

Abstract
Purpose
Age-related macular degeneration (AMD) is a common cause of irreversible vision loss in the elderly. The hypothesis was that in vitro stimulation of RPE cells with Aβ1-40, a constituent of drusen, promotes changes in gene expression and cellular pathways associated with the pathogenesis of AMD, including oxidative stress, inflammation, and angiogenesis.
Methods
Confluent human RPE cells were stimulated with Aβ1-40, or the reverse peptide Aβ40-1, and genome wide changes in gene expression were studied with gene microarrays. Selected genes were verified by qRT-PCR and ELISA. Pathway analysis with gene set enrichment analysis (GSEA) and ingenuity revealed top functional pathways in RPE after Aβ1-40 stimulation.
Results
RPE cells stimulated with Aβ1-40 (0.3 μM) for 24 hours resulted in 63 upregulated and 22 downregulated previously known genes. The upregulated genes were predominantly in inflammatory and immune response categories, but other categories were also represented, including apoptosis, cell signaling, cell proliferation, and signal transduction. Categories of downregulated genes included immune response, transporters, metabolic functions and transcription factors. ELISA confirmed that secreted levels of IL-8 were two times higher than control levels. GSEA and ingenuity analysis confirmed that the top affected pathways in RPE cells after Aβ1-40 stimulation were inflammation and immune response related. Surprisingly, few angiogenic pathways were activated at the doses and exposure times studied.
Conclusions
Aβ1-40 promotes RPE gene expression changes in pathways associated with immune response, inflammation, and cytokine and interferon signaling pathways. Results may relate to in vivo mechanisms associated with the pathogenesis of AMD.
Age-related macular degeneration (AMD) is the most common cause of irreversible vision loss in the elderly. The disease is characterized by extracellular deposits known as drusen, which are found between the basal lamina of the retinal pigment epithelium (RPE) and the inner layer of Bruch’s membrane (BM). Although, the presence of drusen correlates with the development of AMD, the specific mechanisms underlying the biogenesis of drusen and its relationship to the disease remain elusive.1–3 Extensive biochemical analysis of drusen revealed the presence of several proteins linked to inflammation. Such proteins include immunoglobulins, acute-phase molecules (vitronectin, amyloid P, and fibrinogen), complement factors (C5 and C5b-9), and complement regulatory molecules (clusterin and complement receptor 1).4–8 These findings implicate an inflammatory process associated with drusen, but what activates or triggers the inflammation remains unknown.4,9–12
Several lines of evidence suggest that amyloid-β1-40 (Aβ1-40), a known constituent of drusen, is a potential candidate trigger peptide.3,13–15 Genetically modified mice with increased Aβ deposition display many traits consistent with human AMD, such as the accumulation of sub-RPE deposits, microglial activation, and degeneration of the retinal neurons and RPE.16,17 In the retina, cell types including neurons, RPE, and glia may provide a local source of Aβ species; they have been shown to have the cellular machinery for the synthesis of amyloid precursor protein (APP) and cleavage enzymes.10 Sequential APP processing by β- and γ-secretase produces the major Aβ species, Aβ1-40. Recent in vitro studies in which RPE cells are stimulated with Aβ1-40 demonstrated several results that are consistent with a proinflammatory response. Wang et al.18 reported that Aβ1-40 stimulation caused abnormal activity of complement factor I (CFI), an inhibitory regulator of the complement cascade. In another study, stimulation of RPE cells with Aβ1-40 modulated the expression of vascular endothelial growth factor (VEGF), which is known to promote angiogenesis, a late-stage feature of AMD.17 These results prompted us to further elucidate the effects of Aβ1-40 stimulation on RPE cells in vitro by differential microarrays and corresponding functional pathway analysis. We hypothesize that Aβ1-40 stimulation of RPE cells in vitro promotes gene expression changes associated with cellular pathways implicated in the pathogenesis of AMD, including oxidative stress, inflammation, angiogenesis, and apoptosis.
Methods
Aβ1-40 Oligomerization
We chose to focus on the effects of the oligomeric form of Aβ1-40 because, unlike Aβ1-42 (the more toxic form associated with Alzheimer’s plaques), Aβ1-40 was identified in drusen sites.3 Also, a recent study19 using an RPE cell line compared oligomeric Aβ1-40 with Aβ1-42 and demonstrated that their effects on RPE function were comparable, confirming original reports by Kayed et al.20 that Aβ1-40 and Aβ1-42 have similar toxic effects on several cell types tested.
Aβ1-40 peptide and the control reverse peptide Aβ40-1 were purchased from American Peptide (Sunnyvale, CA) as a salt-free, lyophilized powder. The protocol used to synthesize Aβ oligomers was obtained from Invitrogen Canada (Burlington, ON, Canada). Briefly, lyophilized Aβ1-40 (or the reverse peptide Aβ40-1) was dissolved in 200 μL hexafluoroisopropanol (HFIP) and sonicated for 30 seconds. The HFIP solution was then transferred by a 22-gauge syringe to a new 1.4-mL tube (Eppendorf, Fremont, CA) containing 700 μL of distilled water. A Teflon-coated micromagnetic stirring bar was added, and the tube was closed with a perforated cap to allow HFIP to vent after evaporation. The tube was placed in a holder inside a fume hood on top of a stirring plate, and stirred with a Teflon-coated micro stir bar at 300 rpm for 24 to 48 hours. Aliquots of the supernatant were analyzed for confirmation of Aβ oligomers using atomic force microscopy (AFM) and dot blot assay.
Atomic Force Microscopy
AFM was performed as previously described.21,22 Briefly, Aβ1-40 samples were prepared for AFM examination by placing 10 μL of solution onto freshly cleaved mica (Ted Pella, Inc., Redding, CA). The sample was allowed to adhere to the mica surface for 10 minutes at room temperature and was subsequently washed with double-distilled water to eliminate contamination due to buffer and salts and to help reduce background contrast. AFM images were captured with a microscope (BioScope with the Nanoscope IIIa Controller and a G-type scanner; Digital Instruments, Santa Barbara, CA). The contact mode was used for all images with DNP silicon nitride cantilevers (Digital Instruments). A minimum of four random regions of the mica surface was examined to ensure that similar structures existed throughout the sample.
Dot Blot Assay
A dot blot assay was performed to determine the oligomeric form Aβ1-40. Approximately 3 μL of Aβ1-40 solution was spotted onto a nitrocellulose membrane and allowed to dry. The membrane was then blocked with 10% nonfat dry milk in Tris-buffered saline (TBS) containing 0.01% Tween 20 (TBST) solution overnight at 4°C, and washed three times in full-strength TBST for 5 minutes before being incubated for 1 hour at room temperature with the A11 anti-oligomer antibody using the manufacturer’s protocol (Invitrogen Corp., Carlsbad, CA). The membrane was then washed three times in TBST for 5 minutes and incubated for 1 hour at room temperature with a standard horseradish peroxidase–conjugated secondary antibody made in goat against rabbit IgG diluted at 1:2000 (Promega, Madison, WI). The blots were finally washed three times in TBST for 5 minutes and developed for 20 minutes in a solution of tetramethylbenzidine (TMB; Promega).
Cell Culture of Human RPE Cells
Human fetal RPE cells were used in all the experiments. Human RPE cells were isolated from fetal donor eye tissues, as described elsewhere.23,24 Methods for securing human tissue were humane and included proper written informed consent, which complied with the Declaration of Helsinki. Human fetal donor eyes had no known disease and were used under the guidelines and regulation of the IRB at the University of British Columbia, Vancouver, Canada. The eyes were cut circumferentially, the vitreous removed, and the neuroretina gently detached from the RPE cell layer. The choroid/RPE layer was placed in 2% Dispase (Invitrogen) in Hanks’ balanced salt solution (HBSS; Invitrogen) for 25 minutes at 37°C. The RPE layer was then removed in fragments and passed through 70- and 40-μm nylon mesh filters (Falcon Plastics, Oxnard, CA). Only the fragments that were left behind were retained. After centrifugation at 1500 rpm for 5 minutes, the fragments were gently dissociated and seeded on to laminin-coated six-well plates (Falcon Plastics). RPE cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen-Gibco, Grand Island, NY) containing 10% fetal bovine serum (FBS; Invitrogen-Gibco), 100 μg/mL penicillin (Sigma-Aldrich, St. Louis, MO), 100 μg/mL streptomycin (Sigma-Aldrich), and 2 mM L-glutamine (Invitrogen) at 37°C in a humidified atmosphere of 95% air and 5% CO2. At confluence, the cells were detached with 0.05% trypsin/0.02% EDTA (Invitrogen), collected by centrifugation, and expanded. In the following experiments, passage 5 cells were used, and RPE cells from the same donor were used in an individual experiment.
Cell Viability Assay
RPE cells (20 × 103 cells/well) were seeded in 200 μL medium on a 96-well culture plate and incubated with DMEM containing 10% FBS, for 24 hours. The medium was then discarded, and the cells were washed three times with phosphate-buffered saline (PBS). The MTT reduction assay was used as an index of cell viability. Briefly, the oligomeric form of Aβ1-40 (0.01, 0.3, 1.0, 5.0, and 10.0 μM) and the control, reverse peptide, Aβ40-1 (0.3 μM) were used to stimulate RPE cells for various lengths of time (3, 6, 12, or 24 hours). After stimulation, the media were aspirated and the RPE cells were subsequently incubated in serum-free medium containing 0.4 mg/mL MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] for 4 hours. Mitochondrial and cytosolic dehydrogenases of living cells reduced the yellow tetrazolium salt (MTT) to a purple formosan dye that was then detected by spectrophotometry. After 4 hours, the MTT solution was aspirated, and 150 μL of dimethylsulfoxide (DMSO) was added for a period of 20 minutes. Optical densities of the supernatant were read at 550 nm with a microplate spectrophotometer (Bio-Tek Instruments, Inc., Winooski, VT). Absorbances were normalized to those of the untreated control cultures, which were also incubated in serum-free medium, which represented 100% viability. Two independent experiments in quadruplicate were performed in this study. The mean ± SD of the data was analyzed.
Aβ Stimulation
RPE cells (0.3 × 106/well) were seeded in 2 mL of culture medium in a six-well plate for 4 days. Before stimulation, the cells were washed three times with PBS. The RPE cells were then treated with Aβ1-40 (the oligomeric form) or Aβ40-1 (the reverse peptide) at a concentration of 0.3 μM for 3, 6, 12, or 24 hours in 1 mL of serum-free DMEM. Untreated RPE cells containing only serum-free DMEM were used as the negative control. As the chemical constituents of serum often vary, serum-free DMEM was used in all stimulation studies to ensure a controlled experimental design.
Microarray and Data Analysis
The total cellular RNA was isolated from cultured human RPE cells according to manufacturer’s recommendations (TRIzol; Invitrogen). RNA samples were then subsequently treated with DNase treatment and removal (TURBO DNA-free; Ambion, Streetsville, ON, Canada). Quantification of RNA was performed with spectrophotometry (ND-1000; NanoDrop Products, Thermo Fisher Scientific, Wilmington, DE). Total RNA integrity was confirmed with a bioanalyzer (model 2100; Agilent Technologies, Palo Alto, CA) according to the manufacturer’s protocol.
The labeling and hybridization of the cRNA was performed at the Prostate Cancer Microarray Centre, University of British Columbia. One microgram of total RNA from all samples and from human universal reference RNA (Stratagene, La Jolla, CA) was amplified and labeled with fluorescent dyes (Cy3 and Cy5; Low RNA Input Linear Amplification Labeling Kit; Agilent Technologies) according to the manufacturer’s protocol. The amount and specific activity of the resulting fluorescently labeled cDNA was assessed with the spectrophotometer. Equal amounts of Cy3- sample and Cy5-labeled universal human reference cDNA were cohybridized (Whole Human Genome Oligo Microarray; Agilent Technologies), comprising over 41,000 human genes and transcripts, for 18 hours before washing and scanning. Data were extracted from the resulting images (Feature Extraction Software; Agilent Technologies).
Red (Cy5 labeled cRNA) and green (Cy3 labeled cRNA) processed signals were entered into the computer software (GeneSpring 7.3.1; Agilent Technologies) and normalized via manufacturer’s protocols for two-color experiments as follows: per spot, divide by control channel; per chip, normalize to the 50th percentile; per gene, normalized to median. The results represent the mean values from three independent experiments. A list of differentially expressed genes was generated by applying a t-test with P < 0.05 between the Aβ1-40-treated group and the untreated group and by applying a change filter with a cutoff of ±1.5-fold. Linearization of the data was undertaken by log2 transformation.
Real-Time Quantitative (q)RT-PCR
Reverse transcription reactions were performed for each RNA sample (1 μg) using reverse transcription reagents (Superscript III; Invitrogen Canada). Real-time PCR was performed with a sequence-detection system (Prism 7300; Applied Biosystems [ABI], Foster City, CA). SYBR green PCR master mix (ABI) was used on cDNA samples in 96-well optical plates and analyzed by using qRT-PCR methods, as described elsewhere.25 Oligonucleotide primers for all genes of interest and for the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were designed for real-time PCR on computer (Primer Express 2.0 software; ABI). All primers were purchased from Proligo LL (Boulder, CO). Relative quantification of gene expression was performed using the x-fold change method, as published26 and as recommended by the manufacturer of the sequence detection system (ABI). The relative gene expression was represented by the difference between the normalized values of the experimental samples to that of corresponding sham controls (ΔΔCt). 2−ΔΔCt for each product was used to calculate changes. A Pearson’s correlation coefficient was used to calculate the correlation between microarray and qRT-PCR log2-fold changes.
ELISA
Levels of IL-1β, Il-8, and tumor necrosis factor (TNF)–related apoptosis-inducing ligand TNFSF10 (TRAIL) in the supernatant samples were measured using proteome arrays (SearchLight; Thermo Fisher Scientific, Woburn, MA). Samples were incubated for 1 hour on the array plates that were prespotted with capture antibodies specific for each protein biomarker. The plates were emptied and washed three times before the addition of a cocktail of biotinylated detection antibodies to each well and then were incubated for 30 minutes. The plates were then washed three times and incubated again for 30 minutes with streptavidin-horseradish peroxidase. All incubations were at a room temperature with agitation at 200 rpm. The plates were again washed before the addition of chemiluminescent substrate (SuperSignal Femto; Thermo Fisher Scientific) and were immediately imaged (SearchLight; Thermo Fisher Scientific) and the data analyzed (Array Analyst software; Ambion).
Gene Set Enrichment Analysis (GSEA)
To test for sets of related genes that might be systematically altered in Aβ1-40-treated RPE cells, we performed studies of gene sets in functional pathways by analyzing our microarray dataset using GSEA.27 A total of 1892 curated gene sets containing genes whose products are involved in specific metabolic and signaling pathways were obtained from pathway databases or from peer-reviewed published material. The gene sets were screened against the GSEA-ranked microarray data sets to calculate enrichment score (ES) for each gene set. An enrichment score reflects the degree to which a gene set is overrepresented at the extreme top or bottom of the ranked microarray data set list of genes. The curated gene sets were screened against the microarray data set, and enrichment plots were generated for each gene set. An enrichment plot displays a running enrichment score as a function of the rank-ordered probes in the microarray data set. To adjust for multiple hypothesis testing, the ES for each gene was normalized to account for the size of the set, yielding a normalized enrichment score (NES). The proportion of false positives was controlled for by calculating the false discovery rate (FDR) corresponding to each NES.27,28
Pathway Analysis
Differentially expressed genes that met our criteria of selection were also analyzed for global functions, network, and canonical pathways (Ingenuity Pathway Analysis; Ingenuity Systems, Redwood City, CA). A spreadsheet (Excel; Microsoft, Redmond, WA) of our data containing a gene list with gene identifiers and corresponding changes was entered into the system, and patterns of differential gene expression were compared with those in the knowledge base (Pathways Knowledge Base; Ingenuity Systems).
Results
Characterization of Aβ1-40 Oligomers
After incubation for 48 hours, Aβ1-40 oligomers were examined with the use of AFM and immunodot blot assay. AFM analysis showed that the oligomeric preparations were composed of small globular structures with a distinct lack of fibrils (Fig. 1A). These results were consistent with those in previous studies.29–31 Dot blot examination with the anti-oligomeric specific antibody (A11) further confirmed the presence of oligomeric structures as it reacted positively with our Aβ1-40 samples (Fig. 1B). Furthermore, the A11 antibody did not react against a fibrillar standard sample of Aβ1-40, confirming the specificity of the antibody to the oligomeric form.
Figure 1.
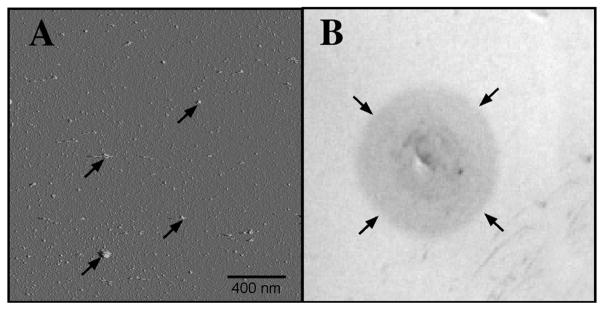
Confirmation of oligomeric form of Aβ1-40. (A) AFM examination of Aβ1-40 oligomeric solution at 48 hours. The Aβ1-40 solution was applied to freshly cleaved mica and imaged according to Chromy et al.48 AFM image shows predominately small globular structures (arrows) and a distinct lack of fibrils. (B) Dot blot with the use of the A11 antibody shows positive staining with the Aβ1-40 sample (arrows) compared to no staining in both negative control and fibrillar form of Aβ samples (Invitrogen). Primary and secondary antibodies were diluted according to manufacturer’s recommendations, 1:1000 and 1:2000, respectively.
RPE Cell Viability after Stimulation with Aβ1-40 Oligomers
We used an MTT reduction assay to examine the effects of Aβ1-40 and the reverse peptide Aβ40-1 on viability of confluent RPE. Figure 2A shows the dose-dependent effects of Aβ1-40 stimulation (0.001–0.3 μM) at 3 hours. Note that the reverse peptide Aβ40-1 had a minor effect on the viability of RPE (97% ± 0.05% SD), compared with the DMEM control (100%). We also tested the effects of several doses of Aβ1-40 (0.01–10.0 μM) on RPE cell viability at the 24-hour exposure time. At 0.01 μM Aβ1-40 oligomers, the lowest dose, mean (SD) cell viability was measured to be 81% (0.08%) and decreased to 25% (0.06%) at 10.0 μM (Fig. 2B). We also examined the effects of 0.3 μM Aβ1-40 on the viability of RPE at 3, 6, 12, and 24 hours. Note that cell survival gradually dropped from 83% (0.03%) at 3 hours to 57% (0.07%) relative to the corresponding control cultures by 24 hours (Fig. 2C).
Figure 2.

MTT reduction assay for cell viability after stimulation with Aβ1-40 or the control reverse peptide Aβ40-1. (A) Confluent RPE cells were subjected to stimulation with 0.001 to 0.3 μM oligomeric Aβ1-40 or the reverse peptide, 0.3 μM Aβ40-1, for 3 hours. Cell viability was significantly reduced at 0.3 μM Aβ1-40 compared with cells incubated in serum-free DMEM or to cells incubated in the control reverse peptide Aβ40-1. Note that lower doses (0.001 to 0.1 μM A β1-40) induced proliferation of the RPE. (B) A dose–response curve for RPE cells stimulated with 0.01- to 10.0-μM solutions of oligomeric Aβ1-40 for 24 hours demonstrates 81% and 57% cell viability at 0.01 and 0.3 μM. Cell viability drops to <30% at concentrations of Aβ1-40 greater than 0.3 μM. All doses resulted in significant differences with cells grown in serum free DMEM only. (C) Confluent RPE cells were stimulated with 0.3 μM solutions of oligomeric Aβ1-40 at 3, 6, 12, and 24 hours. The graph depicts the mean and SD from three independent experiments. RPE cells stimulated with Aβ1-40 at 0.3 μM displayed a time-dependent decrease in viability dropping to 58% cell viability at 24 hours as assessed with the MTT assay. A two-sample t-test was performed to compare the viability of the cells in the control group (cells incubated in serum-free DMEM and not stimulated with Aβ1-40) to that of the cells incubated in serum-free DMEM and stimulated with 0.3 μM Aβ1-40 at each time point. Note that at all time points, the viability of the stimulation group was lower than the control (unstimulated) group (*P < 0.05). The data are shown as the mean and SD of results in three independent experiments.
Gene Expression Analysis of RPE Cells after Aβ1-40 Stimulation
Analysis of the gene expression response of RPE cells after 0.3 μM Aβ1-40 treatment was undertaken by using whole-genome oligo arrays (Agilent). At 24 hours, we observed 63 upregulated and 20 downregulated previously known genes (Tables 1, 2). The upregulated genes were found predominantly in the immune response category, but several other categories were also represented, including apoptosis, transcription factors, transporters, cell proliferation, cell signaling, and signal transduction. The three upregulated genes that exhibited the greatest changes were IL-1β (4.8 ± 0.49-fold), RSAD2 (2.58± 0.57-fold), and IL-8 (2.38 ± 0.42-fold). Relatively few genes were downregulated in response to Aβ1-40 stimulation. Categories of downregulated genes also included immune response, transporters, cell metabolism, and transcription factors.
Table 1.
Upregulation of Differentially Expressed Genes in Aβ-Treated RPE Cells at 24 Hours
| Name | Description | GenBank Accession No. | Change (x-Fold) | P |
|---|---|---|---|---|
| Immune response | ||||
| IL-1β* | Homo sapiens interleukin 1, beta | NM_000576 | 4.80 | 0.0022 |
| RSAD2* | Homo sapiens radical S-adenosyl methionine domain containing 2 | NM_080657 | 2.58 | 0.0256 |
| IL-8* | Homo sapiens interleukin 8 | NM_000584 | 2.38 | 0.00177 |
| LAIR1 | Homo sapiens leukocyte-associated Ig-like receptor 1 | NM_021706 | 2.32 | 0.0332 |
| MX1* | Homo sapiens myxovirus (influenza virus) resistance 1, interferon-inducible protein p78 (mouse) | NM_002462 | 2.18 | 0.0187 |
| OAS3 | Homo sapiens 2′-5′-oligoadenylate synthetase 3, 100kDa | NM_006187 | 1.99 | 0.000495 |
| IF144L* | Homo sapiens interferon-induced protein 44-like | NM_006820 | 1.98 | 0.00842 |
| IFI27* | Homo sapiens interferon, alpha-inducible protein 27 | NM_005532 | 1.81 | 0.00436 |
| OAS1 | Homo sapiens 2′,5′-oligoadenylate synthetase 1, 40/46kDa | NM_002534 | 1.80 | 0.0154 |
| PRKCZ | Homo sapiens mRNA, chromosome 1 specific transcript KIAA0505. | AB007974 | 1.71 | 0.0186 |
| CX3CL1 | Homo sapiens chemokine (C-X3-C motif) ligand 1 | NM_002996 | 1.71 | 0.00906 |
| LSP1 | lymphocyte specific protein 1 | W725T9 | 1.68 | 0.00255 |
| CXCL2* | Homo sapiens chemokine (C-X-C motif) ligand 2 | NM_002089 | 1.68 | 0.023 |
| TNFSF10* | Homo sapiens tumor necrosis factor (ligand) superfamily, member 10 (TRAIL) | NM_003810 | 1.58 | 0.0149 |
| OASL | Homo sapiens 2′-5′-oligoadenylate synthetase-like | NM_003733 | 1.57 | 0.016 |
| IL-33* | Homo sapiens chromosome 9 open reading frame 26 (NF-HEV) (C9orf26) | NM_033439 | 1.55 | 0.00679 |
| GBP4 | Homo sapiens guanylate binding protein 4 | NM_052941 | 1.55 | 0.0332 |
| MX2 | Homo sapiens myxovirus (influenza virus) resistance 2 (mouse) | NM_002463 | 1.54 | 0.0191 |
| CFI* | Homo sapiens I factor (complement) | NM_000204 | 1.54 | 0.0234 |
| SPN | Homo sapiens sialophorin (gpLI15, leukosialin, CD43) | NM_003123 | 1.50 | 0.0138 |
| Apoptosis | ||||
| XAF1* | Homo sapiens XIAP associated factor-1 | NM_017523 | 2.10 | 0.0192 |
| C8orf4 | Homo sapiens chromosome 8 open reading frame 4 | NM_020130 | 1.70 | 0.0152 |
| CECR2 | Homo sapiens cDNA FLJ34435 fis, clone HLUNG2000955 | AK091754 | 1.65 | 0.0156 |
| Transcription regulation | ||||
| ZNF175 | Homo sapiens zinc finger protein 175 | BC007778 | 1.92 | 0.00566 |
| THC2317006 | Q7IRG2(Q71RG2) FP2860, partial (26%) | THC2317006 | 1.89 | 0.0222 |
| FOXA2 | Homo sapiens forkhead box A2 | NM_021784 | 1.76 | 0.0107 |
| TEAD1 | Tea domain family member 1 (SV4O transcriptional enhancement factor) | CR594735 | 1.75 | 0.0169 |
| Transporter | ||||
| NR4A3* | Homo sapiens nuclear RNA export factor 3 | NM_022052 | 1.86 | 0.0198 |
| C14orf68 | Homo sapiens chromosome 14 open reading frame 68 | NM_207117 | 1.68 | 0.00185 |
| SLC30A4* | Homo sapiens solute carrier family 30 (zinc transporter), member 4 | NM_013309 | 1.64 | 0.0303 |
| NPCIL1 | Homo sapiens NPC1 (Niemann-Pick disease, type CI, gene)-like 1 | NM_013389 | 1.64 | 0.00187 |
| PKD2L2 | Homo sapiens polycystic kidney disease 2-like 2 | NM_014386 | 1.55 | 0.0118 |
| Cell proliferation | ||||
| NPY | Homo sapiens neuropeptide Y | NM_000905 | 2.65 | 0.0252 |
| MT3* | Homo sapiens metallothionein 3 (growth inhibitory factor (neurotrophic)) | NM_005954 | 1.71 | 0.0056 |
| Cell signaling | ||||
| STC1 | Homo sapiens stanniocalcin 1 | A1476245 | 1.99 | 4.14E-05 |
| TSHB | Homo sapiens thyroid stimulating hormone, beta | NM_000549 | 1.52 | 0.0498 |
| Metabolism | ||||
| CYP2D6* | Homo sapiens cytochrome P450, family 2, subfamily D, polypeptide 6 | NM_000106 | 1.82 | 0.000452 |
| SULT4A1 | Homo sapiens sulfotransferase family 4A, member 1 | NM_014351 | 2.02 | 0.00517 |
| Signal transduction | ||||
| PMCH | Homo sapiens promelanin-concentrating hormone | NM_002674 | 1.99 | 0.00127 |
| SSTR2* | Homo sapiens somatostatin receptor 2 | NM_001050 | 1.51 | 0.00651 |
| Miscellaneous function | ||||
| EGFL8 | Brain development: EGF-like-domain, multiple 8 | NM_030652 | 1.77 | 0.0248 |
| RCAN3 | Calcium-mediated signaling: RCAN family member 3 | BM457392 | 2.00 | 0.00402 |
| PACRG | Cell death: PARK2 co-regulated | NM_152410 | 1.55 | 0.0222 |
| FLCN | Cell cycle: folliculin | AL831885 | 1.50 | 0.0199 |
| FNDC5* | Extracellular matrix: fibronectin type III domain containing 5 | NM_153756 | 1.52 | 0.0299 |
| LOC221272 | Microtubule cytoskeleton organization: FLJ21659 fis, clone COL08743. | AK025312 | 1.64 | 0.0174 |
| FLJ21272 | Microtubule cytoskeleton organization: FLJ21272 fis, clone COL01753. | AK024925 | 1.56 | 0.0147 |
| MTMR9 | Phospholipid dephosphorylation: myotubularin related protein 9 | AF339813 | 1.55 | 0.0394 |
| DPP6 | Proteolysis: dipeptidyl-peptidase 6 | BX094874 | 1.54 | 0.0403 |
| PMCHL2 | Synaptic transmission: promelanin-concentrating hormone-like 2 | NM_153381 | 2.36 | 0.00696 |
| HERC5* | Ubiqitin cycle: hect domain and RLD 5 | NM_016323 | 1.73 | 0.000338 |
| CLDN6 | Cell-cell adhesion: claudin 6 | NM_021195 | 1.82 | 0.0277 |
| PRRG4 | Calcium-ion binding: proline rich Gla (G-carboxyglutamic acid) 4 (transmembrane) | NM_024081 | 2.19 | 0.00463 |
| SMOC1 | Calcium-ion binding: SPARC related modular calcium binding 1 | NM_022137 | 1.51 | 0.0104 |
| FRMD3 | Protein binding: FERM domain containing 3 | BG216229 | 1.59 | 0.0138 |
| LXN | Enzyme inhibitor: latexin | NM_020169 | 1.57 | 0.00212 |
| CMPK2 | Kinase: cytidine monophosphate(UMP-CMP) kinase 2, mitochondrial | BG547557 | 2.10 | 0.00714 |
| TRIM14 | Metal-ion binding: tripartite motif-containing 14 | NM_014788 | 1.53 | 0.0141 |
| DSCR6 | Protein binding: Down syndrome critical region gene 6 | NM_018962 | 1.87 | 0.0213 |
| CI6ORF70 | Protein binding: chromosome 16 open reading frame 70 | BC008476 | 1.56 | 0.0419 |
| AK123481 | Protein binding: K1AA1377 | AK123481 | 1.56 | 0.0034 |
| BRD4 | Protein binding: bromodomain containing 4 | NM_014299 | 1.64 | 0.0341 |
| RNASE1* | RNA binding: ribonuclease, RNase A family, 1 (pancreatic) | NM_198232 | 2.00 | 0.0026 |
The data are presented as x-fold changes and log-transformed x-fold changes. The expression level of genes greater than 1.5-fold was considered upregulated (P < 0.05).
Genes that are selected for verification by RT-PCR.
Table 2.
Downregulation of Differentially Expressed Genes in Aβ-Treated RPE Cells at 24 Hours
| Name | Description | Gene Bank Accession No. | Change (x-Fold) | P |
|---|---|---|---|---|
| Immune response | ||||
| PSG8 | Homo sapiens pregnancy specific beta-1-glycoprotein 8 | NM_182707 | −1.64 | 0.0123 |
| SEMA4A | Homo sapiens sema domain, immunoglobulin domain (Ig), transmembrane domain (TM) and short cytoplasmic domain, (semaphorin) 4A | NM_022367 | −1.75 | 0.00761 |
| IL-9R | Homo sapiens interleukin 9 receptor (IL-9R) | NM_176786 | −1.85 | 0.0407 |
| Transporter | ||||
| MFSD7 | major facilitator superfamily domain containing 7 | NM_032219 | −1.52 | 0.0141 |
| SLC12A3 | Homo sapiens solute carrier family 12 (sodium/chloride transporter), member 3 | NM_000339 | −1.59 | 0.0158 |
| TRPV5 | Home sapiens transient receptor potential cation channel, subfamily V, member 5 | NM_019841 | −1.61 | 0.0367 |
| GLTPD1 | glycolipid transfer protein domain containing 1 | NM_001029885 | −1.64 | 0.00111 |
| ACCN1 | Homo sapiens amiloride-sensitive cation channel 1, neuronal (degenerin) | NM_183377 | −1.69 | 0.00179 |
| Metabolism | ||||
| LYZL2 | Homo sapiens lysozyme-like 2 | NM_183058 | −1.49 | 0.0139 |
| ALDH1L1 | Homo sapiens aldehyde dehydrogenase 1 family, member L1 | NM_012190 | −2.04 | 0.00838 |
| Transcription regulation | ||||
| MAFA | Homo sapiens v-maf musculoaponeurotic fibrosarcoma oncogene homolog A | NM_201589 | −1.56 | 0.00821 |
| FOXE1 | Homo sapiens HFKH4 mRNA for fork head like protein | X94553 | −1.64 | 0.0499 |
| Miscellaneous function | ||||
| RNF36 | Apoptosis: ring finger protein 36 | NM_182985 | −1.61 | 0.0192 |
| THBS1* | Cell motility: Thrombospondin 1 | N48043 | −1.64 | 0.000305 |
| ACTL6B | Cytoskeleton: actin-like 6B | NM_016188 | −2.22 | 0.00128 |
| PLCXD2 | Intracellular signaling cascade: phosphatidylinositol-specific phospholipase C, X domain containing 2 | NM_153268 | −1.52 | 0.0137 |
| MASTL | Protein phoshorylation: microtubule associated serine/ threonine kinase-like | THC2405366 | −1.56 | 0.0208 |
| GPR78 | Signal transduction: G protein-coupled receptor 78 | NM_080819 | −1.69 | 0.000824 |
| FIBIN | Translation initiation factor: fin bud initiation factor | NM_203371 | −1.52 | 0.00345 |
| RSPO3 | Wnt receptor signaling: R-spondin 3 homolog (Xenopus laevis) | NM_032784 | −1.96 | 0.00811 |
| ACPP | Acid phosphatase activity: acid phosphatase, prostate | NM_001099 | −1.64 | 0.0189 |
| PLEKHA3 | Phospholipid binding: pleckstrin homology domain containing, family A (phosphoinositide binding specific) member 3 | BC035471 | −1.64 | 0.000975 |
The data are presented as x-fold changes and log-transformed x-fold changes. Decreased expression changes are expressed as −(treatment/ control)−1. The expression levels of genes decreased more than −1.5-fold are considered downregulated (P < 0.05).
Genes that are selected for verification by RT-PCR.
Confirmation of Differentially Expressed Genes after Aβ1-40 Stimulation
Real-time qRT-PCR was performed to validate 18 genes of interest that were shown to be differentially regulated by the microarray (black bars, Fig. 3). The gene-specific primer sequences used for qRT-PCR are shown in Table 3. The amplified product for a housekeeping gene, GAPDH, was used as a normalizer and endogenous control. The qRT-PCR-derived validation of the direction and level of expression of the 18 selected genes is displayed in Figure 3 (white bars). The direction of expression was validated by qRT-PCR in 17 of the 18 genes. The Pearson’s correlation coefficient of the PCR and microarray data for the 17 of the 18 genes was 0.65. The single gene that did not validate in the direction of expression with qRT-PCR was thrombospondin, type I, domain containing 1 (THSD1), which the microarray identified as downregulated (−1.64 fold) and RT-PCR identified as upregulated (+1.53-fold).
Figure 3.

Genes of interest from the differential gene expression of RPE cells stimulated by 0.3 μM Aβ1-40 for 24 hours and analyzed by Agilent Oligo microarray were further verified by qRT-PCR. Primers used for each gene product are shown in Table 3. Note that the microarray data shown here and in Tables 1 and 2 all demonstrated a ≥1.5-fold change, and reached significance (P < 0.05). The largest changes were seen for cytokines, IL-1β and IL-8, and RSAD2 (viperin). Values are expressed as relative changes (log2-fold) with respect to the corresponding controls. Data represent the mean ± SEM of three independent experiments for both microarray (P < 0.05) and for qRT-PCR. The Pearson’s correlation coefficient of the microarray data and PCR log2 values for the 17 genes shown in the black and white bars is 0.65. These same 17 genes were also assessed after stimulation with the control reverse peptide (0.3 μM Aβ40-1) for 24 hours by qRT PCR. Note that the changes associated with the reverse peptide were significantly lower than results obtained after 0.3 μM Aβ1-40 stimulation.
Table 3.
RT-PCR Primers
| Gene | Forward Primer | Reverse Primer | Reference |
|---|---|---|---|
| IL-8 | AGGTGCAGTTTTGCCAAGGA | TTTCTGTGTTGGCGCAGTGT | |
| IL-1β | AAGCTGAGGAAGATGCTG | ATCTACACTCTCCAGCTG | |
| CFI | ATTGTGGAGACCAAAGTGATGAAC | TGATACTGGCTTGGAATGCAAA | |
| CXCL2 | GATAGAGGCTGAGGAATCCAAGAA | ACATTTCCCTGCCGTCACA | |
| RSAD2 | ACACAGGAATAATGACCCCAAAA | TGAGCTAAATGTCAGGTCCTGTGTA | |
| MX1 | GCCAGGACCAGGTATACAG | GCCTGCGTCAGCCGTGC | Leong et al.32 |
| IFI44L | CCAATTACACCTGAGCATTCTACTTTT | AGACATAAGCCACACAGTGAATCCT | |
| IFI27 | TGGCTCTGCCGTAGTTTTGC | CACAGCCACAACTCCTCCAAT | |
| HERC5 | GGGATGAAAGTGCTGAGGAG | CATTTTCTGAAGCGTCCACA | Kroismayr et al.33 |
| SLC30A4 | GGCTATCATCAAAATCACCAACCA | CGGTGATGAGCATTATATCTCCATT | Overbeck et al.34 |
| IL-33 | CACCCTCAAATGAATCAGG | GGAGCTCCACAGAGTGTTCC | Carriere et al.35 |
| THSD2 | TGCACGAAGAAGGGAAAAACAT | TCTCGGACCCGTGTTTCAG | |
| TNFSF10 | ATGGCTATGATGGAGGTCCAG | TTGTCCTGCATCTGCTTCAGC | Komatsuda et al.36 |
| XAF1 | CCAGAAAATAAGTATTTCCACC | TTATACTTCTTGTCTTTGGACG | Kempkensteffen et al.37 |
| CYP2D6 | CCTACGCTTCCAAAAGGCTTT | AGAGAACAGGTCAGCCACCACT | Kojima et al.38 |
| SSTR2 | CCCCAGCCCTTAAAGGCATGT | GGTCTCCATTGAGGAGGGTCC | Kumar et al.39 |
| NR4A3 | CAACTAGTCCAACCCCTTAAAATTCA | ACCTTGAGIAACTCTTCACCCTTCA | |
| FN | GACCACACCGCCCACAAC | TCCTACATTCGGCGGGTATG | |
| GAPDH | CATCCATGACAACTTTGGTATCGT | CAGTCTTCTGGGTGGCAGTGA | Wang et al.25 |
In total, 18 genes were selected for validation by RT-PCR. Nearly, all expression patterns were comparable to the microarray data.
We also performed qRT-PCR for selected genes after stimulation of RPE cells with the reverse peptide (Aβ40-1). qRT-PCR was performed for the 17 genes of interest obtained from the microarray analysis (Agilent) that were differentially upregulated after Aβ1-40 stimulation. The amplified product for the same housekeeping gene, GAPDH, was used as a normalizer and endogenous control. As expected, stimulation with the control reverse peptide (Aβ40-1) resulted in changes that were insignificant (Fig. 3, gray bars) relative to those observed after Aβ1-40 stimulation (Fig. 3, white bars).
Supernatant Studies
Analysis of supernatants from the RPE cell culture was performed to identify the secreted levels of selected gene products. Among the three gene products (IL-8, IL-1β, TNFSF10 [TRAIL]) examined, only IL-8 was detected in significant amounts in the supernatant samples. After stimulation with Aβ1-40 for 24 hours, the RPE cells secreted IL-8 at an increased level compared with the untreated RPE cells. As shown in Figure 4, the amount of secreted IL-8 was 91% higher than in the control untreated group.
Figure 4.
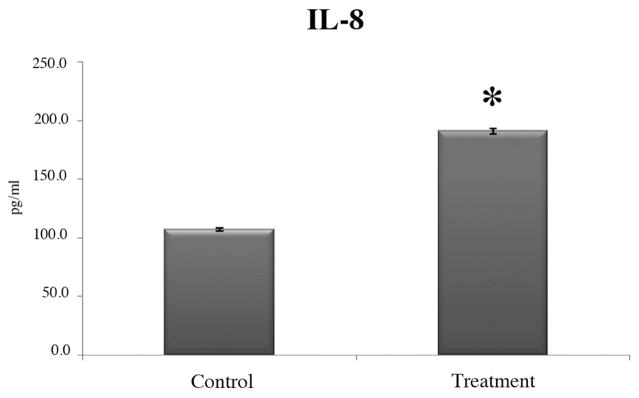
Cytokine levels in cell culture supernatant samples. Human fetal RPE cells challenged with 0.3 μM Ab1-40 oligomers for 24 hours showed increased secretion of IL-8 by ELISA. Compared to control cells, IL-8 secretion was increased, by approximately a factor of 2, in cultures of Ab1-40-treated cells (*significance by t-test, P < 0.05). Mean values from three independent experiments and the SEM are shown.
Pathway Analysis
Using the mRNA expression profiles of human RPE cells treated with Aβ1-40, we sought to determine the functional gene sets that correlated highly with our gene expression data using two types of pathway analysis. GSEA analysis identified 50 significantly enriched gene sets that correlated with the genes induced after Aβ1-40 treatment. Many of these gene sets are highly enriched with genes related to immune response and inflammatory processes. Table 4 lists some of the top gene sets selected by the GSEA analysis. Note that all sets include genes associated with inflammatory processes. After 24 hours, the Aβ1-40 treatment induced strong activation of genes associated with the interferon signaling pathway, immune signaling pathways, and the NF-κB signaling pathway involved in inflammatory processes (NES ≥ 2.03). Leading-edge analysis of gene set 13 in Table 4 within GSEA uncovered a subset of genes, including IL-1β and IL-8, within this gene set that was principally responsible for the total enrichment score (Table 5). As IL-1β and -8 were both upregulated in our data set, the GSEA analysis highlights the significance of inflammatory processes in the major pathways activated after Aβ1-40 stimulation in our study. Complementary GSEA analysis was unable to identify any pathways significantly enriched with Aβ1-40 downregulated genes. Figure 5 lists the top genes that appear across multiple enriched gene sets from the GSEA analysis. The leading-edge analysis of our microarray data identified genes highly correlated with inflammation, immune response, interferon-regulated genes, neurodegeneration, and apoptosis.
Table 4.
Summary of GSEA Analysis
| Gene Set(Pathway) | Genes within Gene Set (Top 20 only) | NES | FDR | |
|---|---|---|---|---|
| 1 | DAC_BLADDER_UP(IFN signaling) | OAS2, MX1, IF144L, OAS1, CXCL2, TNFSF10, STC1, IFIT3, STAT1, ICAM1, MMP1, IRF7, CIS, MX2, C3, IL13RA2, TNFAIP3, HBEGF, NT5E, SLP1 | 2.38 | 0.000 |
| 2 | IFNA_UV-CMV_COMMON_HCMV_6HRS_UP (IFN and inflammatory signaling pathways) | OAS2, RSAD2, MX1, IFI44L, OAS1, IF127, OASL, TNFSF10, IFIT3, INDO, ISG20, CIORF38, TDRD7, STXI1, BAZIA, SAMHD1, TLR3, IRF7, GCH1, MX2 | 2.37 | 0.000 |
| 3 | DAC_IFN_BLADDER_UP (IFN signaling) | OAS2, MX1, OAS1, CXCL2, TNFSF10, IFIT3, STAT1, ICAM1, IRF7, CIS, MX2, C3, ILI3RA2, TNFAIP3, SLP1, CCL5, CCL20 | 2.36 | 0.000 |
| 4 | TAKEDA_NUP8_HOXA9_3D_UP (IFN signaling) | OAS2, RSAD2, MX1, IFI44L, OAS3, OAS1, CTSG, HERC5, IF127, OASL, IFIT3, IFIT1, IFIT2, IFIH1, CCL18, INDO, PLSCR1, DDX58, EPSTI1, DST | 2.35 | 0.000 |
| 5 | IFNA_HCMV_6HRS_UP (IFN and inflammatory signaling pathways) | OAS2, RSAD2, MX1, 1F144L, OAS1, IF127, OASL, TNFSF10, IFIT3, INDO, PLSCR1, ISG20, STAT1, TRIM22, IFIT5, CIORF38, TDRD7, STXI1, ZBTB20, BAZIA | 2.32 | 0.000 |
| 6 | CMV_HCMV_TIMECOURSE_12HRS_UP (IFN and inflammatory signaling_pathways) | OAS2, RSAD2, MX1, OAS1, OASL, TNFSF10, IFIT3, ISG20, CH25H, B4GALT5, IRF7, SOD2, PRPF19, MX2, GBP2, TNFAIP6, RARRES3, RHOB, CCL5, NR4A3 | 2.28 | 0.000 |
| 7 | TAKEDA_NUP8_HOXA9_8D_UP (IFN signaling) | OAS2, RSAD2, MX1, IFI44L, OAS3, OAS1, CTSG, HERC5, IFI27, LXN, NR4A2, IFIT3, IFIT1, IFIT2, DHRS3, C20ORF103, IFIH1, MTSS1, INDO, DDX58 | 2.18 | 0.001 |
| 8 | BENNETT_SLE_UP (IFN signaling) | OAS2, MX1, IFI44L, OAS1, OASL, TNFSF10, IFIT3, PLSCR1, CAMP, STAT1, C2, TAP1, IFI35, TDRD7, IFITM3, IRF7, APOBEC3C, MX2, SERPING1, LY6E | 2.14 | 0.002 |
| 9 | TAKEDA_NUP8_HOXA9_10D_UP (IFN signaling) | OAS2, RSAD2, MX1, IFI44L, OAS3, OAS1, HERC5, DCAL1, IFI27, OASL, NR4A2, IFIT3, IFIT1, IFIT2, DHRS3, CD38, C20ORF103, IFIH1, INDO, DDX58 | 2.12 | 0.003 |
| 10 | IFNALPHA_NL_UP (IFN signaling) | RSAD2, MX1, IFI44L, IF127, TNFSF10, PLSCR1, VCAM1, STAT1, IFITM1, UBE2D3, BST2, IRF7, CALR, MX2, HLA-E, PIAS4, KIAA0409, UBE2L6, AKR7A2, PSME1 | 2.10 | 0.004 |
| 11 | SANA_TNFA_ENDOTHELIAL_UP (IFN, inflammatory, and immune signaling pathways) | IL8, OAS2, MX1, IFI44L, OAS3, OAS1, CXCL2, CX3CL1, IFIT1, CXCL3, IFIH1, FIJ20035, PARP14, VCAM1, BIRC3, UBD, CCL2, GBP1, OXR1, NFKBIA | 2.07 | 0.006 |
| 12 | SANA_IFNG_ENDOTHELIAL_UP (IFN, inflammatory, and immune signaling pathways) | OAS2, MX1, IFI44L, QAS1, TNFSF10, CX3CL1, NOD27, IFIH1, INDO, ILI8BP, FLJ20035, PARP14, TNCRNA, STAT1, UBD, DTX3L, GBP1, LAP3, C17ORF27, CXCL9 | 2.04 | 0.008 |
| 13 | NTHIPATHWAY (NF-κB signaling-related to inflammation) | ILIB, IL8, DUSP1, NFKBIA, NR3C1, EP300, CREBBP, TGFBR1, IKBKB, MAP2K6, TLR2, RELA, MAPK14, MAPK11, TGFBR2, CHUK, MAP3K7, NFKB1, MYD88, MAP2K3 | 2.04 | 0.008 |
| 14 | GALINDO_ACT_UP (Inflammatory and apoptosis signaling) | IL1B, CXCL2, ILI8RAP, ILIRN, PTGS2, PDGFB, LILRB4, DUSP1, NFKB1A, IER3, FOSL1, CSF3, ICAM1, CCL3, RREB1, TNFSF9, BCL3, UBC, TNFAIP2, CCL4 | 2.04 | 0.008 |
| 15 | CMV_UV-CMV_COMMON_HCMV_6HRS_UP (IFN and inflammatory signaling) | RSAD2, MX1, OASL, NR4A2, IFIT3, ILI1, ZC3HAV1, RIPK1, PMAIP1, POLG2, ATF3, CREM, GCH1, C12ORF22, PSCD1, NR4A3, BTRC, SLC745, NR4A1 | 2.03 | 0.007 |
GSEA analysis of the microarray results for Aβ treatment versus control helped in identifying gene sets correlated predominately with inflammation. Pathway 13, NTHIPATHWAY is identified as one of the inflammatory gene sets of particular interest, such as IL-β and IL-8, the top-ranked genes in this set. All gene sets can be accessed at the Molecular Signatures Database, http://www.broad.mit.edu/gsea/msigdb/index.jsp (provided in the public domain by the Broad Institute at Massachusetts Institute of Technology, Cambridge, MA).
Table 5.
Associated Gene Found in the NTHIPATHWAY Gene Set
| Probe | Rank in Gene List | Rank Metric Score | Running Es | Core Enrichment | |
|---|---|---|---|---|---|
| 1 | ILIB* | 0 | 3.264 | 0.3542 | Yes |
| 2 | IL8* | 8 | 2.035 | 0.5747 | Yes |
| 3 | DUSP1 | 961 | 0.461 | 0.5703 | Yes |
| 4 | NFKBIA | 1,052 | 0.439 | 0.6128 | Yes |
| 5 | NR3C1 | 1,116 | 0.428 | 0.6556 | Yes |
| 6 | EF300 | 1,747 | 0.320 | 0.6543 | Yes |
| 7 | CREBBP | 1,812 | 0.311 | 0.6844 | Yes |
| 8 | TGFBR1 | 2,068 | 0.279 | 0.7002 | Yes |
| 9 | IKBKB | 3,348 | 0.180 | 0.6466 | No |
| 10 | MAP2K6 | 3,627 | 0.165 | 0.6486 | No |
| 11 | TLR2 | 4,454 | 0.126 | 0.6151 | No |
| 12 | RELA | 4,626 | 0.119 | 9.6182 | No |
| 13 | MAPK14 | 5,538 | 0.082 | 0.5750 | No |
| 14 | MAPK11 | 5,719 | 0.075 | 0.5729 | No |
| 15 | TGFBR2 | 6,135 | 0.058 | 0.5554 | No |
| 16 | CHUK | 6,171 | 0.057 | 0.5596 | No |
| 17 | MAP3K7 | 6,989 | 0.029 | 0.5160 | No |
| 18 | NFKB1 | 7,241 | 0.020 | 0.5039 | No |
| 19 | MYD88 | 8,062 | −0.006 | 0.4576 | No |
| 20 | MAP2K3 | 10,744 | −0.094 | 0.3146 | No |
| 21 | MAP3K14 | 12,052 | −0.142 | 0.2553 | No |
| 22 | TNF | 16,472 | −0.524 | 0.0597 | No |
A detailed look into this gene set shows that IL-1b and IL-8 are the top-ranked genes in our experimental Aβ dataset. Genes that are found in the leading edge of this gene set are marked “Yes”in the core enrichment column. They are the genes that significantly contribute to the total enrichment score of the NTHIPATHWAY gene set.
Figure 5.

GSEA leading edge gene sets analysis. The analysis selects the genes that appear across multiple enriched gene sets from the public databases. These genes are expected to have higher biological significance. The leading edge analysis of our microarray data identified gene highly correlated with inflammation, immune response, interferon-regulated genes, apoptosis, and neurodegeneration.
Pathway analysis of differentially expressed genes that reached criteria in our Aβ1-40 stimulation studies revealed the top six gene networks that reach threshold (P < 0.05) are leukocyte extravasation signaling, natural killer cell signaling, hepatic cholestasis, FXR/RXR activation, IL-10 signaling, and the complement activation (Fig. 6).
Figure 6.

Pathway analysis by Ingenuity Pathway Analysis (IPA) software program. Note that the six top-ranked pathways, leukocyte extravasation signaling, natural killer cell signaling, hepatic cholestasis, FXR-RXR activation, IL-10 signaling and the complement system, reached the criteria set at P < 0.05. All six are associated with inflammation.
Discussion
Proinflammatory Cytokines and Chemokines
Inflammation has been strongly implicated in the pathogenesis of AMD, but the initiation of the inflammatory processes in AMD is not well understood.4,10–12 In this study, we hypothesized that Aβ1-40, a component of drusen, is a candidate trigger of inflammatory processes in the retina. Aβ is known to activate the classic and alternative complement pathways in neuronal systems.10,14,15,40–43 The results of our study demonstrated that treatment of human RPE cells with the oligomeric form of Aβ1-40 (0.3 μM) for 24 hours resulted in the upregulation of a number of inflammation-associated genes, as shown by microarray and later verified by RT PCR. The top three genes with the greatest increase after Aβ1-40 challenge were IL-1β, RSAD2, and IL-8. Gene set pathway analysis also concurred that the bulk of the gene responses in our study overlap with genes associated with inflammatory processes in the public databases. These results provide further support to the hypothesis that Aβ may trigger inflammatory responses in the RPE/choroidal layers of the eye.
IL-1β is an important proinflammatory cytokine that stimulates the activation of macrophages, and the production of cytokines such as IFN-γ,44–49 which promotes the upregulation of interferon α-inducible protein 27 (IFI27) in several types of cell lines.50 In our studies, IFI27 was upregulated after Aβ1-40 treatment and suggests that activation of pathways associated with IFN-γ are downstream of Aβ1-40 stimulation. Recently, Wu et al.51 showed that IFN-γ, along with oxidative stress, downregulates the complement inhibitor CFH, which could lead to increased complement activation, a known risk factor in AMD. Our studies revealed that Aβ1-40 stimulation of PE cells lead to IL-1β upregulation. In the context of the diseased eye, it is conceivable that interactions between IL-1β and reactive oxygen species (ROS) in the retina induce down-regulation of CFH and thus promote chronic inflammation. More studies are needed to further evaluate the downstream pathways associated with Aβ1-40 stimulation.
IL-1β was also shown to induce ROS in RPE cells.52 It is believed that ROS production may promote secretion of IL-8 in RPE cells.53,54 Thus, it is also possible that IL-1β can regulate the expression of IL-8 in RPE by direct and indirect (via ROS) mechanisms in RPE.52,53,55,56 Because of its chemotaxic abilities, IL-8 release may account for the observed accumulation of inflammatory cells in the regions of drusen formation in patients with AMD. The recruitment of macrophages and neutrophils and other proinflammatory cells that produce and release proteinases and angiogenic factors into drusen could also promote neovascular events associated with the wet form of AMD.57–59
Although, the mRNA of both IL-1β and -8 were significantly expressed, ELISA only detected IL-8 protein in the supernatant, perhaps because the IL-1β precursor protein has been shown to possess a short half-life.60
RSAD2, or viperin, was upregulated 2.58-fold in our studies. RSAD2 is a virus inducible antiviral protein associated with the innate immune response.61 It is upregulated in atherosclerotic plaques and inducible in endothelial cells by stimulation with LPS, IFN-γ, or cytomegalovirus. This is the first report that RPE cells can differentially express RSAD2. Its specific role in AMD pathogenesis is not yet known.
CFI and CFH
In this study, we also observed increased gene expression of complement factor I (CFI), which is a known inhibitor of the complement system. CFI inhibits complement activation by inactivating complement component 3b (C3b); dysregulation of CFI promotes uncontrolled complement activation.18,62,63 Recently, Fagerness et al.64 identified a variant in the CFI gene that is associated with an increased risk of developing AMD. Wang et al.18 discovered that Aβ stimulation of RPE cells resulted in the inability of CFI to cleave active C3b into its inactive form (iC3b). Although the mechanism remains unknown, Aβ is thought to bind to CFI and consequently inhibits its regulatory function and causes unregulated complement activation.18 Future studies are needed to determine the interactions between Aβ and CFI that cause its dysregulation of the complement pathways and the subsequent effects on RPE function. In addition to CFI, there is evidence in the literature supporting involvement of CFH, another inhibitor of the complement cascade, based its known polymorphism in AMD.65–68 However, there was no evidence in our study or in Wang et al.18 of Aβ1-40-induced changes in the expression of CFH in human RPE cells. This finding may relate to the interactions discussed by Wu et al.51 in which IFN-γ, along with oxidative stress, downregulated the complement inhibitor CFH.
Angiogenic Factors
Aβ1-40-treated RPE cells were also observed to express several angiogenesis-related genes. We observed an increased gene expression of somatostatin receptor 2 (SSTR2) and fibronectin (FN) in RPE cells after treatment with Aβ. Somatostatin is a neuropeptide with antiproliferative effects in a variety of normal and cancerous cells.69–73 Along with its receptor SSTR2, somatostatin has been identified in the RPE cells and has been demonstrated to inhibit VEGF mRNA expression,74 a finding that is consistent with our results. On the other hand, Yoshida et al.17 showed that stimulation of human RPE cells with Aβ1-40 resulted in the altered expression of angiogenic genes, specifically vascular endothelial growth factor (VEGF). Although we did not observe changes in the levels of VEGF expression in our study, possibly due to the inhibitory effects of somatostatin on VEGF mRNA expression, the differences may also be due to the specific time courses and doses of Aβ1-40 used in each study.
Another upregulated gene, FN, which codes for an extracellular matrix (ECM) glycoprotein, may also play a role in angiogenesis. It is possible that altered levels of ECM genes, including FN, can lead to abnormal RPE-choriocapillaris behavior.8
Apoptotic Mechanisms
The effect of the oligomeric form of Aβ1-40 on RPE cell viability was assessed by MTT. Several apoptotic genes were upregulated in the microarray study and support the reduction in cell viability observed in the MTT study. qRT-PCR studies showed increased gene expression of two important caspase-independent apoptotic genes, tumor necrosis factor related apoptosis-inducing ligand (TNFSF10 or TRAIL) and X-linked inhibitor of apoptosis associated factor 1 (XAF1). TRAIL, a member of the TNF superfamily, is expressed in several cell types and is activated by Aβ stimulation in neurons. It is believed that TRAIL expression promotes apoptotic cell death, as neutralization of TRAIL protects against Aβ toxicity in a human neuronal cell line.75
XAF1, another gene upregulated in our stimulation studies, interacts with XIAP to block its anticaspase activity.75 In earlier studies XAF1 was identified in several screens as a key mediator of apoptosis and was recently shown to dramatically sensitize cancer cells to apoptotic triggers such as TN-FSF10.76,77 Accordingly, it has been shown that interferon-induced apoptosis is mediated by the overexpression of XAF1, which is believed to increase susceptibility to TRAIL-induced cytotoxicity.77 Thus, in our studies, Aβ1-40 stimulation of RPE cells may have activated cell death in the RPE via XAF1 and TNFSF10 activity, as both genes were significantly upregulated in our study and have been shown to increase apoptosis in cancer cells.78
Our results also showed an increased expression of CYP2D6, a member of the cytochrome P450 family that is involved in the first line of defense against oxidative stress and has been shown to detoxify reactive oxygen species (ROS).79 Aβ1-40 stimulation of RPE cells in our study may also initiate ROS-induced signaling pathways, including stress-activated protein kinases and nuclear transcription factor NF-κB.52,80–82 NF-κB is upregulated by IL-8 and has been implicated in retinal neovascularization.47,83
Proposed Interaction of Gene Products Differentially Expressed in RPE after Aβ1-40 Stimulation
In conclusion, our in vitro studies lend support to the hypothesis that Aβ1-40 promotes local inflammation near drusen sites and within the surrounding RPE layer that may promote the pathogenesis of AMD.3,10,17 Our finding that RPE cells upregulate several genes associated with proinflammatory cytokines (IL-1β, IL-8), interferon-inducible proteins (IFI27, IFI44L), an inhibitor of the complement cascade (CFI), and an inhibitor of oxidative stress (CYP2D6) in response to Aβ1-40 challenge, may present a novel mechanism and partial explanation for the onset of inflammation in the eye near drusen sites. A summary schematic that illustrates the proposed circuitry by which Aβ1-40 may affect RPE gene expression leading to inflammatory events in the eye is shown in Figure 7. We hypothesize that Aβ1-40, known already to be present in drusen of AMD eyes, may induce RPE cells to secrete proinflammatory cytokines in which IL-1β may subsequently further promote overexpression of IL-8 via several known mechanisms, including ROS production.53,54 IL-8, an important chemokine, may promote migration and activation of immune cells (neutrophils, macrophages) toward the RPE.82,85,86 The accumulation of neutrophils, macrophages, and microglial cells may cause further pathologic changes in the RPE and Bruch’s membrane complex.4,8,9,81,87 Our results also suggest that, Aβ1-40 may promote RPE cells to overexpress CFI. Other in vitro studies showed that the interactions between Aβ1-40 and CFI cause CFI to lose its complement regulatory function.18,62 In the absence of CFI, unregulated complement activation would take place resulting in increased inflammation.18 Furthermore, Aβ1-40 may also cause RPE cells to secrete IFN-γ. According to Wu et al.51 IFN-γ (along with oxidative stress) may reduce CFH activity, thus increasing complement activation that may further promote AMD disease progression.
Figure 7.
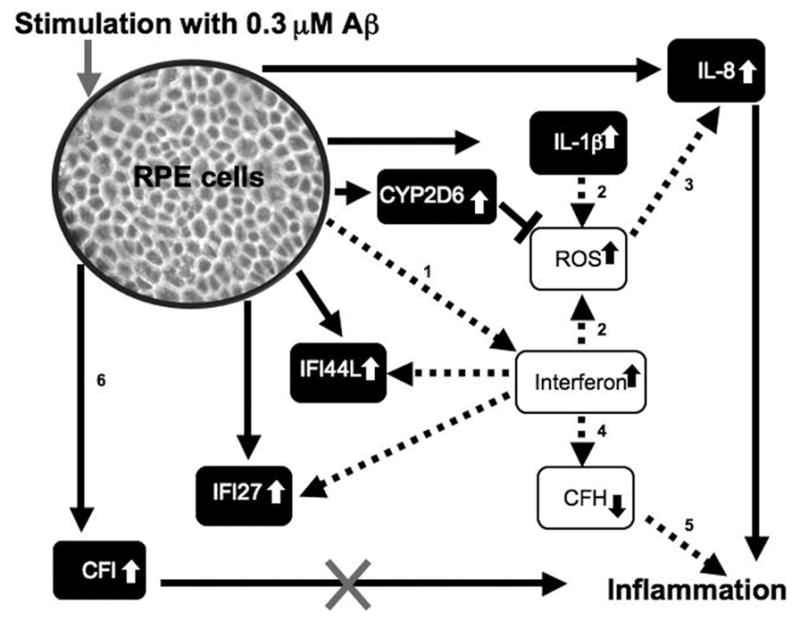
Summary schematic of results obtained from Aβ1-40 stimulation of RPE cells. After 24 hours, genes for IL-1β, IL-8, CFI, CYP2DK, IFI44L and IFI27 were upregulated in response to Aβ1-40 stimulation (represented by black boxes, white text and solid lines). IL-1β is known to also promote generation of ROS, which may further promote IL-8 expression (dashed line). In the context of the eye, IL-8 overexpression by RPE may promote macrophage and microglial migration toward RPE and in drusen containing Aβ. A prolonged macrophage and microglial response in the RPE/choroid layers may further support oxidative injury and inflammatory events through respiratory burst mechanisms. Genes for interferon-induced proteins (IFI44L and IFI27) were also upregulated in response to Aβ1-40 stimulation in our studies. Interferon-γ, in the setting of oxidative stress, was shown to reduce CFH activity, and thus promote inflammation (dashed line). In our study, two of the genes upregulated in response to Aβ1-40 stimulation, CFI and CYP2D6, have protective value. The protective value of CYP2D6 is in its ability to act as a first line of defense against oxidative stress by detoxifying reactive oxygen species (ROS).12 CFI, an inhibitor of the complement cascade, may work to inhibit the complement cascade; however, Aβ was shown to deleteriously interact with CFI causing its regulatory functions to be abolished.18,62 In the absence of CFI, unregulated complement activation would take place resulting in increased inflammation.84 Studies that support the pathways shown: 1, Poleganov et al.49; 2, Yang et al.52; 3, Elner et al.53 and Bian et al.54; 4, Wu et al.51; 5, Chen et al.68; and 6, Wang et al.18
Acknowledgments
Supported by Canadian Institute of Health Research (CIHR) Grant MOP-97806 (JM) and MOP-77736.
The authors thank Li Yu for RT-PCR analysis, Eleanor To for assistance with the illustrations, and Fanny Chu for assistance with AFM measurements.
Footnotes
Disclosure: K.H. Kurji, None; J.Z. Cui, None; T. Lin, None; D. Harriman, None; S.S. Prasad, None; L. Kojic, None; J.A. Matsubara, None
References
- 1.Abdelsalam A, Del Priore L, Zarbin MA. Drusen in age-related macular degeneration: pathogenesis, natural course, and laser photocoagulation-induced regression. Surv Ophthalmol. 1999;44:1–29. doi: 10.1016/s0039-6257(99)00072-7. [DOI] [PubMed] [Google Scholar]
- 2.Johnson PT, Lewis GP, Talaga KC, et al. Drusen-associated degeneration in the retina. Invest Ophthalmol Vis Sci. 2003;44:4481–4488. doi: 10.1167/iovs.03-0436. [DOI] [PubMed] [Google Scholar]
- 3.Luibl V, Isas JM, Kayed R, Glabe CG, Langen R, Chen J. Drusen deposits associated with aging and age-related macular degeneration contain nonfibrillar amyloid oligomers. J Clin Invest. 2006;116:378–385. doi: 10.1172/JCI25843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134:411–431. doi: 10.1016/s0002-9394(02)01624-0. [DOI] [PubMed] [Google Scholar]
- 5.Seth A, Cui J, To E, Kwee M, Matsubara J. Complement-associated deposits in the human retina. Invest Ophthalmol Vis Sci. 2008;49:743–750. doi: 10.1167/iovs.07-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in drusen formation and age related macular degeneration. Exp Eye Res. 2001;73:887–896. doi: 10.1006/exer.2001.1094. [DOI] [PubMed] [Google Scholar]
- 7.Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–732. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- 8.Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol. 2004;122:598–614. doi: 10.1001/archopht.122.4.598. [DOI] [PubMed] [Google Scholar]
- 9.Bok D. Evidence for an inflammatory process in age-related macular degeneration gains support. Proc Natl Acad Sci. 2005;102(20):7053–7054. doi: 10.1073/pnas.0502819102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH. The Alzheimer’s A β-peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci USA. 2002;99:11830–11835. doi: 10.1073/pnas.192203399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGeer PL, McGeer EG. Inflammation and the degenerative diseases of aging. Ann NY Acad Sci. 2004;1035:104–116. doi: 10.1196/annals.1332.007. [DOI] [PubMed] [Google Scholar]
- 12.Donoso LA, Kim D, Frost A, Callahan A, Hageman G. The role of inflammation in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2006;51:137–152. doi: 10.1016/j.survophthal.2005.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000;14:835–846. [PubMed] [Google Scholar]
- 14.Anderson DH, Talaga KC, Rivest AJ, Barron E, Hageman GS, Johnson LV. Characterization of beta amyloid assemblies in drusen: the deposits associated with aging and age-related macular degeneration. Exp Eye Res. 2004;78:243–256. doi: 10.1016/j.exer.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 15.Dentchev T, Milam AH, Lee VM, Trojanowski JQ, Dunaief JL. Amyloid-beta is found in drusen from some age-related macular degeneration retinas, but not in drusen from normal retinas. Mol Vis. 2003;9:184–190. [PubMed] [Google Scholar]
- 16.Ning A, Cui J, To E, et al. Amyloid-beta deposits lead to retinal degeneration in a mouse model of Alzheimer disease. Invest Ophthalmol Vis Sci. 2008;49(11):5136–5143. doi: 10.1167/iovs.08-1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshida T, Ohno-Matsui K, Ichinose S, et al. The potential role of amyloid beta in the pathogenesis of age-related macular degeneration. J Clin Invest. 2005;115:2793–2800. doi: 10.1172/JCI24635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Ohno-Matsui K, Yoshida T, et al. Altered function of factor I caused by amyloid beta: implication for pathogenesis of age-related macular degeneration from drusen. J Immunol. 2008;181:712–720. doi: 10.4049/jimmunol.181.1.712. [DOI] [PubMed] [Google Scholar]
- 19.Bruban J, Glotin AL, Dinet V, et al. Amyloid-β (1-42) alters structure and function of retinal pigmented epithelial cells. Aging Cell. 2009;8:162–177. doi: 10.1111/j.1474-9726.2009.00456.x. [DOI] [PubMed] [Google Scholar]
- 20.Kayed R, Head E, Thompson JL, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 21.Stine WB, Jr, Snyder SW, Ladror US, et al. The nanometer-scale structure of amyloid-beta visualized by atomic force microscopy. J Protein Chem. 1996;15:193–203. doi: 10.1007/BF01887400. [DOI] [PubMed] [Google Scholar]
- 22.Chromy BA, Nowak RJ, Lambert MP, et al. Self-assembly of abeta(1-42) into globular neurotoxins. Biochemistry. 2003;42:12749–12760. doi: 10.1021/bi030029q. [DOI] [PubMed] [Google Scholar]
- 23.Wang XF, Cui JZ, Prasad SS, Matsubara JA. Altered gene expression of angiogenic factors induced by calcium-mediated dissociation of retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2005;46:1508–1515. doi: 10.1167/iovs.04-0951. [DOI] [PubMed] [Google Scholar]
- 24.Gamulescu MA, Chen Y, He S, et al. Transforming growth factor beta2-induced myofibroblastic differentiation of human retinal pigment epithelial cells: Regulation by extracellular matrix proteins and hepatocyte growth factor. Exp Eye Res. 2006;83:212–222. doi: 10.1016/j.exer.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 25.Wang XF, Cui JZ, Nie W, Prasad SS, Matsubara JA. Differential gene expression of early and late passage retinal pigment epithelial cells. Exp Eye Res. 2004;79:209–221. doi: 10.1016/j.exer.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 26.Talaat AM, Howard ST, Hale W, 4th, Lyons R, Garner H, Johnston SA. Genomic DNA standards for gene expression profiling in mycobacterium tuberculosis. Nucleic Acids Res. 2002;30:e104. doi: 10.1093/nar/gnf103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 28.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lambert MP, Viola KL, Chromy BA, et al. Vaccination with soluble abeta oligomers generates toxicity-neutralizing antibodies. J Neurochem. 2001;79:595–605. doi: 10.1046/j.1471-4159.2001.00592.x. [DOI] [PubMed] [Google Scholar]
- 31.Dahlgren KN, Manelli AM, Stine WB, Jr, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 32.Leong WF, Tan HC, Ooi EE, Koh DR, Chow VT. Microarray and real-time RT-PCR analyses of differential human gene expression patterns induced by severe acute respiratory syndrome (SARS) coronavirus infection of vero cells. Microbes Infect. 2005;7:248–259. doi: 10.1016/j.micinf.2004.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kroismayr R, Baranyi U, Stehlik C, Dorfleutner A, Binder BR, Lipp J. HERC5, a HECT E3 ubiquitin ligase tightly regulated in LPS activated endothelial cells. J Cell Sci. 2004;117:4749–4756. doi: 10.1242/jcs.01338. [DOI] [PubMed] [Google Scholar]
- 34.Overbeck S, Uciechowski P, Ackland ML, Ford D, Rink L. Intracellular zinc homeostasis in leukocyte subsets is regulated by different expression of zinc exporters ZnT-1 to ZnT-9. J Leukoc Biol. 2008;83:368–380. doi: 10.1189/jlb.0307148. [DOI] [PubMed] [Google Scholar]
- 35.Carriere V, Roussel L, Ortega N, et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA. 2007;104:282–287. doi: 10.1073/pnas.0606854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Komatsuda A, Wakui H, Iwamoto K, et al. Upregulation of TRAIL mRNA expression in peripheral blood mononuclear cells from patients with active systemic lupus erythematosus. Clin Immunol. 2007;125:26–29. doi: 10.1016/j.clim.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 37.Kempkensteffen C, Hinz S, Schrader M, et al. Gene expression and promoter methylation of the XIAP-associated factor 1 in renal cell carcinomas: Correlations with pathology and outcome. Cancer Lett. 2007;254:227–235. doi: 10.1016/j.canlet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 38.Kojima K, Nagata K, Matsubara T, Yamazoe Y. Broad but distinct role of pregnane x receptor on the expression of individual cytochrome p450s in human hepatocytes. Drug Metab Pharmacokinet. 2007;22:276–286. doi: 10.2133/dmpk.22.276. [DOI] [PubMed] [Google Scholar]
- 39.Kumar M, Liu ZR, Thapa L, Qin RY. Anti-angiogenic effects of somatostatin receptor subtype 2 on human pancreatic cancer xenografts. Carcinogenesis. 2004;25:2075–2081. doi: 10.1093/carcin/bgh216. [DOI] [PubMed] [Google Scholar]
- 40.McGeer PL, Akiyama H, Itagaki S, McGeer EG. Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci Lett. 1989;107:341–346. doi: 10.1016/0304-3940(89)90843-4. [DOI] [PubMed] [Google Scholar]
- 41.Webster S, Lue LF, Brachova L, et al. Molecular and cellular characterization of the membrane attack complex, C5b-9, in Alzheimer’s disease. Neurobiol Aging. 1997;18:415–421. doi: 10.1016/s0197-4580(97)00042-0. [DOI] [PubMed] [Google Scholar]
- 42.Shen Y, Li R, McGeer EG, McGeer PL. Neuronal expression of mRNAs for complement proteins of the classical pathway in Alzheimer brain. Brain Res. 1997;769:391–395. doi: 10.1016/s0006-8993(97)00850-0. [DOI] [PubMed] [Google Scholar]
- 43.Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Planck SR, Huang XN, Robertson JE, Rosenbaum JT. Retinal pigment epithelial cells produce interleukin-1 beta and granulocyte-macrophage colony-stimulating factor in response to interleukin-1 alpha. Curr Eye Res. 1993;12:205–212. doi: 10.3109/02713689308999465. [DOI] [PubMed] [Google Scholar]
- 45.Jaffe GJ, Roberts WL, Wong HL, Yurochko AD, Cianciolo GJ. Monocyte-induced cytokine expression in cultured human retinal pigment epithelial cells. Exp Eye Res. 1995;60:533–543. doi: 10.1016/s0014-4835(05)80068-5. [DOI] [PubMed] [Google Scholar]
- 46.Elner VM, Elner SG, Standiford TJ, Lukacs NW, Strieter RM, Kunkel SL. Interleukin-7 (IL-7) induces retinal pigment epithelial cell MCP-1 and IL-8. Exp Eye Res. 1996;63:297–303. doi: 10.1006/exer.1996.0118. [DOI] [PubMed] [Google Scholar]
- 47.Yoshida A, Yoshida S, Khalil AK, Ishibashi T, Inomata H. Role of NF-kappaB-mediated interleukin-8 expression in intraocular neovascularization. Invest Ophthalmol Vis Sci. 1998;39:1097–1106. [PubMed] [Google Scholar]
- 48.Holtkamp GM, Van Rossem M, de Vos AF, Willekens B, Peek R, Kijlstra A. Polarized secretion of IL-6 and IL-8 by human retinal pigment epithelial cells. Clin Exp Immunol. 1998;112:34–43. doi: 10.1046/j.1365-2249.1998.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poleganov MA, Pfeilschifter J, Muhl H. Expanding extracellular zinc beyond levels reflecting the albumin-bound plasma zinc pool potentiates the capability of IL-1beta, IL-18, and IL-12 to act as IFN-gamma-inducing factors on PBMC. J Interferon Cytokine Res. 2007;27:997–1001. doi: 10.1089/jir.2007.0037. [DOI] [PubMed] [Google Scholar]
- 50.Gjermandsen IM, Justesen J, Martensen PM. The interferon-induced gene ISG12 is regulated by various cytokines as the gene 6–16 in human cell lines. Cytokine. 2000;12:233–238. doi: 10.1006/cyto.1999.0549. [DOI] [PubMed] [Google Scholar]
- 51.Wu Z, Lauer TW, Sick A, Hackett SF, Campochiaro PA. Oxidative stress modulates complement factor H expression in retinal pigmented epithelial cells by acetylation of FOXO3. J Biol Chem. 2007;282:22414–22425. doi: 10.1074/jbc.M702321200. [DOI] [PubMed] [Google Scholar]
- 52.Yang D, Elner SG, Bian ZM, Till GO, Petty HR, Elner VM. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp Eye Res. 2007;85:462–472. doi: 10.1016/j.exer.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elner VM, Strieter RM, Elner SG, Baggiolini M, Lindley I, Kunkel SL. Neutrophil chemotactic factor (IL-8) gene expression by cytokine-treated retinal pigment epithelial cells. Am J Pathol. 1990;136:745–750. [PMC free article] [PubMed] [Google Scholar]
- 54.Bian ZM, Elner SG, Yoshida A, Elner VM. Differential involvement of phosphoinositide 3-kinase/Akt in human RPE MCP-1 and IL-8 expression. Invest Ophthalmol Vis Sci. 2004;45:1887–1896. doi: 10.1167/iovs.03-0608. [DOI] [PubMed] [Google Scholar]
- 55.Baggiolini M, Dewald B, Moser B. Interleukin-8 and related chemotactic cytokines: CXC and CC chemokines. Adv Immunol. 1994;55:97–179. [PubMed] [Google Scholar]
- 56.Hwang YS, Jeong M, Park JS, et al. Interleukin-1beta stimulates IL-8 expression through MAP kinase and ROS signaling in human gastric carcinoma cells. Oncogene. 2004;23:6603–6611. doi: 10.1038/sj.onc.1207867. [DOI] [PubMed] [Google Scholar]
- 57.Higgins GT, Wang JH, Dockery P, Cleary PE, Redmond HP. Induction of angiogenic cytokine expression in cultured RPE by ingestion of oxidized photoreceptor outer segments. Invest Ophthalmol Vis Sci. 2003;44:1775–1782. doi: 10.1167/iovs.02-0742. [DOI] [PubMed] [Google Scholar]
- 58.Killingsworth MC, Sarks JP, Sarks SH. Macrophages related to Bruch’s membrane in age-related macular degeneration. Eye. 1990;4(Pt 4):613–621. doi: 10.1038/eye.1990.86. [DOI] [PubMed] [Google Scholar]
- 59.Heymans S, Luttun A, Nuyens D, et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med. 1999;5:1135–1142. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- 60.Moors MA, Mizel SB. Proteasome-mediated regulation of interleukin-1beta turnover and export in human monocytes. J Leukoc Biol. 2000;68:131–136. [PubMed] [Google Scholar]
- 61.Olofsson PS, Jatta K, Wagsater D, et al. The antiviral cytomegalovirus inducible gene 5/Viperin is expressed in atherosclerosis and regulated by proinflammatory agents. Arterioscler Thromb Vasc Biol. 2005;25(7):e113–6. doi: 10.1161/01.ATV.0000170130.85334.38. [DOI] [PubMed] [Google Scholar]
- 62.Sahu A, Isaacs SN, Soulika AM, Lambris JD. Interaction of vaccinia virus complement control protein with human complement proteins: factor I-mediated degradation of C3b to iC3b1 inactivates the alternative complement pathway. J Immunol. 1998;160:5596–5604. [PubMed] [Google Scholar]
- 63.Tsiftsoglou SA, Willis AC, Li P, et al. The catalytically active serine protease domain of human complement factor I. Biochemistry. 2005;44:6239–6249. doi: 10.1021/bi047680t. [DOI] [PubMed] [Google Scholar]
- 64.Fagerness JA, Maller JB, Neale BM, Reynolds RC, Daly MJ, Seddon JM. Variation near complement factor I is associated with risk of advanced AMD. Eur J Hum Genet. 2008 doi: 10.1038/ejhg.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Edwards AO, Ritter R, 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 67.Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 68.Chen M, Forrester JV, Xu H. Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp Eye Res. 2007;84:635–645. doi: 10.1016/j.exer.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 69.Patel YC. Somatostatin and its receptor family. Front Neuroendocrinol. 1999;20:157–198. doi: 10.1006/frne.1999.0183. [DOI] [PubMed] [Google Scholar]
- 70.Virgolini I, Yang Q, Li S, et al. Cross-competition between vasoactive intestinal peptide and somatostatin for binding to tumor cell membrane receptors. Cancer Res. 1994;54:690–700. [PubMed] [Google Scholar]
- 71.Luo Q, Peyman GA, Conway MD, Woltering EA. Effect of a somatostatin analog (octreotide acetate) on the growth of retinal pigment epithelial cells in culture. Curr Eye Res. 1996;15:909–913. doi: 10.3109/02713689609017634. [DOI] [PubMed] [Google Scholar]
- 72.Albers AR, O’Dorisio MS, Balster DA, et al. Somatostatin receptor gene expression in neuroblastoma. Regul Pept. 2000;88:61–73. doi: 10.1016/s0167-0115(99)00121-4. [DOI] [PubMed] [Google Scholar]
- 73.Sall JW, Klisovic DD, O’Dorisio MS, Katz SE. Somatostatin inhibits IGF-1 mediated induction of VEGF in human retinal pigment epithelial cells. Exp Eye Res. 2004;79:465–476. doi: 10.1016/j.exer.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 74.Lambooij AC, Kuijpers RW, van Lichtenauer-Kaligis EG, et al. Somatostatin receptor 2A expression in choroidal neovascularization secondary to age-related macular degeneration. Invest Ophthalmol Vis Sci. 2000;41:2329–2335. [PubMed] [Google Scholar]
- 75.Cantarella G, Uberti D, Carsana T, Lombardo G, Bernardini R, Memo M. Neutralization of TRAIL death pathway protects human neuronal cell line from beta-amyloid toxicity. Cell Death Differ. 2003;10:134–141. doi: 10.1038/sj.cdd.4401143. [DOI] [PubMed] [Google Scholar]
- 76.Liston P, Fong WG, Kelly NL, et al. Identification of XAFI as an antagonist of XIAP anti-Caspase activity. Nat Cell Biol. 2001;3:128–133. doi: 10.1038/35055027. [DOI] [PubMed] [Google Scholar]
- 77.Leaman DW, Chawla-Sarkar M, Vyas K, et al. Identification of X-linked inhibitor of apoptosis-associated factor-1 as an interferon-stimulated gene that augments TRAIL Apo2L-induced apoptosis. J Biol Chem. 2002;277:28504–28511. doi: 10.1074/jbc.M204851200. [DOI] [PubMed] [Google Scholar]
- 78.Micali OC, Cheung HH, Plenchette S, et al. Silencing of the XAFI gene by promoter hypermethylation in cancer cells and reactivation to TRAIL-sensitization by IFN-β. BMC Cancer. 2007;7:52. doi: 10.1186/1471-2407-7-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Esfandiary H, Chakravarthy U, Patterson C, Young I, Hughes AE. Association study of detoxification genes in age related macular degeneration. Br J Ophthalmol. 2005;89:470–474. doi: 10.1136/bjo.2004.047340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Flohe L, Brigelius-Flohe R, Saliou C, Traber MG, Packer L. Redox regulation of NF-kappa B activation. Free Radic Biol Med. 1997;22:1115–1126. doi: 10.1016/s0891-5849(96)00501-1. [DOI] [PubMed] [Google Scholar]
- 81.Tezel TH, Bora NS, Kaplan HJ. Pathogenesis of age-related macular degeneration. Trends Mol Med. 2004;10:417–420. doi: 10.1016/j.molmed.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 82.Fernandes AF, Zhou J, Zhang X, et al. Oxidative inactivation of the proteasome in RPE: a potential link between oxidative stress and upregulation of IL-8. J Biol Chem. 2008 Jul 25;283(30):20745–20753. doi: 10.1074/jbc.M800268200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Elner SG, Elner VM, Jaffe GJ, Stuart A, Kunkel SL, Strieter RM. Cytokines in proliferative diabetic retinopathy and proliferative vitreoretinopathy. Curr Eye Res. 1995;14:1045–1053. doi: 10.3109/02713689508998529. [DOI] [PubMed] [Google Scholar]
- 84.Jaffe GJ, Van Le L, Valea F, et al. Expression of interleukin-1 alpha, interleukin-1 beta, and an interleukin-1 receptor antagonist in human retinal pigment epithelial cells. Exp Eye Res. 1992;55:325–335. doi: 10.1016/0014-4835(92)90197-z. [DOI] [PubMed] [Google Scholar]
- 85.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 86.Tsai YY, Lin JM, Wan L, et al. Interleukin gene polymorphisms in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49:693–698. doi: 10.1167/iovs.07-0125. [DOI] [PubMed] [Google Scholar]
- 87.Nowak JZ. Age-related macular degeneration (AMD): pathogenesis and therapy. Pharmacol Rep. 2006;58:353–363. [PubMed] [Google Scholar]