

Abstract
Disturbance of T cell homeostasis could lead to intestinal inflammation. Naïve CD4 T cells undergoing spontaneous proliferation, a robust proliferative response that occurs under severe lymphopenic conditions, differentiate into effector cells producing Th1 and/or Th17 type cytokines and induce a chronic inflammation in the intestine that resembles human inflammatory bowel disease. In this study, we investigated key properties of CD4 T cells necessary to induce experimental colitis. α4β7 upregulation was primarily induced by mLN resident CD11b+ dendritic cell subsets via TGFβ/retinoic acid-dependent mechanism. Interestingly, α4β7 expression was essential but not sufficient to induce inflammation. In addition to gut homing specificity, expression of gut Ag specificity was also crucial. T cell acquisition of the specificity was dramatically enhanced by the presence of γδ T cells, a population previously shown to exacerbate T cell mediated colitis. Importantly, IL-23-mediated γδ T cell stimulation was necessary to enhance colitogenicity but not gut antigen reactivity of proliferating CD4 T cells. These findings demonstrate that T cell colitogenicity is achieved through multiple processes, offering a therapeutic rationale by intervening these pathways.
Keywords: α4β7 integrin, CD4 T cells, colitis, gut antigens, IBD
Introduction
Immune reactions at the intestinal mucosal interface are tightly regulated not only to prevent unnecessary activation of immune cells against innocuous commensal antigens (Ag) but also to mount protective immune responses to pathogen associated Ag. Dysregulation of the processes is thought to lead to chronic inflammation seen in inflammatory bowel disease (IBD). However, what initiates the inductive processes and the precise features of effector T cells that mediate the inflammation remain unclear.
Experimentally, T cell-mediated experimental colitis is a widely used murine model to study pathogenesis of IBD 1, 2. Naïve CD4 T cells transferred into lymphopenic recipients undergo spontaneous proliferation and concomitantly differentiate into effector cells producing proinflammatory cytokines, mainly IFNγ and IL-17 2, 3. The proliferation is induced in response to Ag derived from commensal organisms as well as self 4. Gut draining mesenteric LN (mLN) is a major site where the proliferation takes place and potential colitogenic T cells are generated. Expression of gut homing α4β7 integrin, also known as the lymphocyte Peyer's patch adhesion molecule (LPAM1) is thought to be an essential step for effector cells to enter the intestinal mucosa 5. It was reported that dendritic cells (DC) from the gut draining lymphoid tissues including mLN and Peyer's patches, or from small intestine lamina propria (LP) play a key role in generating gut-tropic effector cells 6-8. The vitamin A metabolite, retinoic acid (RA) plays an additional role in upregulating α4β7 on T cells 9. Among DCs in the mLN, CD103+ subsets were shown to express retinal dehydrogenase 2 (RALDH2), a key enzyme to synthesize RA, and to enhance gut homing molecule expression in activated T cells 10. However, mechanisms underlying the generation of gut Ag specific colitogenic T cells in vivo remain unclear.
Both Th1 and Th17 type effector cells have been implicated in the pathogenesis of intestinal inflammation 1, 11, 12. We previously reported that γδ T cells play an additional role in generating colitogenic T cells in part by enhancing Th17 differentiation 3. Thus, lymphopenic mice harboring γδ T cells were highly susceptible while lymphopenic mice deficient in γδ T cells were resistant to the disease despite the fact that the overall T cell proliferation and expansion were comparable. Particularly interesting is that IL-17 production of γδ T cells appears to be directly linked to the Th17-promoting functions 13. IL-17-producing γδ T cell subsets are phenotypically distinct from IFNγ-producing γδ T cell subsets. They express receptor for IL-23, a cytokine known to stimulate γδ T cells to produce IL-17 13-17. Whether IL-23 is important for Th17-promoting γδ T cell functions during T cell-induced colitis has not formally been tested.
Here, we demonstrate that gut homing proinflammatory effector cells are preferentially generated within the gut draining mLN via CD11b+ DCs. Expression of the gut homing molecule, α4β7 integrin, was necessary to induce inflammation, although α4β7 expression itself was not sufficient to do so. Both T cell reactivity against fecal extract Ag and proinflammatory cytokine production were essential features of colitogenic T cells, and the reactivity was enhanced by the presence of γδ T cells stimulated by IL-23. Our results highlight multiple pathways through which Th17 type colitogenic cell generation is regulated.
Results
CD4 T cells undergoing spontaneous proliferation upregulate α4β7 integrin expression in the mLN
α4β7 integrin expression is essential for activated T cells to enter the gut tissues. Targeting α4β7-mediated trafficking has thus been an efficacious approach to attenuate intestinal inflammation in inflammatory bowel disease as well as graft-versus-host diseases 18, 19. Using a model of spontaneous T cell proliferation, a robust T cell response that occurs under lymphopenic conditions leading to a chronic intestinal inflammation we examined the role of α4β7 expression in activated T cells during T cell-mediated intestinal inflammation 20. Naïve CD4 T cells were transferred into T cell-deficient TCRβ-/- recipients and examined for α4β7 expression. Consistent with the previous results 21, α4β7+ CD4 T cells were primarily generated in the gut-draining mesenteric LN (mLN), while the proportion of α4β7+ cells in the peripheral skin-draining peripheral LN (pLN) was very low (Fig 1A). Preferential accumulation of α4β7+ T cells in the mLN was still pronounced 3 weeks post transfer (data not shown). Since activated T cells recirculate, we treated the recipients with FTY720, a S1P1 receptor agonist that blocks lymphocyte egress from the lymphoid tissues 22, to localize the precise location where activated T cells acquire α4β7 expression in vivo. TCRβ-/- mice were treated daily with FTY720 starting at ∼16 hours after T cell transfer. A 16-hour lag was given to allow the T cells to engage with APCs to receive proper stimulation signals 23. As shown in Fig 1B, FTY720 treatment further increased the accumulation of α4β7+ T cells in the mLN, while their numbers in the pLN significantly decreased, a finding consistent with a study recently reported by Ishii and colleagues 21. Therefore, α4β7 acquisition of activated T cells mainly occurs in the mLN, suggesting that APCs residing within the mLN preferentially imprint gut-homing specificity by upregulating α4β7. Of note, the extent of T cell proliferation between the two sites (i.e., pLN vs. mLN) was comparable, indicating that the differential expression of α4β7 in T cells is not due to the level of T cell activation (not shown).
Figure 1. α4β7 expression on CD4 T cells was enhanced in mesenteric lymph node.
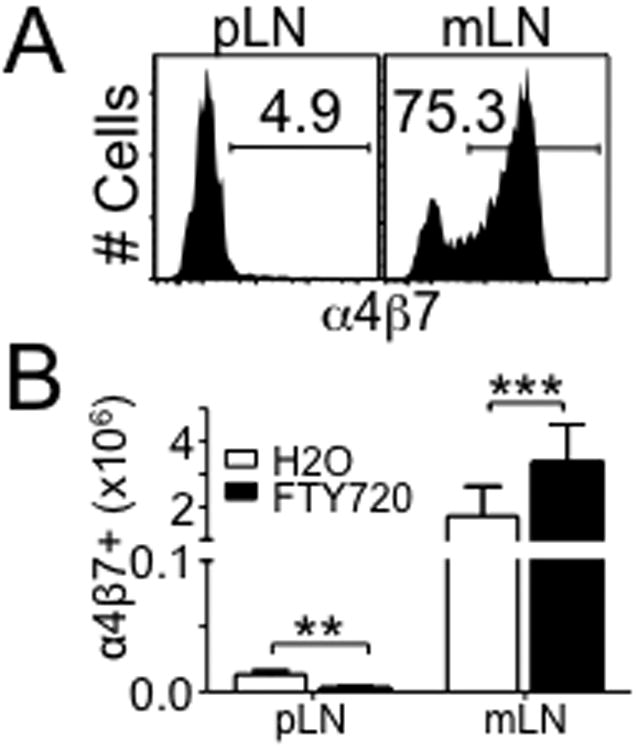
(A) α4β7 expression on CD4 T cells at day 5 following CD4 T cells adoptive transfer to TCRβ-/- mice. Data shown are representative of individually tested recipients. All experiments were repeated more than three times and similar results were observed. (B) Total number of α4β7+ CD4 T cells from the indicated organ after FTY720 treatment. Data shown are the mean ± SD of 6 mice in two independent experiments. **, p<0.01; ***, p<0.001.
CD11b+ DCs within the mLN upregulate α4β7 via TGFβ̃ and retinoic acid-dependent mechanism
To directly examine if APCs residing within the mLN are responsible for α4β7 upregulation, whole pLN and mLN cells isolated from TCRβ-/- mice were used as APCs to stimulate OVA specific OT-II CD4 T cells with OVA peptide in vitro. Consistent with the in vivo results (Fig 1), cells from mLN were highly efficient in generating α4β7+ OT-II T cells (Fig 2A). Specifically, we noticed that adding recombinant TGFβ alone significantly increased α4β7 upregulation (∼30%, Fig 2A), which was further increased to ∼50% by adding TGFβ and IL-6 (Fig 2A). The generation of α4β7+ T cells without these cytokines was very low (Fig 2A). Interestingly, cells from the pLN were still unable to generate α4β7+ OT-II T cells in the presence of both TGFβ and IL-6 (Fig 2A). T cell proliferation and CD44 upregulation were comparable between the conditions, indicating that the differential α4β7 expression is not due to activation status. Importantly, T cell production of IL-17 was efficiently induced regardless of the origin of APCs (data not shown), indicating that the pLN APCs are functionally equivalent to the mLN APCs in activating Ag specific T cells. mLN cells from TCRβ-/- and Rag-/- mice were equivalent in upregulating α4β7 expression in cocultured OT-II cells, suggesting that B cells are dispensable (Fig 2B). Vitamin A metabolite RA has been shown to be critical in inducing α4β7 expression in activated T cells 9, 24. Consistent with this, adding RA receptor antagonist LE540 completely abolished the α4β7 expression (Fig 2C), suggesting that RA produced by mLN DCs plays a key role in mLN APC-mediated expression of α4β7. The level of overall T cell activation was comparable in these conditions (data not shown). We set out to further examine whether there are specific APC subsets among the mLN cells highly specialized in inducing α4β7 expression. Different DC subsets from the mLN were thus isolated and cocultured with OT-II cells. We found that CD11b+ DCs were the major cell type inducing α4β7 expression (Fig 2D). On the other hand, CD11b+ macrophages and other DC subsets including CD8+ DCs or CD11b− CD8− DCs were unable to upregulate α4β7 (Fig 2D). It was previously reported that gut homing α4β7+ CD8 T cells are preferentially generated by CD103+ DCs 25 but that induction of α4β7+ on CD4 T cells is equally induced by both CD103+ and CD103− DCs 26. When CD103 expression of different mLN DCs was compared, the proportion of CD103+ DCs was comparable between the subsets (Fig 2E). Therefore, CD11b+ DC subsets appear to be a unique population that induces gut homing specificity during spontaneous proliferation.
Figure 2. mLN CD11c+ CD11b+ cells induced α4β7 expression on T cells dependent on retinoic acid.
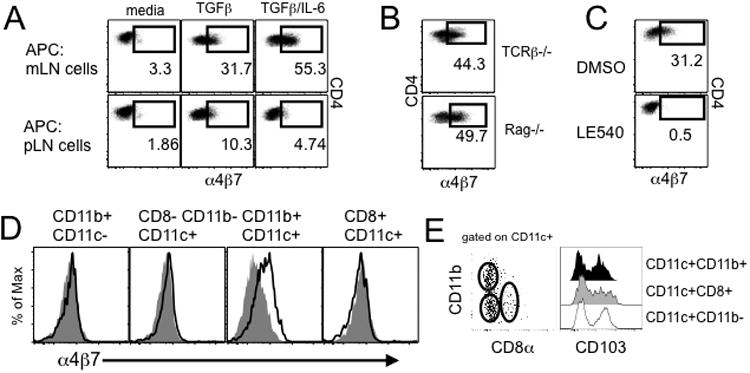
(A) OVA-specific OT-II T cells were cocultured with cells from the indicated tissues in the presence of TGFβ and IL-6. α4β7 expression on OT-II cells was measured after 3 days of culture. The experiments were repeated five times and similar results were observed. (B) OT-II T cells were stimulated with mLN cells from TCRβ-/- or Rag-/- mice. α4β7 expression on OT-II cells was determined. Plots are representative of at least three independent experiments. (C) OT-II cell/mLN cell coculture was repeated in the presence of LE540 or control vehicle. α4β7 expression was similarly measured as above. The experiments were repeated three times and similar results were observed. (D) mLN cells of the indicated phenotypes were FACS sorted and cocultured with OT-II T cells in the presence of Ag. Filled histogram represents α4β7 expression without Ag. The experiments were repeated twice and similar results were observed. *, p<0.05; **, p<0.01. (E) CD103 expression on mLN DC subsets. Data are representative of at least three independent experiments.
α4β7+ CD4 T cells inducing intestinal inflammation display gut Ag reactivity
CD4 T cell expression of α4β7 is essential for activated T cells to adhere MAdCAM (and/or VCAM1) and enter the gut tissues 27. Indeed, α4β7 expression in T cells was directly associated with colitogenic potential. α4β7+ or α4β7− CD4 T cells were isolated from the mLN of TCRβ-/- mice that had received CD4 T cells 3 weeks earlier and subsequently transferred into naïve TCRβ-/- recipients. α4β7+ T cell recipients exhibited severe weight loss and colonic inflammation, while α4β7− T cell recipients did not show any signs of weight loss and intestinal inflammation (Fig 3A and 3B). Consistent with this, the accumulation of α4β7+ T cell subsets in the mLN and colon LP was greater than that of α4β7− cells (Fig 3C), confirming the importance of α4β7 expression in T cell entry to the intestine. Accumulation of IL-17+ donor T cells was significantly higher in both mLN and colon LP of α4β7+ T cell recipients (Fig 3C). Notably, II17a expression of α4β7+ and α4β7− T cells was comparable when measured prior to transfer (Fig 3D). These results suggest that the different susceptibility to the inflammation of α4β7+ and α4β7− T cells is not associated with Th17 effector phenotypes of the donor T cells.
Figure 3. More severe inflammation develops in TCRβ-/- mice by α4β7+ CD4 T cells compare to α4β7− CD4 T cells.
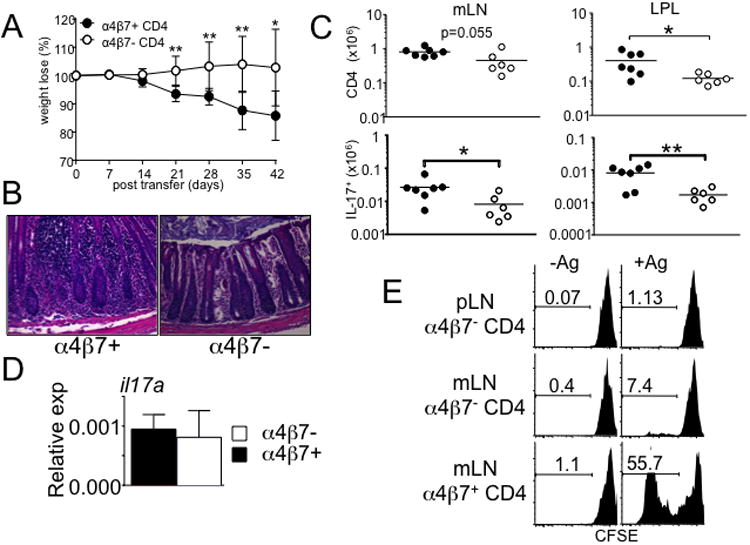
Naïve CD4 T cells were transferred into TCRβ-/- mice. α4β7+ and α4β7− donor T cells were isolated from the mLN 3 weeks post transfer, and retransferred into naïve TCRβ-/- recipients. (A) Weight loss and (B) colon histology in TCRβ-/- mice at 6 weeks post-transfer of α4β7+ or α4β7− CD4 T cells. All images at 20× magnification. Data shown are the mean ± SD. Experiments were repeated twice and similar results were observed. *, p<0.05; **, p<0.01. (C) Absolute number of the total cells and IL-17-producing donor T cells from the indicated organ are shown. Data are mean ± SD of 6-7 mice from two independent experiments. *, p<0.05; **, p<0.01. (D) Real time quantitative PCR analysis of il17a expression on α4β7+ and α4β7− CD4 T cells prior to the transfer. All samples were done in duplicates and normalized to GAPDH. Data shown are representative from 6-7 individually tested mice. (E) α4β7+ and α4β7− donor T cells were reisolated from the mLN and pLN, CFSE labeled, and cocultured with APCs pulsed with fecal extract Ag as described in Methods. CFSE dilution was determined by FACS analysis. Experiments were repeated twice, and similar results were obtained.
Because T cell-induced colitis is attenuated in germ-free conditions 28, gut homing colitogenic T cells are likely reactive to commensal Ag. We thus examined gut Ag reactivity of α4β7+ and α4β7− T cells. We speculated that if α4β7-dependent trafficking is the primary mechanism underlying inflammation, then both α4β7+ and α4β7− T cells would display similar Ag reactivity. It was previously reported that CD4 T cells from SCID mice with colitis proliferate strongly in response to APCs pulsed with fecal extracts 29. α4β7+ and α4β7− cells were thus isolated from the mLN and pLN of TCRβ-/- mice following naïve T cell transfer, CFSE labeled, and cocultured with APCs pulsed with fecal extract Ag. Ag reactivity was determined by proliferative responses. As demonstrated in Fig 3E, mLN α4β7+ T cells strongly proliferated in response to fecal Ag stimulation. To our surprise, α4β7− T cells isolated from the mLN did not exhibit fecal Ag reactivity (Fig 3E). Moreover, α4β7− T cells isolated from the pLN did not respond to the stimulation. All tested cells did not proliferate without fecal Ag pulse, indicating an Ag-induced response (Fig 3E), and the proliferation was blocked by anti-MHCII Ab (data not shown). These results strongly suggest that α4β7 expression appears to be associated with gut Ag reactivity, linking to colitogenicity. Since α4β7 expression is mainly induced by CD11b+ DCs (Fig 2D), these results also suggest that CD11b+ DCs may present gut Ag to T cells, generating colitogenic effector cells.
γδ T cells are not necessary to upregulate α4β7 expression but are important to generate gut Ag reactive colitogenic cells
We previously reported that γδ T cells enhance Th17 differentiation and T cell mediated intestinal inflammation 3. Based on the notion that α4β7 expression and gut Ag reactivity are required for colitogenicity (Fig 3), we next examined whether γδ T cells influence α4β7 expression and gut Ag reactivity of T cells. We first transferred naïve CD4 T cells into TCRβ-/- and TCRβδ-/- recipients, the former develop severe colitis while the latter only develop mild inflammation without overt weight loss 3. Regardless of susceptibility to the colitis, the transferred T cells in the mLN of both TCRβ-/- and TCRβδ-/- recipients efficiently upregulated α4β7 (Fig 4A), indicating that γδ T cells are not necessary for the generation of gut homing effector cells. Consistent with our previous findings 3, α4β7+ T cells isolated from TCRβ-/- mice highly expressed Th17 type cytokines compared to the same phenotype cells isolated from TCRβδ-/- mice (Fig 4B). Other cytokines tested including IL-21, TNFα, GM-CSF, and IFNγ were similarly expressed (Fig 4B). Therefore, T cells primed in the presence of γδ T cells better become IL-17-producing effector cells. α4β7+ T cells were then isolated from the mLN of each group and transferred into naïve ‘colitis-susceptible’ TCRβ-/- mice. α4β7+ T cells isolated from TCRβ-/- mice induced severe weight loss as shown above, whereas α4β7+ T cells isolated from TCRβδ-/- mice were unable to induce noticeable weight loss (Fig 4C). The colon tissue of TCRβ-/- originated α4β7+ T cell recipients was heavily infiltrated with inflammatory cells while that of TCRβδ-/- originated α4β7+ T cell recipients displayed only mild inflammation (Fig 4D). Consistent with the disease progression and intestinal inflammation, donor cell accumulation in both lymphoid and intestinal tissues were significantly greater in recipients of TCRβ-/- derived α4β7+ T cells (Fig 4E). Likewise, the level of IL-17-producing T cells in the mLN and colon was significantly higher in TCRβ-/- originated α4β7+ T cell recipients (Fig 4F).
Figure 4. α4β7+ CD4 cells from TCRβ-/- mice display severe colitogenicity compared to α4β7+ CD4 cells from TCRβδ-/- mice.
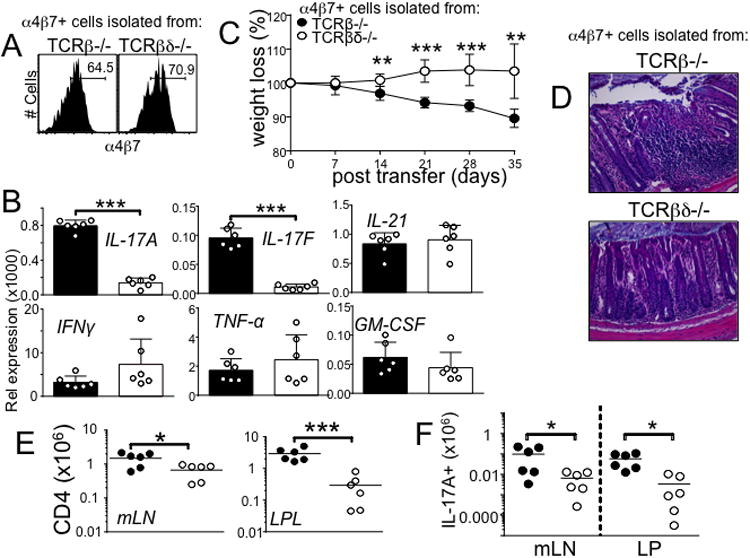
(A) α4β7 expression on CD4 T cells at day 21 following CD4 T cells adoptive transfer to TCRβ-/- and TCRβδ-/- mice. Data shown are representative from 6 individually tested mice in two independent experiments. (B) Real time quantitative PCR analysis of pro-inflammatory gene expression of α4β7+ CD4 T cells. The expression was normalized to GAPDH. Data shown are representative from 6 individually tested mice. ***, p<0.001. (C) Weight loss and (D) colon histology in TCRβ-/- recipient mice at 5 weeks after post-transfer of α4β7+ CD4 T cells from TCRβ-/- or TCRβδ-/- mice. All images at 20× magnification. Data are representative of two independent experiments. **, p<0.01; ***, p<0.001. (E) Absolute number of total cells and (F) IL-17-producing cells from the indicated tissues. Data are mean ± SD of 6 individually tested mice. *, p<0.05; ***, p<0.001.
γδ T cells directly promote the generation of gut Ag reactive effector cells
Our results demonstrate that α4β7+ cells generated without γδ T cells do not express Th17 type cytokines and induce colitis, suggesting a possibility that the lack of Th17 type cytokine expression could be the main reason that these cells are non-colitogenic. Alternatively, γδ T cells may control T cell acquisition of gut Ag reactivity. To test this possibility we harvested α4β7+ T cells from TCRβ-/- and TCRβδ-/- recipients 3 weeks post transfer and cocultured with splenic DCs pulsed with fecal extracts as demonstrated in Fig 3E. CD4 T cells harvested from TCRβ-/- mice dramatically proliferated when cocultured with APCs pulsed with fecal extract Ag, while T cells harvested from TCRβδ-/- mice failed to proliferate in the same conditions (Fig 5A). T cell proliferation was completely blocked by anti-MHCII (Y3P) mAb (Fig 5A). The proliferating T cells produced IL-17A (Fig 5B) and IFNγ (data not shown). Therefore, γδ T cells may enhance generation of gut Ag reactive colitogenic cells.
Figure 5. α4β7+ CD4 T cells from TCRβ-/- recipients strongly respond to fecal extract antigen stimulation ex vivo.
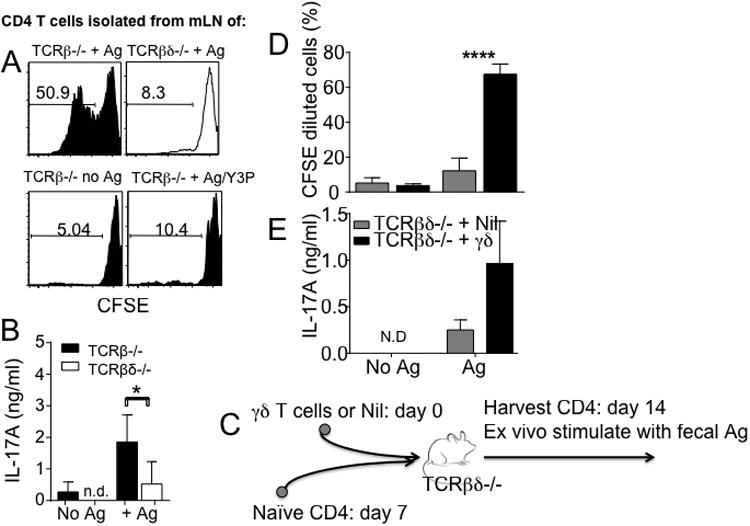
(A) Following naïve CD4 T cell transfer into TCRβ-/- or TCRβδ-/- mice, α4β7+ CD4 cells were isolated from individual recipient mice, CFSE labeled, and cocultured with fecal extract Ag pulsed APCs. CFSE dilution was analyzed at day 5 after stimulation. (B) IL-17 production in the culture supernatant was determined by ELISA. Data are shown of 7-12 individually tested mice in three independent experiments. (C) Experimental model. γδ T cells isolated from the lymphoid tissues were transferred into TCRβδ-/- mice. Seven days later, FACS sorted naïve CD4 T cells were transferred into the same recipients. The donor CD4 T cells were isolated from the mLN 7 days post T cell transfer, and cocultured with fecal extract Ag pulsed APCs. (D) CFSE dilution and (E) IL-17 production in the culture supernatant were determined. Experiments were repeated twice, and similar results were obtained. *, p<0.05; ****, p<0.0001.
To directly test this possibility in vivo we adoptively transferred purified lymphoid γδ T cells (isolated from the secondary lymphoid tissues) into TCRβδ-/- mice. FACS purified naïve CD4 T cells were then transferred into the recipients 7 days post γδ T cell transfer as illustrated in Fig 5C. The donor CD4 T cells were subsequently isolated from the mLN of recipients 7 days following CD4 T cells transfer, and cocultured with fecal extract Ag pulsed APCs. A control group of TCRβδ-/- mice received naïve CD4 T cells without γδ T cell transfer (Nil group in Fig 5C), and CD4 T cells were used for coculture experiments. CD4 T cells activated in the presence of γδ T cells transferred 7 days earlier exhibited a significant gut Ag reactivity based on CFSE dilution (Fig 5D) as well as IL-17 production (Fig 5E). By contrast, CD4 T cells activated without γδ T cells did not proliferate or produce IL-17 (Fig 5D and 5E). Therefore, γδ T cells appear to directly enhance the generation of gut Ag reactive IL-17-producing colitogenic T cells in vivo.
IL-23 mediated stimulation of γδ T cells is important for Th17 differentiation but not for gut Ag reactivity
γδ T cells express IL-23 receptor 30, 31 and are the first cells to respond to IL-23 during EAE 16. IL-23 induces IL-17 production by γδ T cells, amplifying proinflammatory Th17 type immune responses 16, 32. Whether IL-23-induced γδ T cell activation is needed to support colitogenic T cell development was thus examined. TCRβδ-/- mice received naïve CD4 T cells together with wild type or IL-23R-/- γδ T cells. Consistent with the previous report 13, CD4 T cells transferred into TCRβδ-/- mice did not induce weight loss or intestinal inflammation (Fig 6A and data not shown). Wild type γδ T cells cotransferred completely restored T cell induced colitis (Fig 6A). By contrast, transfer of IL-23R-/- γδ T cells did not induce any sign of weight loss (Fig 6C), indicating that IL-23-dependent signal of γδ T cells plays a key role in generating colitogenic T cell responses. Transfer of wild type γδ T cells enhanced expression of Th17 type cytokines as well as cytokines that induce Th17 differentiation such as IL-1β and IL-6 (Fig 6B). On the other hand, the presence of IL-23R-/- γδ T cells completely failed to induce the expression (Supplementary Fig S1).
Figure 6. IL-23-dependent stimulation of γδ T cells is required to enhance colitogenic CD4 T cell development.
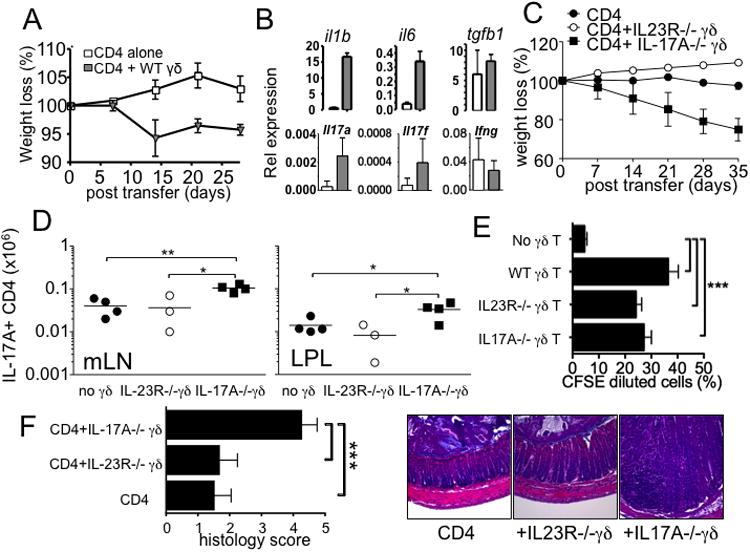
(A and C) FACS sorted CD4 T cells and γδ T cells (wild type, IL-23R-/-, and IL-17A-/-) were cotransferred into TCRβδ-/- mice, and weight loss was weekly monitored. (B) Expression of the indicated genes from CD4 alone (open bar) and CD4 plus wild type γδ T cells (grey bar) was determined by real time PCR analysis. (D) IL-17-producing donor CD4 T cells in the indicated tissues were determined 5 weeks post transfer. (E) Wild type, IL-23R-/-, and IL-17A-/- γδ T cells were transferred into TCRβδ-/- mice followed by naïve CD4 T cell transfer 7 days later as described in Fig 5. Donor CD4 T cells were isolated from the recipients, and cocultured with fecal extract Ag pulsed APCs as described in Fig 5. Experiments were repeated more than twice with similar results. (F) The recipients described above were sacrificed and the colon tissues were H&E stained for histopathology. The colitis score was measured as described in Methods. *, p<0.05; **, p<0.01; ***, p<0.001.
We and others previously reported that IL-17-producing γδ T cell subsets highly express IL-23 receptor 13, 14. Supportingly, IL-17-producing γδ T cells better restored T cell colitogenicity in TCRβδ-/- transfer model 13. To directly test if IL-17 production by γδ T cells is directly involved in Th17 differentiation as well as colitogenicity, we transferred IL-17A-/- γδ T cells into TCRβδ-/- mice together with naïve CD4 T cells. As shown in Fig 6C, we found that TCRβδ-/- mice that received IL-17A-/- γδ T cells rapidly lost body weight, similar to the results obtained from wild type γδ T cell transfer experiments shown in Fig 6A. Moreover, analyzing Th17 differentiation demonstrated that the generation of IL-17-producing CD4 T cells was efficiently enhanced when IL-17A-/- γδ T cells were cotransferred (Fig 6D). By contrast, Th17 differentiation in mice that received IL-23R-/- γδ T cells remained significantly low (Fig 6D). Therefore, IL-23-dependent γδ T cell activation seems critical to restore Th17 differentiation and colitogenic effector cell generation, although IL-17A produced by the activated γδ T cells appears to be dispensable.
We finally determined whether IL-23-dependent γδ T cell activation influences gut Ag reactivity of CD4 T cells. We performed adoptive transfer experiments described in Fig 5C using IL-23R-/- and IL-17A-/- γδ T cells. As demonstrated in Fig 6E, CD4 T cells activated with wild type γδ T cells underwent significant proliferation as determined by CFSE dilution. Likewise, CD4 T cells activated with IL-23R-/- or IL-17A-/- γδ T cells, although they failed to induce colitis, displayed similar gut Ag reactivity (Fig 6E). Consistent with the clinical score, the colon of IL-17A-/- γδ T cells was heavily infiltrated with inflammatory cells, while those recipients of CD4 T cells alone or together with IL-23R-/- γδ T cells exhibited only a mild inflammation (Fig 6F).
Discussion
During spontaneous proliferation, colitogenic effector T cells are generated and severe intestinal inflammation develops. However, the precise features for effector T cells to be colitogenic have not been formally explored. The present study aims to examine characteristics of colitogenic T cells and mechanisms by which they are generated.
α4β7-dependent gut trafficking is essential for colitogenicity. Consistent with a recent report 21, T cell upregulation of α4β7 gut homing integrin was only noted in the gut-draining mLN, whereas T cells similarly proliferating in the peripheral LN failed to do so. The upregulation was TGFβ/RA dependent. The finding that the upregulation is most pronounced when T cells are stimulated by CD11b+ DCs strongly suggests that these DC subsets may be the source of TGFβ and/or RA. It was recently reported that IRF4-dependent CD103+CD11b+ DCs represent the major migratory DCs in the LP that drive Th17 differentiation 33, although no correlation between CD103 expression and DC functions to upregulate α4β7 expression was observed from the current study. Whether CD103+CD11b+ DCs and CD103-CD11b+ DCs function differently in upregulating α4β7 expression and generating colitogenic T cells remains to be examined. It is interesting to note that α4β7 expression on T cells within the draining mLN is not associated with effector phenotype, Th17. When IL-17 expression of CD4 T cells that do or do not acquire α4β7 expression was compared, both populations were equally capable of expressing IL-17, indicating that Th17 differentiation during spontaneous proliferation is independent of α4β7 expression.
Differential ability for α4β7+ T cells to become colitogenic may be linked to their Ag reactivity. We directly tested this question by ex vivo coculturing in vivo activated effector cells with APCs pulsed with fecal extract Ag. Supporting the hypothesis, α4β7+ T cells generated within the mLN strongly reacted to gut Ag stimulation, while α4β7- T cells generated in the same mLN failed to respond to the stimulation despite that their Th17 differentiation is not affected by the lack of α4β7 expression. Based on these findings, we would argue that there are CD11b+ DC subsets preferentially presenting colitogenic gut Ag to naïve T cells and providing necessary signals to generate gut tropic effector cells 34. Indeed, we observed that after intraluminal Ag administration mLN DC subsets that present the Ag were of CD11b+ subsets (Freeman and Min, unpublished observation). Of note, Matsuda et al. previously found a highly restricted repertoire diversity of T cells that expanded within immunodeficient mice, concluding that the expansion of T cells requires the activation of Ag-specific T cells 35. The concept of Ag-specific colitogenic T cell responses agrees with the gut Ag-reactivity reported in this study. When we compared TCRβ repertoire diversity of T cells generated with or without γδ T cells by FACS analysis, we found that the TCR Vβ distribution between the conditions was indistinguishable (Do and Min, unpublished observation). Sequence analysis of responding cells may be necessary to directly compare their clonal diversity.
Of note, gut homing specificity alone was not sufficient to be colitogenic for those effector T cells generated during spontaneous proliferation. Following naïve T cell transfer α4β7+ T cells were equally generated within the mLN of TCRβδ-/- recipients, a condition resistant to T cell mediated colitis 3. When isolated and re-transferred into naïve susceptible TCRβ-/- recipients, α4β7+ T cells generated within TCRβδ-/- mice remained non-colitogenic, which was in good contrast to α4β7+ T cells generated within TCRβ-/- mice. Therefore, gut homing T cells generated in susceptible and resistant lymphopenic conditions appear to be distinct, particularly in regard to Ag specificity, since T cell reactivity to commensal Ag is thought to be essential for colitogenicity 4, 36. In support with this, α4β7+ T cells generated within susceptible TCRβ-/- mice displayed a strong reactivity to ex vivo stimulation with fecal extract Ag, while α4β7+ T cells generated within resistant TCRβδ-/- mice did not exhibit such reactivity. Therefore, resistance vs. susceptibility may be determined at the level of Ag specificity of spontaneously proliferating T cells.
A mechanism behind the induction of effector cells with different Ag specificity is unclear. It is possible that Ags presented possibly by CD11b+ DCs may be different depending on the recipients. Alternatively, stimulatory functions of DCs to prime gut Ag specific T cells could be different in these conditions. Since the presence of γδ T cells is the major difference between these conditions, we propose that γδ T cells may be responsible for enhancing gut Ag uptake and/or migration of gut Ag bearing APCs to the draining mLN. Indeed, γδ T cells pretransferred into TCRβδ-/- mice were sufficient to restore gut Ag specificity of proliferating T cells and colitogenicity. How γδ T cells exert such immunoregulatory functions remains to be determined. It was previously reported that γδ T cells can promote DC maturation via CD40L-CD40 interaction 37. It will be interesting to examine if such cell-to-cell interaction operates in this setting.
It is important to emphasize that IL-23 stimulation of γδ T cells plays an important role in this process. This is based on the observation that CD4 T cells activated in the presence of γδ T cells deficient in IL-23R fail to restore colitis. Interestingly, however, the roles for γδ T cells in inducing gut Ag reactive T cells appear to be dissociated from the role for enhancing generation of colitogenic Th17 type cells. When IL-23R-/- γδ T cells are transferred into TCRβδ-/- mice, gut Ag reactivity is efficiently acquired although both Th17 differentiation and colitogenicity are not observed. IL-23R is highly expressed on IL-17-producing γδ T cell subsets 13, and IL-23 stimulation induces γδ T cell IL-17 production 14, 32. Yet, IL-17A produced by activated γδ T cells is not involved in this process, since both Th17 differentiation and colitis induction efficiently occurred in IL-17A-/- γδ T cell recipients. We previously reported that only CCR6+ but not CCR6- γδ T cells are capable of promoting Th17 differentiation and colitis 13. CCR6+ γδ T cells produce both IL-17A and IL-17F. Moreover, IL-23 stimulation of γδ T cells induce production of both IL-17A and IL-17F (data not shown). Therefore, IL-17F produced by activated γδ T cells may mediate such regulatory functions. It is also possible that IL-17A (or IL-17F) produced by γδ T cells also directly or indirectly supports APC functions to enhance gut Ag reactive T cell generation.
Overall, we identified that T cell expression of the gut tropic adhesion molecule α4β7, gut Ag specificity, and Th1/Th17 phenotype are required to mediate severe colitis and that γδ T cells play a crucial role in this process in part by IL-23. Future investigation should focus on a cellular pathway to develop a better therapeutic strategy to intervene chronic intestinal inflammation such as IBD.
Methods
Mice
C57BL/6, C57BL/6-Rag1-/-, CD45.1 C57BL/6, C57BL/6 TCRβ-/-, and C57BL/6 TCRβδ-/- mice were purchased from the Jackson Laboratory (Bar Harbor, ME). CD45.1 C57BL/6 RAG1-/- OT-II mice were kindly provided from Dr. William Paul (NIH). IL-23R-/- and IL-17-/- mice were previously reported 38, 39. All the mice were maintained under specific pathogen free facility located in the Lerner Research Institute. All animal experiments were performed in accordance with approved protocols for the Cleveland Clinic Foundation Institutional Animal Care and Usage Committee.
Cell sorting and colitis induction
Whole LN naive CD4 T cells were obtained as previously reported 40. In brief, LN cells (axillary, cervical, inguinal, and mesenteric LN) were pooled and total T cells were negatively purified through a magnetic separation. Cells were stained with FITC-conjugated anti-B220, anti-FcγR, anti-NK1.1, and anti-MHC II Abs (all purchased from eBioscience, San Diego, CA). FITC-labeled LN cells were subsequently incubated with anti-FITC-microbeads (Miltenyi, Auburn, CA) and passed through a LS column (Miltenyi). CD25negCD44low naive T cells were further sorted using a FACSAria cell sorter (BD Bioscience, San Jose, CA). 2.5 × 105 naive CD4 T cells were transferred to TCRβ-/- and TCRβδ-/- mice. After T cell transfer, mice were weighed weekly and monitored for signs of disease. In some experiments, α4β7+ CD4 T cells were sorted from TCRβ-/- or TCRβδ-/- recipients at day 7 and 21 after transfer and transferred to naïve TCRβ-/- recipients for colitis induction. γδ T cells were isolated from the secondary lymphoid tissues (LN and spleen) by cell sorting, and adoptively transferred (2 × 106 cells) where indicated.
In vitro OT-II T cell stimulation
Ovalbumin peptide specific TCR transgenic CD4 T cells were obtained from CD45.1 OT-II TCR Tg Rag-/- mice. 2×105 naïve OT-II T cells were cultured with 2 × 105 antigen-presenting cells (APC) in the presence of 1μg/ml OVA peptide323-339. 5 ng/ml human rTGF-β (Peprotech) and 10 ng/ml rIL-6 (Peprotech) were added in the culture. Cultures were incubated for 5 days and cells were analyzed for α4β7 expression. In some in vitro experiments, 1 μM RA receptor antagonist LE540 (Wako Chemical USA) was added.
Fecal antigen stimulation
Fecal extract antigen was prepared from C57BL/6 TCRβ-/- mice as described in previous study 29. Sorted α4β7+ CD4 T cells were labeled with CFSE. T cells were cultured with 0.3mg fecal antigen pulsed APC at a 1:1 ratio. After 5 days, CFSE dilution was assessed by FACS.
FACS Analysis
Cells were stained with anti-CD4 (RM4-5), anti-CD11b (M1/70), anti-CD11c (N418), anti-CD8 (53-6.7), anti-IL-17A (eBio17B7), anti-IFNγ (XMG1.2), anti-I-Ab (AF6-120.1), anti-CD45.1 (A20), anti-CCR9 (ebioCW-1.2), anti-α4β7 (DATK32) (all Abs from eBioscience). Cells were acquired using a FACSCalibur or LSR II (BD Biosciences) and analyzed using a FlowJo software (Treestar, Ashland, OR). For Intracellular staining, cells were separately harvested and ex vivo stimulated with PMA (10 ng/ml) and Ionomycin (1 μM) for 4 hrs in the presence of 2 ⌠M monensin (Calbiochem, San Diego, CA) during the last 2 hrs of stimulation. Cells were immediately fixed with 4% paraformaldehyde, permeabilized, and stained with fluorescence conjugated antibodies.
Real time quantitative PCR
FACS sorted cells or colon tissue was disrupted using a TissueLyser II (Qiagen, Valencia, CA). Total RNA was extracted using an RNeasy column (Qiagen, Valencia, CA). cDNA was subsequently synthesized using a SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA). Real time quantitative PCR was performed using gene specific primers and probe sets (Applied Biosystem, Foster City, CA) and ABI 7500 PCR machine (Applied Biosystem).
Histology
Colon tissues were fixed in 10% acetic acid/60% methanol. Slides were cut and stained with H&E. Colon tissues were scored in a blinded fashion as previously reported41 by two individuals and scores were averaged. In brief, colon tissues were assessed at both low and high magnification to get an overall score using the scoring system: 0: no sign; 1: low infiltration and inflammation; 2: medium infiltration/inflammation; 3: high infiltration/inflammation; 4: severe infiltration with moderate loss of goblet cells and crypt structure; 5: transmural infiltration, loss of goblet cells and crypt structure.
Statistical analysis
Statistical significance was determined by the Student's t-test using the Prism 5 software (GraphPad, La Jolla, CA). A p value of <0.05 was considered statically significant.
Supplementary Material
Acknowledgments
We would like to thank Ms. Jennifer Powers for technical assistance for cell sorting.
This study was supported by NIH grant AI074932 (to B.M.) and T32-GM88088 (A.V.).
Abbreviations used in this article
- Ag
antigens
- DC
dendritic cells
- IBD
inflammatory bowel disease
- LP
lamina propria
- mLN
mesenteric lymph node
- RA
retinoic acid
References
- 1.Powrie F, Leach MW, Mauze S, Menon S, Caddle LB, Coffman RL. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1994;1:553–62. doi: 10.1016/1074-7613(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 2.Ostanin DV, Bao J, Koboziev I, Gray L, Robinson-Jackson SA, Kosloski-Davidson M, et al. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am J Physiol Gastrointest Liver Physiol. 2009;296:G135–46. doi: 10.1152/ajpgi.90462.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Do JS, Visperas A, Dong C, Baldwin WM, 3rd, Min B. Cutting edge: Generation of colitogenic Th17 CD4 T cells is enhanced by IL-17+ gammadelta T cells. J Immunol. 2011;186:4546–50. doi: 10.4049/jimmunol.1004021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kieper WC, Troy A, Burghardt JT, Ramsey C, Lee JY, Jiang HQ, et al. Recent immune status determines the source of antigens that drive homeostatic T cell expansion. J Immunol. 2005;174:3158–63. doi: 10.4049/jimmunol.174.6.3158. [DOI] [PubMed] [Google Scholar]
- 5.Johansson-Lindbom B, Agace WW. Generation of gut-homing T cells and their localization to the small intestinal mucosa. Immunol Rev. 2007;215:226–42. doi: 10.1111/j.1600-065X.2006.00482.x. [DOI] [PubMed] [Google Scholar]
- 6.Johansson-Lindbom B, Svensson M, Wurbel MA, Malissen B, Marquez G, Agace W. Selective generation of gut tropic T cells in gut-associated lymphoid tissue (GALT): requirement for GALT dendritic cells and adjuvant. J Exp Med. 2003;198:963–9. doi: 10.1084/jem.20031244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, et al. Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature. 2003;424:88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- 8.Mora JR. Homing imprinting and immunomodulation in the gut: role of dendritic cells and retinoids. Inflamm Bowel Dis. 2008;14:275–89. doi: 10.1002/ibd.20280. [DOI] [PubMed] [Google Scholar]
- 9.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21:527–38. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 10.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–64. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–6. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elson CO, Cong Y, Weaver CT, Schoeb TR, McClanahan TK, Fick RB, et al. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology. 2007;132:2359–70. doi: 10.1053/j.gastro.2007.03.104. [DOI] [PubMed] [Google Scholar]
- 13.Do JS, Visperas A, Dong C, Baldwin WM, 3rd, Min B. Cutting edge: Generation of colitogenic Th17 CD4 T cells is enhanced by IL-17+ gammadelta T cells. J Immunol. 2011;186:4546–50. doi: 10.4049/jimmunol.1004021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cai Y, Shen X, Ding C, Qi C, Li K, Li X, et al. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity. 2011;35:596–610. doi: 10.1016/j.immuni.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kenna TJ, Davidson SI, Duan R, Bradbury LA, McFarlane J, Smith M, et al. Enrichment of circulating interleukin-17-secreting interleukin-23 receptor-positive gamma/delta T cells in patients with active ankylosing spondylitis. Arthritis Rheum. 2012;64:1420–9. doi: 10.1002/art.33507. [DOI] [PubMed] [Google Scholar]
- 16.Petermann F, Rothhammer V, Claussen MC, Haas JD, Blanco LR, Heink S, et al. gammadelta T cells enhance autoimmunity by restraining regulatory T cell responses via an interleukin-23-dependent mechanism. Immunity. 2010;33:351–63. doi: 10.1016/j.immuni.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sutton CE, Mielke LA, Mills KH. IL-17-producing gammadelta T cells and innate lymphoid cells. Eur J Immunol. 2012;42:2221–31. doi: 10.1002/eji.201242569. [DOI] [PubMed] [Google Scholar]
- 18.Feagan BG, Greenberg GR, Wild G, Fedorak RN, Pare P, McDonald JW, et al. Treatment of ulcerative colitis with a humanized antibody to the alpha4beta7 integrin. N Engl J Med. 2005;352:2499–507. doi: 10.1056/NEJMoa042982. [DOI] [PubMed] [Google Scholar]
- 19.Waldman E, Lu SX, Hubbard VM, Kochman AA, Eng JM, Terwey TH, et al. Absence of beta7 integrin results in less graft-versus-host disease because of decreased homing of alloreactive T cells to intestine. Blood. 2006;107:1703–11. doi: 10.1182/blood-2005-08-3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ostanin DV, Pavlick KP, Bharwani S, D'Souza D, Furr KL, Brown CM, et al. T cell-induced inflammation of the small and large intestine in immunodeficient mice. Am J Physiol Gastrointest Liver Physiol. 2006;290:G109–19. doi: 10.1152/ajpgi.00214.2005. [DOI] [PubMed] [Google Scholar]
- 21.Kawabe T, Sun SL, Fujita T, Yamaki S, Asao A, Takahashi T, et al. Homeostatic proliferation of naive CD4+ T cells in mesenteric lymph nodes generates gut-tropic Th17 cells. J Immunol. 2013;190:5788–98. doi: 10.4049/jimmunol.1203111. [DOI] [PubMed] [Google Scholar]
- 22.Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–60. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 23.Do JS, Foucras G, Kamada N, Schenk AF, Shaw M, Nunez G, et al. Both exogenous commensal and endogenous self antigens stimulate T cell proliferation under lymphopenic conditions. Cell Immunol. 2012;272:117–23. doi: 10.1016/j.cellimm.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim CH. Retinoic acid, immunity, and inflammation. Vitam Horm. 2011;86:83–101. doi: 10.1016/B978-0-12-386960-9.00004-6. [DOI] [PubMed] [Google Scholar]
- 25.Johansson-Lindbom B, Svensson M, Pabst O, Palmqvist C, Marquez G, Forster R, et al. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J Exp Med. 2005;202:1063–73. doi: 10.1084/jem.20051100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Annacker O, Coombes JL, Malmstrom V, Uhlig HH, Bourne T, Johansson-Lindbom B, et al. Essential role for CD103 in the T cell-mediated regulation of experimental colitis. J Exp Med. 2005;202:1051–61. doi: 10.1084/jem.20040662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berlin C, Berg EL, Briskin MJ, Andrew DP, Kilshaw PJ, Holzmann B, et al. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell. 1993;74:185–95. doi: 10.1016/0092-8674(93)90305-a. [DOI] [PubMed] [Google Scholar]
- 28.Powrie F, Mauze S, Coffman RL. CD4+ T-cells in the regulation of inflammatory responses in the intestine. Res Immunol. 1997;148:576–81. doi: 10.1016/s0923-2494(98)80152-1. [DOI] [PubMed] [Google Scholar]
- 29.Brimnes J, Reimann J, Nissen M, Claesson M. Enteric bacterial antigens activate CD4(+) T cells from scid mice with inflammatory bowel disease. Eur J Immunol. 2001;31:23–31. doi: 10.1002/1521-4141(200101)31:1<23::aid-immu23>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 30.Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity. 2009;31:321–30. doi: 10.1016/j.immuni.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 31.Haas JD, Gonzalez FH, Schmitz S, Chennupati V, Fohse L, Kremmer E, et al. CCR6 and NK1.1 distinguish between IL-17A and IFN-gamma-producing gammadelta effector T cells. Eur J Immunol. 2009;39:3488–97. doi: 10.1002/eji.200939922. [DOI] [PubMed] [Google Scholar]
- 32.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–41. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 33.Persson EK, Uronen-Hansson H, Semmrich M, Rivollier A, Hagerbrand K, Marsal J, et al. IRF4 Transcription-Factor-Dependent CD103(+)CD11b(+) Dendritic Cells Drive Mucosal T Helper 17 Cell Differentiation. Immunity. 2013;38:958–69. doi: 10.1016/j.immuni.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 34.Farache J, Koren I, Milo I, Gurevich I, Kim KW, Zigmond E, et al. Luminal bacteria recruit CD103+ dendritic cells into the intestinal epithelium to sample bacterial antigens for presentation. Immunity. 2013;38:581–95. doi: 10.1016/j.immuni.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsuda JL, Gapin L, Sydora BC, Byrne F, Binder S, Kronenberg M, et al. Systemic activation and antigen-driven oligoclonal expansion of T cells in a mouse model of colitis. J Immunol. 2000;164:2797–806. doi: 10.4049/jimmunol.164.5.2797. [DOI] [PubMed] [Google Scholar]
- 36.Cong Y, Brandwein SL, McCabe RP, Lazenby A, Birkenmeier EH, Sundberg JP, et al. CD4+ T cells reactive to enteric bacterial antigens in spontaneously colitic C3H/HeJBir mice: increased T helper cell type 1 response and ability to transfer disease. J Exp Med. 1998;187:855–64. doi: 10.1084/jem.187.6.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inoue S, Niikura M, Takeo S, Mineo S, Kawakami Y, Uchida A, et al. Enhancement of dendritic cell activation via CD40 ligand-expressing gammadelta T cells is responsible for protective immunity to Plasmodium parasites. Proc Natl Acad Sci U S A. 2012;109:12129–34. doi: 10.1073/pnas.1204480109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Awasthi A, Riol-Blanco L, Jager A, Korn T, Pot C, Galileos G, et al. Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J Immunol. 2009;182:5904–8. doi: 10.4049/jimmunol.0900732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, et al. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–87. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- 40.Min B, Foucras G, Meier-Schellersheim M, Paul WE. Spontaneous proliferation, a response of naive CD4 T cells determined by the diversity of the memory cell repertoire. Proc Natl Acad Sci U S A. 2004;101:3874–9. doi: 10.1073/pnas.0400606101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Connor W, Jr, Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y, et al. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol. 2009;10:603–9. doi: 10.1038/ni.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.