

Summary
Background
The development of anti-factor VIII (fVIII) antibodies (inhibitors) is a significant complication in the management of patients with hemophilia A, leading to significant increases in morbidity and treatment cost. Using a panel of anti-fVIII monoclonal antibodies (MAbs) to different epitopes on fVIII, we recently have shown that epitope specificity, inhibitor kinetics, and time to maximum inhibition are more important than inhibitor titer in predicting response to fVIII and the combination of fVIII and recombinant factor VIIa. In particular, a subset of high-titer inhibitors responded to high dose fVIII, which would not be predicted based on their inhibitor titer alone. Thus the ability to quickly map the epitope spectrum of patient plasma using a clinically feasible assay may fundamentally change how clinicians approach the treatment of high-titer inhibitor patients.
Objectives
To map the epitopes of anti-fVIII MAbs, of which 3 are classical inhibitors and one non-classical, using hydrogen-deuterium exchange coupled with liquid chromatography-mass spectrometry (HDX-MS).
Methods
Binding epitopes of 4 MAbs targeting fVIII C2 domain were mapped using HDX-MS.
Results
The epitopes determined by HDX-MS are consistent with those obtained earlier through structural characterization and antibody competition assays. In addition classical and non-classical inhibitor epitopes could be distinguished using a limited subset of C2-derived peptic fragments.
Conclusion
Our results demonstrate the effectiveness and robustness of the HDX-MS method for epitope mapping and suggest a potential role of rapid mapping of fVIII inhibitor epitopes in facilitating individualized treatment of inhibitor patients.
Keywords: Antigen-antibody complex, epitope mapping, factor VIII, hemophilia A, hydrogen deuterium exchange measurement
Introduction
Hemophilia A is an X-linked recessive disorder due to causal mutations in the F8 gene that lead to absent or decreased activity of factor VIII (fVIII). Currently, the most effective treatment for hemophilia A is fVIII replacement therapy, which involves transfusion of plasma-derived or recombinant fVIII [1, 2]. The most significant complication associated with this therapy is an immune response against exogenous fVIII, which can occur in up to 30% of patients [3, 4]. Alloantibodies against fVIII mainly target its A2 and C2 domains [5]. The C2 domain of fVIII is the primary site of interaction for both von Willebrand factor (VWF) and the phosphatidylserine (PS)-containing membrane [6]. VWF and PS-containing membrane are mutually exclusive in binding fVIII, indicating overlapping binding sites in the C2 domain [7]. Anti-C2 domain antibodies have been classified into two types: classical and non-classical inhibitors. Classical inhibitors inhibit fVIII activity by interfering with its binding to VWF and PS-containing membrane [5, 8, 9]. In comparison, non-classical inhibitors were recently shown to inhibit thrombin activation of fVIII [10, 11].
Hemophilia A patients with high-titer inhibitors are routinely treated with bypassing agents including recombinant factor VIIa (rfVIIa) and activated prothrombin complex concentrates (aPCC) [12]. However, both in vitro and in vivo studies suggest that a subset of patients with high-titer inhibitors may respond better to fVIII or a combination of fVIII and bypassing agents than to bypassing agents alone. Using monoclonal antibodies (MAbs) with epitopes to all domains of fVIII in a murine bleeding model and in vitro assays, we recently showed that epitope specificity, inhibitor kinetics, and time to maximum inhibition are more important than inhibitor titer in predicting response to treatment with fVIII and fVIII/rfVIIa combination therapy [11, 13]. For example, non-classical C2 antibodies have ∼20 fold higher titers but better response to fVIII than classical C2 antibodies. Similarly inhibitor kinetics and time to maximum inhibition have been shown to be important in response to fVIII/rfVIIa combination therapy in a pilot study of inhibitor patients [14]. Crystallographic and structural studies have shown the binding sites for both classical and non-classical C2 inhibitors [15, 16]. Due to the variability in fVIII inhibitors and the clinical implications of inhibitory mechanisms, there is a need for a method that can quickly characterize binding epitopes of anti-fVIII antibodies to better predict their activity during fVIII replacement therapy.
Amide hydrogen/deuterium exchange (HDX) is a well-characterized phenomenon in which the amide hydrogen in a protein dissociates and becomes replaced by deuterium [17, 18]. HDX has been used extensively to characterize protein folding and protein-ligand interactions [19, 20]. Pertinent to this paper, binding of an antibody reduces solvent accessibility of antigen residues in the binding interface, thereby decreasing exchange rates and lowering the level of deuterium incorporation in peptic fragments containing the affected residues. Therefore, comparison of the HDX profiles between antibody-free and antibody-bound states can map the antibody epitopes in antigens [21-23]. With recent advances in instrumentation, HDX coupled with mass spectrometry (HDX-MS) has been applied to characterization of large proteins and their complexes [24].
In the present study, we have utilized the HDX-MS method to map the epitopes of several classical and non-classical MAbs, which match those obtained earlier through direct structural characterization and antibody competition assays. In addition, classical and non-classical inhibitor epitopes could be distinguished with the HDX profile of certain sequences in the C2 domain. Our results thus demonstrate the effectiveness and robustness of the HDX-MS method for epitope mapping and suggest a potential role in the rapid mapping of fVIII inhibitors.
Materials and Methods
Materials
Deuterium oxide was purchased from Cambridge Isotope Laboratories (Andover, MA). Porcine pepsin was from Sigma-Aldrich (St. Louis, MO). Recombinant human fVIII C2 domain was produced as described[25], and dialyzed into 10 mM Tris-HCl, 150 mM NaCl, 2.5% (v/v) glycerol, pH 7.4 buffer before storage at -80°C. MAbs BO2C11, I109, 3E6, and G99 were produced as described[26]. MAbs were dialyzed against the deuterium-based exchange buffer (100 mM NaCl, 20 mM Tris·HCl, pH 7.4, 96% D2O) and diluted to 0.8 mg/ml. Protein concentrations were determined using ε280 of 1.37 (mg/ml)-1 ·cm-1.
Hydrogen/deuterium exchange
Recombinant C2 domain at 1.3 mg/ml concentration was diluted in the exchange buffer at a 1:7 ratio to initiate exchange at room temperature. At various time points, an aliquot was removed manually from the exchange reaction and immediately mixed with 5x volume of the pre-chilled quenching buffer (50 mM phosphoric acid, pH 2.45). Subsequently, 1/48 volume of freshly prepared pepsin (2.5 mg/ml in the exchange buffer) was added and incubated for 2 min in ice water. The mixture was analyzed in an Agilent 1260-6130 LC/MS instrument with a reverse-phase C18 column at 4°C. The column was pre-equilibrated with 94.9% H2O/5% acetonitrile/0.1% trifluoroacetic acid (TFA), and subjected to a linear gradient wash to 60% acetonitrile in 13 min at 0.5 ml/min. The mass spectrometer was operated in full scan mode with resolution of 10,000. HDX in the presence of bound MAb was performed following the same procedure except using an antibody-containing exchange buffer to achieve a 1:0.55 molar ratio of C2 to MAb.
Identification of peptic fragments
C2-derived peptic fragments were collected from HPLC as described above and sequenced using an LTQ-Orbitrap-XL instrument. Collected fractions were dried and resuspended in 10 μL of 0.1% TFA aqueous and injected into a C18 trap column running 0.1% formic acid for 5 min at 10 μL/min. The sample was subsequently separated in a C18 column with a linear gradient wash to 90% acetonitrile at 300 nL/min. Eluted peptides were introduced into the mass spectrometer via Michrom Bioresources CaptiveSpray. The mass spectrometer was operated in data-dependent mode in which one cycle of experiments consisted of a full-MS in the Orbitrap (200-1800 m/z) survey scan in profile mode, resolution 30,000, and 5 subsequent MS/MS scans in the LTQ of the most intense peaks in centroid mode using collision-induced dissociation with helium. Each file was searched for protein identification against the FA8_HUMAN sequence in the UniProt database with no enzyme specificity and potential oxidation of methionine or cysteine. DTA generation parameters were peptide tolerance of 20 ppm and fragment ion tolerance of 2 Da. Peptide search results were filtered based on a mass error <5 ppm.
Data analysis and visualization
The centroid mass of each C2-derived peptide was determined using its isotopic distribution [27]. The shift in mass as a function of exchange time was fitted to a single inverse exponential decay. The fitted amplitude represents the extent of deuterium incorporation, which was averaged over three independent experiments and expressed on a per residue basis (D/res). For each peptide, the protective index was calculated as the difference between deuterium incorporation values in the presence and absence of MAb. Statistical significance (p<0.05) was assigned using Student's two-tailed t test.
Deuterium incorporation for an individual residue was calculated as a weighted average from all fragments containing the residue. Due to deuterium scrambling in a peptide [28], the deuterium incorporation for a given residue was calculated as the deuterium incorporation value for the peptide divided by the peptide length. Protective indices of individual residues were likewise generated for each MAb and further converted to a linear color gradient to generate a heat map for the structure of C2 domain.
Results
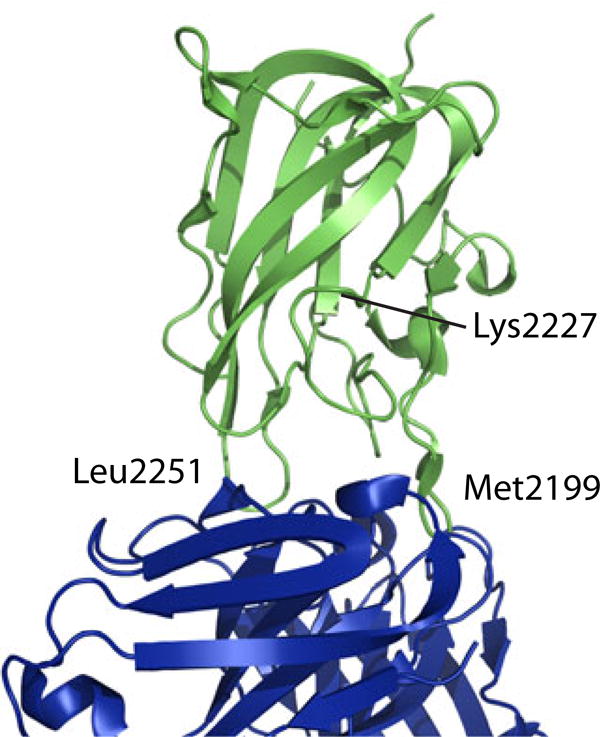
Binding of 4 MAbs to the recombinant C2 domain of human fVIII was analyzed by HDX in this study. BO2C11, I109 and 3E6 are classical inhibitors with overlapping epitopes [9, 26]. The crystal structure of BO2C11 Fab in complex with C2 [15] revealed that its binding epitope in C2 is located on Met2199 and Leu2251 loops (Fig. 1). G99 is a non-classical inhibitor [26]. It does not compete with BO2C11, I109 or 3E6 in competition ELISA. Mutagenesis and biophysical analysis has identified a separate face in C2, including particularly residue Lys2227 (Fig. 1), as the epitope for G99 [16, 26].
Figure 1.

Ribbon diagram showing the structure of fVIII C2 domain (green) in complex with BO2C11 (purple). The primary binding sites around residues 2199 and 2251 are labeled. So is the position of residue 2227.
HDX of C2 domain in the absence of bound antibodies
The HDX profile of recombinant C2 at physiological pH and ionic strengths but in the absence of MAbs was obtained first. Due to the limitation in instrumentation, all mixing steps were accomplished manually. The mass changes of peptic fragments were measured by a single-quadrupole ESI-MS instrument. Concurrently, peptic fragments were collected after the same HPLC separation and sequenced using a tandem mass spectrometer. Careful comparison of the attributes of each mass peak (m/z ratio, charge state, isotopic distribution, HPLC retention time) obtained from two instruments identified 27 C2-derived peptides that could be consistently monitored. These peptides covered 86% of the C2 sequence (Fig. 2). Only N- and C-terminal sequences were not represented, despite the usage of TCEP to reduce the Cys2174-Cys2326 disulfide bond connecting them [29]. Nonetheless, neither sequence comprises an antibody epitope and thus not expected to significantly impact the results here.
Figure 2.
Sequence coverage of peptides generated by pepsin digestion of the recombinant C2 domain.
Time-dependent mass changes for representative peptides are shown in Figure 3; the complete data set is included in Supplement Figure S1. HDX of amide protons occurs by two processes [30-32]. The rate of exchange is affected by local dynamic events, which occur in a time scale of milliseconds to seconds, and limited by global unfolding, which are in a time scale of hours to even days [32, 33]. As a result, the HDX plots displayed a “plateau” of deuterium incorporation around 60 minutes of exchange time (Fig. 3). Each plot was fitted to a single inverse exponential decay, with the amplitude variable representing the extent of deuterium incorporation. For a meaningful comparison among all peptides with different lengths, deuterium incorporation per residue (D/res) was used. Since our measurement was limited in early time points due to manual mixing, the fitted rate constant appeared less consistent than the extent of deuterium incorporation. Thus, the latter was used henceforth to indicate the extent of HDX.
Figure 3.
HDX profiles of five C2-derived peptides in the absence (□) and presence of indicated inhibitors. Deuterium incorporation varies between classical inhibitors (BO2C11, I109, 3E6) and non-classical inhibitor (G99), implying a distinct binding epitope.
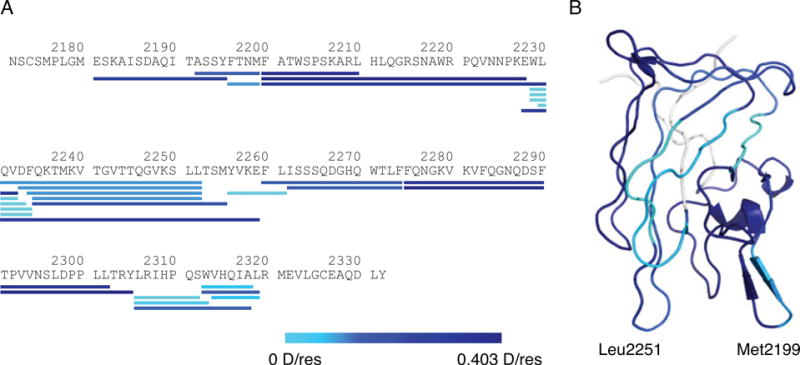
For direct visualization, the extent of HDX for each C2-derived peptide was presented in a color gradient scheme, ranging from light to dark blue (indicating low to high levels of HDX), and aligned with the C2 sequence (Fig. 4A). Deuterium incorporation for each residue was also calculated as described and similarly converted to blue color gradient and mapped onto the C2 structure (Fig. 4B). In general, HDX of C2 peptides correlated well with solvent accessibility of the C2 domain[34]. Peptide 2200-2210, which is highly exposed as a part of the Met2199 loop, exhibited the highest level of HDX. Peptides 2229–2234 and 2256–2261, both of which are largely buried within C2 and therefore the least accessible, exhibited the lowest level of HDX (Fig. 4). Overall, the obtained HDX profile of the C2 domain formed the basis for comparison with that of the MAb-bound C2 domain.
Figure 4.
Deuterium incorporation of C2-derived peptides. Deuterium incorporation values overlaid onto (A) the C2 domain primary sequence and (B) its structure. They are colored based on deuterium incorporation per residue (D/res).
HDX of C2 domain in the presence of bound inhibitors
The HDX profiles of the C2 domain in complex with MAbs were obtained next. Each MAb was dialyzed extensively against the D2O-based exchange buffer before being mixed with C2 as the antibody-free buffer. The MAb:C2 molar ratio in the mixture was 0.55, thus ensuring binding saturation. The extent of HDX was measured for each C2 peptide and compared to that without MAb. Due to the lack of a fully deuterated C2 domain, we did not assess the extent of back exchange in our system. However, as samples were subjected to the same procedure of exchange, quenching, digestion and analysis, back exchange could be considered constant between different samples. Thus, the MAb-conferred reduction or difference in deuterium incorporation, termed the protective index (PI), was calculated and used as the primary indicator for HDX protection (Supplement Figure S2).
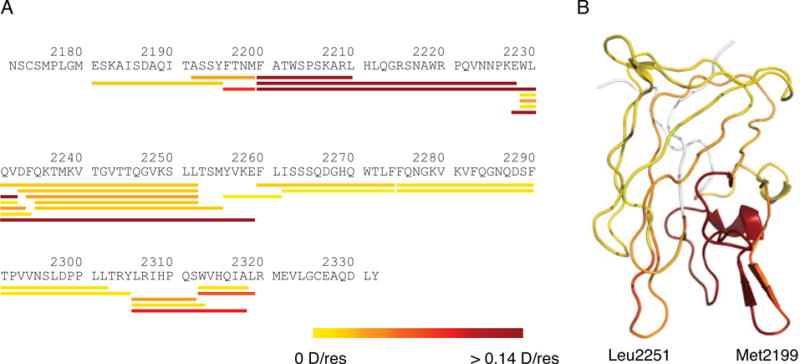
The HDX profile of C2 in complex with BO2C11 showed that several peptides had significantly lower deuterium incorporation than that in the absence of BO2C11 (Fig. 3,5). For instance, two peptides, 2200–2210 and 2228–2259, showed PI of 0.233 and 0.182 D/res, respectively. For direct visualization, PI for each peptide was converted to a color gradient, with yellow representing no HDX protection and red strong protection, and shown in alignment with the C2 sequence (Fig. 5A). Residue-specific PIs were also calculated as described, converted to the yellow-red color gradient and mapped to the C2 structure (Fig. 5B). Sequences with the highest PIs, as a result of BO2C11 binding, are located at Met2199 and Leu2251 loops, matching the epitope elucidated in the C2/BO2C11 complex structure (Fig. 1). Consistently, residues distal from the binding epitope showed little to no change in HDX protection. Thus, the BO2C11 epitope in C2 has been accurately mapped by the HDX-MS method.
Figure 5.
HDX protection of the C2 domain by BO2C11 binding. Measured protective indices were overlaid onto (A) the C2 domain primary sequence and (B) its structure. They are colored based on the protective index, ranging from low (yellow) to high protection (red).
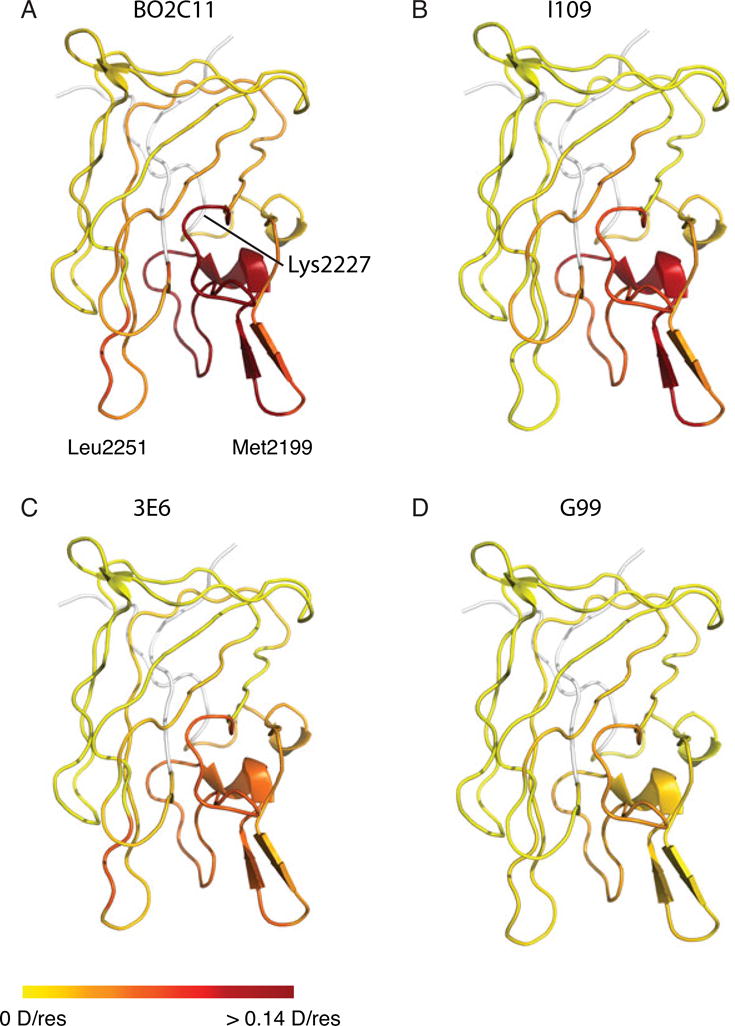
The same procedure was followed to map binding epitopes of the other three MAbs (Fig. S2). Residue-specific PIs were derived for each antibody, enabling a direct comparison of the binding epitopes (Fig. 6). BO2C11, I109, and 3E6 showed remarkable similarity in increased HDX protection around the Met2199 loop (Fig. 6A-C), which is consistent with the overlapping binding epitopes of these classical inhibitors [26]. In contrast, no HDX protection from binding of non-classical inhibitor G99 was observed for peptide 2200-2210, which accounts for half of the Met2199 loop (Fig. 3,6D). Peptide 2196-2199, which constitutes the other half of the Met2199 loop, recorded a much lower PI for G99 than for classical inhibitors. On the other hand, binding of G99 provided moderate protection for peptide 2200-2228, which includes residues 2200-2210 (Fig. S2). This indicates that HDX of residues 2211-2228 was protected by G99, consistent with previous studies identifying a separate binding epitope that does not include the Met2199 loop but critically involves residue Lys2227 [16, 26]. These results show that the HDX protection pattern of the non-classical inhibitor G99 can be clearly distinguished from those of classical inhibitors (Fig. 6).
Figure 6.

Comparison of HDX protection conferred by classical (A-C) and non-classical (D) inhibitors. Structures are colored according to protective indices with red representing stronger protection. Differences in protection pattern are most evident on the Met2199/Phe2200 loop.
In addition to the difference in protection of the Met2199 loop between classical and non-classical inhibitors, small but consistent difference in HDX protection among the three classical inhibitors was observed for certain peptides (Fig. 3A,C). For instance, BO2C11 produced a significantly higher PI for peptide 2231-2252, as a part of the Leu2251 loop, which is consistent with an earlier study [15]. However, I109 and 3E6 exhibited little or no protection on this sequence, agreeing with a prior observation that the L2252F mutation does not interfere with I109 binding to C2 (Meeks, unpublished results). These results indicate that, while PIs of many C2 peptides were low for all tested MAbs, those of several sequences were clearly different, which is the key to the distinction between different classes of fVIII inhibitors. Identification of these key peptides will facilitate future studies of fVIII inhibitors with unknown characteristics and may help future development of the HDX-MS method into a rapid and useful diagnostics for hemophilia treatment.
Discussion
Physicians treating high-titer fVIII inhibitor patients typically base treatment decisions on the inhibitor titer [35]. Patients with inhibitor titers >10 BU/mL are thought to respond poorly to fVIII treatment. They are treated instead with bypassing agents such as rfVIIa and aPCC. However, recent studies have shown that epitope specificity is more important than inhibitor titer in response to fVIII and combinations of fVIII and bypassing agents [13, 14]. Therefore, mapping fVIII inhibitor epitopes in a rapid, reproducible manner will help to formulate personalized treatment options for hemophilia patients with fVIII inhibitors. In this study, as a first step towards automated mapping of fVIII inhibitor epitopes, we have measured the HDX profile of the recombinant C2 domain in the absence and presence of well-characterized fVIII inhibitors by the HDX-MS method. The binding epitopes identified by HDX-MS were consistent with those elucidated by previous structural and mutagenesis studies (Fig. 1,5). Our analysis has identified several C2 peptides as key to distinguish between classical and non-classical inhibitors and further among classical inhibitors (Fig. 3,6). Many of these peptides produced intense MS signals and were readily monitored in HDX-MS (data not shown), which will be helpful in future development of the HDX-MS method to map fVIII inhibitors with overlapping epitopes. Overall, our study indicates that the HDX-MS method is robust and effective in mapping the epitopes of fVIII inhibitors.
The HDX-MS procedure was optimized for the materials and instruments available for this study, and can be further improved with instrument upgrade and sample processing. For instance, usage of a computerized stopped-flow device will enable detection of the early HDX events and improve the detection limit and accuracy. Pre-mixing C2 with the antibody or inhibitor-containing IgG before exchange will allow detection of epitopes that may exchange with deuterium prior to binding the antibody. Moreover, an improved instrument can enable detection of additional fragments for a more informative analysis. For instance, only 3 peptides (2200-2210, 2200-2228, 2200-2232), of which the shortest is 11 residues long, were available in this study to distinguish the epitopes of G99 and 3E6. Additional peptides covering the sequence will provide greater resolution of the mapped epitopes.
Comparing to other epitope-mapping methods such as ortholog sequence swapping, alaninescanning mutagenesis and synthetic peptide mapping [36-38], the HDX-MS method offers several advantages. First, the HDX-MS method uses only the wild-type protein, thereby saving significant efforts and materials in preparing mutant proteins and avoiding potential complications associated with unforeseen or unwanted effects of mutations. This is especially true for fVIII, a large protein that contains more than 2000 residues and does not tolerate mutations very well. Moreover, using the well-folded protein domain rather than short peptide fragments for inhibitor binding will enable detection of conformational sensitive antibody epitopes [39, 40]. Second, the HDX-MS method requires only a small amount of protein. Future improvement in sample handling and signal detection will certainly reduce the sample usage, potentially to a scale that is clinically feasible. Finally, the HDX-MS method is amenable to automation [41] and can be scaled up to full-length fVIII. Unlike other methods, the HDX-MS method is not limited by the protein size. Due to recent technological advances, the HDX-MS method is adequate at monitoring and analyzing a large number of peptic fragments. Our discovery of key sequences in the C2 domain that distinguish between different classes of inhibitors can be potentially incorporated into automated analysis. It is thus possible that in future applications the clinically relevant information be obtained in a rapid and reproducible fashion using a limited number of peptides that reduces cost and simplifies the analysis.
Supplementary Material
Acknowledgments
This work was supported in part by the Aflac Cancer and Blood Disorders Center and Hemophilia of Georgia, as well as by core NIH grants 2R24HD050846-06 (National Center for Medical Rehabilitation Research), 5P30HD040677-10 (Intellectual and Developmental Disabilities Research Center) and UL1RR031988 (CTSI-CN). The authors would like to thank Dr. Kristy J. Brown for her assistance in collection and analysis of LTQ-OrbitrapXL mass spectrometry data and Dr. Pete Lollar for his helpful discussions.
Footnotes
Authorship: A. M. Sevy, R. Li designed research, performed research, analyzed data and wrote the manuscript; J. F. Healey, W. Deng performed research and analyzed data; P. Clint Spiegel provided key reagent; S. L. Meeks designed research, analyzed data and wrote the manuscript.
Disclosure of Conflicts of Interest: The authors state that they have no conflict of interest.
References
- 1.Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in haemophilia A patients--a review of recent studies of recombinant and plasma-derived factor VIII concentrates. Haemophilia. 1999;5:145–154. doi: 10.1046/j.1365-2516.1999.00300.x. [DOI] [PubMed] [Google Scholar]
- 2.Hoots WK. The future of plasma-derived clotting factor concentrates. Haemophilia. 2001;7(Suppl 1):4–9. doi: 10.1046/j.1365-2516.2001.00099.x. [DOI] [PubMed] [Google Scholar]
- 3.Bray GL, Gomperts ED, Courter S, Gruppo R, Gordon EM, Manco-Johnson M, Shapiro A, Scheibel E, White G, 3rd, Lee M. A multicenter study of recombinant factor VIII (recombinate): safety, efficacy, and inhibitor risk in previously untreated patients with hemophilia A. The Recombinate Study Group. Blood. 1994;83:2428–2435. [PubMed] [Google Scholar]
- 4.Lusher JM, Lee CA, Kessler CM, Bedrosian CL. The safety and efficacy of B-domain deleted recombinant factor VIII concentrate in patients with severe haemophilia A. Haemophilia. 2003;9:38–49. doi: 10.1046/j.1365-2516.2003.00708.x. [DOI] [PubMed] [Google Scholar]
- 5.Scandella D, Gilbert GE, Shima M, Nakai H, Eagleson C, Felch M, Prescott R, Rajalakshmi KJ, Hoyer LW, Saenko E. Some factor VIII inhibitor antibodies recognize a common epitope corresponding to C2 domain amino acids 2248 through 2312, which overlap a phospholipid-binding site. Blood. 1995;86:1811–1819. [PubMed] [Google Scholar]
- 6.Lajmanovich A, Hudry-Clergeon G, Freyssinet JM, Marguerie G. Human factor VIII procoagulant activity and phospholipid interaction. Biochim Biophys Acta. 1981;678:132–136. doi: 10.1016/0304-4165(81)90056-8. [DOI] [PubMed] [Google Scholar]
- 7.Saenko EL, Scandella D. A mechanism for inhibition of factor VIII binding to phospholipid by von Willebrand factor. J Biol Chem. 1995;270:13826–13833. doi: 10.1074/jbc.270.23.13826. [DOI] [PubMed] [Google Scholar]
- 8.Shima M, Scandella D, Yoshioka A, Nakai H, Tanaka I, Kamisue S, Terada S, Fukui H. A factor VIII neutralizing monoclonal antibody and a human inhibitor alloantibody recognizing epitopes in the C2 domain inhibit factor VIII binding to von Willebrand factor and to phosphatidylserine. Thromb Haemost. 1993;69:240–246. [PubMed] [Google Scholar]
- 9.Jacquemin MG, Desqueper BG, Benhida A, Vander Elst L, Hoylaerts MF, Bakkus M, Thielemans K, Arnout J, Peerlinck K, Gilles JG, Vermylen J, Saint-Remy JM. Mechanism and kinetics of factor VIII inactivation: study with an IgG4 monoclonal antibody derived from a hemophilia A patient with inhibitor. Blood. 1998;92:496–506. [PubMed] [Google Scholar]
- 10.Nogami K, Shima M, Hosokawa K, Nagata M, Koide T, Saenko EL, Tanaka I, Shibata M, Yoshioka A. Factor VIII C2 domain contains the thrombin-binding site responsible for thrombin-catalyzed cleavage at Arg1689. J Biol Chem. 2000;275:25774–25780. doi: 10.1074/jbc.M002007200. [DOI] [PubMed] [Google Scholar]
- 11.Meeks SL, Healey JF, Parker ET, Barrow RT, Lollar P. Non-classical anti-factor VIII C2 domain antibodies are pathogenic in a murine in vivo bleeding model. J Thromb Haemost. 2009;7:658–664. doi: 10.1111/j.1538-7836.2009.03299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins PW. Treatment of acquired hemophilia A. J Thromb Haemost. 2007;5:893–900. doi: 10.1111/j.1538-7836.2007.02433.x. [DOI] [PubMed] [Google Scholar]
- 13.Doshi BS, Gangadharan B, Doering CB, Meeks SL. Potentiation of thrombin generation in hemophilia A plasma by coagulation factor VIII and characterization of antibody-specific inhibition. PLoS One. 2012;7:e48172. doi: 10.1371/journal.pone.0048172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Livnat T, Martinowitz U, Azar-Avivi S, Zivelin A, Brutman-Barazani T, Lubetsky A, Kenet G. Combined administration of FVIII and rFVIIa improves haemostasis in haemophilia A patients with high-responding inhibitors - a thrombin generation-guided pilot study. Haemophilia. 2013 doi: 10.1111/hae.12181. Epub ehead of print. [DOI] [PubMed] [Google Scholar]
- 15.Spiegel PC, Jr, Jacquemin M, Saint-Remy JM, Stoddard BL, Pratt KP. Structure of a factor VIII C2 domain-immunoglobulin G4kappa Fab complex: identification of an inhibitory antibody epitope on the surface of factor VIII. Blood. 2001;98:13–19. doi: 10.1182/blood.v98.1.13. [DOI] [PubMed] [Google Scholar]
- 16.Walter JD, Werther RA, Polozova MS, Pohlman J, Healey JF, Meeks SL, Lollar P, Spiegel PC., Jr Characterization and solution structure of the factor VIII C2 domain in a ternary complex with classical and non-classical inhibitor antibodies. J Biol Chem. 2013;288:9905–9914. doi: 10.1074/jbc.M112.424564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woodward CK, Hilton BD. Hydrogen exchange kinetics and internal motions in proteins and nucleic acids. Annu Rev Biophys Bioeng. 1979;8:99–127. doi: 10.1146/annurev.bb.08.060179.000531. [DOI] [PubMed] [Google Scholar]
- 18.Englander SW, Mayne L. Protein folding studied using hydrogen-exchange labeling and two-dimensional NMR. Ann Rev Biophys Biomol Struct. 1992;21:243–265. doi: 10.1146/annurev.bb.21.060192.001331. [DOI] [PubMed] [Google Scholar]
- 19.Bai Y, Sosnick TR, Mayne L, Englander SW. Protein folding intermediates: native-state hydrogen exchange. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Englander SW, Sosnick TR, Englander JJ, Mayne L. Mechanisms and uses of hydrogen exchange. Curr Opin Struct Biol. 1996;6:18–23. doi: 10.1016/s0959-440x(96)80090-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coales SJ, Tuske SJ, Tomasso JC, Hamuro Y. Epitope mapping by amide hydrogen/deuterium exchange coupled with immobilization of antibody, on-line proteolysis, liquid chromatography and mass spectrometry. Rapid Commun Mass Spectrom. 2009;23:639–647. doi: 10.1002/rcm.3921. [DOI] [PubMed] [Google Scholar]
- 22.Baerga-Ortiz A, Hughes CA, Mandell JG, Komives EA. Epitope mapping of a monoclonal antibody against human thrombin by H/D-exchange mass spectrometry reveals selection of a diverse sequence in a highly conserved protein. Protein Sci. 2002;11:1300–1308. doi: 10.1110/ps.4670102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Q, Willison LN, Tripathi P, Sathe SK, Roux KH, Emmett MR, Blakney GT, Zhang HM, Marshall AG. Epitope mapping of a 95 kDa antigen in complex with antibody by solution-phase amide backbone hydrogen/deuterium exchange monitored by Fourier transform ion cyclotron resonance mass spectrometry. Anal Chem. 2011;83:7129–7136. doi: 10.1021/ac201501z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Percy AJ, Rey M, Burns KM, Schriemer DC. Probing protein interactions with hydrogen/deuterium exchange and mass spectrometry-a review. Anal Chim Acta. 2012;721:7–21. doi: 10.1016/j.aca.2012.01.037. [DOI] [PubMed] [Google Scholar]
- 25.Spiegel PC, Murphy P, Stoddard BL. Surface-exposed hemophilic mutations across the factor VIII C2 domain have variable effects on stability and binding activities. J Biol Chem. 2004;279:53691–53698. doi: 10.1074/jbc.M409389200. [DOI] [PubMed] [Google Scholar]
- 26.Meeks SL, Healey JF, Parker ET, Barrow RT, Lollar P. Antihuman factor VIII C2 domain antibodies in hemophilia A mice recognize a functionally complex continuous spectrum of epitopes dominated by inhibitors of factor VIII activation. Blood. 2007;110:4234–4242. doi: 10.1182/blood-2007-06-096842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weis DD, Engen JR, Kass IJ. Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J Am Soc Mass Spectrom. 2006;17:1700–1703. doi: 10.1016/j.jasms.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 28.Demmers JA, Rijkers DT, Haverkamp J, Killian JA, Heck AJ. Factors affecting gas-phase deuterium scrambling in peptide ions and their implications for protein structure determination. J Am Chem Soc. 2002;124:11191–11198. doi: 10.1021/ja0125927. [DOI] [PubMed] [Google Scholar]
- 29.McMullen BA, Fujikawa K, Davie EW, Hedner U, Ezban M. Locations of disulfide bonds and free cysteines in the heavy and light chains of recombinant human factor VIII (antihemophilic factor A) Protein Sci. 1995;4:740–746. doi: 10.1002/pro.5560040413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woodward C, Simon I, Tüchsen E. Hydrogen exchange and the dynamic structure of proteins. Mol Cell Biochem. 1982;48:135–160. doi: 10.1007/BF00421225. [DOI] [PubMed] [Google Scholar]
- 31.Li R, Woodward C. The hydrogen exchange core and protein folding. Protein Sci. 1999;8:1571–1590. doi: 10.1110/ps.8.8.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bai Y, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wagner G, Wuthrich K. Amide protein exchange and surface conformation of the basic pancreatic trypsin inhibitor in solution. Studies with two-dimensional nuclear magnetic resonance. J Mol Biol. 1982;160:343–361. doi: 10.1016/0022-2836(82)90180-2. [DOI] [PubMed] [Google Scholar]
- 34.Pratt KP, Shen BW, Takeshima K, Davie EW, Fujikawa K, Stoddard BL. Structure of the C2 domain of human factor VIII at 1.5 A resolution. Nature. 1999;402:439–442. doi: 10.1038/46601. [DOI] [PubMed] [Google Scholar]
- 35.Kasper CK, Aledort L, Aronson D, Counts R, Edson JR, van Eys J, Fratantoni J, Green D, Hampton J, Hilgartner M, Levine P, Lazerson J, McMillan C, Penner J, Shapiro S, Shulman NR. Proceedings: A more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh. 1975;34:612. [PubMed] [Google Scholar]
- 36.Albert T, Egler C, Jakuschev S, Schuldenzucker U, Schmitt A, Brokemper O, Zabe-Kuhn M, Hoffmann D, Oldenburg J, Schwaab R. The B-cell epitope of the monoclonal anti-factor VIII antibody ESH8 characterized by peptide array analysis. Thromb Haemost. 2008;99:634–637. doi: 10.1160/TH07-06-0400. [DOI] [PubMed] [Google Scholar]
- 37.Lollar P. Mapping factor VIII inhibitor epitopes using hybrid human/porcine factor VIII molecules. Haematologica. 2000;85:26–28. [PubMed] [Google Scholar]
- 38.Nguyen PC, Lewis KB, Schuman JT, Meeks SL, Healey JF, Lollar P, Pratt KP. High-resolution mapping of B-cell epitopes on the factor VIII C2 domain using surface plasmon resonance. Blood. 2011;118:203. doi: 10.1182/blood-2013-09-527275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ansong C, Miles SM, Fay PJ. Epitope mapping factor VIII A2 domain by affinity-directed mass spectrometry: residues 497-510 and 584-593 comprise a discontinuous epitope for the monoclonal antibody R8B12. J Thromb Haemost. 2006;4:842–847. doi: 10.1111/j.1538-7836.2006.01831.x. [DOI] [PubMed] [Google Scholar]
- 40.Griffiths AE, Wang W, Hagen FK, Fay PJ. Use of affinity-directed liquid chromatography-mass spectrometry to map the epitopes of a factor VIII inhibitor antibody fraction. J Thromb Haemost. 2011;9:1534–1540. doi: 10.1111/j.1538-7836.2011.04397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kan ZY, Mayne L, Chetty PS, Englander SW. ExMS: data analysis for HX-MS experiments. J Am Soc Mass Spectrom. 2011;22:1906–1915. doi: 10.1007/s13361-011-0236-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.