

Abstract
Treatment of BRAF-mutant melanoma with combined dabrafenib and trametinib, which target RAF and the downstream MEK1 and MEK2 kinases, respectively, improves progression-free survival and response rates compared with dabrafenib monotherapy (1). Mechanisms of clinical resistance to combined RAF/MEK inhibition are unknown. We performed whole exome and transcriptome sequencing on pre-treatment and drug-resistant tumors from five patients with acquired resistance to dabrafenib/trametinib. In three of these patients, we identified additional MAP kinase pathway alterations in the resistant tumor that were not detected in the pre-treatment tumor, including a novel activating mutation in MEK2 (MEK2Q60P). MEK2Q60P conferred resistance to combined RAF/MEK inhibition in vitro, but remained sensitive to inhibition of the downstream kinase ERK. The continued MAP kinase signaling-based resistance identified in these patients suggests that alternative dosing of current agents, more potent RAF/MEK inhibitors, and/or inhibition of the downstream kinase ERK may be needed for durable control of BRAF-mutant melanoma.
INTRODUCTION
Targeted agents that inhibit key effector kinases of the mitogen-activated protein kinase (MAP kinase) signaling cascade, including BRAF (vemurafenib or dabrafenib) and MEK1 and MEK2 (tremetinib) have improved progression-free survival and overall survival when used as monotherapy in BRAF-mutant melanoma (2-5). However, the majority of patients experience disease progression within 6 to 7 months. Many clinical mechanisms of resistance to monotherapy identified in BRAF-mutant melanoma to date result in reactivation of MEK/ERK signaling (6-12). Accordingly recent therapeutic efforts have focused on increased MAP kinase inhibition through combined targeting of BRAF and MEK. In a phase I/II trial of combined dabrafenib and tremetinib, this combination increased progression-free survival, objective response, and duration of response as compared with dabrafenib monotherapy (1). Nonetheless, resistance still developed in most patients after an average of 9.4 months. The mechanisms of resistance to combined RAF/MEK inhibition remain poorly understood.
To begin to investigate clinical mechanisms of resistance to combined RAF/MEK inhibition, we performed whole exome sequencing (WES) and transcriptome sequencing (RNASeq) on tumor samples obtained from five patients with acquired resistance to dabrafenib/trametinib. Here, we describe putative resistance mechanisms to combined RAF/MEK inhibition identified in these patients.
CASE SERIES
Five patients with metastatic BRAF-mutant melanoma were selected from a phase I/II study of first-line dabrafenib and trametinib (1). All five patients experienced a clinical benefit – defined as complete response, partial response, or stable disease for at least 6 months as determined by Response Evaluation Criteria In Solid Tumors (RECIST) (13) – before developing progressive disease. Biopsies were obtained prior to treatment with dabrafenib/trametinib, and at the time of disease progression. Patient characteristics are summarized in Table 1, and the clinical histories are detailed in the Data Supplement.
TABLE 1.
Clinical characteristics and MAP kinase pathway resistance mechanisms in patients with acquired resistance to dabrafenib/trametinib
| Gender | Age | Prior Systemic Therapy for Metastatic Melanoma |
Dabrafenib Dosing Trametinib Dosing |
Best Response |
Duration of Response (months) |
MAP kinase Pathway Candidate Resistance Mechanisms |
||
|---|---|---|---|---|---|---|---|---|
| Start Dose | End Dose | |||||||
| Patient 1 | M | 72 | None | 150mgBID 2mg Daily |
150mgBID 2mg Daily |
PR(−64%) | 3 | MEK2Q60P |
| Patient 2 | M | 48 | None | 150mgBID 2mg Daily |
100mgBID 2mg Daily |
PR(−42%) | 3 | BRAF Splice Isoform |
| Patient 3 | M | 42 | None | 150mgBID 1mg Daily |
150mgBID 1mg Daily |
SD(−19.5%) | 11 | BRAF Amplification |
| Patient 4 | M | 56 | None | 150mgBID 2mg Daily |
75mgBID 0.5mg Daily |
CR(−100%) | 18 | None Identified |
| Patient 5 | M | 49 | None | 150mgBID 2mg Daily |
150mgBID 2mg Daily |
PR (−45%) | 7 | None Identified |
PR, partial response. SD, stable disease. CR, complete response.
RESULTS
Although combined RAF/MEK inhibition is predicted to avoid several known mechanisms of resistance to RAF inhibitor monotherapy through enhanced suppression of MAP kinase signaling, three of the five cases examined nonetheless harbored apparent resistance mechanisms that engage MAP kinase effectors. Results are summarized in Table 1. Below, we describe specific resistance drivers identified in each patient.
An acquired MEK2Q60P mutation confers resistance to RAF/MEK inhibition
WES of the resistant tumor from Patient 1 revealed a mutation in MEK2 (MAP2K2), the downstream kinase from BRAF in the MAP kinase pathway and the target of trametinib (Figure 1A, left panel). This mutation was not detected in the pre-treatment tumor despite robust sequence coverage of this locus (224-fold) (Figure 1A-B; Supplementary Tables 1-3). RNA-Seq data demonstrated that this mutation, MEK2Q60P, was expressed in the resistant tumor but not in the pre-treatment tumor (Figure 1A, right panel). MEK2 mutations have not previously been identified in patients with acquired resistance to RAF or MEK inhibitors, although similar mutations were found to confer resistance to single-agent RAF inhibitors in a companion study (14). MEK2Q60P is homologous to MEK1Q56P, which confers resistance to monotherapy with RAF or MEK inhibitors in vitro (6) and in post-progression tumor samples from patients with acquired resistance to vemurafenib (12).
FIGURE 1. Identification of a MEK2 mutation in a melanoma sample resistant to dabrafenib/trametinib.
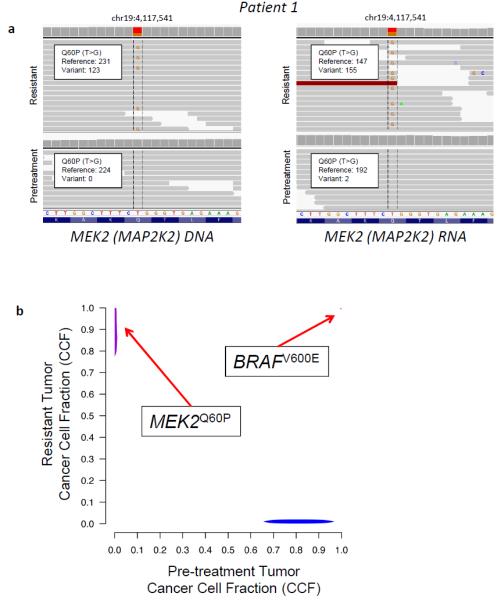
(A) Whole exome sequencing (WES) (left panel) and whole transcriptome sequencing (RNA-Seq) (right panel) of the tumor tissue from Patient 1 both before treatment and after the development of resistance to dabrafenib/trametinib revealed a Q60P mutation in MEK2 in the resistant tumor that was undetectable in the pre-treatment tumor. (B) The fraction of tumor cells (the cancer cell fraction, CCF) harboring each alteration was calculated for the pretreatment and resistant tumor samples. Direct comparison of the CCF for all alterations in the pretreatment and resistant tumor samples demonstrated alterations that occurred in the pretreatment sample only (lower right corner, blue), the resistant sample only (upper left corner, purple), or both samples (upper right corner, red). 15 missense mutations were identified occurring in the resistant sample only (upper left corner, purple), including the MEK2Q60P mutation (see Supplementary Table 3).
To confirm that MEK2Q60P confers resistance to combined RAF/MEK inhibition, the Q60P mutation was introduced into the sequence of wild type MEK2 and the mutant cDNA was expressed in a BRAFV600E melanoma cell line (A375). Compared to parental controls and cells expressing wild type MEK2, the MEK2Q60P mutation conferred profound resistance to the combination of dabrafenib plus trametinib (Figure 2A), as well as to single agent dabrafenib (Figure 2B) and trametinib (Figure 2C). On the other hand, MEK2Q60P did not confer resistance to treatment with an ERK inhibitor (Figure 2D), which targets the MAP kinase pathway downstream of MEK1/2. Cells expressing MEK2Q60P exhibited higher levels of phosphorylated ERK1/2 at baseline and when treated with dabrafenib/trametinib than wild type A375 cells or those expressing MEK2-WT (Figure 2E), indicative of enhanced MAP kinase pathway activation despite combined therapeutic blockade of this pathway.
FIGURE 2. Pharmacologic and biochemical characterization of the MEK2Q60P mutation.

(A-D) Growth inhibition curves are shown for dabrafenib/trametinib (A), as well as monotherapy with dabrafenib (B) or trametinib (C), and the ERK inhibitor VX-11e (D) for A375 (BRAFV600E) melanoma cells (grey) and A375 cells expressing wild type MEK2 (MEK2 WT; blue) or MEK2Q60P (red). (E) The effect of combined dabrafenib/trametinib treatment on ERK1/2 phosphorylation (pERK 1/2) in wild type A375 cells (BRAFV600E) and those expressing wild type MEK2 (MEK2 WT) or MEK2Q60P is shown. The levels of pERK1/2, total ERK1/2, pMEK1/2, total MEK1/2, and vinculin are shown for A375 cells expressing MEK2 mutations after a 16-hour incubation at various drug concentrations as indicated.
Additional MAP kinase pathway resistance effectors in tumors resistant to combined inhibition
Unexpectedly, in 2 patients, WES and RNA-Seq revealed additional alterations in the MAP kinase pathway that have been previously described in patients with acquired resistance to vemurafenib monotherapy (Table 1). RNA-Seq of the resistant tumor from Patient 2 revealed a BRAF variant that lacks exons 2-10, which was not detected in the pre-treatment tumor (Figure 3A). Exons 2-10 did not appear to be deleted in the WES data from the resistant sample based on copy number analysis (see Supplementary Methods), suggesting that this represents an alternative BRAF splice variant. This splice variant had previously been observed in patients with acquired resistance to single-agent vemurafenib (10). Resistance-associated BRAF splice variants lack the RAS binding domain and allow RAS-independent BRAF dimerization, resulting in downstream ERK activation in spite of RAF or MEK inhibition (10). No additional MAP kinase pathway alterations were detected in the resistant tumor from Patient 2 by either WES or RNA-seq.
FIGURE 3. Additional secondary alterations in MAP kinase pathway identified in resistant tumors.

(A) RNA-Seq of the tumor tissue from Patient 2 both before treatment and after the development of resistance to dabrafenib/trametinib revealed a BRAF splice isoform lacking exons 2-10 in the resistant tumor that was undetectable in the pre-treatment tumor. (B). Copy number analysis of whole exome data from Patient 3 demonstrates 2 highly amplified regions in the resistant tumor that are not amplified in the pre-treatment tumor. One of these regions contains the BRAF gene, while the second region contains multiple genes, including SAMD4B.
In Patient 3, WES revealed a focal BRAF chromosomal amplification in the resistant tumor that was absent in the pretreatment tumor (Figure 3B). BRAF amplification has been implicated in acquired resistance to RAF inhibitors (11) or MEK inhibitors (15) as single agents but not in combination.
In an additional patient (Patient 4), WES revealed a novel MEK1 (MAP2K1) mutation (MEK1P162S) in both the pre-treatment and resistant tumor. Certain MEK1 mutations have been shown to confer acquired resistance to monotherapy with RAF or MEK inhibitors both in vitro and in the clinical setting (6, 9, 12). This mutation was detected at high frequency (greater than 50% Cancer Cell Fraction; See Methods) in both the pretreatment and resistant tumors, in spite of the patient’s complete response to therapy (Supplementary Tables 1,2). Moreover, MEK1P162S did not confer resistance to RAF and/or MEK inhibition in vitro (Supplementary Figure 2). This finding is consistent with prior findings that some tumors with MEK1 mutations can respond to RAF inhibition (16).
Additional resistance-associated alterations found by WES and RNA-Seq
Conceivably, drug-resistant tumors may harbor multiple resistance alterations, including new mechanisms discoverable through WES or RNA-Seq. To identify additional putative resistance mechanisms in this cohort, we started with the list of point mutations and small insertions and deletions (indels) found in each tumor sample (Supplementary Table 1). This list was then cross-referenced with a set of 198 genes found to confer resistance to combined RAF/MEK inhibition when overexpressed in vitro (17) or when silenced in a genome-wide RNAi screen (18) (Supplementary Table 4), to generate a list of potential resistance alterations in each patient (Supplementary Table 5). These alterations were further filtered to highlight only those alterations that were significantly enriched in the resistant samples compared to their pretreatment counterparts (Supplementary Table 3).
This approach revealed several alterations that may have contributed to acquired resistance in these patients. For example, a mutation in the ETS transcription factor ETS2 (ETS2P53S) was identified in the resistant tumor from Patient 2 but not in the pretreatment tumor (Supplementary Tables 3). ETS2 is a transcription factor target of the MAP kinase pathway (19, 20) and a validated resistance gene in vitro, as overexpression of wild type ETS2 in BRAF-mutant cell lines resulted in resistance to dabrafenib/trametinib (17). This suggests that dysregulated ETS2 activity might engage a gene expression program capable of promoting resistance to upstream MAP kinase pathway inhibition.
Similarly, in addition to the aforementioned BRAF amplification, the resistant tumor from Patient 3 contained a second highly amplified region consisting of multiple genes, including SAMD4B (Figure 3B). SAMD4B, a poorly characterized gene thought to mediate transcriptional repression (21), was also identified and validated as a resistance gene in vitro (17). SAMD4B was mutated in the corresponding WES data and showed enriched expression of the mutant allele in the resistant tumor (387 variant reads and 39 reference reads in the resistant tumor, as compared to 36 variant reads and 28 reference reads in the pretreatment tumor). Overexpression of wild type SAMD4B in BRAF-mutant cell lines resulted in resistance to combined RAF/MEK inhibition (17) Future functional studies will investigate the mechanism by which mutations in ETS2, SAMD4B, and other candidate genes can contribute to resistance to RAF/MEK inhibition.
In Patients 4 and 5, several alterations were significantly enriched from the pre-treatment to the resistant tumors (Supplementary Tables 2,3,5). In Patient 4, of 543 coding SNVs and indels found in one or both of the tumors (Supplementary Table 2), 49 alterations were found to be significantly enriched from pre-treatment to resistant tumor (Supplementary Table 3). None of these mutations occurred in previously identified resistance genes or known MAPK genes (NRAS, BRAF, CRAF, MEK1/2, ERK1/2), nor were they present in the functional screens described above (Supplementary Table 4). Similarly, in Patient 5, of 212 coding SNVs and indels (Supplementary Table 2), 20 alterations were found to be significantly enriched from pre-treatment to resistant tumor (Supplementary Table 3). None of these were either previously identified resistance genes or present in the functional screens (Supplementary Table 4). Taken together, these results raise the possibility that these tumors may have elaborated as-yet uncharacterized genetic or non-genetic RAF/MEK resistance mechanisms.
DISCUSSION
This case series comprises an initial clinical genomic study of acquired resistance to combined RAF/MEK inhibition in BRAFV600-mutant melanoma. Since mechanisms of resistance may occur through both somatic genetic and transcriptional mechanisms, the use of both WES and RNA-Seq to identify mutations, copy number alterations, fusions, splice isoforms, and allele-specific expression differences between the pretreatment and resistant tumors potentially offers a more comprehensive view of resistance to combined RAF/MEK inhibition than DNA-based characterization alone.
In post-progression tumors from 3 of 5 patients with acquired resistance to dabrafenib/trametinib, we identified alterations in MAP kinase genes that were not detected in the pre-treatment tumors. Two tumors contained MAP kinase alterations that had previously been described in patients with acquired resistance to RAF or MEK inhibitor monotherapy. An additional resistant tumor harbored an activating mutation in MEK2, which has not previously been implicated in resistance to RAF or MEK inhibition, although the homologue MEK1 has been implicated in resistance to RAF inhibitor and MEK inhibitor monotherapy (6, 9, 12). MEK2 represents a logical resistance mechanism: mutations in this kinase may abrogate the effects of dabrafenib (which acts immediately upstream in the RAF-MEK signaling module) while simultaneously overcoming allosteric MEK inhibition by trametinib. We also recently identified additional MEK2 mutations in patients with acquired resistance to RAF inhibitor monotherapy (14).
Novel candidate resistance alterations were also identified in some tumors, including cases with known monotherapy-related resistance mechanisms. This suggests that multiple resistance mechanisms may occur in a single tumor sample. In spite of this, we were not able to identify any obvious candidate mechanisms of resistance in two patients, using validated functional screens and well-known MAPK genes as a primary filter. Additional functional follow-up of genomic or transcriptional alterations arising in resistant tumor samples from all 5 of these patients (Supplementary Table 3) may identify additional resistance effectors. Alternatively, additional mechanisms of resistance in these patients may have occurred through other modes not identifiable in the WES and RNA-Seq data, such as stromally secreted factors (22) or post-translational effects.
This is the first study, to our knowledge, of clinical acquired resistance to combined targeted therapy in cancer. One of the expected advantages of combining targeted therapies in genetically-defined tumor contexts is the theoretical ability to overcome common mechanisms of resistance to monotherapies. In melanoma, targeting the MAPK pathway with dual RAF and MEK inhibition was expected to overcome common MAPK-based resistance mechanisms seen with vemurafenib, dabrafenib, or trametinib alone. While combined RAF/MEK inhibition may indeed prevent resistance due to activating mutations in NRAS, which we did not identify in this study, it was somewhat surprising to find alterations in BRAF, which might have been expected to be overcome by adequate MEK inhibition, emerge in the resistant tumors from 2 patients.
These results indicate that at least some MAP kinase pathway alterations arising in the setting of monotherapy (MEK1/2 mutations, BRAF amplification, BRAF splice isoforms) are also likely to cause cross-resistance to combination therapy. Indeed, several MEK1 and MEK2 mutations (e.g., MEK1C121S, MEK1G128V, MEK2C125S, MEK2L46F) (9, 14) confer resistance to combination dabrafenib/trametinib in vitro even though the patients in whom those mutations were identified had never been exposed to that combination (Supplementary Figure 3). This result may help to provide a mechanistic basis for the much higher proportion of patients with intrinsic resistance to dabrafenib/trametinib when this combination is used following single-agent RAF or MEK inhibitors (23) – further supporting the use of combined RAF/MEK inhibition as first-line therapy.
As with resistance to single-agent RAF inhibition (7-12), the prevalence of MAP kinase pathway alterations in these resistant tumors indicates that BRAF-mutant melanomas remain dependent on MEK/ERK signaling despite combined pathway inhibition. Conceivably, more potent MEK inhibition might circumvent some of these resistance mechanisms—although toxicity concerns have constrained the dosing of MEK inhibitor monotherapy. In the future, small-molecule ERK inhibitors, now in clinical trials, may provide an additional avenue for overcoming RAF- or MEK-centered resistance mechanisms.
Finally, the identification of somatic mutations in genes such as ETS2 and SAMD4B is noteworthy for two reasons. First, these observations highlight the potential value of integrating systematic functional data pertaining to drug resistance derived from preclinical studies with the results of deep genomic characterization of clinical tumor specimens obtained prior to treatment with targeted therapies and following relapse. This type of integrative approach is likely to provide a valuable means for cross filtering and prioritization of candidate mechanisms identified from preclinical or clinical analyses individually.
Second, these findings raise the possibility that mechanisms outside the canonical MAP kinase pathway may also emerge as RAF/MEK resistance effectors in the future. Of course, the specific functional effects of these mutations will need to be examined mechanistically to clarify their importance in driving resistance phenotypes. Nonetheless, the observation supports the notion that clinical testing of higher-order therapeutic combinations directed against other signaling pathways as well as immunotherapy should be prioritized in addition to MAP kinase-directed therapy. The use of serial biopsies and genomic/molecular profiling at the time of resistance may ultimately improve the care of patients with resistant BRAF-mutant melanoma through tailored targeted combinations to overcome specific resistance mechanisms.
METHODS
Patients and Tumor Samples
We obtained pre-treatment and drug-resistant tumor specimens along with normal blood samples from 5 patients with acquired resistance to dabrafenib/trametinib. All patients provided written informed consent to genomic profiling of tumor and normal DNA/RNA, as approved by the Dana-Farber/Harvard Cancer Center Institutional Review Board (DF/HCC Protocol 11-181).
Whole Exome and Whole Transcriptome Sequencing and Analysis
Whole exome sequencing was performed on pre-treatment tumors, resistant tumors, and normal samples from all 5 patients, as detailed in the Data Supplement. The mean depth of coverage for the tumor samples was 255X (Range 125X – 338X) (Supplementary Table 6). Sequencing data was analyzed using tools to identify somatic point mutations, small insertions/deletions (indels), and copy number alterations (see Data Supplement).
Whole transcriptome sequencing was performed on pre-treatment and resistant tumors from 4 patients, as detailed in the Data Supplement and Supplementary Table 6. RNA-Seq of the pre-treatment samples from Patients 4 and 5 could not be completed due to poor-quality RNA. For Patient 4, RNA was obtained from a second biopsy taken one week after the start of therapy, and this was used as a substitute for the pre-treatment sample in the RNA-Seq analysis for this patient. Transcriptome data was analyzed for rearrangements/fusions that were enriched in the resistant tumor as compared to the pre-treatment tumor. In addition, transcriptome data was analyzed specifically for alternatively spliced isoforms from the MAP kinase pathway genes BRAF, NRAS, MEK1, and MEK2.
To prioritize candidate resistance alterations, we highlighted those somatic point mutations or indels that were novel or significantly enriched in the resistant samples as compared to each matched pre-treatment sample (24). This was done by estimating the fraction of tumor cells (the cancer cell fraction, CCF) harboring a given alteration in each pair of samples using the ABSOLUTE algorithm (see Data Supplement). Transcriptome data was queried to determine if these specific DNA alterations were expressed in the pre-treatment and resistant tumors. ABSOLUTE for Patient 3 could not be completed for technical reasons; for this patient, the pre-treatment and resistant WES and RNA-Seq data were manually compared.
Detailed analyses of all sequencing results are available in the Data Supplement (Supplementary Tables 1-6; Supplementary Figure 1).
Experimental Analysis
Expression plasmids containing MEK1 and MEK2 cDNA were generated and site-directed mutagenesis was performed as detailed in the Data Supplement. Viral infections, cell growth inhibition analysis and immunoblot studies were performed using standard protocols (see Data Supplement). Cell lines were obtained from ATCC, which verifies identity by short tandem repeat profiling, and were passaged less than 6 months following receipt. Physical and biologic containment procedures for recombinant DNA followed institutional protocols in accordance with the National Institutes of Health Guidelines for Research Involving Recombinant DNA Molecules.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
This study represents an initial clinical genomic study of acquired resistance to combined RAF/MEK inhibition in BRAFV600-mutant melanoma, using whole exome and whole transcriptome sequencing. The presence of diverse resistance mechanisms suggests that serial biopsies and genomic/molecular profiling at the time of resistance may ultimately improve the care of patients with resistant BRAF-mutant melanoma by specifying tailored targeted combinations to overcome specific resistance mechanisms.
Acknowledgments
Financial Support: This work was supported by the Conquer Cancer Foundation (N.W., E.M.V), the Dana-Farber Leadership Council (E.M.V.), the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (K.T.F., L.A.G.), the Melanoma Research Alliance (L.A.G), the Starr Cancer Consortium (L.A.G.), the National Human Genome Research Institute 5U54HG003067-11 (G.G., S.A.G., L.A.G.), and the National Cancer Institute P01 CA163222 01A1 and P50CA93683 (L.A.G.).
Footnotes
Disclosures: Consultant/Advisory Role: Foundation Medicine (N.W., L.A.G.), Novartis (L.A.G.), Daiichi Sankyo (L.A.G.), Millennium/Takeda (L.A.G.), Boehringer Ingelheim (L.A.G.). Ownership Interest: Foundation Medicine (N.W., L.A.G.). Research Support: Glaxo-Smithkline (K.T.F.), Novartis (L.A.G.). Patents: None. Honoraria: None.
REFERENCES
- 1.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. The New England journal of medicine. 2012 Nov;367(18):1694–703. doi: 10.1056/NEJMoa1210093. PubMed PMID: 23020132. Pubmed Central PMCID: 3549295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. The New England journal of medicine. 2010 Aug 26;363(9):809–19. doi: 10.1056/NEJMoa1002011. PubMed PMID: 20818844. Pubmed Central PMCID: 3724529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. The New England journal of medicine. 2011 Jun 30;364(26):2507–16. doi: 10.1056/NEJMoa1103782. PubMed PMID: 21639808. Pubmed Central PMCID: 3549296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. The New England journal of medicine. 2012 Feb 23;366(8):707–14. doi: 10.1056/NEJMoa1112302. PubMed PMID: 22356324. Pubmed Central PMCID: 3724515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. The New England journal of medicine. 2012 Jul 12;367(2):107–14. doi: 10.1056/NEJMoa1203421. PubMed PMID: 22663011. [DOI] [PubMed] [Google Scholar]
- 6.Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2009 Dec 1;106(48):20411–6. doi: 10.1073/pnas.0905833106. PubMed PMID: 19915144. Pubmed Central PMCID: 2777185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010 Dec 16;468(7326):968–72. doi: 10.1038/nature09627. PubMed PMID: 21107320. Pubmed Central PMCID: 3058384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010 Dec 16;468(7326):973–7. doi: 10.1038/nature09626. PubMed PMID: 21107323. Pubmed Central PMCID: 3143360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011 Aug 1;29(22):3085–96. doi: 10.1200/JCO.2010.33.2312. PubMed PMID: 21383288. Pubmed Central PMCID: 3157968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011 Dec 15;480(7377):387–90. doi: 10.1038/nature10662. PubMed PMID: 22113612. Pubmed Central PMCID: 3266695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nature communications. 2012;3:724. doi: 10.1038/ncomms1727. PubMed PMID: 22395615. Pubmed Central PMCID: 3530385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trunzer K, Pavlick AC, Schuchter L, Gonzalez R, McArthur GA, Hutson TE, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013 May 10;31(14):1767–74. doi: 10.1200/JCO.2012.44.7888. PubMed PMID: 23569304. [DOI] [PubMed] [Google Scholar]
- 13.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) European journal of cancer. 2009 Jan;45(2):228–47. doi: 10.1016/j.ejca.2008.10.026. PubMed PMID: 19097774. [DOI] [PubMed] [Google Scholar]
- 14.Van Allen EM, Wagle N, Sucker A, Treacy D, Goetiz EM, Place CS, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Submitted. 2013 doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corcoran RB, Dias-Santagata D, Bergethon K, Iafrate AJ, Settleman J, Engelman JA. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Science signaling. 2010;3(149):ra84. doi: 10.1126/scisignal.2001148. PubMed PMID: 21098728. Pubmed Central PMCID: 3372405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi H, Moriceau G, Kong X, Koya RC, Nazarian R, Pupo GM, et al. Preexisting MEK1 exon 3 mutations in V600E/KBRAF melanomas do not confer resistance to BRAF inhibitors. Cancer discovery. 2012 May;2(5):414–24. doi: 10.1158/2159-8290.CD-12-0022. PubMed PMID: 22588879. Pubmed Central PMCID: 3594852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johannessen CM, Johnson LA, Piccioni F, Frederick DT, Donahue MK, Narayan R, et al. A cyclic AMP-regulated melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature. 2013 doi: 10.1038/nature12688. Manuscript Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whittaker SR, Theurillat JP, Van Allen E, Wagle N, Hsiao J, Cowley GS, et al. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer discovery. 2013 Mar;3(3):350–62. doi: 10.1158/2159-8290.CD-12-0470. PubMed PMID: 23288408. Pubmed Central PMCID: 3606893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foulds CE, Nelson ML, Blaszczak AG, Graves BJ. Ras/mitogen-activated protein kinase signaling activates Ets-1 and Ets-2 by CBP/p300 recruitment. Molecular and cellular biology. 2004 Dec;24(24):10954–64. doi: 10.1128/MCB.24.24.10954-10964.2004. PubMed PMID: 15572696. Pubmed Central PMCID: 533975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seth A, Watson DK. ETS transcription factors and their emerging roles in human cancer. European journal of cancer. 2005 Nov;41(16):2462–78. doi: 10.1016/j.ejca.2005.08.013. PubMed PMID: 16213704. [DOI] [PubMed] [Google Scholar]
- 21.Luo N, Li G, Li Y, Fan X, Wang Y, Ye X, et al. SAMD4B, a novel SAM-containing protein, inhibits AP-1-, p53- and p21-mediated transcriptional activity. BMB reports. 2010 May;43(5):355–61. doi: 10.5483/bmbrep.2010.43.5.355. PubMed PMID: 20510020. [DOI] [PubMed] [Google Scholar]
- 22.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012 Jul 26;487(7408):500–4. doi: 10.1038/nature11183. PubMed PMID: 22763439. Pubmed Central PMCID: 3711467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sosman J, Daud A, Weber J, Kim K, Kefford R, Flaherty KT, et al. BRAF inhibitor (BRAFi) dabrafenib in combination with the MEK1/2 inhibitor (MEKi) trametinib in BRAFi-naive and BRAFi-resistant patients (pts) with BRAF mutation-positive metastatic melanoma (MM) Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31 suppl; abstr 9005. [Google Scholar]
- 24.Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013 Feb 14;152(4):714–26. doi: 10.1016/j.cell.2013.01.019. PubMed PMID: 23415222. Pubmed Central PMCID: 3575604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.