

Abstract
Purpose
Here, we describe a novel interplay between NAD synthesis and degradation involved in pancreatic tumor growth.
Experimental Design
We used human pancreatic cancer cells both in vitro (cell culture experiments) and in vivo (xenograft experiments) to demonstrate the role of NAD synthesis and degradation in tumor cell metabolism and growth.
Results
We demonstrated that pharmacological and genetic targeting of Nampt, the key enzyme in the NAD salvage synthesis pathway, inhibits cell growth and survival of pancreatic cancer cells. These changes were accompanied by a reduction of NAD levels, glycolytic flux, lactate production, mitochondrial function, and levels of ATP. The massive reduction in overall metabolic activity induced by Nampt inhibition was accompanied by a dramatic decrease in pancreatic tumor growth. The results of the mechanistic experiments showed that neither the NAD-dependent enzymes PARP-1, nor SIRT1 play a significant role on the effect of Nampt inhibition on pancreatic cancer cells. However, we identified a role for the NAD degradation pathway mediated by the NADase CD38 on the sensitivity to Nampt inhibition. The responsiveness to Nampt inhibition is modulated by the expression of CD38; low levels of this enzyme decrease the sensitivity to Nampt inhibition. In contrast, its overexpression decreased cell growth in vitro and in vivo and further increases the sensitivity to Nampt inhibition.
Conclusions
Our study demonstrates that NAD metabolism is essential for pancreatic cancer cell survival and proliferation and that targeting NAD synthesis via the Nampt pathway could lead to novel therapeutic treatments for pancreatic cancer.
Introduction
In a series of seminal studies in the early 1900’s, Otto Warburg defined unique metabolic features of cancer cells (1–4). These metabolic changes are critical fortumor cell survival, proliferation, and metastatic potential (1–5). However, it was not until recently that cancer cell metabolism became the focus of intense investigation (1–11).
Nicotinamide adenine dinucleotide (NAD) is a crucial co-factor in redox reactions in metabolic pathways of nearly every cell (7, 12). It has been shown that NAD participates in multiple physiological processes (7, 13–20). In addition, NAD metabolism appears to have a crucial role in fate of tumor cells (21–24). Cellular NAD levels are maintained at stable levels via equilibrium between NAD degradation and NAD synthesis. NAD synthesis is mediated by two distinct mechanisms, the salvage and the de novo pathway (7, 12). NAD degradation is mainly regulated by CD38 (13–18), with other enzymes including sirtuins, Poly (ADP-ribose) polymerases (PARPs), and ADP-ribosyl-transferases (ARTs) playing a complementary role.
In this study, we investigated a novel hypothesis that the interplay between (NAD) synthetic and degrading pathways was involved in the regulation of pancreatic tumorigenesis. We studied how inhibition of Nampt, the rate limiting enzyme of the salvage pathway, affects NAD levels, metabolism, cellular energy production, and tumorigenesis. We also studied the role of NAD degrading enzymes in modulating this response.
Material and methods
Cell lines
PaTu8988t, Panc-1, SU86.86, Panc04.03 and HPDE cells were provided by Dr. D. Billadeau or from ATCC. Cultures used for experiments were reinitiated every 4–6 months from the cryopreserved stocks. The pancreatic cancer cells lines possess K-ras and/or p53 mutations that were validated by DNA sequence analysis using published primers flanking each mutated exon. PaTu8988t and Panc-1 cells were maintained in high-glucose DMEM supplemented with 10% FBS and penicillin/streptomycin (Invitrogen, Eugene, OR, USA). SU86.86 and Panc04.03 cells were grown in RPMI medium supplemented with 10% FBS and penicillin/streptomycin. HPDE cells were grown in SFM-keratinocyte medium supplemented with 5 ng/ml of EGF and 50 μg/ml of bovine pituitary extract. For all the experiments, cells were maintained in media containing 1% FBS for at least 48 hours unless specified.
Reagents and antibodies
Except when specified, all reagents and chemicals were purchased from Sigma Chemical. Antibodies were from: CD38 (Epitomics), Nampt (Bethyl), NaprT1 (Proteintech), P21 (Santa Cruz Biotechnology). EX527 was from Cayman. PARP-1 inhibitor (4-amino-1,8-naphthalimide) was from Enzo Life Sciences.
MTT assay and trypan blue dye exclusion assay
Cells were plated in 96 well plates (3–5×103/well) and treated with the drugs for 48–72 hours at 37 °C. Cell viability was determined by the standard MTT assay or trypan blue assay. IC50 were calculated using CalcuSyn software (Biosoft, Cambridge, UK). The values represent the mean ± SD from 3 independent experiments.
Short interfering RNA
Non-targeting siRNA (Dharmacon # D001210-03-20) was used as control. For CD38 siRNAs IDT (HSC.RNAI.N001775.12.2) and Dharmacon (J-004581-06) were used. Nampt siRNAs were a pool of 3 target-specific siRNAs (sc-45843, Santa Cruz), and a human on-target plus probe (J-009222-05, Dharmacon). Transfections were performed with 50 nM of siRNA using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instruction.
Transfection and western-blots
Panc-1 cells were transfected with Flag or Flag-CD38 vector using lipofectamine 2000 (Invitrogen). For stable transfections Panc-1 cells were co-transfected with Flag-CD38/puromycin vector or Flag-vector/puromycin vector and selected with 4 μg/ml of puromycin. Western-blots were performed using standard laboratory techniques as described before (14, 16).
β-Galactosidase staining
Cells were washed in PBS, fixed for 10 min with 3% formaldehyde, washed and incubated for 24 hours at 37°C with β-Gal staining solution: X-Gal 1 mg/ml, 40 mM citric acid, sodium phosphate (pH 6.0), 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, 2 mM MgCl2.
Soft agar colony formation assay
Cells were seeded at a density of 10,000/well in 6-well plates in 0.35% agar over 0.6% bottom agar layer in growth media containing 5% FBS and increasing concentrations of FK866. Cell colonies were grown in a humidified 5% CO2 incubator at 37 °C. Colonies measuring ≥ 50 μm were counted after 7–10 days of culture using a cell colony counter (Gelcount, Oxford Optronix). Experiments were repeated 3 times, each in triplicates.
NAD quantification and NADase activity
NAD was measured by an enzymatic cycling assay as extensively described by us (14, 16, 18). NADase activity was determined by us using etheno-NAD as a substrate (14, 16–18).
Determination of glycolytic intermediates
Nuclear magnetic resonance (NMR) metabolomic analysis was performed with Glucose C13 as a tracer. Briefly, culture media of non-treated and FK866-treated cells were replaced by glucose-free DMEM supplemented with D-[U-13C] 5 mM glucose and cells were incubated for 1 hour. Metabolic analysis was by one-dimensional 13C spectra of media and cell extracts. Spectra of Panc-1 cells metabolites were acquired with a Brucker DRX 400 MHz using a triple resonance probe (TXI). Spectra processing and analysis were performed using Topspin 2.0 and metabolite assignment was done by chemical shift comparison of known metabolites deposited in the Human Metabolome Database v 1.0.
ATP measurements
ATP levels were measured in tumor tissues using the Aposensor ATP luminescence assay from BioVision, and in cells using the ATPlite Luminescence assay system from PerkinElmer.
Lactate production
Lactate assay was performed using hydrazine/glycine buffer (pH 9.2), 5mg/mL β-NAD+ and 15 units/mL lactate dehydrogenase. NADH formation was monitored at 340nm (25).
Oxygen Consumption of intact cells
O2 consumption rates were measured polarographically using high-resolution respirometry (Oroboros-O2K, Insbruck, Austria). FK886 or vehicle treatment cells were used to measure oxygen consumption at the same time at 37 °C (26). After recording routine (basal) oxygen consumption in DMEM-serum free, oligomycin 2μg/mL was added to inhibit ATP synthesis (leak respiration), followed by titration with the uncoupler FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone) until it reached maximum uncoupled respiration (0,2 – 2,5μM). 2μM Rotenone was added to inhibit complex I. Oxygen consumption after rotenone (not-mitochondria related) was subtracted from all other oxygen consumption measurements. Coupled respiration was calculated as the difference between routine and leak respiration.
Tumor xenograft study
Female athymic nu/nu mice were obtained from the National Cancer Institute (NCI). The experimentswere performed under the supervision and approval of the Institutional Animal Care and Use Committee at Mayo Clinic (protocol A39511).
Subconfluent Panc-1 cells were harvested, and suspensions consisting of single cells with 90% viability were used for subcutaneous injections in both flanks of 5–6 week mice (4×106 cells in 100 μl of PBS:matrigel (1:1)/site).11 days after implantation, (tumor volume ~60 mm3), mice were randomized in two groups: (i) untreated control (vehicle; PBS containing 1% Hydroxypropyl)-β-cyclodextrine and 12% propylenglycol); (ii) FK866 (15mg/kg, twice daily by i.p. injection). Tumor volumes were measured weekly with a caliper and calculated using the formula V=4/3π(l × w × d), were l is the length, w is the width and d is the depth.
Quantification of mRNA
mRNAs from tissue samples were prepared from biospecimens obtained from the Mayo Clinic Tissue Registry under an approved Institutional review board protocol. RNA was isolated from a set of pancreatic adenocarcinoma patient samples for which frozen, paired tumor and non-tumor pancreas tissue was available. mRNA abundance was analyzed by quantitative PCR analysis using on an Applied Biosystems 7900HT thermal cycler. TaqMan Gene Expression Assay probe sets (Applied Biosystems) were used for analysis of 18s (Hs99999901_s1), Nampt (Hs00237184_m1) and CD38 (Hs01120071_m1). The data was analyzed using the standard curve method. Gene expression was normalized to 18S.
RNA was isolated from pancreatic cell lines using the RNeasy kit (Qiagen). TaqMan (Applied Biosystems) gene expression probes for human Nampt, Naprt1 (Hs00376971_g1), CD38, and GAPDH (Hs02758991_g1). The relative of target genes was calculated by normalizing GAPDH expression. The expression changes were calculated relative to control pancreatic cells (HPDE).
Statistical analysis
Data are expressed as means ± SD from at least 3 independent experiments. Data was analyzed using unpaired t-test, one-way and two-way ANOVA. Significance was set at p < 0.05.
Results
Nampt plays a key role in maintaining NAD levels in pancreatic cancer cells
We evaluated Nampt expression in a panel of pancreatic cancer cell lines and a normal pancreatic cell line (HPDE). We found that Nampt was expressed in all cell lines (Fig. 1A), with a higher expression in the cell lines Panc04.03 and SU86.86. Next, we examined if inhibition of Nampt using FK866 resulted in a reduction in cellular NAD levels (Fig. 1B). The effect of FK866 on intracellular NAD levels was variable between the cells tested, with some cells (e.g. PaTu8988t) very sensitive to Nampt inhibition and other cells like Su86.86 showing a significant lower sensitivity to FK866 (Fig. 1B). To confirm the role of Nampt in the regulation of cellular NAD levels, we tested the effect of an acute reduction in the enzyme expression via knock-down with siRNA in PaTu8988t and Panc-1 cells. Treatment with Nampt siRNA reduced Nampt expression and NAD levels in these cells (Fig. 1C and D), indicating that the salvage pathway of NAD synthesis plays a key role in maintaining NAD levels in these pancreatic cancer cells.
Figure 1. Nampt inhibition by FK866 or Nampt knockdown by siRNA reduces NAD levels in pancreatic cancer cells.

A, Different pancreatic cancer cell lines were imunoblotted for Nampt and Actin. B, NAD levels were measured in pancreatic cancer cells that were maintained in growth medium containing 1% FBS for 48 hours and then treated with FK866 or vehicle for 24 hours. C and D, Cells were transfected with Nampt siRNA or control siRNA analyzed by immunoblotting 72 hours later (insets). 24 hours after transfection cells were re-plated in growth medium containing 1% FBS and NAD levels were determined 48 hours later. * indicates p<0.05.
To determine if FK866 can cause a decrease in overall metabolism in pancreatic cancer cells, we measured several metabolic parameters. First, we observed that glucose consumption was severely impaired in cells treated with FK866 (Fig. 2A). NAD is important for several biochemical processes including oxi-reduction reactions of the glycolytic pathway (27). We observed an accumulation of the glycolytic intermediary Fructose 1,6 biphosphate in cells treated with FK866 (Figure 2A), indicating that FK866 inhibits glycolytic fluxes and may cause cellular metabolic collapse. To confirm these effects, we measured lactate production in FK866 treated cells and found thatFK866 inhibited lactate release, in both PaTu8988t cells Panc-1 cells (Figure 2B). We further investigated the effect of FK866 on mitochondrial oxygen consumption, ATP levels, and the activation of AMP-activated protein kinase (AMPK). FK866 treatment decreased mitochondrial maximum respiratory capacity (Fig. 2C and Supplementary Fig. S1), ATP levels (Fig. 2D), and induced phosphorylation/activation of AMPK in pancreatic cancer cells (Fig. 2D), confirming that Nampt inhibition promotes energy collapse in pancreatic cancer cells.
Figure 2. FK866 produces metabolic collapse in pancreatic cancer cells.

A, Panc-1 cells were kept in 1% FBS for 48 hours and then treated with FK866 (10 nM) or vehicle. 48 hours later media was changed to glucose-free DMEM supplemented with 5 mM D-[C13] glucose. Glucose consumption and glycolytic intermediates were determined using NMR spectroscopy, and glycolytic intermediates determined. B, cells were kept in 1% FBS for 48 hours and then treated with FK866 (10 nM) or vehicle for 48 hours. The medium was replaced by RPMI phenol red and serum-free and aliquots of cultured medium were collected for lactate release evaluation. C, O2 consumption rates were measured by high resolution respirometry. After 48 hours treatment with 50 nM of FK866 or vehicle, the cells resuspended in serum-free DMEM, and ETS-stimulated respiration (maximum respiration) was measured. Results represent the mean ± SD of three independent experiments. D, Cells were kept in 1% FBS for 48 hours then treated with FK866 or vehicle for 48 hours before ATP measurements. Values are means ± SD of three independent experiments. Cell lysates of PaTu8988t cells treated with 10nM FK866 for 24 hours were immunoblotted with anti-p-AMPK and AMPK antibodies. * indicates p<0.05
Inhibition of Nampt decreases pancreatic cancer cell growth and survival
We examined the role of Nampt in the growth and survival of the pancreatic cancer cells. FK866 cause a dose-dependent inhibition of viability in Panc-1 and PaTu8988t cells (Fig. 3A). We observed a significant difference in the sensitivity of cells to Nampt inhibition, with PaTu8988t cells being nearly 10 times more sensitive than other cells. The least sensitive was the normal pancreatic cell HPDE (Fig. 3 and Supplementary Fig. S2). Since the MTT assay determines cell viability via an NAD-dependent mechanism we further confirmed our findings with two other complementary assays to measure cell growth and viability (Fig. 3).
Figure 3. Nampt inhibition decreased pancreatic cancer cell survival and growth.

A, Cells were treated with vehicle or FK866 and submitted to MTT analysis 72 hours later. Values are means ± SD of three independent experiments. B, Cells were plated and treated with vehicle or FK866 24 hours later. Cells were counted by trypan blue dye exclusion assay 72 hours after treatment. C, Cells were grown in triplicates in soft agar containing different concentrations of FK866 for 7 days. Results represent the mean ± SD of three independent experiments. D, Cells were transfected with Nampt siRNA or a non-target siRNA (Control). 24 hours after transfection the cells were re-plated in growth medium containing 1% FBS and viable cells were counted by Trypan blue exclusion assay 48 hours later. Efficiency of knockdown was determined by immunoblotting. * indicates p<0.05
Panc-1 and PaTu8988t cells were treated with 50 nM FK866 for 72 hours and viable cells were counted by the Trypan blue exclusion assay. Treatment with FK866 decreased the number of viable cells in both cells lines (Fig. 3B).
We determined whether Nampt can regulate anchorage-independent cell growth, an indicator of malignant behavior. Pancreatic cancer cells were treated with FK866 and colony formation in soft agar was measured. Colony formation was decreased by treatment with FK866 in PaTu8988t cells and to a lesser extent in Panc-1 cells (Fig. 3C). Furthermore, we knocked-down Nampt using two different siRNAs. Nampt siRNAs inhibited cell growth in both Panc-1 and PaTu8988t cell lines (Fig3D and Supplementary Fig. S3).
NAMPT inhibition impairs pancreatic tumor growth
We further tested the effect of FK866 in a xenograft animal model of pancreatic tumor. Nude mice were injected with Panc-1 cells and 10 days after implantation the animals were treated with daily intraperitoneal injections of vehicle or 15mg/kg of FK866. FK866 treatment decreased tumor size when compare with vehicle (Fig. 4A). Tumors from mice treated with FK866 had lower cellular NAD and ATP levels (Fig. 4B), and increased phosphorylation of AMPK (Fig. 4C). During the treatment, no obvious treatment-related toxicity was observed. Body weight and food intake of both groups of animals remained similar (Fig. 4D). We conclude that FK866 inhibits pancreatic tumor growth in vivo, by inducing tumoral metabolic collapse.
Figure 4. NAMPT inhibition prevents pancreatic tumor growth in vivo.

Panc-1 cells were used in an in vivo xenograft mouse model. Starting at 10 days after implantation the animals were treated with daily intraperitoneal injections of vehicle or FK866. Tumors were measured for an additional 40 days. A, Graph shows the average size of 16 vehicle and 16 FK866-treated tumors measured during the experiment. Picture shows size of representative tumors. B, presents NAD and ATP levels from 6 vehicle and 6 FK866-treated tumors. C, immunostaining for p-AMPK (Thr172) shows activation of AMPK (p-AMPK) in tumors collected from vehicle and FK866-treated animals. D, mice were weighted during the treatment period.* indicates p<0.05
The effect of FK866 in pancreatic cancer cell viability is reversed by NMN and Nicotinic Acid
Because in cells NAD can be synthesized by both the salvage and the de novo pathway, we explored whether the two pathways were involved in the regulation of pancreatic cancer cell growth. In the salvage pathway, Nampt produces nicotinamide mononucleotide (NMN) from nicotinamide. In the de novo pathway, Nicotinic acid phosphoribosyltransferase (Naprt1) produces nicotinic acid mononucleotide (NaMN) from nicotinic acid. The effect of FK866 on cell viability of PaTu8988t was completely reversed by treatment with the NAD precursors NMN and Nicotinic Acid (Fig. 5A). In addition, the pattern of expression of Naprt1, the enzyme involved in the de novo pathway, was similar to the expression of Nampt in the different pancreatic cell lines (Fig 5A). These data suggest that both pathways are involved in the modulation of pancreatic cancer cell growth. However, inhibition of the salvage pathway is sufficient to promote energy collapse and cell death in pancreatic cancer cells.
Figure 5. FK866 effect in pancreatic cancer cell viability is blocked by NMN and Nicotinic Acid, but not by SIRT1 or PARP-1 inhibitors.

A, PaTu8988t cells were treated with vehicle (control), 25 μm NMN or 25 μM Nicotinic Acid (NA) for 6 hours before addition of 2 nM FK866. Cells were submitted to MTT analysis 72 hours later. Values are mean ± SD of three independent experiments. Different pancreatic cell lines were imunoblotted for NAPRT1 and actin. B, PaTu8988t cells were treated with vehicle (control), 10 μM of the SIRT1 inhibitor (EX527) or 10 μM of the PARP inhibitor (4-Amino-1,8-naphthalimide) for 6 hours before addition of 2 nM FK866. Cells were submitted to MTT analysis 72 hours after treatment. Values are mean ± SD of three independent experiments. * indicates conditions that are significantly different than control, but are not significantly different from each other. C, Cells were transfected with siRNA specific for SIRT1 or a non-target siRNA. 24 hour after transfection the cells were re-plated and viable cells were counted at different time points. Values are mean ± SD of triplicates. Plot is representative of three independent experiments. D, PaTu8988t and SU86.86 cells were treated with the SIRT1 inhibitor EX527 (10 μM) or vehicle. Cells were counted 72 hours after treatment by trypan blue dye exclusion assay. Values are mean ± SD of three independent experiments.* indicates p < 0.05
SIRT1 and PARP-1 are not involved on the effect of FK866 in pancreatic cancer cells
Since NAD is necessary for both oxi-reduction and non-oxi-reduction reactions, we further explored whether inhibition of some of the non-oxidative NAD-dependent cellular reactions were involved on the decrease in cellular viability induced by Nampt inhibition. Two enzymes, PARP-1 and SIRT1 catalyze crucial non-oxidative NAD-dependent reactions. Surprisingly, neither PARP1 nor SIRT1 inhibition recapitulated or modified the effects of Nampt inhibition on cell viability (Fig. 5B). In addition, transient SIRT1 knockdown increased cell proliferation of PaTu8988T cells (Figure 5C). The difference in cell proliferation is shown by the decrease in doubling time in SIRT1 siRNA treated cells compared to non-target siRNA transfected cells. The doubling time for non-target siRNA transfected cells was 21.7 ± 1.2 hours and for SIRT1 siRNA transfected cells was 19.2 ± 0.6 hours (p value = 0.015). Moreover, pharmacological inhibition of SIRT1 using the synthetic inhibitor EX527 resulted in a small induction of cell growth of both PaTu8988t and SU86.86 cells, by 22 and 35 % respectively (Figures 5D). Taken together, these results indicate that neither PARP-1 nor SIRT1-inhibiton can explain the effects of Nampt inhibition in the pancreatic cancer cells.
Sensitivity of cultured pancreatic cancer cells in vitro to FK866 is modulated by CD38 expression
Decreases in cellular NAD can be achieved by inhibition of NAD synthesis or by an increase in its degradation. The enzyme CD38 is the main NADase in many normal mammalian tissues (13–18). Interestingly, it has been proposed that CD38 expression may be lost during the development of prostate cancer (28). However, the functional role of CD38 in pancreatic cancer cells or any other solid tumor has not been explored. Here we investigated the role of CD38 in pancreatic NAD metabolism and its role in the effect of the Nampt inhibitor FK866 on cell viability and cell growth. Most cultured pancreatic cancer cells tested had low levels of CD38 expression (Fig. 6A), except for PaTu8988t, that expresses high amounts of CD38. When we knocked-down CD38 in PaTu8988t cells, there was an increase in NAD levels and a decrease in NADase activity (Fig. 6B). Although no significant difference on cell viability was observed after cell knockdown of CD38 (data not shown), the cells that were knocked down for CD38 showed lower sensitivity to FK866 than control cells (Fig. 6B, and Supplementary Fig. S4).
Figure 6. Sensitivity of pancreatic cancer cells to FK866 depends on CD38 levels.
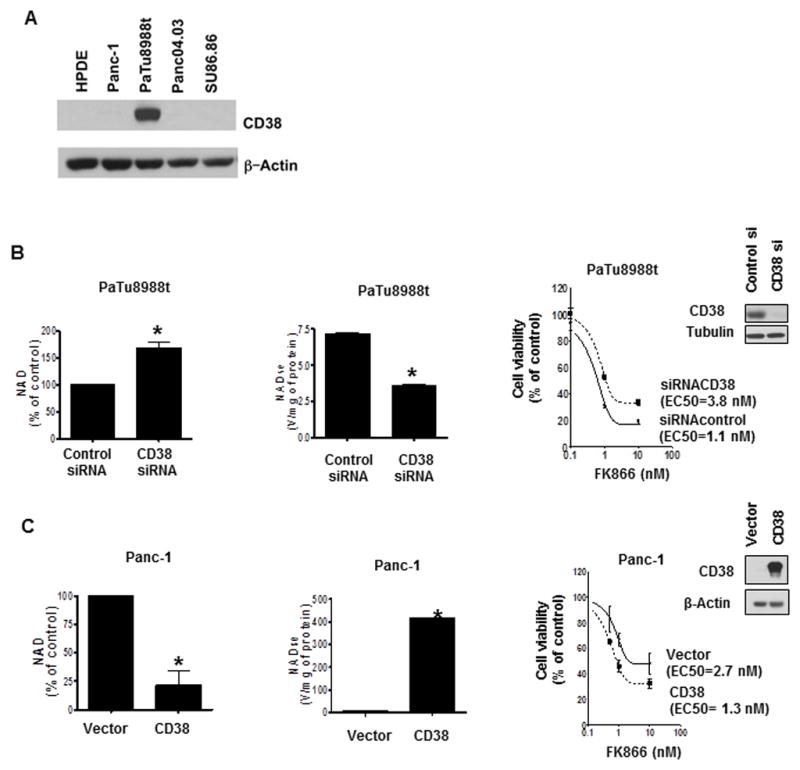
A, Different pancreatic cell lines were imunoblotted for CD38 and actin. B, PaTu8988t cells were transfected with CD38 siRNA or a non-targeting siRNA. 72 hours after transfection, we measured NAD levels and NADase activity. For MTT analysis, cells were re-plated 24 hours after transfection, and treated with vehicle control or FK866 24 hours later. MTT analysis was performed 48 hours after treatment. C, Panc-1 cells were transfected with a Flag-CD38 or empty vector. NAD levels and NADase activity were measured 72 hours later. For MTT analysis cells were re-plated 24 hours after transfection, and treated with vehicle control or FK866 24 hours later. MTT analysis was performed 48 hours after treatment. Efficiency of transfections was determined by immunoblotting 72 hours after transfection (insets). Values are mean ± SD of three independent experiments. * indicates p<0.05
In contrast, Panc-1 cells have nearly undetectable levels of CD38 (Fig. 6A). Transient expression of CD38 increased NADase activity, decreased NAD levels, and increased sensitivity to the effect of FK866 on cell viability (Fig. 6C). Expression of CD38 in Panc-1 cells did not affect mitochondrial respiration by itself, but it sensitized the cells to the inhibitory effect of FK866 in mitochondrial function (Supplementary Fig. S5A). Also, overexpression of CD38 in SU86.86 promoted a decrease in NAD levels (data not shown) and a significant decrease in cell viability compared to vector-transfected cells (Supplementary Fig. S5B and C).
To further explore the role of CD38 in NAD metabolism and cell growth we generated a Panc-1-CD38 stable cell line. This cell line has increased NADase activity and lower NAD levels than the control cell line (Fig. 7A). In addition, the Panc-1-CD38 cells showed cell growth arrest both in vitro and also in an in vivo xenograft mouse model (Fig. 7A and B). These cells also exhibit senescent markers, as measured by β-galactosidase staining and increased p21 protein levels (Fig. 7C). Moreover, in colony formation assays, Panc-1-CD38 cells showed an increase in sensitivity to the FK866 effect in comparison to the control cells (Fig. 7D), confirming that CD38 has an important effect in regulating the sensitivity to Nampt inhibition.
Figure 7. CD38 stable overexpression inhibits anchorage-dependent and anchorage-independent cell growth of Panc-1 cells and increases the inhibitory effect of FK866.

A, intracellular NAD levels, NADase activity, and cell growth were determined in Panc-1 control stable clone and a Panc-1-CD38 clone. The expression of CD38 was evaluated by immunoblotting. B, Panc-1 cells and Panc-1-CD38 cells were used in an in vivo xenograft mouse model and the size of the tumors was measured during a three week period (n=5). * indicates p<0.05. C, Panc-1 control cells and Panc-1-CD38 cells were stained for β-Galactosidase. The expression of CD38 and P21 was evaluated in these cells by immunoblotting for Flag and P21. D, Panc-1 control cells and Panc-1-CD38 cells were grown in soft agar containing 1 nM FK866 for 7–10 days in triplicates. Results represent the mean ± SD of three independent experiments. Quantitative analysis of colony formation shows significant reduction (* p< 0.05, n=3) in colony number upon CD38 overexpression in comparison to empty vector; and (# p< 0.05, n=3) upon FK866 treatment in Panc-1-CD38 but not in Panc-1 control cells.
Heterogeneous expression of NAD metabolizing enzymes in pancreatic tumor tissues
We further explored the expression of Nampt and CD38 in pancreatic cancer tumor samples from patients. There was a significant variability in the expression of CD38 and Nampt in pancreatic cancer samples, but, in general, the expression of both Nampt and CD38 were higher in samples from pancreatic tumors than in normal tissue (Supplementary Fig. 6A and B). To compare these results with data from the pancreatic cancer cell lines, we performed real time PCR in samples from pancreatic cancer cell lines and the control pancreatic cell HPDE. All cells express Nampt and Naprt1, but the expression levels differ between cell lines, with some cancer cells expressing similar levels and others lower levels than the control pancreatic cell (Supplementary Fig. 6C and D). In contrast, the only pancreatic cancer cell that expresses high amounts of CD38 is PaTu8988t (Supplementary Fig. 6E). For all the cell lines, the patterns of expression of protein and mRNA had a high correlation with all genes tested (Fig. 1A, 5B, 6A).
Since we found that the expression of CD38 is low in pancreatic cancer cells in culture, the relatively high expression of CD38 in the pancreatic tumor tissues may be mediated by either inflammatory cell infiltration or by stromal cells. However, it is also possible that pancreatic cancer cells in vivo may express CD38 differently than in vitro.
Discussion
Pancreatic cancer is one of the top five causes of cancer-related deaths around the globe, with an extremely poor prognosis (about 5% survival in 5 years) and a median survival for metastatic disease of about 6 months (29). New and effective therapies are urgently needed for this disease. The studies described here clearly show a role for the salvage pathway as potential therapeutic target in pancreatic cancer and also identified NAD catabolic pathways that modulate the sensitivity to Nampt inhibitors in cancer models. Recently, it has been proposed that NAD metabolism may be a potential target for the treatment of cancers (6, 12, 21–24, 30). NAD is a crucial co-factor in redox reactions in metabolic pathways of nearly every cell (6, 12). NAD can be found in two states, oxidized (NAD+) or reduced (NADH). Oxidized NAD is necessary for the initial steps of the glycolytic pathway that is important for cancer cell survival and growth (6, 12). Intracellular NAD metabolism in cancer cells can be manipulated in several different ways. For example, the equilibrium between the oxi-reduction states of NAD (the NAD/NADH ratio) can be shifted one way or the other to either support or inhibit glycolysis (31). Recently, Lu et al have elegantly shown that the enzyme NADP(H) oxidase (NOX), plays a key role in supporting increased glycolysis in pancreatic cancer cells by oxidizing NAD (31).
Yet, another way to modulate NAD-dependent reactions in cancer cells is to modify its total pool via its anabolism and catabolism. To date, the functional role of the NAD anabolic pathway in pancreatic cancer cells has not been investigated. In fact, the only information published about NAD synthetic pathways in pancreatic cancer cells is a study that showed that colo357 pancreatic cancer cells express Nampt in response to IL-1 treatment (9). In this regard, characterization of the metabolic pathways for synthesis was one of the goals of our study. We describe for the first time that pancreatic cancer cells use the salvage pathway and rely on the enzyme Nampt for NAD synthesis, and that inhibition of the Nampt enzyme causes pancreatic cancer cell death. The mechanism by which NAD collapse mediated by Nampt inhibition causes cancer cell death has been proposed to be mediated by inhibition of the glycolytic pathway and /or inhibition of the NAD-dependent deacetylase SIRT1 (7, 8, 22, 27, 32). In our experiments, inhibition of SIRT1 was neither necessary nor sufficient to explain the cellular effects of Nampt-inhibition in pancreatic cancer cells.
Tumor cells have a highly active “aerobic glycolysis” known as the “Warburg effect” (1–2). Mechanistically, the first reaction in the glycolytic pathway that is dependent on NAD(H) is the conversion of glyceraldehyde 3 phosphate to 1.3 biphosphoglycerate catalyzed by GAPDH (27). However, the reaction preceding this step, the interconversion of glyceraldehyde 3 phosphate and Fructose-1,6 BP is reversible, and we expected that inhibition of the GAPDH reaction could lead to accumulation of one of these metabolites. Treatment with FK866 leads to decrease in lactate production, metabolic fluxes and accumulation of Fructose-1,6 BP in pancreatic cancer cells. Our data supports a model where Nampt inhibition promotes cell death via a decrease in NAD that leads to inhibition of glycolytic metabolism, ATP depletion, and energy collapse, and not via SIRT1 dependent mechanisms.
Another goal of our study was to characterize the NAD catabolizing pathways in pancreatic cancer cells. To our knowledge the mechanisms of NAD degradation in tumor cells have not been well described. In particular, given the emerging interest in developing NAD targeted therapies for human cancers, it is important to determine the mechanisms that modulate NAD-degradation in the setting of inhibition of NAD synthetic pathways.
We observed in cultured pancreatic cancer cells that expression of the NADase CD38 played a key role on the sensitivity of cells to the Nampt inhibitor FK866. In addition, we observed that CD38 and Nampt expression are quite variable between tumors from pancreatic cancer patients (Supplemental Figure 6), and we propose that the relative expression of Nampt and CD38 may play a key role on the response to salvage pathway-targeted therapy and may serve as potential biomarkers for the cellular response to Nampt inhibition.
In conclusion, our data provides the first evidence of the role of NAD anabolism and catabolism in pancreatic cancer cells. Specifically, we demonstrated that pancreatic cancer cells rely on the salvage pathway and that inhibition of the enzyme Nampt causes metabolic collapse and cell death both in vitro and in vivo. Furthermore, we provided one of the first analyses of NAD catabolism in cancer cells. We believe that further systematic characterization of the anabolism and catabolism of NAD in cancer cells may have potential implications for the development of novel and more rational NAD-targeted therapy for cancer.
Supplementary Material
Statement of translational relevance.
Pancreatic cancer is one of the top five causes of cancer-related deaths around the globe. No effective therapies are available for pancreatic cancer and new and effective therapies are urgently needed. The role of NAD metabolism in pancreatic cancer has never been studied. NAD homeostasis is maintained by equilibrium between synthesis and degradation. In this study, we demonstrated for the first time that pancreatic cancer cells use the salvage pathway for NAD synthesis and that Nampt inhibition is effective both in vitro and in vivo to decrease tumor cell growth. Furthermore, we identified the NADase CD38 as a key factor in the responsiveness of pancreatic cancer cells to NAD synthesis inhibition in vitro. Our data provides extremely novel pre-clinical knowledge in pancreatic cancer NAD metabolism and opens a new avenue for further clinical studies of manipulating NAD metabolism for the treatment of this deadly disease.
Acknowledgments
The project described was supported by Award Number P50CA102701 (Mayo Clinic SPORE in Pancreatic Cancer) from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Abbreviations
- CD38
Cluster of Differentiation 38 (glycoprotein)
- Nampt
Nicotinamide phosphoribosyltransferase
- NAD
Nicotinamide adenine dinucleotide
- MTT
(3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
Footnotes
Authors have no conflict of interest.
The authors declare no conflict of interest related to the current study.
References
- 1.Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Targeting Glucose Metabolism: An Emerging Concept for Anticancer Therapy. Am J Clin Oncol. 2011;34:628–35. doi: 10.1097/COC.0b013e3181e84dec. [DOI] [PubMed] [Google Scholar]
- 2.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 3.Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009;15:6479–83. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fritz V, Fajas L. Metabolism and proliferation share common regulatory pathways in cancer cells. Oncogene. 2010;29:4369–77. doi: 10.1038/onc.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107:2037–42. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nahimana A, Attinger A, Aubry D, Greaney P, Ireson C, Thougaard AV, et al. The NAD biosynthesis inhibitor APO866 has potent antitumor activity against hematologic malignancies. Blood. 2009;113:3276–86. doi: 10.1182/blood-2008-08-173369. [DOI] [PubMed] [Google Scholar]
- 7.Garten A, Petzold S, Körner A, Imai S, Kiess W. Nampt: linking NAD biology, metabolism and cancer. Trends Endocrinol Metab. 2009;20:130–38. doi: 10.1016/j.tem.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang B, Hasan MK, Alvarado E, Yuan H, Wu H, Chen WY. NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene. 2011;30:907–21. doi: 10.1038/onc.2010.468. [DOI] [PubMed] [Google Scholar]
- 9.Bauer L, Venz S, Junker H, Brandt R, Radons J. Nicotinamide phosphoribosyltransferase and prostaglandin H2 synthase 2 are up-regulated in human pancreatic adenocarcinoma cells after stimulation with interleukin-1. Int J Oncol. 2009;35:97–07. doi: 10.3892/ijo_00000317. [DOI] [PubMed] [Google Scholar]
- 10.Okumura S, Sasaki T, Minami Y, Ohsaki YJ. Nicotinamide Phosphoribosyltransferase: A Potent Therapeutic Target in Non-small Cell Lung Cancer with Epidermal Growth Factor Receptor-Gene Mutation. Thorac Oncol. 2012;7:49–56. doi: 10.1097/JTO.0b013e318233d686. [DOI] [PubMed] [Google Scholar]
- 11.Shackelford RE, Bui MM, Coppola D, Hakam A. Over-expression of nicotinamide phosphoribosyltransferase in ovarian cancers. Int J Clin Exp Pathol. 2010;3:522–527. [PMC free article] [PubMed] [Google Scholar]
- 12.Khan JV, Forouhar F, Tao X, Tong L. Nicotinamide adenine dinucleotide metabolism as an attractive target for drug discovery. Expert Opin Ther Targets. 2007;11:695–05. doi: 10.1517/14728222.11.5.695. [DOI] [PubMed] [Google Scholar]
- 13.Chini EN. CD38 as a regulator of cellular NAD: a novel potential pharmacological target for metabolic conditions. Curr Pharm Des. 2009;15:57–63. doi: 10.2174/138161209787185788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barbosa MT, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P, et al. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007;21:3629–39. doi: 10.1096/fj.07-8290com. [DOI] [PubMed] [Google Scholar]
- 15.Malavasi F, Deaglio S, Zaccarello G, Horenstein AL, Chillemi A, Audrito V, et al. The hidden life of NADC-consuming ectoenzymes in the endocrine system. Journal of Molecular Endocrinology. 2010;45:183–91. doi: 10.1677/JME-10-0082. [DOI] [PubMed] [Google Scholar]
- 16.Aksoy P, White TA, Thompson M, Chini EN. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem Biophys Res Commun. 2006;345:1386–92. doi: 10.1016/j.bbrc.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 17.Hartman WR, Pelleymounter LL, Moon I, Kalari K, Liu M, Wu TY, et al. CD38 expression, function, and gene resequencing in a human lymphoblastoid cell line-based model system. Leuk Lymphoma. 2010;51:1315–25. doi: 10.3109/10428194.2010.483299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sahar S, Nin V, Barbosa MT, Chini EN, Sassone-Corsi P. Altered behavioral and metabolic circadian rhythms in mice with disrupted NAD+ oscillation. Aging (Albany NY) 2011;3:794–02. doi: 10.18632/aging.100368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–07. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van der Veer E, Ho C, O’Neil C, Barbosa N, Scott R, Cregan SP, et al. Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J Biol Chem. 2007;282:10841–845. doi: 10.1074/jbc.C700018200. [DOI] [PubMed] [Google Scholar]
- 21.Olesen UH, Thougoord AV, Josen PB, Sehested M. A Preclinical Study on the Rescue of Normal Tissue by Nicotinic Acid in High-Dose Treatment with APO866, a Specific Nicotinamide Phosphoribosyltransferase. Inhibitor Mol Cancer Ther. 2010;9:1609–17. doi: 10.1158/1535-7163.MCT-09-1130. [DOI] [PubMed] [Google Scholar]
- 22.Boniface JJ. MPC-9528, a cancer metabolism inhibitor, demonstrates greater therapeutic index in a Naprt1 deficient cancer xenograft model with co-administration of nicotinic acid. Cancer and Metabolism: Pathways to the Future Symposium; September 19–21, 2010; Edinburgh, Scotland. [Google Scholar]
- 23.Holen K, Saltz LB, Hollywood E, Burk K, Hanauske AR. The pharmacokinetics, toxicities, and biological effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Invest New Drugs. 2008;26:45–51. doi: 10.1007/s10637-007-9083-2. [DOI] [PubMed] [Google Scholar]
- 24.von Heideman A, Berglund A, Larsson R, Nygren P. Safety and efficacy of NAD depleting cancer drugs: results of a phase I clinical trial of CHS 828 and overview of published data. Cancer Chemother Pharmacol. 2010;65:1165–72. doi: 10.1007/s00280-009-1125-3. [DOI] [PubMed] [Google Scholar]
- 25.Hamilton SD, Pardue HL. Quantitation of lactate by a kinetic method with an extended range of linearity and low dependence on experimental variables. Clin Chem. 1984;30:226–29. [PubMed] [Google Scholar]
- 26.Hütter E, Renner K, Pfister G, Stöckl P, Jansen-Dürr P, Gnaiger E. Senescence-associated changes in respiration and oxidative phosphorylation in primary human fibroblasts. Biochem J. 2004;380:919–28. doi: 10.1042/BJ20040095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan B, Young DA, Lu ZH, Wang T, Meier TI, Shepard RL, et al. Pharmacological inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD+ biosynthesis, in human cancer cells: metabolic basis and potential clinical implications. J Biol Chem. 2013;288:3500–11. doi: 10.1074/jbc.M112.394510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kramer G, Steiner G, Födinger D, Fiebiger E, Rappersberger C, Binder S, et al. High expression of a CD38-like molecule in normal prostatic epithelium and its differential loss in benign and malignant disease. J Urol. 1995;154:1636–41. [PubMed] [Google Scholar]
- 29.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 30.Chiarugi A, Dölle C, Felici R, Ziegler M. The NAD metabolome — a key determinant of cancer cell biology. Nature Reviews Cancer. 2012;12:741–52. doi: 10.1038/nrc3340. [DOI] [PubMed] [Google Scholar]
- 31.Lu W, Hu Y, Chen G, Chen Z, Zhang H, Wang F, et al. Novel Role of NOX in Supporting Aerobic Glycolysis in Cancer Cells with Mitochondrial Dysfunction and as a Potential Target for Cancer Therapy. PLoS Biol. 2012;10:e1001326. doi: 10.1371/journal.pbio.1001326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bowlby SC, Thomas MJ, D’Agostino RB, Jr, Kridel SJ. Nicotinamide phosphoribosyl transferase (Nampt) is required for de novo lipogenesis in tumor cells. PLoS One. 2012;7:e40195. doi: 10.1371/journal.pone.0040195. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.