

Abstract
We present a model of interaction of Gi protein with activated rhodopsin (R*) which pin-points energetic contributions to activation and reconciles the β2AR–Gs crystal structure with new and previously published experimental data. In silico analysis demonstrated energetic changes when the Gα C-terminal helix (α5) interacts with the R* cytoplasmic pocket, leading to displacement of the helical domain and GDP release. The model features a less dramatic domain opening than the crystal structure. The α5 helix undergoes a 63º rotation, accompanied by a 5.7Å translation, which reorganizes interfaces between α5 and α1 helices and between α5 and β6–α5. Changes in the β6–α5 loop displace αG. All of these movements lead to opening of the GDP binding pocket. The model creates a roadmap for experimental studies of receptormediated G protein activation.
Introduction
G protein coupled receptors catalyze GDP (guanosine diphosphate) release on cognate G proteins through a mechanism that is not fully elucidated, however studies released in the last several years have greatly accelerated our understanding of this process. Previously, numerous structural and functional studies demonstrated the key roles that regions such as the C terminus and the α4–β6 loop of Gα play in receptor-mediated G protein activation1–7. However, it was not until the crystal structure of the β2AR (adrenergic receptor)–Gs complex was determined in 2011 (ref. 7) that the extent of these G protein-receptor interactions could be fully appreciated. This structure provides a stunning picture of the G protein-activated receptor complex (R*–G). What the structure alone cannot tell us is the allosteric mechanism that links interaction of a G protein with the receptor to GDP release — the R* and GDP binding sites are separated by 39Å. We first predicted8 and later demonstrated using DEER (double electron electron resonance) experiments9 that receptor-mediated GDP release is accompanied by opening the interface between the GTPase (guanosine triphosphatase) and helical domains in the Gαi subunit. While the loss of interaction between the domains is confirmed by the crystal structure of the β2AR–Gs complex, the authors suggested that the exact location of the helical domain may be influenced by the process of crystallization. To better understand receptor mediated G protein activation, we combined DEER data with the structure of the β2AR-Gs complex to construct a unified model of the complex of activated rhodopsin with heterotrimeric Gαiβγ (R*–Gi). The model proposes the C terminus of Gα triggers conformational changes leading to GDP release and concomitant domain opening. This unified model is consistent with published EPR (electron paramagnetic resonance), deuterium exchange, and electron microscopy data. The current study has resulted in the development of a structural hypothesis for the receptor–Gi complex, supported by experimental data. From this structural model, we performed energetic analysis using the Rosetta force fields and identified residues that show marked energetic changes between the free G protein and G protein bound to activated receptor. Based on the energetic analysis, we propose a mechanism for receptor-mediated GDP release from the G protein. Finally, this hypothesis was validated with DEER, CW (continuous wave)-EPR, fluorescence, mutagenesis, and was consistent with previous electron microscopy and H/D (hydrogen deuterium) exchange experimental data.
Results
Our strategy included construction of a comparative model for the interaction of activated rhodopsin with Gi (R*–Gi) that unifies available experimental data with crystallographic data (Figure 1, Supplemental Movie 1). The receptor unbound model of Gαiβγ was constructed using Rosetta, based on the PDB coordinates 1GOT10 which provides a higher resolution than any other Gi family member structure9,10 (alignment shown in Supplemental Figure 1). The receptor-bound model of R*–Gαiβγ is based on the crystal structure of the β2AR–Gs complex (PDB 3SN67; alignment shown in Supplemental Figure 2). Energetic minimization of the structure utilized Rosetta’s relaxation protocol with full atom energy potentials, including membrane specific terms to accommodate the receptor11,12. Rosetta’s refinement and force fields are capable of i’dentifying native structures and recovering protein backbone and side chain conformations at atomic detail accuracy13. The purpose was to allow the sequence dependent interactions to transition from the template structure to the interactions defined by the sequence of the target (Supplemental Figure 3d). The model with lowest Rosetta energy was the starting point for several simulations that maximize consistency with all experimental data. We systematically compared free heterotrimeric Gαiβγ to the receptor-bound form and analyzed amino acid interactions across key interfaces between and within the two proteins. Thereby we identified residues that contribute to stabilizing both states. We additionally mapped how these key interactions are altered when Gαi interacts with R*.
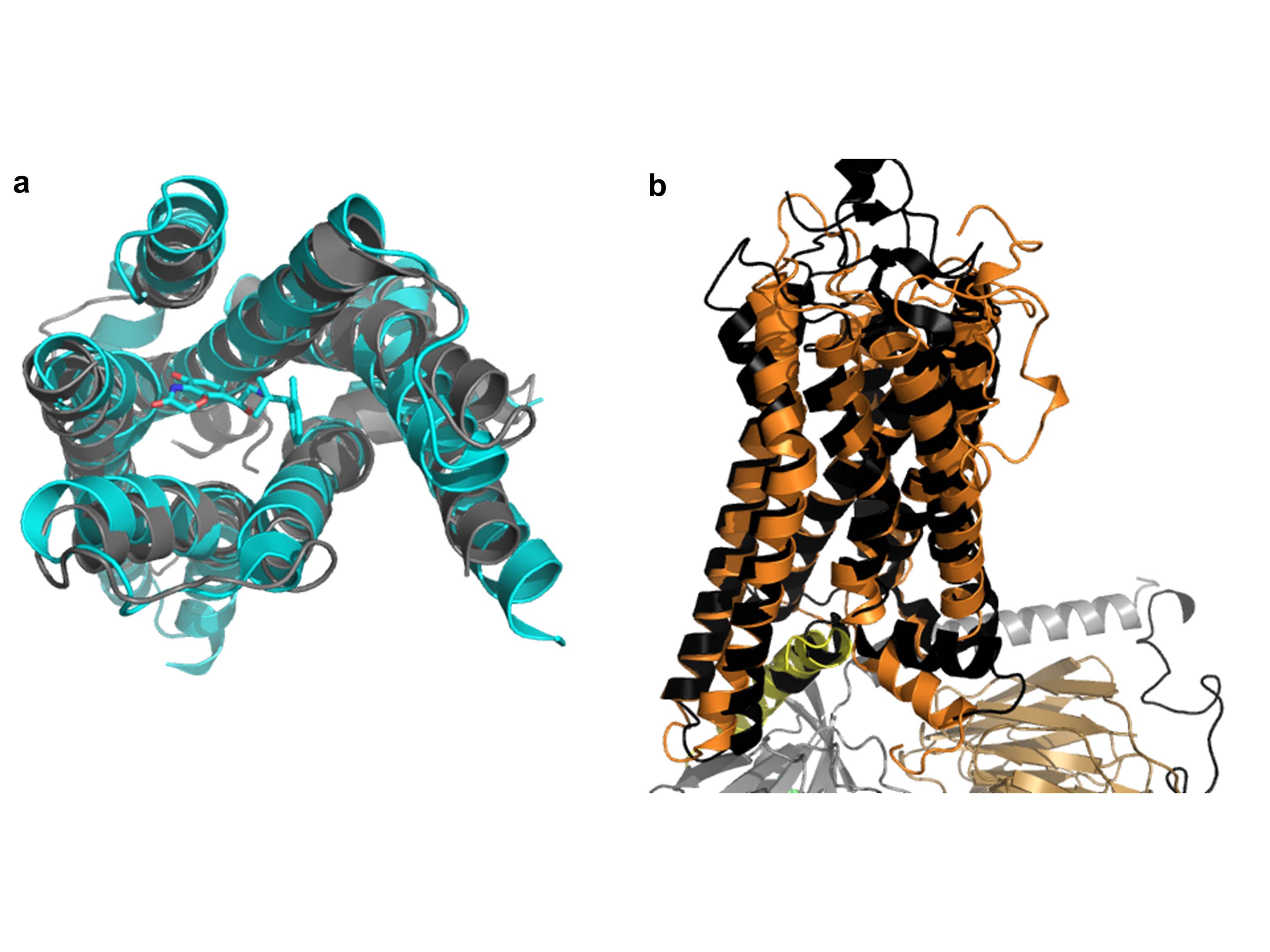
Figure 1.
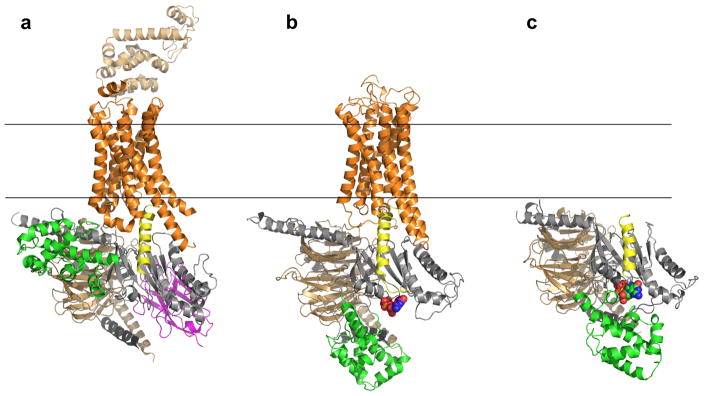
Overall structure of β2AR–Gs complex, our model of the R*–Gi complex, and the unbound Gi heterotrimer. (a) Crystal structure of β2AR–Gs complex (PDB 3SN611). The α5 helix of Gas is displaced 6Å towards the receptor and the helical domain (green) is displaced towards the membrane interface. (b) Unified model of the R*–Gi complex: According to DEER measurements, the displacement of helical domain (green) is on average a 15Å translation and 62º rotation after receptor binding. (c) Gi heterotrimer constructed as comparative model from Gt (PDB 1GOT10) structure. Receptor (orange), Gα GTPase domain (grey), Gα helical domain (green), Gβ (light brown), Gγ (black), Nanobody (magenta), T4L (sand), GDP (in spheres).
Gα C-terminal helix interactions triggers domain opening
We observed a 5.7Å translation and 63º rotation of the α5 helix. Our energetic analysis of this conformational change linked receptor-mediated changes in the α5 helix to the β6–α5 loop, the α1 and αG helices, and the GDP binding site. We hypothesize that disruption of contacts between these entities and the helical domain leads to domain separation. We determined an ensemble of models of the open state that match published data, and the ensemble reflects a wider space sampled by the helical domain than that presented in our recent work,9 which was published prior to the crystallographic structure of the complex. This unified model is overall consistent with the structure of the complex, with differences in the magnitude of domain separation.
Exploring possible locations of the helical domain
While qualitatively consistent with the β2AR–Gs complex, the placement of the helical domain in the unified model is less dramatic than that seen in the crystal structure, based on our DEER experiments for the R*–Gi complex9 (Supplemental Table 1, Supplemental Table 2). Average distances between residues in the helical and GTPase domain are less than the distances observed in the β2AR–Gs complex crystal structure. While the average interdomain distance is less than that seen in the crystal structure, the distribution of these distances is wide, consistent with a highly flexible helical domain that explores a range of conformations in the nucleotide-free state, as observed with electron crystallography13. Crystallization may stabilize a conformation that is not well populated in solution studies, whereas DEER captures an ensemble of conformations. We explored the possible positions of the helical domain of Gαi upon receptor binding through rigid body docking with subsequent reconstruction of loop regions and energy minimization. This protocol resulted in a pool of 739 models of the receptor-bound state with different positions of the helical domain.
Helical domain positions consistent with EPR distances
From this pool of docked complexes we selected an ensemble of nine models that collectively best reproduced the distance probability distributions of five different DEER distance measurements9 between pairs of spin-labeled residues (Figure 2b, Figure 2b bottom). In comparison, the ensemble of models for the basal state generated from Rosetta relaxation is less variable (Figure 2a, Figure 2a bottom). We converted distances between Cβ atoms (Cβ-Cβ distances) measured in the models to DEER distance probability distributions14,15. For a given ensemble of models, these probability distributions were compared with the DEER measurement (Supplemental Table 2)16. We compared the experimentally observed distance distributions with the distance distributions of the final ensemble model of the R*–Gi complex (Figure 3a).
Figure 2.

Placement of helical domain and rotation of α5 as observed by EPR measurements. (a) Gi in the basal state. (b) Gi bound to activated receptor R*. To illustrate motion, landmark residues are colored: L092 (red), E122 (green), D158 (yellow), V335 (cyan), I343 (blue). In both cases we show an ensemble of models that collectively fits the experimental data best. (a bottom, b bottom) Space-filled representations of the helical domain illustrate its positions for the respective states.
Figure 3.

Agreement of unified model with available experimental data. (a) Comparison of experimental distance distribution as observed in DEER measurements (blue) with the predicted distribution computed from the unified model of the R*–Gi complex (red). (b) Representation of the agreement with changes in accessibility observed in CW-EPR experimental data in C terminus | Gαi interface. Experimentally observed changes were classified into five groups from strong decrease (-2) to strong increase (+2). Average amino acid accessibility changes were classified likewise into five groups from strong decrease (-2) to strong increase (+2). Plotted is the difference, i.e. yellow and green colors indicate good agreement of model and experiment. (c) Agreement of unified model with changes in accessibility observed in deuterium exchange measurements using the same color scale as panel (b). (d) Agreement of unified model with single particle EM class averages.
Superimposing Gα of the generated conformations with the crystal structure of β2AR indicated that there are structures that agree to within a RMSD (root mean square deviation) of 2.2Å. This demonstrated that the location of the helical domain seen in the crystal structure was sampled, because there are Gα conformations which are similar to the Gα of the β2AR structure. This is important as these conformations could have been selected for the model ensemble if needed for agreement with the EPR data. The fact that these conformations were not selected i.e. needed for good agreement with the EPR data, suggests that they were not appreciably contributing to the conformational space sampled in our experiments.
The ensemble is consistent with single particle EM data
Westfield and co-workers13 performed single-particle electron microscopy (EM) analysis to examine the architecture of agonist-occupied β2-adrenoceptor (β2AR) in complex with the heterotrimeric G protein Gαsβγ. In their experiments, the location of the nanobody (Nβ37)-bound helical domain is variable, occupying a conformational space similar to that sampled by the helical domain in our ensemble (Figure 3d, Supplemental Movie 2). The space sampled by the helical domain overlaps to a large part with the region occupied by the helical domain and nanobody in the EM study13. The slight deviations observed can perhaps be attributed to the negative stain EM sample preparation, which may restrict the motion of the helical domain. Regardless, there is overall agreement between the EM structure and our unified model built on DEER restraints.
Agreement of model with accessibility data
To compare the unified model with accessibility information derived from CW-EPR and H/D exchange experiments, we computed the relative solvent accessible surface area for unbound and receptor-bound states of Gi. The amplitude and direction of this change in exposure was compared to the experimental values which had been classified into five bins (large increase, small increase, neutral, small decrease, large decrease, Supplemental Table 3 and Supplemental Table 4). As expected, we generally found that the predicted changes in accessibility exhibit similar trends to those seen in the experimental data (Figure 3b, c). The correlation coefficients are 0.33 for the CW EPR measurements and 0.56 for the H/D-exchange data. Note that no perfect correlation is expected as (1) experiments capture additional aspects beyond amino acid exposure and (2) exposure is estimated from the Cβ position alone. Small deviations from perfect agreement were expected as the experimental data depend not only on solvent accessibility but also on side chain and backbone dynamics only incompletely considered in this model.
Energetic analysis of inter- and intra-domain interfaces
We examined the stabilizing interactions between key interfaces in Gαi using Rosetta before and after receptor binding. Specifically, we studied four interfaces: Gαi-helical domain|Gαi-GTPase domain interface, GDP| Gαi-GTPase domain interface, C-terminal helix α5|Gαi-GTPase, and R*|Gαi-GTPase domain interface. We determined interactions that stabilize these regions before and after receptor activation17.
Basal Gαi-helical domain|Gαi-GTPase and GDP|Gαi-GTPase interfaces
The helical domain is held in place by interactions of α1 (E043, T048, K051, K054, I055) with αA (E65) and αF (Q171, L175, 5.5 Rosetta Energy Units (REUs) which correlate with kcal per mol18, Figure 4a, Supplemental Table 5, Supplemental Movie 3). The helical domain is also fixed by electrostatic interactions of αG (K270, K277) and β4–α3 (V233, E238) loops with αA (R090), αD–αE loop (R144, Q147, D150) and αF–β2 loop (R178, 4.3 REU). Lastly, the interface is stabilized by a contact between GDP and αD–αE loop (Y154, 2.0 REU). The total interaction energy is approximately 10.1 REU. GDP is stabilized through interactions with α1 (S044, S047, T048, 3.1 REU), the helical domain (Y154, 0.8 REU), and β6–α5 loop (T327, 0.9 REU). The total interaction energy is approximately 5.1 REU (Figure 4b, Supplemental Table 5, Supplemental Movie 4).
Figure 4.

Rosetta energetic analysis. (a) Analysis of energetics of helical domain|Gαi interface in free Gαi. The thickness of arrows in the top panel corresponds to the strength of the interaction in Rosetta Energy Units (REU, see legend). Residues in the bottom panel are colored by the interaction energy REU from red (repulsive) over white (neutral) to blue (attractive). Residues that contribute more than 0.5 REU are displayed as sticks and the three residues with the largest contributions are labeled. (b) Energetics of the GDP|Gαi interface in free Gαi. (c) Energetics of R*|Gαi interface in the R*-Gαi complex.
Receptor-bound R*|Gαi-GTPase domain interface
The Gα C-terminal peptide (I344, N347, L348, D350, C351, L353, F354) binds to the receptor through TM3 (V138, V139, K141), TM6 (E249, V250), and TM7–αC loop (K311, Q312, 8.2 REU, Figure 4c, Supplemental Table 5, Supplemental Movie 5). Further, intracellular loop 2 (F146) is interacting with αN–β1 loop at R32 (2.2 REU). The extended intracellular loop 3 (Q237, S240, T242, T243) interacts with α4 (E308), α4–β6-loop (D315, K317), and β6 (T321, 5.6 REU). The total interaction energy was approximately 17.2 REU. Comparison of residue distances for this interface with the coordinates of the β2 adrenergic receptor–Gs complex structure indicated residue E249 changes interactions most drastically, while the model ensemble showed small variation in the interface distances (Supplemental Table 6).
α5|Gαi-GTPase interface rewiring upon receptor interaction
In the basal state the C-terminal helix α5 of Gαi (N331, V332, Q333, V335, F336, A338, V339, T340, V342, I343) interacts favorably with β2, β3, β5, and β6 (F191, F196, I265, F267, Y320, H322, 6.4 REU) and α1 (T048, Q52, M053, I056, 5.0 REU, Figure 5a, Supplemental Table 5, Supplemental Movie 6). The β6–α5 loop (A326, T327, T329) interacts with α1 (T048, Q052, 2.5 REU) and GDP (1.4 REU).
Figure 5.

Rosetta energetic analysis of the interface between α5|Gαi-GTPase. (a) Basal state energetics. (b) Energetics of the R*–Gαi complex. Residues are colored by the interaction energy REU from red (repulsive) over white (neutral) to blue (attractive). Residues that contribute more than 0.5 REU are displayed as sticks and the three residues with the largest contributions are labeled. (c) Energy change (ΔREU) of C-terminal residues (β6–α5 loop and α5 helix) upon receptor binding. A blue color indicates stabilization, a red color indicates destabilization.
Upon interaction with the activated receptor (Figure 5b, Supplemental Movie 7), the α5 helix (I344, N347, L348, K349, D350, C351, G352, L353, F354) experiences an attraction to the receptor of 8.6 REU. This attractive interaction moves the α5 helix 5.7Å towards the receptor and triggers a rotation of the α5 helix by 63º. This is accompanied by a loss of helicity at the base of the α5 helix, which is in close proximity to bound nucleotide in the inactive heterotrimer. Thus, the base of the α5 helix appears to “melt” in the nucleotide-free state. The interaction of the α5 helix with β2, β3, β5, and β6 is modified and strengthened (F191, K192, L194, F196, I265, F267, E318, Y320, H322, 10.3 REU) upon interaction with activated receptors. At the same time, interactions of the α5 helix with α1 (T048, Q52, M053, I056, 2.2 REU) and GDP (0.2 REU) are substantially weakened. This was accompanied by loss of helical structure at the top of the α1 helix, effectively elongating the linker region between the GTPase domain and the helical domain, which may facilitate domain separation. A summary of residue stabilizations and destabilizations is shown in Figure 5c.
Interactions of residues E249 and E311 of R* changed most drastically from the coordinates of the β2AR–Gs complex structure, as measured by the change in distance to other residues in the interface. Also, the model ensemble showed small variation in the interface distances indicating the interactions were consistently predicted (Supplemental Table 7).
Helical domain position verified by DEER distances
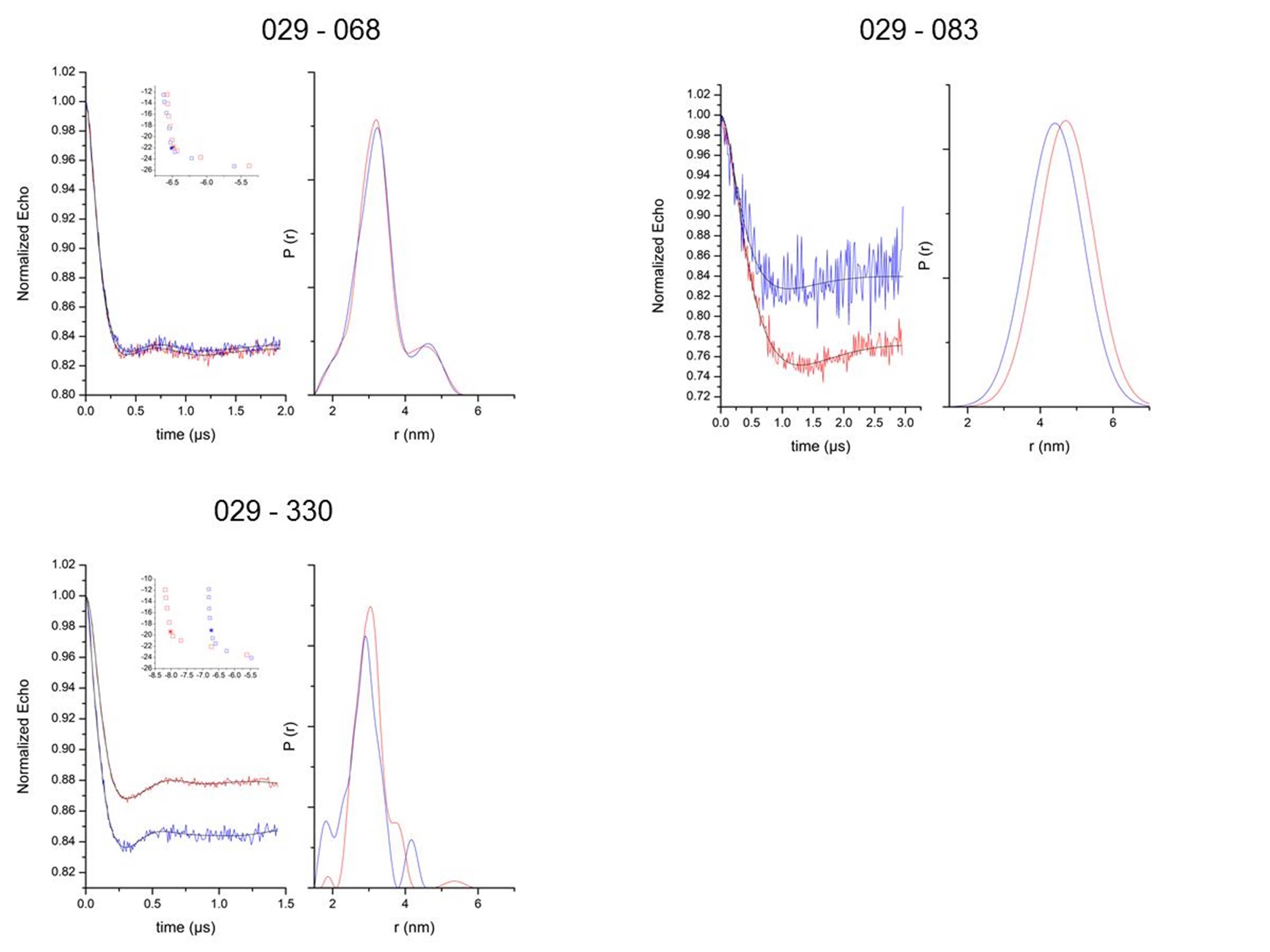
Double cysteine mutants in positions 29(αN)-68(αA) and 29(αN)-83(αA) were prepared to independently verify the position of the helical domain with respect to the GTPase domain in the unified model, and to differentiate it from the β2AR–Gs crystal structure. A cysteine depleted Gαi parent protein was used as a starting point for these studies. The cysteine mutants were labeled with a thiol-selective nitroxide probe, tested for functionality, and distances were determined by DEER19,20,21. Before receptor activation, the major populations in the distribution of 29-68 and 29-83 were centered at ~31Å and ~49Å, respectively. This was consistent with the model for the unbound state (Supplemental Figure 4a, Supplemental Table 1). Upon receptor activation, the distribution was centered at ~32 Å and ~45 Å, respectively. These results were in agreement with the receptor-bound ensemble in the unified model (Figure 6a), but different than that seen in the β2AR–Gs crystal structure, which predicts a substantial reduction of these distances (Supplemental Figure 4b). These results suggest that the helical domain may have been stabilized in an extreme orientation in the crystal structure. Nevertheless, the loss of interdomain contacts observed in the crystal structure is in overall agreement with our model. Our model supports a range of motion for the helical domain upon receptor activation, and the crystal structure may represent an extreme value along the continuum of possible orientations for the helical domain during signaling.
Figure 6.

Agreement of unified model with new structural data. (a) Comparison of the experimental distance distribution as observed in DEER measurements (blue) with the predicted distribution computed from the ensemble mode of the R*–Gi complex (red). (b–d) Comparison of accessibility of residues 171 and 191 in Gαi in the basal (black) and activated state (red). (b) Residues 171 and 191 in a Gαi protein were specifically modified with Alexa-fluor (A1), and emission (Em) was scanned at A1-specific wavelengths. (c) Measured fluorescence of cysteine mutants labeled with a fluorescent probe. Data represent the mean of a minimum of three independent experiments. Error bars show standard error of the mean. (d) Predicted burial as indicated by neighbor count based on the unified model. Bars show mean, and error bars show standard deviation.
Verification of the α5 helix rotation and translation
We prepared one double mutant in positions 29(αN)-330(α5) in order to test the intramolecular rearrangement of α5 after receptor activation. Both unified model and crystal structure predict a contraction of this distance. The observed distance distributions were consistent with this prediction, although the reduction was not as pronounced as in the model (compare Figure 6a, Supplemental Figure 4a). The ensemble of models gives a reduction of 5.0 Å, which is in agreement with the 2.2 Å experimental distance change. Specifically, the DEER distance distributions showed a change from 30.7 Å to 28.5 Å, as calculated from their weighted averages. The ensemble of models shows a change from 31.3 Å to 26.3 Å going from the receptor unbound to receptor-bound states.
We measured the number of nearest neighbors in our model to predict changes in solvent accessibility in the β2 strand and the linker between the α-helical and GTPase domain. Using this method, the solvent accessibility of F191, located in the β2 strand, is predicted to decrease upon activation, while solvent accessibility of Q171, located in one of the linkers between the α-helical and GTPase domain, is predicted to increase (Figure 6d). As an independent verification of our model, we individually mutated each of these residues to Cys in a Gαi protein lacking solvent exposed cysteines. We then labeled each mutant protein with a fluorescent probe, and examined the polarity of the environment of the labeled residues before and after receptor activation. Increases in solvent exposure increase the polarity reported by the probe, reflected by a reduction in the fluorescence emission. An increase in the hydrophobicity of the probe’s environment is typically reflected by an increase in emission from the labeled residue. As predicted by our model, residue 171 exhibited a decreased fluorescence upon receptor activation, as compared to the inactive state (Figure 6d, red vs black, respectively), suggesting a more solvent exposed environment for this residue upon domain separation, this is consistent with its location in the linker region between the helical and GTPase domains. On the other hand, F191 is located in the GTPase domain, packed between the β2-sheet and α5. We observed an increased emission from labeled residue 191 upon receptor activation (Figure 6b, red vs black), consistent with the increase in nearest neighbors predicted by our model. The increase and decrease in solvent exposure we observed for residues 171 and 191, respectively, were also consistent with mobility data previously reported for these residues (Supplemental Table 3, Supplemental Table 4). This is also consistent with a recent study identifying a more solvent excluded environment for the β2–β3 loop upon receptor activation 22,23. Thus, these new data provided independent validation of the predictions from the current model, in regions which are predicted to show both increases and decreases in solvent accessibility upon receptor activation.
Four critical interface regions were used to test our model experimentally, in both basal and receptor-activated state. M53 is in the interface of the α1 helix and the α5 helix and was predicted to stabilize this interaction. F196 is in the β3-sheet and was also predicted to stabilize the interaction with the α5 helix. E308 is in the α4 helix and critical for interaction with the receptor. All four residues were predicted by the model to be critical to stabilize the stimulated state; all but E308 were predicted to be critical residues stabilizing the basal state. We mutated each residue to a cysteine, and tested basal and receptor-mediated GTP and GDP exchange for each of the mutants relative to wild type (Figure 7b), and compared them to the predicted Rosetta interface energies (Figure 7a). Calculations were conducted using the ensemble of models, which contain the native sequence for heterotrimer and receptor. The predicted and experimental values were consistent (Supplemental Table 8), further supporting the predictive ability of our model for identifying residues critical for receptor interaction and nucleotide exchange.
Figure 7.
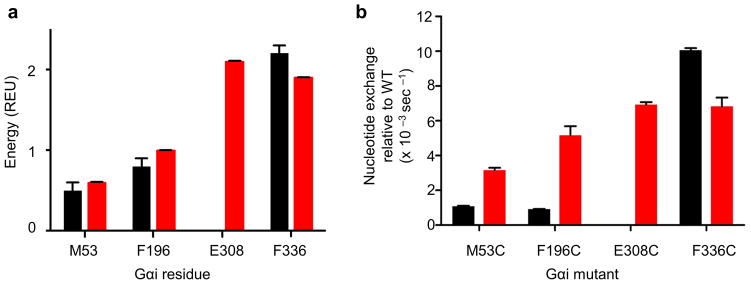
Validation of the model energetic predictions. (a) The predicted energetic contribution to a given residue’s corresponding interface is plotted for basal (black) and stimulated (red) states. Residues M53, F196, and F336 are within the α5 | Gαi interface. Residue E308 is within the R* | Gαi interface, and therefore no interface contributions are predicted in the basal state. Energy is given in Rosetta Energy Units (REU). Bars show mean, and error bars show standard deviation. (b) Basal and receptor-mediated nucleotide exchange rates. Gαi mutant exchange rates were compared, taking the absolute value of the difference of the nucleotide exchange rate relative to wild type Gαi in both basal and receptor mediated state. Data represent between 4 – 8 independent experiments, and error bars show standard error of the mean. The basal exchange of E308C was determined experimentally but was not significantly different from WT (wildtype), as was predicted by the model.
Discussion
In the current study, we highlighted changes in the orientation of the C-terminal α5 helix relative to its orientation in inactive heterotrimer, prior to binding to receptor. The energy associated with the interface of the α5 helix and surrounding regions are critically important for GDP binding and receptor-mediated GDP release. The β2AR–Gs complex was used as a template for creating a homology model of the rhodopsin–Gi heterotrimer complex that is the focus of our current model. Important interactions of Gα within the rhodopsin-Gi complex were compared to the interactions that the same regions exhibit in the inactive heterotrimer, in the absence of activated receptor. We then compared the orientation of the helical domain in the rhodopsin–Gi complex to that of the helical domain in the β2AR–Gs crystal structure in order to better understand the similarities and differences between the orientations afforded by the two different systems and methodologies involved.
There are some potential drawbacks inherent in our approach, such as perturbations of the system by the introduction of spin-labels or fluorescent probes. These can potentially perturb the biologically relevant conformation on a local or global level. Each experimental approach is aimed at a particular system under unique conditions. Coverage of experimental data is nonuniform, resulting in regions of high-confidence supported by multiple datasets and regions of low-confidence where data are sparse and/or affiliated with large error. Because of this, observations using different approaches and systems are not likely to be identical, nor do we expect them to be. Therefore, the hybrid model presented herein, like all models, is not likely to be correct in every detail, but is consistent with the current state of existing knowledge. The power of such a model is that it presents an atomic-detail hypothesis of the structure and energetics, thereby creating a roadmap for future experimental studies that can verify or reject parts of the model. In an iterative fashion, a completely verified atomic-detail model of the system can then be constructed.
The present analysis is specific for the rhodopsin–Gαiβγ complex. Gi is a close Gt family member which also couples to rhodopsin24. Gi was used for all experiments and modeling instead of Gt, as Gt does not express well. In result, the experimental EPR data used as restraints during modeling were specific for the rhodopsin–Gαiβγ complex. The energetic analysis which is sequence dependent was also specific for the rhodopsin–Gαiβγ complex. Mutational studies conducted on this specific system confirm our model.
To what extent the findings can be generalized to other GPCR–G protein systems is an important question that remains to be determined. The location of the helical domain as described by the structural ensemble is likely to be sampled in other GPCR–G protein systems. The mechanistic model resulting from using the crystal structure (β2AR–Gs) as a template, as was used here, would be expected to be similar to the extent that all GPCR–G protein systems exhibit some degree of similarity. However, specific, sequence-dependent differences likely contribute to the differences we observe, at both the G protein and GPCR level. A more rigorous and experimentally dense study focused on the individual proteins of interest will be required to study the same interactions in the β2AR–Gs or other GPCR–G protein systems.
The mechanism of receptor mediated G protein activation has been previously investigated. A “sequential release mechanism” proposes that binding of the C terminus of Gα allosterically causes the release of GDP25,26. This qualitative observation agrees with our model, which quantitatively describes the importance of the various interactions leading to GDP release. Another previous study used molecular modeling to investigate the mechanism of GDP release from Gα upon receptor binding. Consistent with our results, they propose that a rotation of α5 is a critical step towards GDP release, and implicate the β6–α5 loop as playing a key role in propagating the signal to GDP25, which is supported in a mutational study examining rates of nucleotide release27. This study implicated an interaction between the IL2 of the receptor and the N-terminus of Gα, an interaction which our energetic analysis independently identifies as an important interaction between R* IL 2 with the αN–β1 loop28. Molecular dynamics investigations of GDP release from Gαiβγ conducted in the absence of receptors suggested that several residues may be important for in interactions with GDP, including S44, S47, and T327 (ref. 29). Molecular dynamics were also used to look at the structural changes that the Gα subunit of transducin (Gαt) undergoes to release GDP30, again in the absence of receptors. Thus, the inclusion of activated receptor in the current study presents a major advance in efforts to model the changes in Gα which occur upon receptor activation.
The relative conformational space sampled by the helical domain within the ensemble to the GTPase domain of Gα was determined using DEER. Given the small number and large uncertainty of the EPR distance measurements, the nine conformations represented in the stimulated, receptor-bound state formed a representative ensemble of conformations sampled. Furthermore, the relative conformational space sampled by the helical domain within the ensemble was wider than in our previous model, which does not take into account the distribution of distances between labeled residues upon receptor activation. This relatively wide distribution resulted in an ensemble of models which may represent the dynamic changes in the orientation of the helical domain which accompanies receptor-mediated GDP release in a physiologically relevant environment.
Other regions of the model that were derived primarily from the crystallographic template are necessarily less flexible. Since our modeling template was based on the crystal structure, the model accuracy in these regions was sufficiently high to approach atomic detail. Therefore, precise values for the 5.7 Å shift and 63° rotation of the Gα C terminus with respect to its orientation in the inactive heterotrimer are reported. In these regions, our analysis of the energetic contributions to the stability of specific interfaces between regions of Gα in the inactive heterotrimer and receptor-bound activated complex lead to the current model of the mechanism of receptor-mediated nucleotide release.
The recent determination of the crystal structure of the β2AR–Gs protein complex provides the atomic-detail insight into the interaction of a G protein with an active GPCR that was required in order to complete the present study. The availability of this experimental structure is a milestone which greatly advanced our understanding the structural determinants of the receptor–G protein complex. Using primary data and computational modeling, and taking into account the crystal structure of the β2AR–Gs complex, we obtained an ensemble of structurally dynamic states, consistent with mutational, biophysical, and structural studies that are currently available. In our model, the average interdomain separation is less dramatic than that seen in the crystal structure, but it is in qualitative agreement with it, as well as cryo-EM studies. This model integrates multiple published data and provides a detailed energetic pathway for signal transduction between activated receptor and Gi protein. It thereby creates a pathway to elucidate the structural and energetic determinants of signal transduction between activated receptor R* and Gi.
In summary, based on DEER distance measurements and the hybrid model, the rhodopsin–Gi complex is best represented as a structural ensemble allowing GDP release and opening of the interdomain cleft, and the Gα helical domain to sample multiple orientations. The hybrid model here represents elements from both the β2AR–Gs crystal structure and dynamic conformational changes which occur in solution as the G protein interacts with activated receptor to catalyze the release of GDP. Thus, this work provides a framework and a roadmap for future experiments including high resolution modeling of the receptor-G protein complex.
Online Methods
Receptor unbound model of Gαiβγ
The model of Gαiβγ was constructed based on the PDB coordinates 1GOT9,10. Missing residues were reconstructed using kinematic loop closure31. The model of the receptor unbound state was then subjected to 100 independent relaxation trajectories that iterate between backbone perturbation, fast side chain optimization using a rotamer library32, and all atom gradient minimization in Rosetta full-atom force field33. The ten models with lowest Rosetta energy form the conformational ensemble representing Gαiβγ in the receptor unbound state (structures available in Supplemental Data 1). GDP was present throughout all steps of the protocol.
Receptor-bound Gαiβγ model consistent with experimental data
The crystal structure of the β2AR–Gs complex (PDB 3SN67) was used as the template for constructing a comparative model for the rhodopsin bound state of Gαiβγ. The sequence of metarhodopsin, bovine Gβ1 and Gγ1, and Gαi were threaded on the 3SN67 crystal structure. The receptor sequence was aligned using structure-structure alignment of 3SN67 with the structure of metarhodopsin 3PQR34. A blast sequence alignment was used to align Gβγ. For the alpha subunit, the published sequence alignment between Gαs and Gαi was used35. For each chain, Rosetta kinematic loop closure31 was used to construct missing coordinates. After loop construction, the model was relaxed in Rosetta 46 times. To accommodate the receptor, the relaxation utilized Rosetta’s full atom membrane potential11,12. The model with lowest Rosetta energy was used as the starting point for the comparative model of the R*–Gi complex.
No agonist was present during model construction. However, comparison of the crystal structure of activated opsin (3DQB36) with the β2AR–Gs complex crystal structure shows that the presence or absence of an agonist has only a small effect on the structure of the TM domain (Supplemental Figure 5a). The two receptor structures can be superimposed with an RMSD of 2.0 Å. The agonist likely stabilizes the active conformation of the β2AR, whereas our goal is to model the G-protein bound activated state of native rhodopsin.
Our best scoring model of activated rhodopsin aligned structurally to 2.5 Å RMSD to the β2AR– Gs over the entire complex. The receptor in our model agreed with the crystal structure of activated rhodopsin (PDBID 3DQB36) to an RMSD of 2.5 Å (Supplemental Figure 5b). Importantly, the crystal structure of activated rhodopsin (PDBID 3DQB36) could be superimposed with the β2AR to 2.0 Å RMSD. This indicates that the TM domain in the model remains in an active conformation during comparative modeling even though the agonist has not been explicitly added.
No regions of the model were assumed to be correct a priori. The goal was to refine the model with as much experimental data as is available. However, different parts of the model were influenced by different sets of data, and the backbone conformation of the receptor and G-protein complex was only slightly refined in some regions but sampled more exhaustively in others. Portions of the model were based on (a) the crystal structure template and refinement, (b) reconstructed through comparative modeling, and (c) positioned through EPR restraints and refinement (Supplemental Figure 3a, b, c).
Additionally, multiple experimental data were used to validate the model for specific residues: CW EPR (Supplemental Table 3, Figure 3b); DEER measurements (Supplemental Table 1); H/D exchange data (Supplemental Table 4, Figure 3c).
Exploring possible locations of the helical domain
The helical domain (residues 63 to 177) was separated from the rest of the nucleotide binding domain by removing linking residues 58–62 and 178–185. Possible placements of the helical domain were explored in 1,000 independent docking simulations. Both linker regions were reconstructed31 after docking and before each of these models was relaxed in the Rosetta full atom energy membrane potential11,12. This protocol resulted in a pool of 739 non-clashing models of the receptor-bound state with different positions of the helical domain. Detailed computational and experimental protocols are given in the supplementary note.
Helical domain positions consistent with DEER distances
A subset of models was selected that optimally reproduces the DEER distances and signal shapes. DEER data were simulated for each model using the knowledge-based potential14,15. The overall score of a given ensemble of models was the sum of the scores for the five previously published DEER distance measurements9. An ensemble of nine structures was selected from 1000 independent Monte Carlo simulations. This ensemble gave the best agreement between experiment and model (Supplemental Table 2). It constitutes the ensemble of the R*–Gi complex (structures available in Supplemental Data 2).
The distance distributions seen were in most cases too large to be explained with intrinsic flexibility of the label37. Therefore, an implicit model of the spin label is used to describe the conformational distribution of the spin label, as detailed previously16. We used this method to distinguish label distribution from backbone conformational changes. Distance K29-K330 in Figure 6a is an example of a distribution that is dominated by the spin label conformational distribution, with very little contribution by backbone changes in the ensemble. Distance K29-A83 in Figure 6a is an example with a distribution too wide to result from label conformational changes only.
Inter- and intra-domain interface energetic analysis
The energy values are reported in Rosetta Energy Units (REU) which correlate with kcal per mol 18. Energies are broken down on a per-residue basis to identify positions with changing interactions upon complex formation (Figure 4, Figure 5, Supplemental Table 5).
Materials for experimental studies
GDP and GTPγS were purchased from Sigma-Aldrich (Milwaukee, WI), and the cysteine reactive probe Alexa Fluor 595 C5 maleimide was purchased from Invitrogen (Madison, WI). All other reagents and chemicals were of the highest available purity. ROS membranes containing rhodopsin and Gβ1γ1 were prepared as described in6.
Protein expression and purification
Gαi and Gαi HI proteins were expressed and purified as described previously6, 24, 40. Both proteins were stored at −80 °C in 50 mM Tris, 100 mM NαCl, 2 mM MgCl2, 1 mM DTT, 10 μM GDP and 10% glycerol (pH. 7.5).
Intrinsic Trp fluorescence and AlF4 activation
Intrinsic tryptophan fluorescence was measured as describe previously 38. Gα (200 nM) subunits are monitored (ex/em 280/340 nm) before and after activation with 10 μM AlF4 in 50 mM Tris, 100 mM NαCl, 2 mM MgCl2, and 10 μM GDP, pH 7.5. Evaluation of the ability of selected Gαi proteins to undergo activation-dependent changes as a result of basal nucleotide exchange of GDP for BD-GTPγS was measured as described previously 39, with Gαi HI proteins exhibiting a 10-times higher rate of exchange than wild-type proteins due to removal of solvent-exposed cysteines required for site-specific fluorescent labeling. Briefly, emission intensity of Gαi protein (200 nM) was monitored at ex/em 280/340 nm before and after addition of GTPγS (10 μM). Exchange of GDP for GTPγS was determined by monitoring relative increase intrinsic Trp fluorescence, as described above. Nucleotide exchange assays are performed in buffer containing 50 mM Tris, 100 mM NαCl, 1 mM MgCl2, pH 7.5 at 18 ºC. Changes in fluorescence emission were determined from a minimum of three independent experiments, +SEM. Time-dependent fluorescence changes were fit to an exponential association curve using Prism 4.0 (GraphPad Software).
Protein labeling
Gαi HI proteins24 were labeled at a concentration of approximately 1 mg/mL in buffer free of reducing agent with a 5:1 probe:protein molar ratio in 50 mM Tris, 130 mM NαCl, 2 mM MgCl2 and 100 μM GDP, pH 7.5, followed by quenching with β-mercaptoethanol and removal of unbound probe with HPLC by size exclusion using a SW2000 column (Sigma-Aldrich, St. Louis, MO). Efficiency of labeling was between 25-40%. Chromatography was carried out in the same buffer supplemented with 10 μM GDP and 1 mM DTT. Monodispersity and molecular weight of the monomeric, labeled proteins was confirmed after purification by gel filtration HPLC comparing peak retention times and peak shape to results from column calibration performed with a broad range of molecular weight standards run on the same day as the purified samples (BioRad, Hercules, CA). The monomeric, labeled, purified proteins were pooled based on their ability to undergo activation-dependent changes as measured by intrinsic Trp211 activation (described above). Proteins with mutation of Trp211 were assayed by BD-GTPγS binding (described below) to ensure functional integrity of the labeled proteins.
Extrinsic fluorescence assays
For fluorescence studies of A1-labeled proteins, the emission maxima of labeled Gi protein (400 nM) was determined by scanning emission between 590-750 nm, with excitation at 580 nm after reconstitution of labeled Gα subunit with equimolar Gβ1γ1 subunit in buffer consisting of 50 mM Tris, 100 mM NαCl, 2 mM MgCl2, and 1 mM DTT, pH 7.5 at 18 ºC. All fluorescence data were analyzed as described under intrinsic Trp fluorescence.
A decrease in fluorescence after receptor activation indicates an increase in the polarity of the environment of the labeled residue, as compared to the environment in the inactive heterotrimer. A decrease in emission upon receptor activation is consistent with a more solvent exposed environment for the labeled residue. An increase in fluorescence is likewise correlated with a more hydrophobic environment, consistent with an increase in packing for the residue upon receptor activation.
Membrane binding assay
Membrane binding assay was evaluated as described previously40. Briefly, Gαi (5 μM) subunits were preincubated with Gβγ (10 μM) subunits on ice for 10 min. Then, in the dark, rhodopsin (50 μM) within ROS membranes was added to the heterotrimetric G protein in a buffer containing 50 mM Tris (pH 8.0), 100 mM NαCl, 2 mM MgCl2 and incubated on ice for 5 min. For dark measurements, reaction mixtures were protected from light for the rest of the procedure. Light activated samples, as well as light activated samples with GTPγS (100 μM), were incubated on ice for 30 min. Membranes and supernatant were separated by centrifugation and samples were resolved by SDS-PAGE and visualized with Coomassie blue and quantified by densitometry using a BioRad Multimager. The data represent the average of three independent experiments (Supplemental Figure 6a).
Spin Labeling and DEER measurement
Spin label (S-(1-oxy-2,2,5,5,-tetramethylpyrroline-3-methyl)-methanethiosulfonate, 200mM) in DMF was mixed with Gα subunit in a 2:1 molar ratio with buffer containing 50 mM Tris (pH 7.4), 100 mM NαCl, 2 mM MgCl2 and 50 μM GDP. The reaction mixture was shaken gently for 16 h at 4 ºC. Unreacted spin label was removed from sample by gel filtration chromatography or extensive washing with labeling buffer by using centrifugal concentrator with a molecular mass cutoff of 10 kDa. The final labeled protein was determined by Bradford assay using bovine serum albumin as standard. All of the spin-labeled mutants showed basal and receptor mediated tryptophan fluorescent increases in the presence of GTPγS with comparable level of unlabeled GαiHI protein (Supplemental Figure 6b). In addition to nucleotide exchange, they all showed the ability to form stable receptor–G protein complexes in the absence of guanine nucleotide. Double electron electron resonance (DEER) measurements were performed on a Bruker 580 pulsed EPR spectrometer operating at Q-band (33.5 GHz) using a standard four-pulse protocol41,42. Glycerol (30% w/w) was added to the samples prior to cooling. All experiments were carried out at 83 K. Analysis of the DEER data to determine the distance distributions, P(r), was carried in DeerAnalysis 2011 (ref. 43). The data was fit with Tikhonov regularization and L-curve determination of the optimal regularization parameter44 (Supplemental Figure 7). Some data was fitted with gaussians when the data was not adequately fit with Tikhonov regularization. For example, there are situations where the assumptions of Tikhonov regularization may not be suitable, as in the case of very broad distributions. These very broad distributions tend to have poorly defined L-curves in the typical range used to fit most data. It is in these cases that the Gaussian distributions were used to fit the data. The parameters derived from the Gaussian distribution overlap with the distribution obtained using Tikhonov regularization omitting the uncertainty in the fine structure of the distribution. To test our assay system we measured the distance between 90–238 residues before and after receptor activation and we found comparable distance distribution with the previous study9.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Work in the Meiler laboratory is supported through US National Institutes of Health (NIH) (R01 GM080403, R01 MH090192, R01 GM099842) and US National Science Foundation (Career 0742762) (J.M.). NIH National Research Service Award (MH086222) provided additional support (N.S.A). Work in the Hamm laboratory is supported through NIH (EY006062; A.M.P., A.I.K., and H.E.H.). NIH provided additional support (U54 GM084757; R.A.S.). The authors would like to thank S. Deluca of Vanderbilt University for implementing the Rosetta per-residue interface energy tool. The authors would like to thank H. Mchaourab of Vanderbilt University for his support with the DEER measurements (support to NIH S10 RR027091).
Footnotes
Author Contributions
N.S.A, A.M.P, A.I.K., H.E.H, and J.M. designed the experiments. N.S.A., A.M.P., A.I.K. and R.A.S. collected data. All authors contributed analysis. N.S.A, A.M.P, H.E.H, and J.M. wrote the manuscript with input from A.I.K. and R.A.S.
References
- 1.Onrust R, et al. Receptor and βγ Binding Sites in the α Subunit of the Retinal G Protein Transducin. Science. 1997;275:381–384. doi: 10.1126/science.275.5298.381. [DOI] [PubMed] [Google Scholar]
- 2.Cai K, Itoh Y, Khorana HG. Mapping of contact sites in complex formation between transducin and light-activated rhodopsin by covalent crosslinking: Use of a photoactivatable reagent. Proceedings of the National Academy of Sciences. 2001;98:4877–4882. doi: 10.1073/pnas.051632898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mazzoni MR, Hamm HE. Interaction of Transducin with Light-activated Rhodopsin Protects It from Proteolytic Digestion by Trypsin. Journal of Biological Chemistry. 1996;271:30034–30040. doi: 10.1074/jbc.271.47.30034. [DOI] [PubMed] [Google Scholar]
- 4.Slessareva JE, et al. Closely Related G-protein-coupled Receptors Use Multiple and Distinct Domains on G-protein α-Subunits for Selective Coupling. Journal of Biological Chemistry. 2003;278:50530–50536. doi: 10.1074/jbc.M304417200. [DOI] [PubMed] [Google Scholar]
- 5.Marin EP, Krishna AG, Sakmar TP. Disruption of the alpha5 helix of transducin impairs rhodopsin-catalyzed nucleotide exchange. Biochemistry. 2002;41:6988–94. doi: 10.1021/bi025514k. [DOI] [PubMed] [Google Scholar]
- 6.Oldham WM, Van Eps N, Preininger AM, Hubbell WL, Hamm HE. Mechanism of the receptor-catalyzed activation of heterotrimeric G proteins. Nat Struct Mol Biol. 2006;13:772–7. doi: 10.1038/nsmb1129. [DOI] [PubMed] [Google Scholar]
- 7.Rasmussen SG, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–55. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noel JP, Hamm HE, Sigler PB. The 2.2 A crystal structure of transducin-alpha complexed with GTP gamma S. Nature. 1993;366:654–63. doi: 10.1038/366654a0. [DOI] [PubMed] [Google Scholar]
- 9.Van Eps N, et al. Interaction of a G protein with an activated receptor opens the interdomain interface in the alpha subunit. Proc Natl Acad Sci U S A. 2011;108:9420–4. doi: 10.1073/pnas.1105810108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lambright DG, et al. The 2.0 angstrom crystal structure of a heterotrimeric G protein. Nature. 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- 11.Barth P, Wallner B, Baker D. Prediction of membrane protein structures with complex topologies using limited constraints. Proc Natl Acad Sci U S A. 2009;106:1409–14. doi: 10.1073/pnas.0808323106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barth P, Schonbrun J, Baker D. Toward high-resolution prediction and design of transmembrane helical protein structures. Proc Natl Acad Sci U S A. 2007;104:15682–7. doi: 10.1073/pnas.0702515104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Westfield GH, et al. Structural flexibility of the G alpha s alpha-helical domain in the beta2-adrenoceptor Gs complex. Proc Natl Acad Sci U S A. 2011;108:16086–91. doi: 10.1073/pnas.1113645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirst SJ, Alexander N, McHaourab HS, Meiler J. RosettaEPR: an integrated tool for protein structure determination from sparse EPR data. J Struct Biol. 2011;173:506–14. doi: 10.1016/j.jsb.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexander N, Al-Mestarihi A, Bortolus M, Mchaourab H, Meiler J. De Novo High-Resolution Protein Structure Determination from Sparse Spin-Labeling EPR Data. Structure. 2008;16:181–95. doi: 10.1016/j.str.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamarainen JK, et al. Improving similarity measures of histograms using smoothing projections. Pattern Recogn Lett. 2003;24:2009–2019. [Google Scholar]
- 17.Kortemme T, Kim DE, Baker D. Computational alanine scanning of protein-protein interfaces. Sci STKE. 2004:pl2. doi: 10.1126/stke.2192004pl2. [DOI] [PubMed] [Google Scholar]
- 18.Kellogg EH, Leaver-Fay A, Baker D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins: Structure, Function, and Bioinformatics. 2011;79:830–838. doi: 10.1002/prot.22921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeschke G, Bender A, Paulsen H, Zimmermann H, Godt A. Sensitivity enhancement in pulse EPR distance measurements. Journal of Magnetic Resonance. 2004;169:1–12. doi: 10.1016/j.jmr.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 20.Borbat PP, McHaourab HS, Freed JH. Protein structure determination using long-distance constraints from double-quantum coherence ESR: study of T4 lysozyme. J Am Chem Soc. 2002;124:5304–14. doi: 10.1021/ja020040y. [DOI] [PubMed] [Google Scholar]
- 21.Jeschke G, Polyhach Y. Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Physical Chemistry Chemical Physics. 2007;9:1895–1910. doi: 10.1039/b614920k. [DOI] [PubMed] [Google Scholar]
- 22.Hamm HE, Kaya AI, Gilbert JA, Iii, Preininger AM. Linking receptor activation to changes in Sw I and II of Gα proteins. Journal of Structural Biology. doi: 10.1016/j.jsb.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Preininger AM, Meiler J, Hamm HE. Conformational Flexibility and Structural Dynamics in GPCR-Mediated G Protein Activation: A Perspective. Journal of Molecular Biology. 2013;425:2288–2298. doi: 10.1016/j.jmb.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Medkova M, Preininger AM, Yu NJ, Hubbell WL, Hamm HE. Conformational changes in the amino-terminal helix of the G protein alpha(i1) following dissociation from Gbetagamma subunit and activation. Biochemistry. 2002;41:9962–72. doi: 10.1021/bi0255726. [DOI] [PubMed] [Google Scholar]
- 25.Scheerer P, et al. Structural and kinetic modeling of an activating helix switch in the rhodopsintransducin interface. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:10660–10665. doi: 10.1073/pnas.0900072106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kapoor N, Menon ST, Chauhan R, Sachdev P, Sakmar TP. Structural Evidence for a Sequential Release Mechanism for Activation of Heterotrimeric G Proteins. Journal of Molecular Biology. 2009;393:882–897. doi: 10.1016/j.jmb.2009.08.043. [DOI] [PubMed] [Google Scholar]
- 27.Chung KY, et al. Conformational changes in the G protein Gs induced by the [bgr]2 adrenergic receptor. Nature. 2011;477:611–615. doi: 10.1038/nature10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fanelli F, Dell’Orco D. Dark and photoactivated rhodopsin share common binding modes to transducin. Febs Letters. 2008;582:991–996. doi: 10.1016/j.febslet.2008.02.041. [DOI] [PubMed] [Google Scholar]
- 29.Louet M, Perahia D, Martinez J, Floquet N. A Concerted Mechanism for Opening the GDP Binding Pocket and Release of the Nucleotide in Hetero-Trimeric G-Proteins. Journal of Molecular Biology. 2011;411:298–312. doi: 10.1016/j.jmb.2011.05.034. [DOI] [PubMed] [Google Scholar]
- 30.Ceruso MA, Periole X, Weinstein H. Molecular Dynamics Simulations of Transducin: Interdomain and Front to Back Communication in Activation and Nucleotide Exchange. Journal of Molecular Biology. 2004;338:469–481. doi: 10.1016/j.jmb.2004.02.064. [DOI] [PubMed] [Google Scholar]
- 31.Mandell DJ, Coutsias EA, Kortemme T. Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat Methods. 2009;6:551–2. doi: 10.1038/nmeth0809-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bower MJ, Cohen FE, Dunbrack RL., Jr Prediction of protein side-chain rotamers from a backbone-dependent rotamer library: a new homology modeling tool. J Mol Biol. 1997;267:1268–82. doi: 10.1006/jmbi.1997.0926. [DOI] [PubMed] [Google Scholar]
- 33.Bradley P, Misura KM, Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309:1868–71. doi: 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- 34.Choe HW, et al. Crystal structure of metarhodopsin II. Nature. 2011;471:651–5. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- 35.Johnston CA, Siderovski DP. Receptor-mediated activation of heterotrimeric G-proteins: Current structural insights. Molecular Pharmacology. 2007;72:219–230. doi: 10.1124/mol.107.034348. [DOI] [PubMed] [Google Scholar]
- 36.Scheerer P, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 37.McHaourab Hassane S, Steed PR, Kazmier K. Toward the Fourth Dimension of Membrane Protein Structure: Insight into Dynamics from Spin-Labeling EPR Spectroscopy. Structure. 2011;19:1549–1561. doi: 10.1016/j.str.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mazzoni MR, Hamm HE. Tryptophan207 is involved in the GTP-dependent conformational switch in the alpha subunit of the G protein transducin: chymotryptic digestion patterns of the GTP gamma S and GDP-bound forms. J Protein Chem. 1993;12:215–21. doi: 10.1007/BF01026043. [DOI] [PubMed] [Google Scholar]
- 39.Preininger A, et al. Helix dipole movement and conformational variability contribute to allosteric GDP release in Gi subunits. Biochemistry. 2009;48:2630–2642. doi: 10.1021/bi801853a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Preininger AM, et al. Myristoylation exerts direct and allosteric effects on Galpha conformation and dynamics in solution. Biochemistry. 2012;51:1911–24. doi: 10.1021/bi201472c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pannier M, Veit S, Godt A, Jeschke G, Spiess HW. Dead-time free measurement of dipole-dipole interactions between electron spins. Journal of Magnetic Resonance. 2000;142:331–340. doi: 10.1006/jmre.1999.1944. [DOI] [PubMed] [Google Scholar]
- 42.Zou P, McHaourab HS. Increased Sensitivity and Extended Range of Distance Measurements in Spin-Labeled Membrane Proteins: Q-Band Double Electron-Electron Resonance and Nanoscale Bilayers. Biophysical Journal. 2010;98:L18–L20. doi: 10.1016/j.bpj.2009.12.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeschke G, et al. DeerAnalysis2006 - a comprehensive software package for analyzing pulsed ELDOR data. Applied Magnetic Resonance. 2006;30:473–498. [Google Scholar]
- 44.Chiang YW, Borbat PP, Freed JH. The determination of pair distance distributions by pulsed ESR using Tikhonov regularization. Academic Press Inc Elsevier Science; 2005. pp. 279–295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.