

Abstract
Each individual carries thousands of non-synonymous single nucleotide variants (nsSNVs) in their genome, each corresponding to a single amino acid polymorphism (SAP) in the encoded proteins. It is important to be able to directly detect and quantify these variations at the protein level in order to study post-transcriptional regulation, differential allelic expression, and other important biological processes. However, such variant peptides are not generally detected in standard proteomic analyses, due to their absence from the generic databases that are employed for mass spectrometry searching. Here, we extend previous work that demonstrated the use of customized SAP databases constructed from sample-matched RNA-Seq data. We collected deep coverage RNA-Seq data from the Jurkat cell line, compiled the set of nsSNVs that are expressed, used this information to construct a customized SAP database, and searched it against deep coverage shotgun MS data obtained from the same sample. This approach enabled detection of 421 SAP peptides mapping to 395 nsSNVs. We compared these peptides to peptides identified from a large generic search database containing all known nsSNVs (dbSNP) and found that more than 70% of the SAP peptides from this dbSNP-derived search were not supported by the RNA-Seq data, and thus are likely false positives. Next, we increased the SAP coverage from the RNA-Seq derived database by utilizing multiple protease digestions, thereby increasing variant detection to 695 SAP peptides mapping to 504 nsSNV sites. These detected SAP peptides corresponded to moderate to high abundance transcripts (30+ transcripts per million, TPM). The SAP peptides included 192 allelic pairs; the relative expression levels of the two alleles were evaluated for 51 of those pairs, and found to be comparable in all cases.
Keywords: single nucleotide variants, single nucleotide polymorphisms, RNA-Seq, mass spectrometry, amino acid polymorphism, variant peptides, multiple proteases, SIFT, PolyPhen-2, differential allelic expression, allele-specific expression, proteoforms, Bowtie, Tophat, SAMtools
INTRODUCTION
DNA sequencing technologies have allowed researchers to uncover an astounding amount of genetic variation in humans, including a multitude of single nucleotide variations, insertions, deletions, tandem repeats, inversions, translocations, and duplications.1 Among these variations, single nucleotide variants (SNVs), the single nucleotide differences between two genomes that occur on average about once every 860 base pairs, have been the most intensely researched, mainly through genome-wide association studies that seek to uncover the sets of causative SNVs that are responsible for a disease or trait.1–3 Advances in sample preparation, sequencing instrumentation, and computational data analysis have made it easier for researchers to rapidly sequence and discover the millions of SNVs found within a genome, and thus the challenge today is not how to discover these variations but how to sift through them to find those with functional significance.4
One way to simplify the study of SNVs is to focus on those SNVs that lie within coding regions, because these SNVs can cause a change in the protein amino acid sequence and are thus most likely to modify the function of a protein. Coding SNVs can be classified into three types: (1) synonymous, which does not change the corresponding amino acid, (2) nonsense, which introduces a premature stop codon, and (3) non-synonymous, also called missense, which changes the corresponding amino acid. While it is well accepted that synonymous SNVs do not affect the protein function, and nonsense SNVs usually cause a loss of function (because the protein is truncated)5, it is harder to determine what effect a non-synonymous SNV (nsSNV) has on a given protein’s function.6
Current strategies employed to study the functional effects of nsSNVs include determining statistical associations between well phenotyped populations (i.e. genome-wide association studies), computationally predicting the functional effect of an SNV using programs like SIFT and PolyPhen-27, 8, and, most recently, evaluating the nsSNV within the biological system, such as in a protein-protein interaction or regulatory network9. These approaches guide the prioritization of nsSNVs for subsequent validation and hypothesis testing using in vitro and in vivo functional assays. Though these statistical and bioinformatic strategies have aided the study of nsSNVs, another valuable piece of information is the direct measurement of the variant-containing protein.
The direct detection of proteins containing single amino acid polymorphisms (SAPs) encoded by an nsSNV can aid researchers in studying the functional significance of these variants. Directly measuring these SAP-containing proteoforms10 is essential to understanding how an SNV influences a variety of processes at the protein-level such as post-translational regulation of protein expression (e.g. protein degradation and stability), localization of the protein, modulation of protein-protein interactions, and influence of the SAP on patterns of post-translational modifications (PTMs). Furthermore, understanding the influence of SAPs across various cell states would be very difficult without technologies to measure these protein variations. Fortunately, mass spectrometry-based proteomics has undergone remarkable development in the past decade and can now be used to comprehensively identify and quantify large portions of the proteome.11–13 MS-based proteomics has tremendous potential to detect SAPs on a large scale, providing researchers with valuable information regarding the relationship between genomic variations and the ultimate protein products they encode.
The main impediment to the wide-spread adoption of variant peptide detection using mass spectrometry has been the lack of proteomic databases that include sample-specific variant sequences. The current practice in proteomics to identify peptides or proteins is to search the mass spectra against the sequences contained in a reference proteomic database, which is derived from either the human reference genome or cDNA sequence repositories14–17. Since the reference protein sequences do not contain the amino acid variations specific to a sample, a mass spectrum produced from a variant-containing peptide will not correctly match to a sequence and, therefore, will fail to be detected.
Several researchers have addressed this problem by constructing proteomic databases that include SAPs and then searching these databases against tandem mass spectra to detect SAP peptides. One approach relies on the construction of an exhaustive SAP database which includes amino acid changes resulting from every hypothetical nucleotide change in the genome.18–20 Another approach relies on the construction of a database that includes every SAP found within SNV or cancer mutation repositories, such as dbSNP or COSMIC.21–34 Both of these approaches successfully allowed the detection of SAP peptides that are absent from the reference proteome and thus show the potential of proteomics to characterize variant peptides. However, the databases are greatly increased in size by tens of thousands of SAP-containing sequences, many of which are not expressed in the sample. This results in a concomitant increase in the false positive rate and a decrease in peptide identification sensitivity18, 21, 22. These problems were overcome in two studies that used RNA-Seq data to build SAP databases customized for a sample, enabling the detection of dozens of SAP peptides, including peptides containing novel variants resulting from either rare SNVs or de novo mutations.35, 36 These studies showed how rapid advances in next generation sequencing technologies and the ease with which scientists can empirically measure all the coding SNVs in a sample can be harnessed to expand the detection of SAPs on a proteome-wide scale.
Here, we build upon those studies by comprehensively investigating SAP peptide detection in the Jurkat human cell line. This study follows from previous work in which we used RNA-Seq data to detect novel splice-junction peptides.37 We collected deep coverage RNA-Seq data from the Jurkat cell line, compiled the set of nsSNVs that are expressed, used this information to construct a customized SAP database, and searched it against deep coverage shotgun MS data obtained from the same sample. The SAP peptides identified from this customized database workflow were of much higher quality as compared to those identified using a larger aggregate database that incorporates all known nsSNVs (dbSNP). We employed multiple protease digestions to increase proteomic coverage and, thus, the number of SAP peptide identifications. These detected SAP peptides represent the most comprehensive study to date. Using this dataset, we describe various characteristics of the detected SAP peptides, including their corresponding transcriptional abundance, SNV functional effect scores, and degree of allele-specific expression.
EXPERIMENTAL PROCEDURES
Mammalian cell culture
Jurkat cells (TIB-152) were grown in 10% Fetal Bovine Serum and 90% RPMI-1640 buffer at 37°C to a concentration of ~1.3×106 cells/mL (cell line and media were purchased from ATCC, Manassas, VA). In total, there were 12 flasks each containing 25 mL of Jurkat cell suspension. Upon harvesting, cell viability for each flask was determined with the trypan blue assay and cells were counted on a TC10 Automated Cell Counter system (BioRad, Hercules, CA). All cell cultures had 95%+ viability.
Mass spectrometry sample preparation and data collection
The proteomic sample preparation has been described previously in detail.37 Briefly, Jurkat cell suspension was pelleted and rinsed twice in cold PBS buffer before storage at −80°C. Cell lysis was performed by following the FASP protocol.38 Pellets were solubilized in SDT lysis buffer (4% w/v SDS, 100 mM DTT, 50 mM Tris-HCl), heated, sonicated, and 150 μg aliquots of protein were transferred to a 100K MW Amicon Ultra filter (Millipore, Billerica, MA). For this study, the FASP protocol was slightly modified to allow for multiple enzymatic digestions. The FASP method was followed for initial wash steps, alkylation, and the last three wash steps, which employed 50 mM ammonium bicarbonate. Then, each filter was washed with two additional rounds of buffer compatible with a protease and the enzyme was added directly to the filter as listed here: 3 μg of trypsin (50:1 protein to enzyme ratio) in 50 mM ammonium bicarbonate at 37°C for 16 hours (Promega, Madison, WI); 1.5 μg of rLysC (100:1) in 25 mM Tris-HCl pH 8.5, 1 mM EDTA, 4 M urea at 37°C for 16 hours (Promega, Madison, WI); 1.5 μg of ArgC (100:1) in 270 μL of 50 mM Tris-HCl pH 7.6, 5 mM CaCl2, and 2 mM EDTA and 30 μL of 50 mM Tris-HCl pH 7.6, 50 mM DTT, and 2 mM EDTA at 37°C for 16 hours (Promega, Madison, WI); 1.5 μg of AspN (100:1) in 50 mM sodium phosphate pH 8.0 at 25°C for 16 hours (Roche, Indianapolis, IN); 1.5 μg of GluC (100:1) in 25 mM ammonium bicarbonate at 25°C for 16 hours (Roche, Indianapolis, IN); and 1.5 μg of chymotrypsin (100:1) in 100 mM Tris-HCl pH 8.0 and 10 mM CaCl2 at 25°C for 4 hours (Promega, Madison, WI). At the end of the incubation time, each filter was centrifuged at 14,000 g for 15 minutes and the amount of peptide recovered was quantified via the Nanodrop UV-Vis spectrometer (Thermo Fisher Scientific, Wilmington, DE).
At least 100 μg of peptide digest was fractionated on a Shimadzu HPLC system (LC-10AD, SCL-10A VP, SPD-10A VP, Shimadzu, Columbia, MD) using a Phenomenex C18 Gemini 3μ, 110Å, 3.0×150mm column (Phenomenex, Torrance, CA) and high pH mobile phases. Mobile phase A (MPA) was aqueous 20 mM ammonium formate pH 10, and B (MPB) was 20 mM ammonium formate pH 10, in 70% acetonitrile. The HPLC flow was 0.5 mL/min and the gradient was as follows: 0% MPB isocratic for 15 minutes (trapping step), linear ramp to 100% MPB over 60 minutes, hold at 100% MPB for 5 minutes, to 0% MPB over 2 minutes, and equilibration at 0% MPB for 20 minutes. A Gilson 203 fraction collector (Gilson, Middleton, WI) was used to collect 28 fractions for the tryptic digest and 11 fractions for each of the LysC, ArgC, AspN, GluC, and chymotrypsin digests during detected (214 nm UV absorbance) peptide elution. Fractions were dried down using vacuum centrifugal concentration (Savant SpeedVac, Thermo, Pittsburgh, PA) and stored at −80°C.
Each of the dried down fractions were reconstituted in 2% acetonitrile and 0.2% formic acid in water and then chromatographically separated on a nanoAquity LC system (Waters, Milford, MA) using a 20 cm reverse phase capillary column (100 μm i.d.) packed with 3 μm MAGIC aqC18 beads (Bruker-Michrom, Auburn, CA). Mobile phase A was 0.2% formic acid in water and B was 0.2% formic acid in acetonitrile. The full HPLC method was 180 minutes long and included online trapping, a 90 minute gradient, and re-equilibration time. A Velos-Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) was programmed to collect a full scan (300–1500 m/z) at a resolution of 60,000 followed by the top ten precursor HCD fragmentation spectra at a resolution of 7,500. Precursor fragmentation repeat count was set to two and the dynamic exclusion was set to 60 seconds. XCalibur software version #2.1.0 was used for data collection.
RNA-Sequencing
The RNA-Seq data collection has previously been described in detail.37 Briefly, total RNA was extracted from a 2 mL aliquot of each Jurkat culture (~2.6×106 cells) using the TRIzol® Reagent (Life Technologies, Grand Island, NY) and the RNA integrity was evaluated on a 2100 Agilent Bioanalyzer (Agilent, Santa Clara, CA). Illumina paired-end libraries were prepared for each of 12 samples using the TruSeq RNA Sample Prep Rev. A (kit lot #6849988, Illumina, San Diego, CA). Briefly, mRNA was isolated with poly dT beads, fragmented, reverse transcribed to cDNA, and then cDNA ends were repaired, adenylated, and ligated to Illumina adapters. The cDNA library was run on an Invitrogen 2% Size Select Gel (Lot# R19090-01) and a ~350 base pair band was excised and sequenced on an Illumina HiSeq 2000 in paired-end mode (2×100bp). An average of 12 million reads were generated per sample, and some samples were run multiple times, resulting in a total of ~300 million reads.
RNA-Seq data analysis
Bowtie/Tophat RNA-Seq read alignment
RNA-Seq reads were aligned to the human reference genome (hg19) using Bowtie (v0.12.7) and Tophat (v1.4.0).39, 40 Alignments were performed within Tophat, which uses Bowtie. The Tophat mate inner distance was set to 150. All other parameters were default. RefSeq gene models were supplied in GTF format and reads were aligned to both RefSeq genes and novel genes (option –G). RefSeq is NCBI’s curated, non-redundant reference sequence database and includes DNA, RNA, and protein sequences and annotations.41 The binary alignment or BAM file was used for subsequent SNV calling.
SAMtools SNV calling
SAMtools (v0.1.18) was used to call SNVs, nucleotide differences between the aligned RNA-Seq reads and the human reference genome. The mpileup command was used with the –u and –D options. Bcftools was then used (-bvcg options) to format the binary call format or BCF file. Finally, the SAMtools vcfutils.pl script was used to create a variant call format or VCF file. Only SNVs with a read depth (DP) higher than 10 and a quality score (QUAL) higher than 10 were used for subsequent analysis. QUAL is a phred-scaled score that reflects the confidence of the SNV call.
All RNA-Seq data processing was performed on the Phoenix cluster at the University of Wisconsin-Madison Chemistry department.
Retrieval of amino acid polymorphisms
The variant_effect_predictor.pl Perl script (version 2.7) downloaded from Ensembl along with the human annotation file (Ensembl v72) was used to convert the SNVs to amino acid coordinates and retrieve the calculated SIFT and PolyPhen-2 scores.42 Only SNVs passing the DP and QUAL filters were used. Each SNV coordinate contained the chromosome, chromosome position, forward strand reference nucleotide, and forward strand alternative nucleotide. After analysis, the program output a variant effect predictor (VEP) formatted file containing all the non-synonymous SNVs, and each entry included the corresponding amino acid change, the amino acid index within a RefSeq protein sequence, and the associated SIFT and PolyPhen-2 score.
Construction of a customized SAP database
SAP coordinate information was converted into a customized SAP FASTA database. Within the VEP file output from the previous step, SNVs that resided within RefSeq protein coding regions were retrieved. The RefSeq protein FASTA file was downloaded from NCBI’s FTP site (ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/mRNA_Prot/human.protein.faa.gz, release 59).41 For each coding SNV, the reference and alternative nucleotide and its position within the genome was listed, as well as the reference and alternative amino acid and position within a RefSeq protein entry (NP accession). An in-house perl script was used to extract an 80 aa substring containing the SAP and change the reference aa to the variant aa. A FASTA header including the amino acid change and position within the RefSeq NP entry was linked to each SAP-containing sequence and all these sequences were appended to the RefSeq protein and cRAP FASTA file. cRAP or the common Repository of Adventitious Proteins is a database of protein sequences that are found as contaminants in proteomics experiments (http://www.thegpm.org/crap/).
Construction of a SAP database from the dbSNP repository
For comparison purposes, a FASTA file containing SAPs derived from NCBI dbSNP repository was constructed. The ASN-1 flat file containing all 53,233,155 dbSNP rs entries for human was downloaded from NCBI’s ftp site (/snp/organisms/human_9606, build 137) and the 691,356 rs entries representing missense mutations (fxn-class = missense) were retrieved. Each rs entry lists the reference and alternative amino acid position within a RefSeq protein entry. An in-house perl script was used to extract an 80 aa substring containing the SAP and change the reference aa to the variant aa. A FASTA header including the amino acid change and its position within the RefSeq NP entry was added to each dbSNP-SAP-containing sequence and all these sequences were appended to the RefSeq protein and cRAP FASTA file.
Mass spectrometry searching
Raw mass spectrometry files were searched against the customized SAP+RefSeq+cRAP and the dbSNP-SAP+RefSeq+cRAP FASTA files using the SEQUEST/Percolator search algorithm within ProteomeDiscoverer (v1.3.0.339, Thermo Fisher Scientific, San Jose, CA). Default peaklist-generating parameters were used. Precursor m/z tolerance was set to 10 ppm and product m/z tolerance was set to 0.05 Da. Peptides with up to two missed cleavages (proteolytic) were permitted. Variable methionine oxidation and static carbamidomethylation were used. Using reversed sequences as a decoy database, peptides passing both a 1% and 5% global FDR were used for downstream analysis. Validation was based on q-values generated by Percolator. For identification of a protein using ProteomeDiscoverer, protein grouping and strict parsimony principle was enabled, leucine and isoleucine were considered equal, and only peptides passing a 1% FDR and having a delta Cn higher than 0.15 were used. Each peptide identification counted only if that peptide had a unique primary sequence. A minimum of two peptides per protein was required for identification. MS data collected from alternative enzymatic digests were separately searched against the customized SAP+RefSeq+cRAP FASTA file with identical parameters to the trypsin search except with the relevant enzyme specificity.
Estimation of allele-specific protein expression
Using Skyline software (v1.4)43, MS1 extracted ion chromatograms were integrated for heterozygous peptide pairs that had a high degree of structural similarity (same length, only one amino acid difference). Only peaks that overlapped a target peptide MS2 identification, contained minimal background interference, and had an appropriate chromatographic peak shape were accepted. Default Skyline parameters for peak integration were used.
RESULTS
Overview
Each human cell line or tissue sample contains thousands of non-synonymous SNVs (nsSNVs) that give rise to single amino acid polymorphisms (SAPs); however, these variations are typically absent from generic proteomic databases. Therefore, sample-specific peptides containing these SAPs fail to be identified during mass spectrometry searching. Fortunately, RNA-Seq can be used to experimentally detect the nsSNVs in a sample, which allows for the creation of a customized SAP database, thereby enabling identification of SAP peptides.36
Here, we describe the comprehensive detection and evaluation of SAP peptides from a human cell line. We created a customized SAP database using RNA-Seq data collected from Jurkat cells that enabled the detection of 421 SAP peptides mapping to 395 nsSNV sites. For comparison purposes, we constructed an all-inclusive SAP database derived from all known human nsSNVs (NCBI’s dbSNP) leading to the identification of 891 SAP peptides. Though there were a higher number of SAP peptides passing a 1% FDR using this all-inclusive database, we show that the peptide spectral matches (PSMs) were of much lower quality, indicating a false positive issue. After this finding, we proceeded to determine the extent of SAP peptide detection using the customized database. We employed multiple protease digestions to increase proteomic coverage and thus identified 695 SAP peptides mapping to 504 nsSNV sites (9% of total nsSNVs, 504/5755). These SAP peptides corresponded to transcripts with a median of 44 transcripts per million, indicating that they are derived from moderate to high abundance transcripts. For all the SAP peptides, we report the computationally predicted functional effect scores (SIFT, PolyPhen-2). And last, the detected SAP peptides included 192 allelic pairs, in which the reference and SAP peptide were both detected; we measured the relative allele-specific expression for 51 of these pairs.
Construction and use of the customized RNA-Seq database
RNA-Seq data was collected from Jurkat cell culture and used to create a customized SAP database used for MS searching. The detection of variant peptides from SAP databases is shown in Figure 1 and the bioinformatic workflow numbers are shown in Figure 2.
Figure 1.

Overview of sample-specific SAP peptide detection from custom databases. Single nucleotide variants (SNVs) are detected directly from RNA-Seq reads by finding differences between the transcript and human reference genome nucleotide sequences. The set of non-synonymous SNVs are converted into amino acid sequences that are consolidated into a customized protein database that is used for MS searching. Here, both the reference and variant (SAP) peptides are detected, demonstrating that both allelic forms are expressed at the protein level.
Figure 2.
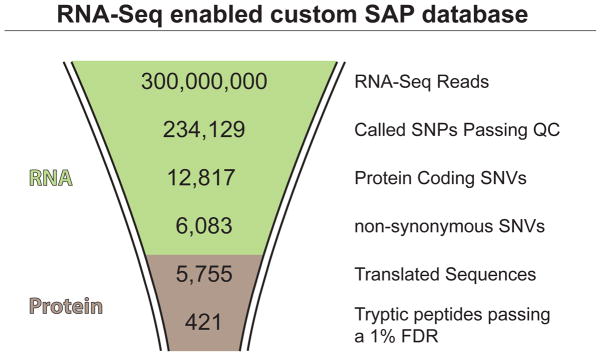
Bioinformatic workflow numbers for customized SAP database construction and subsequent MS search results.
First, RNA-Seq and MS data was collected from Jurkat human cell culture. Total RNA was extracted from several Jurkat cultures (95%+ viability, trypan blue) using the TRIzol® method and each sample was used to create a barcoded Illumina cDNA library using the TruSeq protocol. Each library was sequenced at least once on an Illumina HiSeq 2000, resulting in a total of ~300 million paired end reads (350bp, 2×100bp). Protein was extracted and digested from the Jurkat cultures using the FASP method and the resulting peptides were fractionated via a high pH HPLC and run on a nanoLC-Velos Orbitrap operating in data-dependent mode. Approximately 500,000 mass spectra were collected.
The RNA-Seq data were analyzed to find Jurkat cell-specific SNVs. Bowtie and Tophat were used to align the RNA-Seq reads to the human reference genome (hg19). RefSeq gene models were used to guide alignment, but reads that aligned to novel genes were also allowed. 82.8% of the singletons (one member of the read pair) and 67.7% of the full read pair were successfully aligned. All read alignments were stored within a binary alignment (BAM) file (61.41 GB). Next, SAMtools (mpileup command) was used to call SNVs. Here, the genome is traversed one nucleotide at a time and for each nucleotide position, the reads overlapping a nucleotide is examined. If there is evidence that the nucleotide sequence within the RNA-Seq reads differ from the nucleotide in the reference genome with statistical significance, an SNV is “called” or reported. After SNV calling, several quality metrics are used to filter SNVs, including the quality of the nucleotides at the SNV site, the score of the read alignment, and the depth (i.e. coverage) of the reads. From the mapped reads in this study, a total of 473,868 SNVs were called while 234,129 SNVs passed quality filters—read depth (DP) of 10 or higher and quality score (QUAL) of 10 or higher. Figure 3 shows the distribution of read depth versus quality score for all the SNVs called, with filtered out SNVs shaded in gray.
Figure 3.
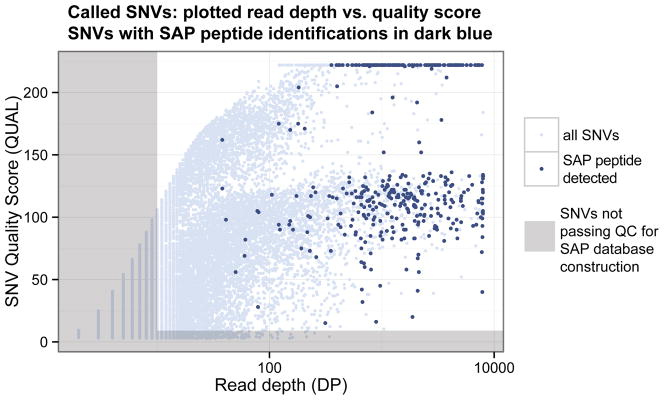
Plot of RNA-Seq read depth versus quality score for each called SNV. This graph shows the distribution of depth and quality scores for the SNVs called using SAMtools, with discarded SNVs highlighted in gray. The bimodal shape is due to the presence of homozygous (top portion) and heterozygous (bottom portion) alleles. The nsSNVs that resulted in a SAP peptide identification are dark blue. These nsSNVs tend to be of higher read depth and quality.
Of the 234,129 SNVs that passed quality filters, 12,817 SNVs were found to reside within RefSeq protein coding regions. 6,535 (52%) were synonymous SNVs and 6,083 (47%) were non-synonymous SNVs (nsSNVs). These percentages are similar to percentages reported by the 1000 Genomes Project (55% synonymous, 45% non-synonymous; average values from 1,092 individuals).1 The high proportion (94.5%) of SNVs that did not reside in coding regions were predominantly located within UTRs or introns, and this was especially true for nucleotides near the 3′ end of the transcript. This suggests that many untranslated SNVs are detected from incompletely spliced mRNAs that were isolated during the polydT bead enrichment step of the Illumina library preparation protocol.
The set of nsSNVs found in the RNA-Seq data was used to derive all SAP-containing polypeptide sequences in RefSeq. To accomplish this, each amino acid position and index within the RefSeq protein sequence (NP accession number) was retrieved. For each SAP, a custom Perl script was used to extract an 80 aa subsequence containing the SAP position, and the amino acid at that position was changed to the variant form. In a few cases (5%) the RefSeq protein sequence corresponded to the SAP encoded by the nsSNV. This is because of minor discrepancies between the RefSeq and hg19 sequence data, due to their different origins—hg19 is the product of genome sequencing efforts, whereas RefSeq is derived from cDNA sequencing data. 5,755 SAP-containing sequences mapping to 3,837 distinct NP accessions were extracted and appended to the RefSeq protein (35,930 entries) and cRAP (155 entries) databases to create a customized SAP database. The SAP entries marginally increased the size of the database by 2.2% (442,740 aa added to 19,899,407 aa). 38% (2,162 entries out of 5,755) of the SAPs were not present in dbSNP and are likely to represent undocumented variations, including somatic mutations, rare variants, and variations exclusively in the RNA from RNA editing or RNA polymerase nucleotide misincorporations.
The RefSeq+cRAP+SAP database was searched against the MS data using the Percolator/SEQUEST algorithm. 73,552 peptides (each with unique sequences) were identified at a 1% FDR. From these, there were a total of 421 SAP peptides mapping to 395 unique SNVs, corresponding to 0.6% of all peptides. This percentage, representing the proportion of SAP peptides detected in a shotgun proteomics experiment, is similar to previous findings36; however, the present study identified over ten times the number of SAP peptides. The significantly higher number of SAP peptides identified is likely due to the deep proteomic sampling achieved in this study. This suggests that even more SAP peptides could be discovered by the collection of deeper-coverage proteomics data. A list of the SAP peptide identifications may be found in Supplementary Table S1 in the Supplementary Information (SI).
The relative quality of peptide spectral matches (PSMs) was compared between RefSeq and SAP peptides. When MS searches are performed against proteomic databases that are augmented with putative sequences (e.g. splice junction sequences), there is an increased chance of false positives37. A typical indication that there are false positive issues is when peptides matching the non-canonical database (e.g. SAP peptide) have lower than expected MS search scores. Therefore, the average MS search scores—in this case, the SEQUEST XCorr score that represents the degree of match (via the cross-correlation function) between the theoretical and experimental MS2 spectra—were compared between RefSeq and SAP peptides. Surprisingly, the SAP peptide XCorr scores, on average, were actually higher than the RefSeq peptide scores, indicating that the SAP peptide identifications are of high quality. Figure 4 shows these comparisons.
Figure 4.

Comparison of average XCorr scores for peptides matching the RefSeq protein, dbSNP-SAP, or custom (RNA-Seq) SAP database. SAP peptides identified from the custom database tended to have higher XCorr scores than those identified from the dbSNP database. Score distributions for peptides passing a 1% FDR (A) and 5% FDR (B) are shown.
Construction and use of the dbSNP database
The nsSNVs listed in dbSNP were used to create an exhaustive SAP database, which was then used for MS searching. Key bioinformatic workflow numbers describing this process are shown in Figure 5.
Figure 5.

Bioinformatic workflow numbers for the dbSNP-derived SAP database construction and MS search. Though more SAP peptides were detected using the dbSNP database, the peptide identifications had low peptide spectral match (PSM) scores, indicating a false positive issue.
NCBI’s dbSNP is one of the largest repositories of known SNVs consolidated from various sources of data such as sequence tagged sites, Genbank, and the 1000 genomes project33. dbSNP was used to create an exhaustive SAP database for proteomic searching. A human dbSNP ANS-1 flat file containing all 53,555,486 entries was downloaded from NCBI’s FTP site (May 3rd, 2013). Of those entries, 679,490 were classified as non-synonymous SNVs (fxn-class=missense) and 378,986 as synonymous (fxn-class=synonymous). The 679,490 non-synonymous SNVs mapped to 33,557 distinct RefSeq NP sequences and, therefore, the dbSNP nsSNVs covered nearly all RefSeq protein sequences.
A SAP-containing polypeptide sequence was created from the SNV coordinate information listed in each dbSNP entry. Using the dbSNP nsSNV coordinate information, a custom Perl script was used to extract, from the RefSeq protein entry, the 80 amino acid stretch of protein sequence containing the SAP and to change the amino acid to reflect the variant form. Each entry was created in FASTA format and the header included the chromosome and protein position of the nucleotide and amino acid change, respectively. In total, 691,356 dbSNP-SAP entries were created. Some dbSNP entries contained two or more alternative alleles, thereby generating multiple SAP entries from a single dbSNP. The dbSNP-SAP entries were appended to the RefSeq protein (35,930 entries) and cRAP (155 entries) databases to create the dbSNP-SAP database. The dbSNP-SAP entries drastically increased the size of the database by 268% (53,233,115 aa added to 19,899,407 aa).
The RefSeq+cRAP+dbSNP-SAP database was searched against the MS data using the Percolator/SEQUEST algorithm. 72,250 RefSeq peptides (each with unique sequences) were identified at a 1% FDR. A total of 891 dbSNP-SAP peptides were identified. An additional 652 dbSNP-SAP peptides were identified at a 5% FDR threshold. A list of the dbSNP-SAP peptide identifications may be found in Supplementary Table S2 in the SI. Though at first glance it may seem that more SAP peptides were identified with the dbSNP-SAP database, there were false positive issues that bring into question the quality of these peptide identifications. This topic is discussed in the next section.
Comparing RNA-Seq and dbSNP-derived SAP peptides
The dbSNP-SAP database represents all the nsSNVs found in any number of different human cell and tissue types, whereas the custom SAP database derived herein is from a single sample-matched RNA-Seq dataset and represents the set of nsSNVs that exist in this particular single cell-line. Although use of an aggregate database, such as the set of dbSNP-derived SAPs, obviates the need to collect sample-specific RNA-Seq data, these databases contain an extremely large number of polypeptide sequences that do not exist in the sample. Inclusion of a large number of extraneous sequences in proteomics databases increases the probability that a theoretical mass spectrum derived from an extraneous peptide sequence falsely matches to an experimental mass spectrum by mere chance, a well-known phenomenon.44
A strong disadvantage of using an aggregate database, like the dbSNP-derived SAP database, is that there are many false positives in the set of SAP peptides identified. Evidence for this phenomenon can be seen in the comparison of MS search score distributions of the RefSeq and SAP peptides. Figure 4A shows that for peptides passing a 1% FDR, the median XCorr score for RefSeq (canonical) peptides was 3.0: The custom SAP peptides had a median value of 3.6, which was even better than the RefSeq median, but, notably, the dbSNP-SAP peptides had lower XCorr scores, a median of 2.8. These trends for RefSeq, custom SAP, and dbSNP-SAP were even more pronounced when comparing median XCorr scores for peptides passing a 5% FDR, that is, 2.9, 3.6, and 1.8, respectively (Figure 4B), underscoring both the high quality of RNA-Seq derived custom SAP peptide identifications, and the low quality and higher number of false positives within the dbSNP-SAP peptide identifications. Note that the peptide posterior error probabilities (PEP) and q-values for the peptide groups also showed similar trends (Figures S1 and S2).
We examined the extent of overlap in peptide identifications between RNA-Seq versus dbSNP-derived SAP peptides. Venn diagrams are shown in Figure 6. A large fraction of the RNA-Seq SAP peptides (42% of peptides passing a 1% FDR) were not present in the dbSNP database, showing that despite dbSNP’s large size, it still does not include every SNV in this particular human cell line. Moreover, it is reasonable to assume that aggregate databases, as they stand today, would fail to detect a number of variants in other cell or tissue types, as many SNVs are yet to be documented. Conversely, a large fraction of dbSNP-SAP peptides (73% of peptides passing a 1% FDR, and 84% passing a 5% FDR) lacked evidence of expression in the deep coverage RNA-Seq data and, hence, are most likely false positives. This would suggest that the nominal false discovery rates for 1% and 5% FDR passing dbSNP-SAP peptides are actually 73% and 84%, respectively. While the total number of dbSNP-SAP peptides identified is greater than the number of RNA-Seq SAP peptides identified, the exceedingly high actual false positive rate compromises their utility.
Figure 6.

Comparison of dbSNP versus RNA-Seq derived SAP peptide identifications. Venn diagrams show the overlap of SAP peptides identified from MS searching. For example, 245 SAP peptides passing a 1% FDR were identified in both the dbSNP and RNA-Seq SAP database searches. (A) dbSNP-SAP and RNA-Seq SAP peptides passing global FDRs, (B) dbSNP-SAP peptides re-analyzed to pass a local FDR and then compared to the same RNA-Seq SAP peptides. The terms “local” and “global” FDR are explained by Käll, et al. 57.
Next, we asked if the dbSNP-SAP peptide false positive issue could be remedied by applying more stringent peptide identification thresholds. It is well known that MS searches against extremely large databases tend to produce many false positive peptide identifications, and various strategies have been developed to reduce the incidence of false positives, including sequential (multi-tiered) MS searches and calculation of local FDRs44, 45. We calculated a local FDR for the dbSNP-SAP peptides by utilizing posterior error probability (PEP) values (see Supplemental Table S2)37, 46. We found that even with the application of a local FDR threshold, the dbSNP-SAP peptide score distributions were still slightly shifted to lower values (Figure S3). And, more importantly, applying the local FDR cut-off did not eliminate many false positive dbSNP-SAP peptides, as shown in the Venn diagrams in Figure 6B, where more than 70% of dbSNP-SAP peptides were not present in the RNA-Seq data and are therefore likely to be false positives.
The coverage and accuracy of the SAP peptide identifications must be high to be of use in biological applications such as the confirmation of nsSNV translation. These results show that utilizing sample-matched RNA-Seq data to identify SAP peptides offers significant advantages in these respects.
Multiple protease digests to expand SAP peptide detection
It was shown above that 395 SNV sites were detected at the protein level from searching the custom (RNA-Seq derived) SAP database against MS data collected on tryptically-digested lysate. As far as we know, this is the largest number of SAP peptides detected for a single human cell line. However, these SAP peptides represent only 6.9% (395/5755) of all possible translated nsSNVs. Of the 5755 total SAP sequences, 4325 contain SAP peptides that are between 6 and 39 amino acids, the typical range of peptide lengths that are identified in shotgun proteomics studies. Using this reduced number, a larger fraction of length-filtered SAP peptides were identified, specifically 9.7% (395/4325). Assuming that the nsSNVs detected at the RNA level are indeed translated into protein, these results provide a good estimate of the proportion of nsSNVs corresponding to detectable SAPs.
We asked what fraction of nsSNVs could be detected at the protein-level with shotgun proteomics. To explore this question, we collected high coverage proteomics data by employing multiple protease digestions. Jurkat cell lysate was separated into five aliquots and was digested with either LysC, ArgC, AspN, GluC, or chymotrypsin. Each of the five peptide digests were fractionated on a high pH HPLC and analyzed on a Velos-Orbitrap mass spectrometer in data dependent mode, and each dataset was searched against the RefSeq+cRAP+SAP database. Similarly to the trypsin-derived SAP peptides, the SAP peptides had higher XCorr distributions than RefSeq peptides on average (Figure S4). Figure 7 shows the peptide and SNV site identification results. Note that the trypsin dataset was based on 28 high pH HPLC fractions whereas the datasets for the other enzymes were based on 11. The number of SAP peptides with unique sequences was calculated for cumulative combinations of proteolytic search results. For example, 508 unique SAP sequences were found with combined trypsin and LysC data and 547 unique SAP sequences were found with combined trypsin, LysC, and ArgC data. When the multiple protease data was compared with the original tryptic dataset, the number of unique SAP peptides increased by 65% while the number of unique nsSNV sites for which there was direct peptide evidence increased by 28%. In other words, while data from all six enzymes detected 695 unique SAP peptide sequences, these peptides corresponded to only 504 unique nsSNV sites. These results suggest that higher coverage shotgun proteomics data increases the number of identified SAP peptides with unique sequences, but that many of these SAP peptides are repeatedly sampling the same set of SNVs. All multiple protease SAP peptide search results may be found in Supplementary Table S3 in the SI.
Figure 7.

Cumulative number of identified SAP peptide and nsSNV sites with consolidated protease digest data. The enhanced protein coverage afforded by multiple protease digestions increased the number of translated nsSNVs detected by 28%.
Transcript abundances for detected SAP peptides
With high coverage proteomic data, 8.8% (504/5755) of the total number of possible nsSNVs were identified at the protein level. This represents a much higher fraction of detected SAP peptides as compared to previous studies21, 22, 24, 27, 36, but it lags in comparison to the SNV detection sensitivity afforded by next generation sequencing technologies. MS-based proteomics can only detect a small fraction of all possible protein-level variants within a sample. To understand why, the abundance distribution, in transcripts per million (TPM), was plotted for all transcripts and for transcripts in which the corresponding protein was identified (Figure 8). The median TPM for transcripts with a protein identification was much higher than the median TPM for all transcripts. Two reasons for this are: first, some lower abundance transcripts are not translated, especially for transcripts that are stochastically expressed, and, second, mass spectrometry is not as sensitive as RNA-Seq and the sampling depth of peptides is limited by many factors such as peptide ionization efficiency, sample complexity, and the MS duty cycle. The abundance distribution for transcripts in which there was a detected SNV was also plotted and compared to the abundances of transcripts for which there was a detected SAP peptide (Figure 8B). This plot shows that SAP peptides are primarily detected from highly expressed transcripts and suggests that as MS sensitivity and sampling depth increases, the number of SAP peptides detected will also increase.
Figure 8.

Distribution of transcript abundances for transcripts encoding detected proteins and transcripts encoding detected SAP peptides. (A) The abundance distribution for all transcripts (light blue) versus just those transcripts with a protein identification (dark blue). (B) The abundance distribution for transcripts with an nsSNV (light blue) versus just those transcripts with a detected SAP peptide (dark blue).
The transcript versus protein abundance was plotted for all genes detected in the Jurkat cell line (Figure S5). The degree of transcript ~ protein correlation (Spearman’s rank correlation coefficient = 0.62) was similar to those reported in previous studies.11–13
Computationally predicted functional effect scores
The functional consequence of a given SNV can be computationally predicted using a variety of tools such as SIFT and PolyPhen-27, 8. SIFT examines the degree of evolutionary conservation of the nucleotide polymorphism and depends on the assumption that an SNV found in a highly conserved genomic region is more likely to affect the function of the protein. PolyPhen-2 examines the physicochemical properties of the amino acid change and how much this change affects conserved protein domains. Because the number of discovered SNVs far exceeds the number of SNVs that can be biologically validated, both SIFT and PolyPhen-2 are ubiquitously used to analyze and rank SNVs discovered in genome research.
We were interested in evaluating the functional predictive scores for both the RNA and protein-level SNVs. We used Ensembl’s Variant Effect Predictor (VEP) program to retrieve the SIFT and PolyPhen-2 scores for each nsSNV (see Supplementary Table S4 in SI). The distribution of SIFT and PolyPhen-2 scores for nsSNVs detected at the RNA level and the subset of nsSNVs that was detected at the protein level, as evidenced by a SAP peptide ID, were similar. Figure 9 shows histograms of both SIFT and PolyPhen-2 score distributions. 27% of all nsSNVs and 29% of nsSNVs with peptide evidence had a SIFT score less than 0.05, which is categorized as “deleterious”. 16% nsSNVs and 14% of nsSNVs with peptide evidence had a PolyPhen-2 scores greater than 0.903, which is categorized as “probably damaging”.
Figure 9.

Comparing SIFT and PolyPhen-2 functional effect prediction scores between all nsSNVs and nsSNVS with a SAP peptide ID. The distributions were similar between the two groups.
RNA and protein allele-specific expression
In diploid organisms such as human, there are two copies of each chromosome, and thus each RNA or protein is derived from one of two alleles. When the gene is homozygous, the sequence of the allelic pair is identical and there is no way to distinguish which chromosomes the gene products come from. But when the gene is heterozygous, the sequences of the allelic pair are different and it is possible to track which gene an RNA or protein arose from by detecting the RNA-Seq read or SAP peptide containing the SNV or SAP, respectively. Additionally, it is possible to quantify allele-specific expression (ASE). The ASE at the RNA-level can be estimated by comparing the depth of reads mapping to the reference and alternative SNVs47. Analogously, the ASE at the protein-level can be estimated by quantifying the amount of reference and SAP peptide48. Previously, a SILAC-based approach was developed that allowed global quantification of ASE in yeast49.
We examined the RNA-Seq and mass spectrometry datasets to identify, at the protein-level, the number of detected allelic pairs and to measure ASE. At least one SAP peptide was detected for each of 504 nsSNV sites, as shown in an earlier section of this paper. Both the reference and SAP peptides were detected for 38% (192 out of 504) of those nsSNV sites showing that a significant number of heterozygous peptide pairs are readily detected by shotgun proteomics. The amino acid sequences of the heterozygous peptide pairs were either significantly different (e.g. the SAP introduces a lysine causing the SAP peptide to be much shorter than the reference peptide) or highly similar (e.g. the SAP is a single amino acid change in the middle of the peptide sequence). 74 heterozygous peptide pairs were found in the latter category. The peptides in these pairs have highly similar sequences (i.e. a difference of only one amino acid). They could be considered structural analogues of each other; the predicted HPLC retention time using SSRCalc50 and the predicted ionization efficiency using ESPPredictor51 between these pairs were found to be near-identical. We estimated the relative SAP to reference peptide concentrations by integrating the area of MS1 extracted ion chromatograms using the Skyline program43.
Figure 10 displays a plot of the estimated allelic expression for peptide and RNA-level heterozygous pairs. The reference to alternative peptide ratio was distributed around 1:1, for both the nsSNVs (RNA-Seq reads) or SAPs (peptide) measured. As expected, allele-specific peptide expression shows greater variability than allele-specific RNA expression due to MS variables such as electrospray current and complexity of the sample matrix (i.e. co-eluting peptides). Future work could utilize heavy-labeled internal standards and employ more precise methods of quantification to further explore allele-specific expression. All ASE results can be found in Supplementary Table S5 in the SI.
Figure 10.

RNA and protein-level allele-specific expression. A line corresponding to 1:1 allelic expression has been overlaid. For both RNA (A) and protein (B), the expression levels for allelic pairs are roughly the same. Protein-level expression had higher variability.
DISCUSSION
The full repertoire of SNVs expressed in RNA can be detected using the latest sequencing technologies but the power to detect the corresponding SAPs at the protein level has been lagging. Direct detection of the SAP within a peptide (or protein) is important for understanding how variants influence biological phenomena such as post-transcriptional regulation and differential allelic expression. Little work has been done to date to measure SAP peptides on a large-scale using mass spectrometry because the conventional strategy for identifying peptides is through database searches against a generic proteomic database that does not include the variant sequences.
We have described the large-scale detection of SAP peptides made possible through the construction of a customized SAP database from sample-matched RNA-Seq data. With the customized database, we confirmed the translation of hundreds of non-synonymous SNVs that were specific to the Jurkat cell line, representing the most comprehensive set of SAP peptide identifications to date. To determine how many SAP peptides are detectable by shotgun proteomics, we employed multiple protease digestions and collected even higher coverage proteomics data, allowing us to detect 695 sequence-unique SAP peptides corresponding to 504 unique nsSNV sites, or ~10% of all RNA-level nsSNVs (504/5755). These results illustrate that a significant number of SAP peptides are detectable through shotgun proteomics, but also indicate that further improvements in proteomics technologies are needed for them to equal the coverage of variants that can be obtained at the RNA level with next generation sequencing technologies.
The unusually high number of SAP peptides identified in this work along with the sample-matched RNA-Seq data provided us with the opportunity to analyze properties of nsSNVs and the SAP peptides identified via mass spectrometry. The SAP peptides, similarly to all peptides identified, corresponded to moderate to high abundance transcripts (30+ transcripts per million, TPM). The distribution of these detected SAP peptides’ computationally predicted functional effects (e.g. SIFT, PolyPhen-2) was similar to the distribution for the complete set of all possible SAPs, indicating no selection of particular SAP types. Finally, for 192 out of the 504 SNVs, we detected both the reference and SAP peptides, confirming that a significant fraction of heterozygous alleles are expressed at the protein level. Related to this finding, we also investigated the feasibility of quantifying differential allelic expression on a large scale. Previously, SRM methods employing stable isotope labeled peptide standards were developed to quantify three allelic peptide pairs52 and a small number of related mutant peptides48, 53. Here, we presented preliminary label-free quantification of allele-specific expression based on the integrated MS1 extracted ion chromatograms from 51 allelic peptide pairs.
We compared the number and quality of SAP peptide identifications resulting from MS searches against (1) an aggregate SAP database derived from NCBI’s dbSNP repository and (2) a customized SAP database derived from sample-specific RNA-Seq data—which contained only those nsSNVs detected in the human cell line of study (Jurkat cells). There were many clear advantages to using a customized database, including its smaller size (reducing the incidence of false positive peptide IDs), inclusion of nsSNVs not yet in public SNV repositories, and the ability to compare RNA and protein nsSNV expression. The aggregate database may be an option in the case that NGS data cannot be collected, but we found that the large database size (over 100 times larger than the customized database) caused the identification of many false positive SAP peptides, a problem not remedied by application of stringent MS search cut-offs (e.g. local FDR). In light of these findings, it is recommended to use some strategy for condensing or customizing proteomic databases when searching for novel protein variations.
An issue that will become important as methodology for the detection of sample-specific SAP peptides is adopted is that the various genetic, transcriptomic, and proteomic databases have discrepancies in sequence. These sequence discrepancies make it difficult to assess the incidence and extent of protein variations in samples. The genomics community has solved this problem by calling an SNV when there is a nucleotide that is different from the human reference genome that is maintained by the Genome Reference Consortium54. No such convention has yet been implemented in the area of proteomics. For example, many proteomics researchers use protein databases containing sequences that are not derived from the human reference genome, such as UniProt, so the set of SAPs called from the reference genome will be different from those called from UniProt.
As outlined in the introduction, it would be beneficial if MS-based proteomics could detect and quantify all the translated nsSNVs in a human sample. In this study, we show that up to ~10% of nsSNVs identified in RNA were also detected at the protein level, meaning that there are many SAP peptides that are not presently detected. Two factors could improve SAP detection: higher proteomic coverage and increased MS sampling sensitivity. With high proteomic coverage, there is a better chance of detecting a peptide corresponding to an nsSNV. In this study, we used multiple proteases and increased the number of detected SNV sites by ~25%. With increasing sensitivity, there are improved chances of detecting SAPs that are expressed at lower levels. Whereas MS instrument sensitivity is an inherent feature of each MS platform, another factor affecting sensitivity that we can control is the sampling depth, that is, the ability for the instrument to choose precursor peptides of low ion intensity (within a complex matrix) for subsequent MS2 fragmentation. For example, one solution to increase the number of SAP peptides detected would be to employ a targeted approach by using selected reaction monitoring (SRM) assays55, SAP peptide inclusion lists during data dependent acquisition, or even intelligent data acquisition (IDA) strategies56. These SAP peptide targeting approaches could be employed in future work to detect a larger fraction of translated nsSNV sites.
Supplementary Material
Acknowledgments
We thank Gergana Hinrichs and William Horvat for technical assistance with the cell culture and proteomics sample preparation. This work was supported by NIH grants 1P01GM081629 and 1P50HG004952. This research was also supported in part by National Science Foundation Grant CHE-0840494 through use of the University of Wisconsin-Madison chemistry computing resources. RNA-Sequencing work was performed at the University of Wisconsin—Madison Biotechnology Center. GMS was supported by the NIH Genomic Sciences Training Program 5T32HG002760.
ABBREVIATIONS
- ASE
allele-specific expression
- ATCC
American Type Culture Collection
- BAM
binary alignment file
- BCF
binary call format
- cDNA
complementary DNA
- COSMIC
catalogue of somatic mutations in cancer
- cRAP
The common Repository of Adventitious Proteins
- DAE
differential allelic expression
- DP
read depth
- DTT
dithiothreitol
- EST
expressed sequence tags
- FASP
filter-aided sample preparation
- FDR
false discovery rate
- GTF
gene transfer format
- IDA
intelligent data acquisition
- MPA
mobile phase A
- NCBI
National Center for Biotechnology Information
- NGS
next generation sequencing
- NP
RefSeq protein sequence accession
- nsSNV
non-synonymous single nucleotide variant
- PBS
phosphate buffered saline
- PolyPhen-2
polymorphism phenotyping version 2
- PSM
peptide spectral match
- SAP
single amino acid polymorphism
- SDT
SDS/DTT buffer (see other abbreviations)
- SIFT
Sorting Intolerant From Tolerant
- SNP
single nucleotide polymorphism
- SNV
single nucleotide variant
- SRM
selected reaction monitoring
- TPM
transcripts per million
- VCF
variant call format
- VEP
variant effect predictor
- XIC
extracted ion chromatogram
References
- 1.The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1, 092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JPA, Hirschhorn JN. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9(5):356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 3.Wellcome Trust Case Control Consortium. Genome-wide association study of 14, 000 cases of seven common diseases 3, 000 shared controls. Nature. 2007;447(7145):661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TFC, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, Jostins L, Habegger L, Pickrell JK, Montgomery SB, Albers CA, Zhang ZDD, Conrad DF, Lunter G, Zheng HC, Ayub Q, DePristo MA, Banks E, Hu M, Handsaker RE, Rosenfeld JA, Fromer M, Jin M, Mu XJ, Khurana E, Ye K, Kay M, Saunders GI, Suner MM, Hunt T, Barnes IHA, Amid C, Carvalho-Silva DR, Bignell AH, Snow C, Yngvadottir B, Bumpstead S, Cooper DN, Xue YL, Romero IG, Wang J, Li YR, Gibbs RA, McCarroll SA, Dermitzakis ET, Pritchard JK, Barrett JC, Harrow J, Hurles ME, Gerstein MB, Tyler-Smith C Genomes Project C. A Systematic Survey of Loss-of-Function Variants in Human Protein-Coding Genes. Science. 2012;335(6070):823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sunyaev SR. Inferring causality and functional significance of human coding DNA variants. Human Molecular Genetics. 2012;21:R10–R17. doi: 10.1093/hmg/dds385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ng PC, Henikoff S. Predicting Deleterious Amino Acid Substitutions. Genome Research. 2001;11(5):863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khurana E, Fu Y, Chen JM, Gerstein M. Interpretation of Genomic Variants Using a Unified Biological Network Approach. Plos Computational Biology. 2013;9(3) doi: 10.1371/journal.pcbi.1002886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith LM, Kelleher NL. Proteoform: a single term describing protein complexity. Nat Meth. 2013;10(3):186–187. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beck M, Schmidt A, Malmstroem J, Claassen M, Ori A, Szymborska A, Herzog F, Rinner O, Ellenberg J, Aebersold R. The quantitative proteome of a human cell line. Mol Syst Biol. 2011:7. doi: 10.1038/msb.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lundberg E, Fagerberg L, Klevebring D, Matic I, Geiger T, Cox J, Algenas C, Lundeberg J, Mann M, Uhlen M. Defining the transcriptome and proteome in three functionally different human cell lines. Molecular Systems Biology. 2010:6. doi: 10.1038/msb.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Paabo S, Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol. 2011:7. doi: 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang HZ, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, Yeh LSL. The universal protein resource (UniProt) Nucleic Acids Research. 2005;33:D154–D159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benson DA, Karsch-Mizrachi I, Clark K, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Research. 2012;40(D1):D48–D53. doi: 10.1093/nar/gkr1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, Durbin R, Eyras E, Gilbert J, Hammond M, Huminiecki L, Kasprzyk A, Lehvaslaiho H, Lijnzaad P, Melsopp C, Mongin E, Pettett R, Pocock M, Potter S, Rust A, Schmidt E, Searle S, Slater G, Smith J, Spooner W, Stabenau A, Stalker J, Stupka E, Ureta-Vidal A, Vastrik I, Clamp M. The Ensembl genome database project. Nucleic Acids Research. 2002;30(1):38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, Barnes I, Bignell A, Boychenko V, Hunt T, Kay M, Mukherjee G, Rajan J, Despacio-Reyes G, Saunders G, Steward C, Harte R, Lin M, Howald C, Tanzer A, Derrien T, Chrast J, Walters N, Balasubramanian S, Pei BK, Tress M, Rodriguez JM, Ezkurdia I, van Baren J, Brent M, Haussler D, Kellis M, Valencia A, Reymond A, Gerstein M, Guigo R, Hubbard TJ. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Research. 2012;22(9):1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Creasy DM, Cottrell JS. Error tolerant searching of uninterpreted tandem mass spectrometry data. Proteomics. 2002;2(10):1426–1434. doi: 10.1002/1615-9861(200210)2:10<1426::AID-PROT1426>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 19.Hyatt D, Pan CL. Exhaustive database searching for amino acid mutations in proteomes. Bioinformatics. 2012;28(14):1895–1901. doi: 10.1093/bioinformatics/bts274. [DOI] [PubMed] [Google Scholar]
- 20.Gatlin CL, Eng JK, Cross ST, Detter JC, Yates JR. Automated identification of amino acid sequence variations in proteins by HPLC/microspray tandem mass spectrometry. Analytical Chemistry. 2000;72(4):757–763. doi: 10.1021/ac991025n. [DOI] [PubMed] [Google Scholar]
- 21.Bunger MK, Cargile BJ, Sevinsky JR, Deyanova E, Yates NA, Hendrickson RC, Stephenson JL. Detection and validation of non-synonymous coding SNPs from orthogonal analysis of shotgun proteomics data. Journal of Proteome Research. 2007;6(6):2331–2340. doi: 10.1021/pr0700908. [DOI] [PubMed] [Google Scholar]
- 22.Li J, Su ZL, Ma ZQ, Slebos RJC, Halvey P, Tabb DL, Liebler DC, Pao W, Zhang B. A Bioinformatics Workflow for Variant Peptide Detection in Shotgun Proteomics. Molecular & Cellular Proteomics. 2011;10(5) doi: 10.1074/mcp.M110.006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schandorff S, Olsen JV, Bunkenborg J, Blagoev B, Zhang Y, Andersen JS, Mann M. A mass spectrometry-friendly database for cSNP identification. Nature Methods. 2007;4(6):465–466. doi: 10.1038/nmeth0607-465. [DOI] [PubMed] [Google Scholar]
- 24.Chen M, Yang B, Ying WT, He FC, Qian XH. Annotation of Non-Synonymous Single Polymorphisms in Human Liver Proteome by Mass Spectrometry. Protein and Peptide Letters. 2010;17(3):277–286. doi: 10.2174/092986610790780242. [DOI] [PubMed] [Google Scholar]
- 25.Chernobrovkin AL, Mitkevich VA, Popov IA, Indeikina MI, Ilgisonis EV, Lisitsa AV, Archakov AI. Identification of Single Amino Acid Polymorphisms in MS/MS Spectra of Peptides. Doklady Biochemistry and Biophysics. 2011;437(1):90–93. doi: 10.1134/S1607672911020098. [DOI] [PubMed] [Google Scholar]
- 26.Alves G, Ogurtsov AY, Yu YK. RAId_DbS: mass-spectrometry based peptide identification web server with knowledge integration. Bmc Genomics. 2008:9. doi: 10.1186/1471-2164-9-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mathivanan S, Ji H, Tauro BJ, Chen YS, Simpson RJ. Identifying mutated proteins secreted by colon cancer cell lines using mass spectrometry. Journal of Proteomics. 2012;76:141–149. doi: 10.1016/j.jprot.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 28.Xi H, Park JS, Ding GH, Lee YH, Li YX. SysPIMP: the web-based systematical platform for identifying human disease-related mutated sequences from mass spectrometry. Nucleic Acids Research. 2009;37:D913–D920. doi: 10.1093/nar/gkn848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nijveen H, Kester MGD, Hassan C, Viars A, de Ru AH, de Jager M, Falkenburg JHF, Leunissen JAM, van Veelen PA. HSPVdb-the Human Short Peptide Variation Database for improved mass spectrometry-based detection of polymorphic HLA-ligands. Immunogenetics. 2011;63(3):143–153. doi: 10.1007/s00251-010-0497-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawabata T, Ota M, Nishikawa K. The protein mutant database. Nucleic Acids Research. 1999;27(1):355–357. doi: 10.1093/nar/27.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR. Current Protocols in Human Genetics. John Wiley & Sons, Inc; 2001. The Catalogue of Somatic Mutations in Cancer (COSMIC) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Duncan DT, Zhang B. CanProVar: A Human Cancer Proteome Variation Database. Human Mutation. 2010;31(3):219–228. doi: 10.1002/humu.21176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Research. 2001;29(1):308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yip YL, Famiglietti M, Gos A, Duek PD, David FPA, Gateau A, Bairoch A. Annotating single amino acid polymorphisms in the UniProt/Swiss-Prot knowledgebase. Human Mutation. 2008;29(3):361–366. doi: 10.1002/humu.20671. [DOI] [PubMed] [Google Scholar]
- 35.Evans VC, Barker G, Heesom KJ, Fan J, Bessant C, Matthews DA. De novo derivation of proteomes from transcriptomes for transcript and protein identification. Nat Meth. 2012 doi: 10.1038/nmeth.2227. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Slebos RJC, Wang D, Halvey PJ, Tabb DL, Liebler DC, Zhang B. Protein Identification Using Customized Protein Sequence Databases Derived from RNA-Seq Data. Journal of Proteome Research. 2011 doi: 10.1021/pr200766z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheynkman GM, Shortreed MR, Frey BL, Smith LM. Discovery and mass spectrometric analysis of novel splice-junction peptides using RNA-Seq. Molecular & Cellular Proteomics. 2013 doi: 10.1074/mcp.O113.028142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nature Methods. 2009;6(5):359–U60. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 39.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10(3) doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Research. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26(16):2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schilling B, Rardin MJ, MacLean BX, Zawadzka AM, Frewen BE, Cusack MP, Sorensen DJ, Bereman MS, Jing EX, Wu CC, Verdin E, Kahn CR, MacCoss MJ, Gibson BW. Platform-independent and Label-free Quantitation of Proteomic Data Using MS1 Extracted Ion Chromatograms in Skyline APPLICATION TO PROTEIN ACETYLATION AND PHOSPHORYLATION. Molecular & Cellular Proteomics. 2012;11(5):202–214. doi: 10.1074/mcp.M112.017707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Castellana N, Bafna V. Proteogenomics to discover the full coding content of genomes: A computational perspective. Journal of Proteomics. 2010;73(11):2124–2135. doi: 10.1016/j.jprot.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jagtap P, Goslinga J, Kooren JA, McGowan T, Wroblewski MS, Seymour SL, Griffin TJ. A two-step database search method improves sensitivity in peptide sequence matches for metaproteomics and proteogenomics studies. PROTEOMICS. 2013;13(8):1352–1357. doi: 10.1002/pmic.201200352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi H, Ghosh D, Nesvizhskii AI. Statistical validation of peptide identifications in large-scale proteomics using the target-decoy database search strategy and flexible mixture modeling. Journal of Proteome Research. 2008;7(1):286–292. doi: 10.1021/pr7006818. [DOI] [PubMed] [Google Scholar]
- 47.Yan H, Yuan WS, Velculescu VE, Vogelstein B, Kinzler KW. Allelic variation in human gene expression. Science. 2002;297(5584):1143–1143. doi: 10.1126/science.1072545. [DOI] [PubMed] [Google Scholar]
- 48.Wang Q, Chaerkady R, Wu JA, Hwang HJ, Papadopoulos N, Kopelovich L, Maitra A, Matthaei H, Eshleman JR, Hruban RH, Kinzler KW, Pandey A, Vogelstein B. Mutant proteins as cancer-specific biomarkers. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(6):2444–2449. doi: 10.1073/pnas.1019203108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khan Z, Bloom JS, Amini S, Singh M, Perlman DH, Caudy AA, Kruglyak L. Quantitative measurement of allele-specific protein expression in a diploid yeast hybrid by LC-MS. Molecular Systems Biology. 2012:8. doi: 10.1038/msb.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krokhin OV. Sequence-specific retention calculator. Algorithm for peptide retention prediction in ion-pair RP-HPLC: Application to 300-and 100-angstrom pore size C18 sorbents. Analytical Chemistry. 2006;78(22):7785–7795. doi: 10.1021/ac060777w. [DOI] [PubMed] [Google Scholar]
- 51.Fusaro VA, Mani DR, Mesirov JP, Carr SA. Prediction of high-responding peptides for targeted protein assays by mass spectrometry. Nature Biotechnology. 2009;27(2):190–198. doi: 10.1038/nbt.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Su ZD, Sun L, Yu DX, Li RX, Li HX, Yu ZJ, Sheng QH, Lin X, Zeng R, Wu JR. Quantitative detection of single amino acid polymorphisms by targeted proteomics. Journal of Molecular Cell Biology. 2011;3(5):309–315. doi: 10.1093/jmcb/mjr024. [DOI] [PubMed] [Google Scholar]
- 53.Ruppen-Cañás I, López-Casas PP, García F, Ximénez-Embún P, Muñoz M, Morelli MP, Real FX, Serna A, Hidalgo M, Ashman K. An improved quantitative mass spectrometry analysis of tumor specific mutant proteins at high sensitivity. PROTEOMICS. 2012;12(9):1319–1327. doi: 10.1002/pmic.201100611. [DOI] [PubMed] [Google Scholar]
- 54.Nielsen R, Paul JS, Albrechtsen A, Song YS. Genotype and SNP calling from next-generation sequencing data. Nature Reviews Genetics. 2011;12(6):443–451. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nature Methods. 2012;9(6):555–566. doi: 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- 56.Bailey DJ, Rose CM, McAlister GC, Brumbaugh J, Yu PZ, Wenger CD, Westphall MS, Thomson JA, Coon JJ. Instant spectral assignment for advanced decision tree-driven mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(22):8411–8416. doi: 10.1073/pnas.1205292109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Käll L, Storey JD, MacCoss MJ, Noble WS. Posterior Error Probabilities and False Discovery Rates: Two Sides of the Same Coin. Journal of Proteome Research. 2007;7(1):40–44. doi: 10.1021/pr700739d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.