed] [Google Scholar]
- 289.Harder E, MacKerell AD, Jr, Roux B. J Am Chem Soc. 2009;131:2760. doi: 10.1021/ja806825g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Vladimirov E, Ivanova A, Roesch N. J Phys Chem B. 2009;113:4425. doi: 10.1021/jp809774q. [DOI] [PubMed] [Google Scholar]
- 291.Cieplak P, Dupradeau FY, Duan Y, Wang J. J Phys: Condens Matter. 2009;21:333102. doi: 10.1088/0953-8984/21/33/333102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Yan T, Wang Y, Knox C. J Phys Chem B. 2010;114:6905. doi: 10.1021/jp9089112. [DOI] [PubMed] [Google Scholar]
- 293.Baugh L, Le Trong I, Cerutti DS, Guelich S, Stayton PS, Stenkamp RE, Lybrand TP. Biochemistry. 2010;49:4568. doi: 10.1021/bi1005392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Lamoureux G, Roux B. J Phys Chem B. 2006;110:3308. doi: 10.1021/jp056043p. [DOI] [PubMed] [Google Scholar]
- 295.Yu H, Whitfield TW, Harder E, Lamoureux G, Vorobyov I, Anisimov VM, MacKerell AD, Jr, Roux B. J Chem Theory Comput. 2010;6:774. doi: 10.1021/ct900576a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.McMurchie L, Davidson E. J Comput Phys. 1978;26:218. [Google Scholar]
- 297.Ren P, Ponder JW. J Comput Chem. 2002;23:1497. doi: 10.1002/jcc.10127. [DOI] [PubMed] [Google Scholar]
- 298.Ponder JW, Case DA. Adv Protein Chem. 2003;66:27. doi: 10.1016/s0065-3233(03)66002-x. [DOI] [PubMed] [Google Scholar]
- 299.Ren P, Ponder JW. J Phys Chem B. 2004;108:13427. [Google Scholar]
- 300.Dinpajooh M, Keasler SJ, Truhlar DG, Siepmann JI. Theo Chem Acc. 2011;130:83. [Google Scholar]
- 301.Weis J, Levesque D. Advanced Computer Simulation Approaches for Soft Matter Sciences II. In: Holm C, Kremer K„, editors. Advances in Polymer Science. Vol. 185. Springer Verlag; 2005. p. 163. [Google Scholar]
- 302.Qian W, Krimm S. J Phys Chem A. 2005;109:5608. doi: 10.1021/jp040683v. [DOI] [PubMed] [Google Scholar]
- 303.Wheatley R. Mol Phys. 2011;7:761. [Google Scholar]
- 304.Hayes I, Stone A. Mol Phys. 1984;53:83. [Google Scholar]
- 305.Gavezzotti A. J Phys Chem B. 2002;106:4145. [Google Scholar]
- 306.Eckhardt CJ, Gavezzotti A. J Phys Chem B. 2007;111:3430. doi: 10.1021/jp0669299. [DOI] [PubMed] [Google Scholar]
- 307.Politzer P, Ma Y. Int J Quantum Chem. 2004;100:733. [Google Scholar]
- 308.Volkov A, Li X, Koritsanszky T, Coppens P. J Phys Chem A. 2004;108:4283. [Google Scholar]
- 309.Volkov A, Coppens P. J Comp Chem. 2004;25:921. doi: 10.1002/jcc.20023. [DOI] [PubMed] [Google Scholar]
- 310.Coppens P, Volkov A. Acta Cryst A. 2004;60:357. doi: 10.1107/S0108767304014953. [DOI] [PubMed] [Google Scholar]
- 311.Hansen N, Coppens P. Acta Cryst A. 1978;34:909. [Google Scholar]
- 312.Morokuma K. J Chem Phys. 1971;55:1236. [Google Scholar]
- 313.Ziegler T, Rauk A. Theo Chem Acc. 1977;46:1. [Google Scholar]
- 314.Volkov A, Koritsanszky T, Coppens P. Chem Phys Lett. 2004;391:170. [Google Scholar]
- 315.Sundholm D. J Chem Phys. 2005;122:194107. doi: 10.1063/1.1898206. [DOI] [PubMed] [Google Scholar]
- 316.Spackman MA. Chem Phys Lett. 2006;418:158. [Google Scholar]
- 317.Tafipolsky M, Engels B. J Chem Theory Comput. 2011;7:1791. doi: 10.1021/ct200185h. [DOI] [PubMed] [Google Scholar]
- 318.Tabacchi G, Mundy CJ, Hutter J, Parrinello M. J Chem Phys. 2002;117:1416. [Google Scholar]
- 319.Car R, Parrinello M. Phys Rev Lett. 1985;55:2471. doi: 10.1103/PhysRevLett.55.2471. [DOI] [PubMed] [Google Scholar]
- 320.Gordon RG, Kim YS. J Chem Phys. 1972;56:3122. [Google Scholar]
- 321.Tabacchi G, Hutter J, Mundy CJ. J Chem Phys. 2005;123:074108. doi: 10.1063/1.2001637. [DOI] [PubMed] [Google Scholar]
- 322.Paricaud P, Predota M, Chialvo AA, Cummings PT. J Chem Phys. 2005;122:244511. doi: 10.1063/1.1940033. [DOI] [PubMed] [Google Scholar]
- 323.Dyer PJ, Cummings PT. J Chem Phys. 2006;125:144519. doi: 10.1063/1.2355485. [DOI] [PubMed] [Google Scholar]
- 324.Chialvo AA, Horita J. J Chem Phys. 2010;133:074504. doi: 10.1063/1.3469769. [DOI] [PubMed] [Google Scholar]
- 325.Cisneros GA, Piquemal JP, Darden TA. J Chem Phys. 2005;123:044109. doi: 10.1063/1.1947192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Piquemal JP, Cisneros GA, Reinhardt P, Gresh N, Darden TA. J Chem Phys. 2006;124:104101. doi: 10.1063/1.2173256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Godbout N, Andzelm J. DGauss Version 2.0, 2.1, 2.3, 4.0: the file that contains the A1 and P1 auxiliary basis sets can be obtained from the CCL WWW. Computational Chemistry List, Ltd; Ohio: 1998. site at http://ccl.osc.edu/cca/data/basis-sets/DGauss/basis.v3.html. [Google Scholar]
- 328.Bagus PS, Hermann K, Bauschlicher CW., Jr J Chem Phys. 1984;80:4378. [Google Scholar]
- 329.Piquemal JP, Marquez A, Parisel O, Giessner-Prettre C. J Comp Chem. 2005;26:1052. doi: 10.1002/jcc.20242. [DOI] [PubMed] [Google Scholar]
- 330.Cisneros GA. J Chem Theory Comput. 2012;12:5072. doi: 10.1021/ct300630u. [DOI] [PubMed] [Google Scholar]
- 331.Wheatley RJ, Price SL. Mol Phys. 1990;69:507. [Google Scholar]
- 332.Domene C, Fowler PW, Wilson M, Madden P, Wheatley RJ. Chem Phys Lett. 2001;333:403. [Google Scholar]
- 333.Darden TA. Dual bases in crystallographic computing. In: Shmueli U, editor. International Tables of Chrystallography. B Kluwer Academic Publishers; Dordrecht, The Netherlands: 2007. [Google Scholar]
- 334.Cisneros GA, Elking DM, Piquemal JP, Darden TA. J Phys Chem A. 2007;111:12049. doi: 10.1021/jp074817r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Elking DM, Cisneros GA, Piquemal JP, Darden TA, Pedersen LG. J Chem Theo Comp. 2010;6:190. doi: 10.1021/ct900348b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Jensen L, Åstrand PO, Osted A, Kongsted J, Mikkelsen KV. J Chem Phys. 2002;116:4001. [Google Scholar]
- 337.Masia M, Probst M, Rey R. J Chem Phys. 2005;123:164505. doi: 10.1063/1.2075107. [DOI] [PubMed] [Google Scholar]
- 338.Giese TJ, York DM. J Chem Phys. 2005;123:164108. doi: 10.1063/1.2080007. [DOI] [PubMed] [Google Scholar]
- 339.Giese TJ, York DM. J Chem Phys. 2007;127:194101. doi: 10.1063/1.2778428. [DOI] [PubMed] [Google Scholar]
- 340.Barker JA. Proc R Soc A. 1953;219:367. [Google Scholar]
- 341.Buckingham AD, Pople JA. Trans Faraday Soc. 1955;51:1173. [Google Scholar]
- 342.Buckingham AD, Pople JA. Transactions of the Faraday Society. 1955;51:1179. [Google Scholar]
- 343.Vesely FJ. J Comp Phys. 1977;24:361. [Google Scholar]
- 344.Stillinger FH, David CW. J Chem Phys. 1978;69:1473. [Google Scholar]
- 345.Barnes P, Finney JL, Nicholas JD, Quinn JE. Nature. 1979;282:459. [Google Scholar]
- 346.Warshel A. J Phys Chem. 1979;83:1640. [Google Scholar]
- 347.Lybrand TP, Kollman PA. J Chem Phys. 1985;83:2923. [Google Scholar]
- 348.Vanbelle D, Froeyen M, Lippens G, Wodak SJ. Mol Phys. 1992;77:239. [Google Scholar]
- 349.Kutteh R, Nicholas J. Comput Phys Comm. 1995;86:227. [Google Scholar]
- 350.Kutteh R, Nicholas JB. Comp Phys Comm. 1995;86:236. [Google Scholar]
- 351.Guillot B. J Mol Liq. 2002;101:219. [Google Scholar]
- 352.Ahlström P, Wallqvist A, Engström S, Jonsson B. Mol Phys. 1989;68:563. [Google Scholar]
- 353.Sprik M, Klein M. J Chem Phys. 1988;89:7556. [Google Scholar]
- 354.Cieplak P, Kollman P, Lybrand T. J Chem Phys. 1990;92:6755. [Google Scholar]
- 355.Wallqvist A, Ahlström P, Karlström G. J Phys Chem. 1990;94:1649. [Google Scholar]
- 356.Niesar U, Corongiu G, Clementi E, Kneller GR, Bhattacharya DK. J Phys Chem. 1990;94:7949. [Google Scholar]
- 357.Dang LX. J Chem Phys. 1992;96:6970. [Google Scholar]
- 358.Dang LX, Chang TM. J Chem Phys. 1997;106:8149. [Google Scholar]
- 359.Rappe AK, Goddard WA. J Phys Chem. 1991;95:3358. [Google Scholar]
- 360.WJM, SKG, SS jacs. 1986;108:4315. [Google Scholar]
- 361.Kusalik PG, Svishchev IM. Science. 1994;265:1219. doi: 10.1126/science.265.5176.1219. [DOI] [PubMed] [Google Scholar]
- 362.Stern HA, Rittner F, Berne BJ, Friesner RA. J Chem Phys. 2001;115:2237. [Google Scholar]
- 363.Drude P. Annalen der Physik. 1900;306:566. [Google Scholar]
- 364.Drude P, Mann CR, Millikan RA. The theory of optics. Longmans, Green, and Co; New York etc: 1902. p. 1.p. 1.p. xxi.p. 546. [Google Scholar]
- 365.Bade WL. J Chem Phys. 1957;27:1280. [Google Scholar]
- 366.Dick BG, Overhauser AW. Phys Rev. 1958;112:90. [Google Scholar]
- 367.Yu HB, van Gunsteren WF. J Chem Phys. 2004;121:9549. doi: 10.1063/1.1805516. [DOI] [PubMed] [Google Scholar]
- 368.Bernardo DN, Ding YB, Kroghjespersen K, Levy RM. J Phys Chem. 1994;98:4180. [Google Scholar]
- 369.Burnham C, Li J, Xantheas S, Leslie M. J Chem Phys. 1999;110:4566. [Google Scholar]
- 370.Burnham CJ, Xantheas SS. J Chem Phys. 2002;116:1479. [Google Scholar]
- 371.Burnham CJ, Xantheas SS. J Chem Phys. 2002;116:1500. [Google Scholar]
- 372.Burnham CJ, Xantheas SS. J Chem Phys. 2002;116:5115. [Google Scholar]
- 373.Xantheas SS, Burnham CJ, Harrison RJ. J Chem Phys. 2002;116:1493. [Google Scholar]
- 374.Kong Y. PhD thesis. Washington University Medical School; 1997. Multipole Electrostatic Methods for Protein Modeling with Reaction Field Treatment. [Google Scholar]
- 375.Thole BT. Chem Phys. 1981;59:341. [Google Scholar]
- 376.Donchev AG, Ozrin VD, Subbotin MV, Tarasov OV, Tarasov VI. Proc Natl Acad Sci USA. 2005;102:7829. doi: 10.1073/pnas.0502962102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Donchev AG, Galkin NG, Illarionov AA, Khoruzhii OV, Olevanov MA, Ozrin VD, Pereyaslavets LB, Tarasov VI. J Comp Chem. 2008;29:1242. doi: 10.1002/jcc.20884. [DOI] [PubMed] [Google Scholar]
- 378.Defusco A, Schofield DP, Jordan KD. Mol Phys. 2007;105:2681. [Google Scholar]
- 379.Kumar R, Wang FF, Jenness GR, Jordan KD. J Chem Phys. 2010;132:014309. doi: 10.1063/1.3276460. [DOI] [PubMed] [Google Scholar]
- 380.Lefohn AE, Ovchinnikov M, Voth GA. J Phys Chem B. 2001;105:6628. [Google Scholar]
- 381.Jeon J, Lefohn AE, Voth GA. J Chem Phys. 2003;118:7504. [Google Scholar]
- 382.Rick SW, Stuart SJ. Rev Comp Ch. 2002;18:89. [Google Scholar]
- 383.Applequist J, Carl JR, Fung KK. J Am Chem Soc. 1972;94:2952. [Google Scholar]
- 384.Stout JM, Dykstra CE. J Am Chem Soc. 1995;117:5127. [Google Scholar]
- 385.Stout JM, Dykstra CE. J Phys Chem A. 1998;102:1576. [Google Scholar]
- 386.Olson ML, Sundberg KR. J Chem Phys. 1978;69:5400. [Google Scholar]
- 387.Applequist J. J Phys Chem. 1993;97:6016. [Google Scholar]
- 388.Stern H, Kaminski G, Banks J, Zhou RH, Berne B, Friesner R. J Phys Chem B. 1999;103:4730. [Google Scholar]
- 389.Karlström G. Theor Chim Acta. 1982;60:535. [Google Scholar]
- 390.Stone AJ. Chem Phys Lett. 1993;211:101. [Google Scholar]
- 391.Mo Y, Gao J, Peyerimhoff SD. J Chem Phys. 2000;112:5530. [Google Scholar]
- 392.Mo Y, Bao P, Gao J. Phys Chem Chem Phys. 2011;13:6760. doi: 10.1039/c0cp02206c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Xie W, Orozco M, Truhlar DG, Gao J. J Chem Theory Comput. 2009;5:459. doi: 10.1021/ct800239q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Isegawa M, Gao J, Truhlar DG. J Chem Phys. 2011;135:084107. doi: 10.1063/1.3624890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Khaliullin RZ, Cobar EA, Lochan RC, Bell AT, Head-Gordon M. J Phys Chem A. 2007;111:8753. doi: 10.1021/jp073685z. [DOI] [PubMed] [Google Scholar]
- 396.Bagus PS, Hermann K, Bauschlicher CW. J Chem Phys. 1984;80:1966. [Google Scholar]
- 397.Stevens WJ, Fink WH. Chem Phys Lett. 1987;139:15. [Google Scholar]
- 398.Wu JC, Piquemal JP, Chaudret R, Reinhardt P, Ren PY. J Chem Theory Comput. 2010;6:2059. doi: 10.1021/ct100091j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Zhao Z, Rogers DM, Beck TL. J Chem Phys. 2010;132:014502. doi: 10.1063/1.3283900. [DOI] [PubMed] [Google Scholar]
- 400.Clavaguera-Sarrio C, Brenner V, Hoyau S, Marsden CJ, Millie P, Dognon JP. J Phys Chem B. 2003;107:3051. [Google Scholar]
- 401.Hagberg D, Karlström G, Roos BO, Gagliardi L. J Am Chem Soc. 2005;127:14250. doi: 10.1021/ja0526719. [DOI] [PubMed] [Google Scholar]
- 402.Uchimaru T, Korchowiec J, Tsuzuki S, Matsumura K, Kawahara S. Chem Phys Lett. 2000;318:203. [Google Scholar]
- 403.Cohen AJ, Mori-Sánchez P, Yang W. Science. 2008;321:792. doi: 10.1126/science.1158722. [DOI] [PubMed] [Google Scholar]
- 404.Mori-Sánchez P, Cohen AJ, Yang W. Phys Rev Lett. 2008;100:146401. doi: 10.1103/PhysRevLett.100.146401. [DOI] [PubMed] [Google Scholar]
- 405.Jorgensen W, Chandrasekhar J, Madura J, Klein M. J Chem Phys. 1983;79:926. [Google Scholar]
- 406.Abascal JL, Vega C. J Chem Phys. 2005;123:234505. doi: 10.1063/1.2121687. [DOI] [PubMed] [Google Scholar]
- 407.Lee AJ, Rick SW. J Chem Phys. 2011;134:184507. doi: 10.1063/1.3589419. [DOI] [PubMed] [Google Scholar]
- 408.Norberg J, Nilsson L. Q Rev Biophys. 2003;36:257. doi: 10.1017/s0033583503003895. [DOI] [PubMed] [Google Scholar]
- 409.McDowell SE, Nadia S, Sponer J, Walter NG. Biopolymers. 2007;85:169. doi: 10.1002/bip.20620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Orozco M, Noy A, Perez A. Curr Opin Struct Bio. 2008;18:185. doi: 10.1016/j.sbi.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 411.Mackerell AD. J Comp Chem. 2004;25:1584. doi: 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- 412.Horn HW, Swope WC, Pitera JW, Madura JD, Dick TJ, Hura GL, Head-Gordon T. J Chem Phys. 2004;120:9665. doi: 10.1063/1.1683075. [DOI] [PubMed] [Google Scholar]
- 413.Mahoney M, Jorgensen W. J Chem Phys. 2000;112:8910. [Google Scholar]
- 414.Perera L, Berkowitz ML. J Chem Phys. 1991;95:1954. [Google Scholar]
- 415.Perera L, Berkowitz ML. J Chem Phys. 1992;96:8288. [Google Scholar]
- 416.Perera L, Berkowitz ML. J Chem Phys. 1993;99:4236. [Google Scholar]
- 417.Perera L, Berkowitz ML. J Chem Phys. 1993;99:4222. [Google Scholar]
- 418.Perera L, Essmann U, Berkowitz ML. J Chem Phys. 1995;102:450. [Google Scholar]
- 419.Yeh IC, Perera L, Berkowitz ML. Chem Phys Lett. 1997;264:31. [Google Scholar]
- 420.Tobias DJ, Jungwirth P, Parrinello M. J Chem Phys. 2001;114:7036. [Google Scholar]
- 421.Valiev M, D’Auria R, Tobias DJ, Garrettt BC. J Phys Chem A. 2009;113:8823. doi: 10.1021/jp903625k. [DOI] [PubMed] [Google Scholar]
- 422.D’auria R, Tobias DJ, Gerber RB, Miller Y, Xantheas SS. Abstracts of Papers of the American Chemical Society. 2006:232. [Google Scholar]
- 423.Annapureddy HVR, Dang LX. J Phys Chem B. 2012;116:7492. doi: 10.1021/jp301859z. [DOI] [PubMed] [Google Scholar]
- 424.Markovich G, Giniger R, Levin M, Cheshnovsky O. J Chem Phys. 1991;95:9416. [Google Scholar]
- 425.Markovich G, Pollack S, Giniger R, Cheshnovsky O. J Chem Phys. 1994;101:9344. [Google Scholar]
- 426.Raugei S, Klein ML. J Chem Phys. 2002;116:196. [Google Scholar]
- 427.Berendsen HJC, Grigera JR, Straatsma TP. J Chem Phys. 1987;91:6269. [Google Scholar]
- 428.Grossfield A, Ren PY, Ponder JW. J Am Chem Soc. 2003;125:15671. doi: 10.1021/ja037005r. [DOI] [PubMed] [Google Scholar]
- 429.Jiao D, King C, Grossfield A, Darden TA, Ren PY. J Phys Chem B. 2006;110:18553. doi: 10.1021/jp062230r. [DOI] [PubMed] [Google Scholar]
- 430.Piquemal JP, Perera L, Cisneros GA, Ren PY, Pedersen LG, Darden TA. J Chem Phys. 2006;126:054511. doi: 10.1063/1.2234774. [DOI] [PubMed] [Google Scholar]
- 431.Sponer J, Sabat M, Burda JV, Doody AM, Leszczynski J, Hobza P. J Biomol Struc Dynamics. 1998;16:139. doi: 10.1080/07391102.1998.10508235. [DOI] [PubMed] [Google Scholar]
- 432.Zhang J, Yang W, Piquemal JP, Ren P. J Chem Theory Comput. 2012;8:1314. doi: 10.1021/ct200812y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Clavaguera C, Sansot E, Calvo F, Dognon JP. J Phys Chem B. 2006;110:12848. doi: 10.1021/jp062277c. [DOI] [PubMed] [Google Scholar]
- 434.Marjolin A, Gourlaouen C, Clavaguera C, Ren P, Wu J, Gresh N, Dognon JP, Piquemal JP. Theor Chem Acc. 2012;131:1. [Google Scholar]
- 435.Jiao D, Golubkov PA, Darden TA, Ren P. Proc Natl Acad Sci USA. 2008;105:6290. doi: 10.1073/pnas.0711686105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Talhout R, Villa A, Mark AE, Engberts JBFN. J Am Chem Soc. 2003;125:10570. doi: 10.1021/ja034676g. [DOI] [PubMed] [Google Scholar]
- 437.Mobley DL, Graves AP, Chodera JDC, MA, Shoichet BK, Dill KA. J Mol Biol. 2007;371:1118. doi: 10.1016/j.jmb.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Oostenbrink C, Villa A, Mark AE, Van Gunsteren WF. J Comp Chem. 2004;25:1656. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- 439.Halgren TA, Damm W. Curr Opin Struct Bio. 2001;11:236. doi: 10.1016/s0959-440x(00)00196-2. [DOI] [PubMed] [Google Scholar]
- 440.Lopes PEM, Lamoureux G, Roux B, MacKerell AD. J Phys Chem B. 2007;111:2873. doi: 10.1021/jp0663614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Warshel A, Kato M, Pisliakov AV. J Chem Theory Comput. 2007;3:2034. doi: 10.1021/ct700127w. [DOI] [PubMed] [Google Scholar]
- 442.Warshel A, Levitt M. J Mol Biol. 1976;103:227. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 443.Gresh N, Pullman A, Claverie P. Int J Quantum Chem. 1985;28:757. [Google Scholar]
- 444.Gresh N. J Chim Phys Phys- Chim Biol. 1997;94:1365. [Google Scholar]
- 445.Piquemal JP, Williams-Hubbard B, Fey N, Deeth RJ, Gresh N, Giessner-Prettre C. J Comp Chem. 2003;24:1963. doi: 10.1002/jcc.10354. [DOI] [PubMed] [Google Scholar]
- 446.Guo H, Gresh N, Roques BP, Salahub DR. J Phys Chem B. 2000;104:9746. [Google Scholar]
- 447.Gresh N, Policar C, Giessner-Prettre C. J Phys Chem B. 2002;106:11A. [Google Scholar]
- 448.Ledecq M, Lebon F, Durant F, Giessner-Prettre C, Marquez A, Gresh N. J Phys Chem B. 2003;107:10640. [Google Scholar]
- 449.Roux C, Gresh N, Perera LE, Piquemal JP, Salmon L. J Comp Chem. 2007;28:938. doi: 10.1002/jcc.20586. [DOI] [PubMed] [Google Scholar]
- 450.Roux C, Bhatt F, Foret J, de Courcy B, Gresh N, Piquemal JP, Jeffery CJ, Salmon L. Proteins: Struct Funct Bioinf. 2011;79:203. doi: 10.1002/prot.22873. [DOI] [PubMed] [Google Scholar]
- 451.de Courcy B, Pedersen LG, Parisel O, Gresh N, Silvi B, Pilme J, Piquemal JP. J Chem Theory Comput. 2010;6:1048. doi: 10.1021/ct100089s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Gresh N, Audiffren N, Piquemal JP, de Ruyck J, Ledecq M, Wouters J. J Phys Chem B. 2010;114:4884. doi: 10.1021/jp907629k. [DOI] [PubMed] [Google Scholar]
- 453.de Courcy B, Piquemal JP, Garbay C, Gresh N. J Am Chem Soc. 2010;132:3312. doi: 10.1021/ja9059156. [DOI] [PubMed] [Google Scholar]
- 454.Engkvist O, Åstrand PO, Karlström G. Chem Rev. 2000;100:4087. doi: 10.1021/cr9900477. [DOI] [PubMed] [Google Scholar]
- 455.Carignano MA, Karlström G, Linse P. J Phys Chem B. 1997;101:1142. [Google Scholar]
- 456.Brdarski S, Åstrand PO, Karlström G. Theo Chem Acc. 2000;105:7. [Google Scholar]
- 457.Stenhammar J, Linse P, Malmqvist PA, Karlström G. J Chem Phys. 2009;130:124521. doi: 10.1063/1.3100312. [DOI] [PubMed] [Google Scholar]
- 458.Dang LX, Rice JE, Caldwell J, Kollman PA. J Am Chem Soc. 1991;113:2481. [Google Scholar]
- 459.Wang J, Cieplak P, Cai Q, Hsieh MJ, Wang JM, Duan Y, Luo R. J Phys Chem B. 2012;116:7999. doi: 10.1021/jp212117d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Wang JM, Cieplak P, Li J, Cai Q, Hsieh MJ, Luo R, Duan Y. J Phys Chem B. 2012;116:7088. doi: 10.1021/jp3019759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Wang JM, Cieplak P, Li J, Hou TJ, Luo R, Duan Y. J Phys Chem B. 2011;115:3091. doi: 10.1021/jp112133g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Wang JM, Cieplak P, Li J, Wang J, Cai Q, Hsieh MJ, Lei HX, Luo R, Duan Y. J Phys Chem B. 2011;115:3100. doi: 10.1021/jp1121382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Wang ZX, Zhang W, Wu C, Lei HX, Cieplak P, Duan Y. J Comp Chem. 2006;27:994. doi: 10.1002/jcc.20386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Kaminski GA, Stern HA, Berne BJ, Friesner RA, Cao YXX, Murphy RB, Zhou RH, Halgren TA. J Comp Chem. 2002;23:1515. doi: 10.1002/jcc.10125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Kaminski GA, Stern HA, Berne BJ, Friesner RA. J Phys Chem A. 2004;108:621. [Google Scholar]
- 466.Kaminski GA, Ponomarev SY, Liu AB. J Chem Theory Comput. 2009;5:2935. doi: 10.1021/ct900409p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Ponomarev SY, Kaminski GA. J Chem Theory Comput. 2011;7:1415. doi: 10.1021/ct1007197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Rasmussen TD, Ren PY, Ponder JW, Jensen F. Int J Quantum Chem. 2007;107:1390. [Google Scholar]
- 469.Ren P, Wu C, Ponder JW. J Chem Theory Comput. 2011;7:3143. doi: 10.1021/ct200304d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Shi Y, Wu C, Ponder JW, Ren P. J Comp Chem. 2011;32:967. doi: 10.1002/jcc.21681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Wu JC, Chattre G, Ren P. Theor Chem Acc. 2012;131:1138. doi: 10.1007/s00214-012-1138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Jiao D, Zhang JJ, Duke RE, Li GH, Schnieders MJ, Ren PY. J Comp Chem. 2009;30:1701. doi: 10.1002/jcc.21268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Yang T, Wu JC, Yan C, Wang Y, Luo R, Gonzales MB, Dalby KN, Ren P. Proteins-Structure Function and Genetics. 2011;79:1940. doi: 10.1002/prot.23018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Shi Y, Zhu CZ, Martin SF, Ren P. J Phys Chem B. 2012;116:1716. doi: 10.1021/jp210265d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Zhang J, Shi Y, Ren P. Protein-Ligand Interactions. Wiley-VCH Verlag GmbH and Co. KGaA; 2012. p. 99. [Google Scholar]
- 476.Clavaguera C, Pollet R, Soudan JM, Brenner V, Dognon JP. J Phys Chem B. 2005;109:7614. doi: 10.1021/jp051032h. [DOI] [PubMed] [Google Scholar]
- 477.De Miranda Tomasio S, Walsh TR. Mol Phys. 2007;105:221. [Google Scholar]
- 478.Jiang H, Jordan KD, Taylor CE, Sk J Phys Chem B. 2007;111:6486. doi: 10.1021/jp068505k. [DOI] [PubMed] [Google Scholar]
- 479.Cui J, Liu H, Jordan KD. J Phys Chem B. 2006;110:18872. doi: 10.1021/jp056416m. [DOI] [PubMed] [Google Scholar]
- 480.Liang T, Walsh TR. Phys Chem Chem Phys. 2006;8:4410. doi: 10.1039/b608672a. [DOI] [PubMed] [Google Scholar]
- 481.Liang T, Walsh TR. Mol Sim. 2007;33:337. [Google Scholar]
- 482.Kaminsky J, Jensen F. J Chem Theory Comput. 2007;3:1774. doi: 10.1021/ct700082f. [DOI] [PubMed] [Google Scholar]
- 483.Bostick DL, Brooks rCL. Proc Natl Acad Sci USA. 2007;104:9260. doi: 10.1073/pnas.0700554104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Bostick DL, Brooks rCL. J Am Chem Soc. 2010;132:13185. doi: 10.1021/ja106197e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Jiang JL, Wu YB, Wang ZX, Wu C. J Chem Theory Comput. 2010;6:1199. [Google Scholar]
- 486.Ponder JW. TINKER: Software Tools for Molecular Design, Version 4.2. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Friedrichs MS, Eastman P, Vaidyanathan V, Houston M, Legrand S, Beberg AL, Ensign DL, Bruns CM, Pande VS. J Comp Chem. 2009;30:864. doi: 10.1002/jcc.21209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Whitfield TW, Varma S, Harder E, Lamoureux G, Rempe SB, Roux B. J Chem Theory Comput. 2007;3:2068. doi: 10.1021/ct700172b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Anisimov VM, Vorobyov IV, Roux B, MacKerell AD. J Chem Theory Comput. 2007;3:1927. doi: 10.1021/ct700100a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Lopes PEM, Zhu X, Lau A, Roux B, MacKerell AD. Biophys J. 2011;100:612. [Google Scholar]
- 491.Baker CM, Lopes PEM, Zhu X, Roux B, MacKerell AD. J Chem Theory Comput. 2010;6:1181. doi: 10.1021/ct9005773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Jiang W, Hardy DJ, Phillips JC, MacKerell AD, Schulten K, Roux B. J Phys Chem Lett. 2011;2:87. doi: 10.1021/jz101461d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Patel S, Brooks CL. J Comp Chem. 2004;25:1. doi: 10.1002/jcc.10355. [DOI] [PubMed] [Google Scholar]
- 494.Patel S, Mackerell AD, Brooks CL. J Comp Chem. 2004;25:1504. doi: 10.1002/jcc.20077. [DOI] [PubMed] [Google Scholar]
- 495.Warren GL, Patel S. J Phys Chem B. 2008;112:11679. doi: 10.1021/jp8038835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Patel S, Davis JE, Bauer BA. J Am Chem Soc. 2009;131:13890. doi: 10.1021/ja902903m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Bauer BA, Patel S. J Chem Phys. 2009;131:084709. doi: 10.1063/1.3200869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Bauer BA, Lucas TR, Meninger DJ, Patel S. Chem Phys Lett. 2011;508:289. doi: 10.1016/j.cplett.2011.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Gao J, Habibollazadeh D, Shao L. J Phys Chem. 1995;99:16460. [Google Scholar]
- 500.Xie WS, Pu JZ, MacKerell AD, Gao JL. J Chem Theory Comput. 2007;3:1878. doi: 10.1021/ct700146x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Xie W, Song L, Truhlar DG, Gao J. J Chem Phys. 2008;128:234108. doi: 10.1063/1.2936122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Xie W, Gao J. J Chem Theory Comput. 2007;3:1890. doi: 10.1021/ct700167b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Song L, Han J, Lin YL, Xie W, Gao J. J Phys Chem A. 2009;113:11656. doi: 10.1021/jp902710a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Borodin O, Smith GD. J Phys Chem B. 2003;107:6801. [Google Scholar]
- 505.Borodin O, Douglas R, Smith GA, Trouw F, Petrucci S. J Phys Chem B. 2003;107:6813. [Google Scholar]
- 506.Borodin O, Smith GD, Douglas R. J Phys Chem B. 2003;107:6824. [Google Scholar]
- 507.Chen B, Xing JH, Siepmann JI. J Phys Chem B. 2000;104:2391. [Google Scholar]
- 508.Chen B, Siepmann JI, Klein ML. J Phys Chem A. 2005;109:1137. doi: 10.1021/jp0463722. [DOI] [PubMed] [Google Scholar]
- 509.Rivera JL, Starr FW, Paricaud P, Cummings PT. J Chem Phys. 2006;125:094712. doi: 10.1063/1.2345063. [DOI] [PubMed] [Google Scholar]
- 510.Tao Z, Cummings PT. Mol Sim. 2007;33:1255. [Google Scholar]
- 511.Borodin O. J Phys Chem B. 2009;113:11463. doi: 10.1021/jp905220k. [DOI] [PubMed] [Google Scholar]
- 512.Maple JR, Cao YX, Damm WG, Halgren TA, Kaminski GA, Zhang LY, Friesner RA. J Chem Theory Comput. 2005;1:694. doi: 10.1021/ct049855i. [DOI] [PubMed] [Google Scholar]
- 513.Schnieders MJ, Baker NA, Ren P, Ponder JW. J Chem Phys. 2007;126:124114. doi: 10.1063/1.2714528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Schnieders MJ, Ponder JW. J Chem Theory Comput. 2007;3:2083. doi: 10.1021/ct7001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Constanciel R, Contreras R. Theor Chim Acta. 1984;65:1. [Google Scholar]
- 516.Truchon JF, Nicholls A, Iftimie RI, Roux B, Bayly CI. J Chem Theory Comput. 2008;4:1480. doi: 10.1021/ct800123c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Truchon JF, Nicholls A, Roux B, Iftimie RI, Bayly CI. J Chem Theory Comput. 2009;5:1785. doi: 10.1021/ct900029d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Hu H, Yang W. Ann Rev Phys Chem. 2008;59:573. doi: 10.1146/annurev.physchem.59.032607.093618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Senn HM, Thiel W. Angew Chem Int Ed Engl. 2009;48:1198. doi: 10.1002/anie.200802019. [DOI] [PubMed] [Google Scholar]
- 520.Alden RG, Parson WW, Chu ZT, Warshel A. J Am Chem Soc. 1995;117:12284. [Google Scholar]
- 521.Warshel A, Papazyan A. Curr Opin Struct Bio. 1998;8:211. doi: 10.1016/s0959-440x(98)80041-9. [DOI] [PubMed] [Google Scholar]
- 522.Kritayakornupong C, Plankensteiner K, Rode BM. Chem Phys Lett. 2003;371:438. [Google Scholar]
- 523.Schaefer P, Riccardi D, Cui Q. J Chem Phys. 2005;123:014905. doi: 10.1063/1.1940047. [DOI] [PubMed] [Google Scholar]
- 524.Benighaus T, Thiel W. J Chem Theory Comput. 2009;5:3114. doi: 10.1021/ct900437b. [DOI] [PubMed] [Google Scholar]
- 525.Benighaus T, Thiel W. J Chem Theory Comput. 2011;7:238. doi: 10.1021/ct1005455. [DOI] [PubMed] [Google Scholar]
- 526.Aleksandrov A, Field M. Phys Chem Chem Phys. 2011;13:10503. doi: 10.1039/c0cp02828b. [DOI] [PubMed] [Google Scholar]
- 527.Gao J, Alhambra C. J Chem Phys. 1997;107:1212. [Google Scholar]
- 528.Gao J. J Phys Chem. 1992;96:6432. [Google Scholar]
- 529.Walker RC, Crowley MF, Case DA. J Comp Chem. 2008;29:1019. doi: 10.1002/jcc.20857. [DOI] [PubMed] [Google Scholar]
- 530.Zhang P, Truhlar DG, Gao J. Phys Chem Chem Phys. 2012;14:7821. doi: 10.1039/c2cp23758j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Dehez F, Martins-Costa M, Rinaldi D, Millot C. J Chem Phys. 2005;122:234503. doi: 10.1063/1.1931667. [DOI] [PubMed] [Google Scholar]
- 532.Laino T, Mohamed F, Laio A, Parrinello M. J Chem Theo Comp. 2006;2:1370. doi: 10.1021/ct6001169. [DOI] [PubMed] [Google Scholar]
- 533.Laino T, Mohamed F, Laio A, Parrinello M. J Chem Theo Comp. 2005;1:1176. doi: 10.1021/ct050123f. [DOI] [PubMed] [Google Scholar]
- 534.Das D, Eurenius KP, Billings EM, Sherwood P, Chatfield DC, Hodoscek M, Brooks BR. J Chem Phys. 2003;117:10534. [Google Scholar]
- 535.Wang B, Truhlar DG. J Chem Theory Comput. 2010;6:359. doi: 10.1021/ct900366m. [DOI] [PubMed] [Google Scholar]
- 536.Lin H, Truhlar DG. J Phys Chem A. 2005;109:3991. doi: 10.1021/jp0446332. [DOI] [PubMed] [Google Scholar]
- 537.Wang B, Truhlar DG. Phys Chem Chem Phys. 2011;13:10556. doi: 10.1039/c0cp02850a. [DOI] [PubMed] [Google Scholar]
- 538.Zhang Y, Lin H, Truhlar DG. J Chem Theory Comput. 2007;3:1378. doi: 10.1021/ct7000107. [DOI] [PubMed] [Google Scholar]
- 539.Biswas PK, Gogonea V. J Chem Phys. 2008;129:154108. doi: 10.1063/1.2992527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Cisneros GA, Piquemal JP, Darden TA. J Phys Chem B. 2006;110:13682. doi: 10.1021/jp062768x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Fujimoto K, Yang W. J Chem Phys. 2008;129:054102. doi: 10.1063/1.2958257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Hu X, Jin Y, Zeng X, Hu H, Yang W. Phys Chem Chem Phys. 2012;14:7700. doi: 10.1039/c2cp23714h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Wesolowski TA, Warshel A. J Phys Chem. 1993;97:8050. [Google Scholar]
- 544.Wesolowski T, Muller RP, Warshel A. J Phys Chem. 1996;100:15444. [Google Scholar]
- 545.Dulak M, Kaminski JW, Wesolowski TA. J Chem Theo Comp. 2007;3:735. doi: 10.1021/ct600367t. [DOI] [PubMed] [Google Scholar]
- 546.Wesolowski TA. Phys Rev A. 2008;77:012504. [Google Scholar]
- 547.Fradelos G, Wesolowski TA. J Chem Theo Comp. 2011;7:213. doi: 10.1021/ct100415h. [DOI] [PubMed] [Google Scholar]
- 548.Zhou X, Kaminski JW, Wesolowski TA. Phys Chem Chem Phys. 2011;13:10565. doi: 10.1039/c0cp02874f. [DOI] [PubMed] [Google Scholar]
- 549.Ma Q, Izaguirre JA. Multiscale Model Simul. 2004;2:1. [Google Scholar]
- 550.Venturoli M, Sperotto M, Kranenburg M, Smit B. Phys Rep. 2006;437:1. [Google Scholar]
- 551.Voth GA, editor. Coarse-Graining of Condensed Phase and Biomolecular Systems. CRC Press; Boca Raton, FL: 2008. [Google Scholar]
- 552.Praprotnik M, Site LD, Kremer K. Ann Rev Phys Chem. 2008;59:545. doi: 10.1146/annurev.physchem.59.032607.093707. [DOI] [PubMed] [Google Scholar]
- 553.Murtola T, Bunker A, Vattulainen I, Deserno M, Karttunen M. Phys Chem Chem Phys. 2009;11:1869. doi: 10.1039/b818051b. [DOI] [PubMed] [Google Scholar]
- 554.Peter C, Kremer K. Faraday Disc. 2010;144:9. doi: 10.1039/b919800h. [DOI] [PubMed] [Google Scholar]
- 555.Lyman E, Zuckerman DM. J Chem Theory Comput. 2006;2:656. doi: 10.1021/ct050337x. [DOI] [PubMed] [Google Scholar]
- 556.Liu P, Shi Q, Lyman E, Voth GA. J Chem Phys. 2008;129:114103. doi: 10.1063/1.2976663. [DOI] [PubMed] [Google Scholar]
- 557.Rzepiela AJ, Schäfer LV, Goga N, Risselada HJ, De Vries AH, Marrink SJ. J Comp Chem. 2010;31:1333. doi: 10.1002/jcc.21415. [DOI] [PubMed] [Google Scholar]
- 558.Poblete S, Praprotnik M, Kremer K, Delle Site L. J Chem Phys. 2010;132:114101. doi: 10.1063/1.3357982. [DOI] [PubMed] [Google Scholar]
- 559.Stansfeld PJ, Sansom MS. J Chem Theory Comput. 2011;7:1157. doi: 10.1021/ct100569y. [DOI] [PubMed] [Google Scholar]
- 560.Riniker S, van Gunsteren WF. J Chem Phys. 2012;137:044120. doi: 10.1063/1.4739068. [DOI] [PubMed] [Google Scholar]
- 561.Henderson RL. Phys Lett. 1974;49A:197. [Google Scholar]
- 562.Chayes JT, Chayes L, Lieb EH. Comm Math Phys. 1984;93:57. [Google Scholar]
- 563.Chayes JT, Chayes L. J Stat Phys. 1984;36:471. [Google Scholar]
- 564.Hohenberg P, Kohn W. Phys Rev. 1964;136:B864. [Google Scholar]
- 565.Lyubartsev AP, Laaksonen A. Phys Rev E. 1995;52:3730. doi: 10.1103/physreve.52.3730. [DOI] [PubMed] [Google Scholar]
- 566.Reith D, Meyer H, Mu F, Plathe FM. Macromolecules. 2001;34:2335. [Google Scholar]
- 567.Izvekov S, Voth GA. J Chem Phys. 2005;123:134105. doi: 10.1063/1.2038787. [DOI] [PubMed] [Google Scholar]
- 568.Izvekov S, Swanson JMJ, Voth GA. J Phys Chem B. 2008;112:4711. doi: 10.1021/jp710339n. [DOI] [PubMed] [Google Scholar]
- 569.Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH. J Phys Chem B. 2007;111:7812. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- 570.Ollila OHS, Louhivuori M, Marrink SJ, Vattulainen I. Biophys J. 2011;100:1651. doi: 10.1016/j.bpj.2011.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Baoukina S, Monticelli L, Risselada HJ, Marrink SJ, Tieleman DP. Proc Natl Acad Sci USA. 2008;105:10803. doi: 10.1073/pnas.0711563105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572.Murtola T, Vuorela TA, Hyvönen MT, Marrink SJ, Karttunen M, Vattulainen I. Soft Matter. 2011;7:8135. [Google Scholar]
- 573.Hoogerbrugge PJ, Koelman JMVA. Europhys Lett. 1992;19:155. [Google Scholar]
- 574.Español P, Warren P. Europhys Lett. 1995;30:191. [Google Scholar]
- 575.Forrest BM, Suter UW. J Chem Phys. 1995;102:7256. [Google Scholar]
- 576.González-Melchor M, Mayoral E, Velázquez ME, Alejandre J. J Chem Phys. 2006;125:224107. doi: 10.1063/1.2400223. [DOI] [PubMed] [Google Scholar]
- 577.Yesylevskyy SO, Schäfer LV, Sengupta D, Marrink SJ. PLoS Comput Biol. 2010;6:e1000810. doi: 10.1371/journal.pcbi.1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Dias CL, Ala-Nissila T, Grant M, Karttunen M. J Chem Phys. 2009;131:054505. doi: 10.1063/1.3183935. [DOI] [PubMed] [Google Scholar]
- 579.Wu Z, Cui Q, Yethiraj A. J Phys Chem B. 2010;114:10524. doi: 10.1021/jp1019763. [DOI] [PubMed] [Google Scholar]
- 580.Nistor RA, Polihronov JG, Müser MH, Mosey NJ. J Chem Phys. 2006;125:094108. doi: 10.1063/1.2346671. [DOI] [PubMed] [Google Scholar]
- 581.Mortier WJ, Van Genechten K, Gasteiger J. J Am Chem Soc. 1985;107:829. [Google Scholar]
- 582.Rappe AK, Casewit CJ, Colwell KS, Goddard WA, Skiff WM. J Am Chem Soc. 1992;114:10024. [Google Scholar]
- 583.van Duin ACT, Baas JMA, van de Graaf B. J Chem Soc, Faraday Trans. 1994;90:2881. [Google Scholar]
- 584.York D, Yang W. J Chem Phys. 1996;104:159. [Google Scholar]
- 585.Yang ZZ, Wu Y, Zhao DX. J Chem Phys. 2004;120:2541. doi: 10.1063/1.1640345. [DOI] [PubMed] [Google Scholar]
- 586.Zhao DX, Yu L, Gong LD, Liu C, Yang ZZ. J Chem Phys. 2011;134:194115. doi: 10.1063/1.3590718. [DOI] [PubMed] [Google Scholar]
- 587.Lucas TR, Bauer BA, Davis JE, Patel S. J Comp Chem. 2012;33:141. doi: 10.1002/jcc.21927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Chelli R, Procacci P, Righini R, Califano S. J Chem Phys. 1999;111:8569. [Google Scholar]
- 589.Verstraelen T, Van Speybroeck VMW. J Chem Phys. 2009;131:044127. doi: 10.1063/1.3187034. [DOI] [PubMed] [Google Scholar]
- 590.Mongan J, Case D, McCammon J. J Comp Chem. 2004;25:2038. doi: 10.1002/jcc.20139. [DOI] [PubMed] [Google Scholar]
- 591.Mongan J, Case D. Curr Opin Struct Bio. 2005;15:157. doi: 10.1016/j.sbi.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 592.Khandogin J, Brooks C. Biophys J. 2005;89:141. doi: 10.1529/biophysj.105.061341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Vorobjev YN, Vila JA, Scheraga HA. J Phys Chem B. 2008;112:11122. doi: 10.1021/jp709969n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594.Williams SL, Blachly PG, McCammon JA. Proteins: Struct Funct Bioinf. 2011;79:3381. doi: 10.1002/prot.23136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595.Schnieders MJ, Fenn TD, Pande VS, Brunger AT. Acta Cryst D. 2009;65:952. doi: 10.1107/S0907444909022707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Fenn TD, Schnieders MJ, Brunger AT, Pande VS. Biophys J. 2010;98:2984. doi: 10.1016/j.bpj.2010.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Schnieders MJ, Fenn TD, Pande VS. J Chem Theory Comput. 2011;7:1141. doi: 10.1021/ct100506d. [DOI] [PubMed] [Google Scholar]
- 598.Fenn TD, Schnieders MJ. Acta Cryst D. 2011;67:957. doi: 10.1107/S0907444911039060. [DOI] [PubMed] [Google Scholar]
- 599.Drew H, Dickerson R. J Mol Biol. 1981;151:535. doi: 10.1016/0022-2836(81)90009-7. [DOI] [PubMed] [Google Scholar]
- 600.Shaw DE, Maragakis P, Lindorff-Larsen K, Piana S, Dror RO, Eastwood MP, Bank JA, Jumper JM, Salmon JK, Shan Y, Wriggers W. Science. 2010;330:341. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- 601.Stone JE, Phillips JC, Freddolino PL, Hardy DJ, Trabuco LG, Schulten K. J Comp Chem. 2007;28:2618. doi: 10.1002/jcc.20829. [DOI] [PubMed] [Google Scholar]
- 602.Raval A, Piana S, Eastwood MP, Dror RO, Shaw DE. Proteins: Struct Funct Genet. 2012;80:2071. doi: 10.1002/prot.24098. [DOI] [PubMed] [Google Scholar]
- 603.Huber T, Torda AE, van Gunsteren WF. J Comput Aided Mol Des. 1994;8:695. doi: 10.1007/BF00124016. [DOI] [PubMed] [Google Scholar]
- 604.Darve E, Pohorille A. J Chem Phys. 2001;115:9169. [Google Scholar]
- 605.Wang F, Landau D. Phys Rev Lett. 2001;115:2050. doi: 10.1103/PhysRevLett.86.2050. [DOI] [PubMed] [Google Scholar]
- 606.Laio A, Parrinello M. Proc Natl Acad Sci USA. 2002;99:12562. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Iannuzzi M, Laio A, Parrinello M. Phys Rev Lett. 2003;90:238302. doi: 10.1103/PhysRevLett.90.238302. [DOI] [PubMed] [Google Scholar]
- 608.Ensing B, Laio A, Parrinello M, Klein M. J Phys Chem B. 2005;109:6676. doi: 10.1021/jp045571i. [DOI] [PubMed] [Google Scholar]
- 609.Lelièvre T, Rousset M, Stoltz G. J Chem Phys. 2007;126:134111. doi: 10.1063/1.2711185. [DOI] [PubMed] [Google Scholar]
- 610.Babin V, Roland C, Sagui C. J Chem Phys. 2008;128:134101. doi: 10.1063/1.2844595. [DOI] [PubMed] [Google Scholar]
- 611.Hansmann U. Chem Phys Lett. 1997;281:140. [Google Scholar]
- 612.Sugita Y, Okamoto Y. Chem Phys Lett. 1999;314:141. [Google Scholar]
- 613.Sugita Y, Kitao A, Okamoto Y. J Phys Chem. 2000;113:6042. [Google Scholar]
- 614.Raiteri P, Laio A, Gervasio FL, Micheletti C, Parrinello M. J Phys Chem. 2006;110:3533. doi: 10.1021/jp054359r. [DOI] [PubMed] [Google Scholar]
- 615.Babin V, Sagui C. J Chem Phys. 2010;132:104108. doi: 10.1063/1.3355621. [DOI] [PubMed] [Google Scholar]
- 616.Rosso L, Tuckerman M. Mol Sim. 2002;28:91. [Google Scholar]
- 617.Abrams JB, Rosso L, Tuckerman ME. J Chem Phys. 2006;125:074115. doi: 10.1063/1.2232082. [DOI] [PubMed] [Google Scholar]