

Abstract
In the present work, a series of small molecules were designed and synthesized based on structural optimization. Significant improvement in the enzymatic inhibition activity of the synthesized compounds was discovered. Moreover, tested compounds have moderate preference for class I HDACs over HDAC6 proved by enzymatic selectivity assay. The in vitro anti-proliferation assay reveals that representative compounds can selectively inhibit the growth of the non-solid lymphomatous cells and leukemic cells such as U937, K562 and HL60 cell lines. In the in vivo anti-tumor assay, molecule D17 showed better performance than SAHA in inhibition of U937 tumor growth. The western blot analysis revealed that representative molecules can block the function of both class I HDACs and HDAC6. More importantly, our western blot results revealed that the levels of some oncogenic proteins (p-Akt in the PI3K/AKT/mTOR signal pathway, c-Raf and p-Erk in the MAPK signal pathway) were dramatically down-regulated by our compounds in U937 cell line rather than MDA-MB-231 cells. This cellular mechanism difference might be an important reason why U937 cell line was more sensitive to our HDACs inhibitors than MDA-MB-231 cell line.
Keywords: histone deacetylase, inhibitors, anti-tumor, structural optimization
Introduction
Histone deacetylases (HDACs) are a class of enzymes responsible for removing acetyl groups from the ε-N-acetyl lysine on a histone. 18 members of HDACs have been discovered until now, and they are divided into 4 classes. [1] Class I HDACs include HDAC 1, 2, 3 and 8, within this class, HDAC 1, 2 and 8 are primarily found in nucleus, HDAC 3 is found in both nucleus and cytoplasm. Class II HDACs (IIa: HDAC4, 5, 7 and 9; IIb: HDAC6 and 10) are able to shuttle in and out of the nucleus. Class I and II HDACs are also called “classical zinc-dependent” HDACs. Class III HDACs (known as sirtuins, Sirt1–7) are a set of NAD-dependent enzymes. Class IV HDAC (HDAC11) which also exists in both nucleus and cytoplasm is considered as an atypical category of its own based on sequence similarity to the others.
Acetylation and deacetylation of histones and non-histone proteins by histone acetylases (HATs) and HDACs play important role in the regulation of gene expression and transcription. [2] The effects include signal transduction, protein phosphorylation, cell cycle, proliferation, apoptosis, cardiac development and so on. [3] Overexpression and aberrant recruitment of HDACs have a significant role in tumorigenesis.[4]
Histone deacetylase inhibitors (HDACIs) have been proved to alter gene expression by blocking the action of HDACs and inducing hyperacetylation of histones.[5] A number of structurally diverse HDACIs have shown potent antitumor efficacy in various stages of clinical trials. Suberoyl anilide hydroxamic acid (SAHA) [6] and FK228[7] have been approved by FDA for treatment of advanced cutaneous T-cell lymphoma (CTCL).
In order to develop novel HDACIs as antitumor agents, a series of small molecules were designed and synthesized based on virtual screening results. [8] A phenylglycine containing structure (4h) was discovered to have good performance in the enzyme inhibition assays and tumor cell growth inhibition assays. Structural modifications were performed in the present work in discovery of more potent molecules. To improve the activities of synthesized compounds, aromatic groups introduced to substitute with the Boc group of 4h, were designed to interact with hydrophobic residues (Phe155, Phe210 and Tyr308) in the active site of HDAC2 (Figure 1 and 2).
Figure 1.
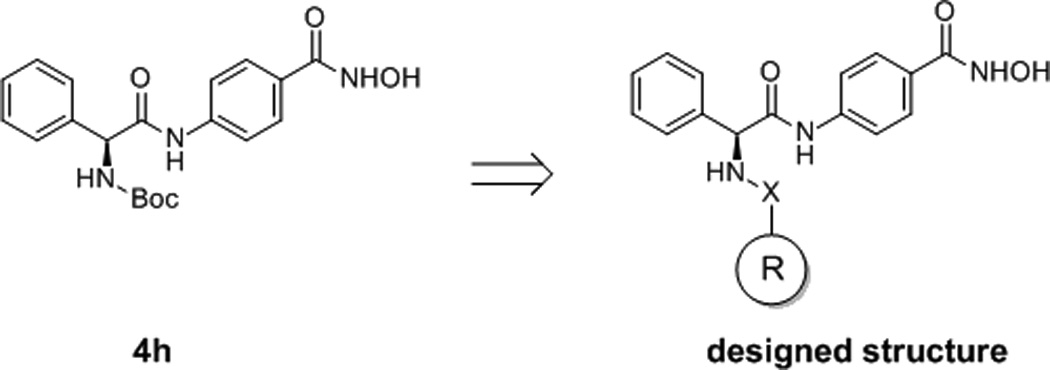
Design of new molecules based on the structure of 4h. X group is CO, SO2 or CONH, R group is substituted aromatic rings.
Figure 2.

a: molecule D5, D16 and D17 in the surface of HDAC2, the phenyl ring of D16 was colored cyan, the naphthalene ring of D16 was colored green; the Binding pattern of molecule D5 (b), D16 (c) and D17 (d) in the active site of HDAC2.
Chemistry
Based on the lead structure 4h, a series of phenylglycine containing hydroximic acid derivatives were synthesized according to the procedures described in scheme 1. The starting material (S)-2-amino-2-phenylacetic acid was protected by Boc group; the other material 4-aminobenzoic acid was protected by methyl ester. Intermediate (S)-methyl 4-(2-((tert-butoxycarbonyl)amino)-2-phenylacetamido)benzoate (a) was derived by coupling these two products. subsequent N-deprotection of intermediate a and condensation with various acyl chloride sulfonyl chloride and arylamines afforded c1-c19. Target compounds D1-D19 were derived by treating the corresponding intermediates (c1-c19) with NH2OK in methanol.
Results and Discussion
The enzymatic inhibition assay was performed against Hela cell nucleus extract which is a mixture of HDAC isoforms. Significant improvement in the inhibitory activity of the derived molecules (compared with 4h), indicates that aromatic R groups enhance the inhibitor-receptor interactions by binding to hydrophobic residues (Phe155, Phe210 and Tyr308) (Table 1). A carbamido group containing molecule D18 has the best performance in the enzymatic inhibition assay with IC50 value of 7.5 nM. Molecules with bigger aromatic groups (naphthalene ring) have predominant inhibitory activities such as D3 (14.3 nM), D5 (15.6 nM), D16 (23.5 nM) and D17 (9.7 nM). The chlorine substituted in the phenyl ring reduces the activity apparently compared with D7, such as D1 (348 nM), D2 (95.5 nM) and D10 (125 nM). However, the bromine substituted in the phenyl group can improve the activity obviously (D13: 13.0 nM). The fatty group (such as tert-butyl group) and strong electrophilic group (such as trifluoro-methyl group) make negative effect to the inhibitory activity (D6: 105 nM, D8: 85.5 nM; D18: 7.5 nM, D19: 47.7 nM). As to fluorine and methoxy group substituents, the para position is better than the ortho position (D4 vs D9, D11 vs D12), although the difference is not significant. The position also plays important role in the naphthalene sulfonyl groups, the 1-position (D5) has comparative better performance than the 2-position (D16). Furthermore, polar group in the naphthalene ring improves the enzymatic inhibitory activity obviously (D17). From the above findings, we can conclude that bigger aromatic ring in the R group is beneficial to the HDAC inhibitory activity. And to the substituent groups of the aromatic ring, polar groups (not strong electrophilic groups) are favored and the positions of the substituent groups also affect the activity.
Table 1.
The enzyme inhibitory activities of the derived molecules
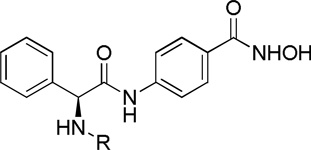 | ||
|---|---|---|
| Compound | R | IC50 (nM)a |
| D1 |  |
348.6 ± 21.2 |
| D2 |  |
95.5 ± 4.6 |
| D3 | 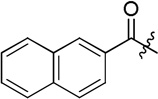 |
14.3 ± 0.9 |
| D4 | 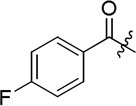 |
25.5 ± 1.6 |
| D5 | 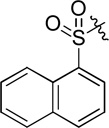 |
15.6 ± 1.1 |
| D6 | 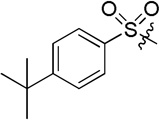 |
105.5 ± 9.7 |
| D7 | 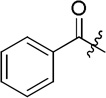 |
28.7 ± 2.2 |
| D8 | 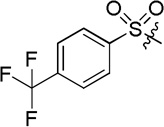 |
85.5 ± 6.7 |
| D9 | 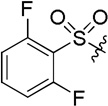 |
60.2 ± 4.7 |
| D10 | 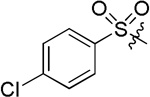 |
125.5 ± 10.6 |
| D11 | 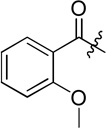 |
33.6 ± 2.3 |
| D12 | 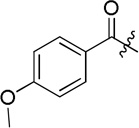 |
20.5 ± 1.1 |
| D13 | 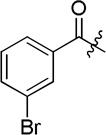 |
13.7 ± 0.7 |
| D14 | 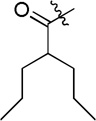 |
22.8 ± 1.8 |
| D15 | 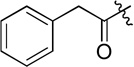 |
34.6 ± 2.4 |
| D16 | 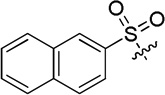 |
23.5 ± 1.7 |
| D17 | 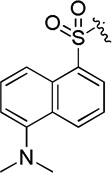 |
9.7 ± 0.3 |
| D18 | 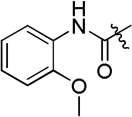 |
7.5 ± 0.2 |
| D19 | 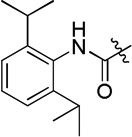 |
47.7 ± 3.3 |
| 4h | 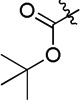 |
144.4 ± 9.7 |
| SAHA | 145.6 ± 7.6 | |
Each value is the mean of at least three experiments.
To characterize the isoform selectivity of these molecules, representative compounds D3, D5, D13, D17 and D18 were tested against HDAC1, HDAC2, HDAC3 and HDAC6 (Table 3). The result shows that these four molecules display preference for HDAC1, HDAC2 and HDAC3 over HDAC6. As to class I HDACs, the tested molecules show modest selectivity for HDAC1 and HDAC3 over HDAC2. Molecule D18 has the most potent activity in the selectivity assay with IC50 values of 4.8 nM, 19.5 nM, 5.9 nM and 294.2 nM against HDAC1, HDAC2, HDAC3 and HDAC6, respectively.
Table 3.
HDACs isoform selectivity of representative molecules (IC50, nM) a
| Compound | HDAC1 | HDAC2 | HDAC3 | HDAC6 |
|---|---|---|---|---|
| D3 | 28.7 ± 1.9 | 243.5 ± 11.7 | 16.8 ± 1.2 | 375.8 ± 19.7 |
| D5 | 30.7 ± 1.3 | 170.6 ± 9.7 | 23.1 ± 1.1 | 294.5 ± 11.7 |
| D13 | 11.8 ± 0.9 | 89.2 ± 4.7 | 8.5 ± 0.3 | 195.8 ± 11.9 |
| D17 | 24.7 ± 1.4 | 106.7 ± 8.9 | 24.4 ± 1.5 | 365.9 ± 20.6 |
| D18 | 4.8 ± 0.3 | 19.5 ± 1.4 | 5.9 ± 0.4 | 294.2 ± 22.3 |
| SAHA | 76.1 ± 2.6 | 256.3 ± 11.4 | 28.1 ± 1.8 | 118.2 ± 10.8 |
Each value is the mean of three experiments.
Docking process was performed to predict the binding patterns of derived molecules in the active site of HDAC2 (Figure 2). There is a narrow tunnel in the active site of HDAC2, and the zinc ion locates at the end of the tunnel. An open pocket which was surrounded by hydrophobic residues (Phe155, Phe210 and Tyr308) lies under the opening of the gap (Figure 2a). Molecules D5 (Figure 2b) and D17 (Figure 2d) have similar binding patterns in the active site of HDAC2. The hydroximic acid group chelating to the zinc ion makes the molecule bind tightly to the receptor. The linker locating in the tunnel has strong hydrophobic interactions with residues around the gap. The benzene ring of the phenylglycine group interacting with the hydrophobic area of the opening also makes contributions to the binding. The external motif binding to the pocket under the opening has good spatial matching with the surface of the active site. All these effects in combination enhanced the ligand-receptor interactions. However, molecule D16 has a different binding mode (Figure 2c); both the phenyl ring and the naphthalene ring in the external motif locate in the opening of the gap. The H-bond interactions formed between the ligands and HDAC2 also have a significant role in the binding. The NH group of the hydroximic acid group of all these molecules has H-bond interaction with NE2 of His146. Asp104 which is also a key residue for H-bond binding has H-bond interaction with NH1 of D5 and NH1 of D17. Molecule D16 has no H-bond interactions with Asp104. Lack of interactions with the pocket and H-bond bindings was supposed to be the reason for comparative low activity of D16 in the enzyme inhibition assay.
To evaluate the anti-proliferation activity of the synthesized molecules, MTT assays were performed using U937, K562, HL60, MCF7 PC3 and MDA-MB-231 cell lines. The derived compounds were firstly screened by U937 cells at the concentration of 2 µM, molecules have >50% inhibition were selected for the anti-proliferation assay and their IC50 values will be calculated (Table 2). On the whole, lymphomatous and leukemic cells lines (U937, K562 and HL60) are more sensitive to these molecules than the solid tumor cell lines (MCF7, PC3 and MDA-MB-231), which is in line with the published preclinical and clinical research results that the therapeutic efficacy of HDACIs is restricted predominantly to hematological malignancies.[9] Inhibition the growth of U937 cells, D3 (1.32 µM) and D17 (1.24 µM) have smaller IC50 values than SAHA. As to K562 cells most of the derived compounds (except D11 and D12) have better performances than SAHA, and D17 is the most active molecule (IC50=0.70 µM). Only D11 has better IC50 values than SAHA in inhibition HL60 cell growth (D11: 0.78; SAHA: 1.44). Molecule D3 (2.92 µM) and D4 (2.04 µM) have better performance than SAHA (5.09 µM) in the MCF7 cell growth inhibition assays. Only three compounds have bigger IC50 values than SAHA in the PC3 cell assays, and D18 (IC50=1.51 µM) is the most active molecule. Molecule D3 (IC50=4.23 µM) has the best performance in inhibition the growth of MDA-MB-321 cells.
Table 2.
The tumor cell growth inhibitory activities of synthesized molecules (IC50, µM)a
| Compounds | U937 | K562 | HL60 | MCF7 | PC3 | MDA- MB-231 |
|---|---|---|---|---|---|---|
| D3 | 1.32 ± 0.07 | 1.19 ± 0.04 | 3.15 ± 0.13 | 2.92 ± 0.15 | 3.77 ± 0.27 | 4.23 ± 0.33 |
| D4 | 1.64 ± 0.08 | 1.59 ± 0.06 | 4.55 ± 0.21 | 2.04 ± 0.17 | 5.88 ± 0.36 | 4.78 ± 0.44 |
| D5 | 1.63 ± 0.09 | 0.93 ± 0.02 | 2.88 ± 0.16 | 8.66 ± 0.73 | 10.1 ± 0.57 | 5.12 ± 0.34 |
| D7 | 1.77 ± 0.06 | 1.47 ± 0.07 | 2.02 ± 0.15 | 10.8 ± 0.93 | 5.67 ± 0.35 | 6.22 ± 0.24 |
| D11 | 1.71 ± 0.03 | 3.16 ± 0.11 | 0.78 ± 0.02 | 8.23 ± 0.32 | 20.5 ± 1.73 | 6.67 ± 0.25 |
| D12 | 1.55 ± 0.04 | 1.93 ± 0.12 | 1.80 ± 0.09 | 6.28 ± 0.33 | 3.00 ± 0.15 | 4.65 ± 0.24 |
| D13 | 1.39 ± 0.03 | 1.58 ± 0.06 | 1.53 ± 0.04 | 9.71 ± 0.44 | 31.6 ± 2.33 | 4.47 ± 0.31 |
| D17 | 1.24 ± 0.02 | 0.70 ± 0.01 | 3.66 ± 0.13 | 5.34 ± 0.22 | 3.31 ± 0.16 | 4.46 ± 0.28 |
| D18 | 1.31 ± 0.07 | 1.55 ± 0.05 | 3.83 ± 0.29 | 5.14 ± 0.43 | 1.51 ± 0.05 | 4.34 ± 0.26 |
| Av.b | 1.51 ± 0.05 | 1.57 ± 0.06 | 2.69 ± 0.14 | 6.57 ± 0.41 | 9.48 ± 0.66 | 4.99 ± 0.30 |
| SAHA | 1.38 ± 0.09 | 1.86 ± 0.07 | 1.44 ± 0.06 | 5.09 ± 0.21 | 6.33 ± 0.22 | 4.70 ± 0.35 |
Each value is the mean of three experiments.
The mean IC50 value of tested molecules in each column.
Human leukemic monocyte lymphoma cell line (U937) was the most sensitive to the tested molecules (mean IC50 value of 1.51 µM). Therefore, U937 cell line was selected for the in vivo anti-tumor assay. The most effective U937 cell growth inhibitor D17 which also has the best water solubility was selected for the in vivo anti-proliferation assay. To preliminarily investigate the in vivo anti-proliferation ability of molecule D17, only one concentration (100 mg/kg/d) was applied and SAHA was used as a positive control. After 16 days of intragastric administration, the mice were executed, and the tumor weight was measured. The mean weight of tumors in the negative control group is 2.66 g (SD 37%), and the value of SAHA groups and D17 group is 2.00 g (SD 26%) and 1.24 g (SD 18%), respectively (Figure 3a). The inhibitory ratio of molecule D17 is 53.57% compared with SAHA with a value of 24.97% (Figure 3b). The result showed that D17 exhibited improved anti-tumor activity compared with SAHA. The tumor volume was measured every two days, and the result has correlation with the tumor weight (Figure 3c and Figure 4). All these results confirm molecule D17 is more statistically significant than SAHA. No evidence of mice body weight (measured every two days) loss reveals that molecule D17 is safe (Figure 3d). Moreover, no obvious toxic signs in liver and spleen were detected.
Figure 3.

a: tumor weight plot of negative control group, positive control group (SAHA) and D17 group; b: inhibitory ratio plot of SAHA and D17; c: tumor volume plot of these experimental groups (negative control, SAHA and D17); d: mice body weight plot of these experimental groups (negative control, SAHA and D17).
Figure 4.

Picture of dissected tumor tissues.
Western blot studies were performed to evaluate the intracellular functions of representative molecules (Figure 5). The results show that these investigated molecules (D3, D5, D13, D17 and D18) are cell permeable and can inhibit intracellular and even nuclear HDACs by monitoring the acetylation levels of tubulin (target of HDAC6), histone H3 and H4 (targets of HDAC1 and HDAC2), and p21 which is substrate of class I HDACs. All the tested molecules increase the acetylated level of tubulin in the 6h treatment, indicating that the tested molecule can efficiently block the function of HDAC6 in U937 cells. After 24h, the effect of D3, D5 and D13 in the increase of acetylated tubulin level is still significant, but the action of D17 and D18 reduced. All the tested molecules have stronger abilities than SAHA in the increase of the cyclin-dependent kinase (CDK) inhibitor p21 levels in both time points. It indicates that the representative molecules are effective and stable class I HDAC inhibitor. The tested molecules have a manner of more effective in the induction of the acetylated levels of histone H3 than histone H4 which correlates with the enzymatic selectivity assay results.
Figure 5.
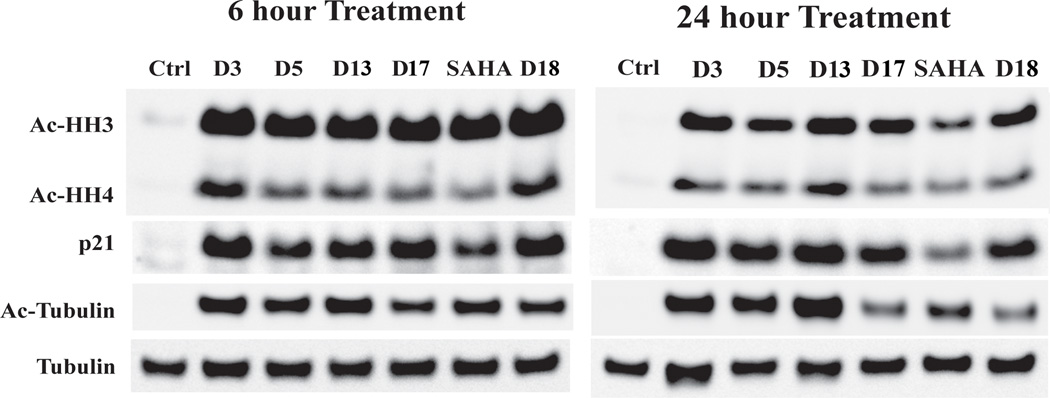
Western blot analysis of acetylated histone H3, acetylated histone H4, p21, and acetylated tubulin in U937 cell line after 6h and 24h of treatment with compounds at 1 µM. Tubulin was used as a loading control.
HDAC inhibitors have the limitation of ineffective low concentrations in solid tumors. [9] The US FDA approved drug SAHA and FK228 have no obvious efficacy in treating relapsed or refractory breast, colorectal, non-small cell lung cancer, metastatic head and neck cancer and so on proved by various clinical trials. [10] In the present work, in order to explore the reason why hematological tumor cell lines were more sensitive to HDACIs than solid tumor cell lines, we performed a series of western blot analysis to investigate the different effects of molecules D3 and D17 on the signaling cascades between non-solid (U937) and solid (MDA-MB-231) tumor cell lines (Figure 6). The c-Raf and Erk proteins are two crucial kinases in the activation of MAPK pathway. The phosphorylated Erk (p-Erk) is the active form of Erk. MDA-MB-231 has hyper-active MAPK pathway and the selected molecules cannot lower the signal. However, the tested molecules can obviously decrease the levels of c-Raf and p-Erk protein without reduction the expression of Erk protein in U937 cells. For the PI3K/AKT/mTOR cell proliferation pathway, although MDA-MB-231 cells have high-level of Akt protein, the p-Akt level is very low, which indicates that MDA-MB-231 cells do not depended on PI3K/AKT/mTOR signal pathway to grow and to survive. On the contrary, although U937 cells have lower Akt level, the p-Akt level is very high, which could be significantly decreased by compounds D3 and D17. Our results contribute the sensitivity of U937 cells to the tested inhibitors to their effects on these two oncogenic pathways, which are critical for cancer cell survivals. To the best of our knowledge, the present work constitutes the first report of HDACs inhibitors antitumor mechanism difference between non-solid (U937) and solid (MDA-MB-231) tumor cell lines. Further detailed mechanism studies which are performed to support our findings are still in progress.
Figure 6.

Western blot analysis of c-Raf, p-Erk, Erk, p-Akt, Akt and histone H3 in U937 and MDA-MB-231 cell line after 24h of treatment with compounds (D3 and D17) at 1 µM. Histone H3 was used as a loading control.
Conclusions
A series of HDAC inhibitors were synthesized by structural modification of lead compound and evaluated by enzymatic inhibition assays, in vitro and in vivo anti-proliferation assays, isoform selectivity test and western blot analysis. Significant improvement in the enzymatic inhibition activity was discovered by changing the Boc group to be substituted aromatic groups. The MTT assays show that the derived molecules are potent anti-proliferation agents especially against non-solid tumor cell lines such as U937, K562, and HL60 cells. In the in vivo anti-tumor assays using athymic nude mice inoculated with U937 cells, molecule D17 shows better performance than the positive control SAHA. The evidence indicates molecule D17 is valuable for further development. The isoform selectivity study shows that the representative molecules are relatively more prone to bind to class I HDACs (HDAC1, HDAC2 and HDAC3) rather than HDAC6. The western blot assay reveals that the representative molecules can block the action of both class I HDACs and HDAC6. The anti-tumor mechanism difference between non-solid (U937) and solid tumor cells (MDA-MB-231) was investigated by western blot analysis. The MAPK and PI3K/AKT/mTOR signal pathways were discovered to make U937 cells rather than MDA-MD-231 cells sensitive to the tested molecules.
EXPERIMENT SECTION
Materials and methods
All commercially available starting materials, reagents and solvents were used without further purification. All reactions were monitored by TLC with 0.25 mm silica gel plates (60GF-254). UV light and ferric chloride were used to visualize the spots. 1H NMR spectra were recorded on a Bruker DRX spectrometer at 600 MHz, using TMS as an internal standard. High-resolution mass spectra were performed in Shandong Analysis and Test Center in Ji’nan, China. Melting points were determined uncorrected on an electrothermal melting point apparatus. Optical rotation values were measured at room temperature using a modular circular polarimeter 200 operating at λ = 589 nm. Maximum absorption wave lengths of the target compounds were detected for HPLC analysis. The molecules were dissolved in methanol at a concentration of 1 mg per 100 mL and scanned from 500 nm to 200 nm using a UV-2102PCS spectrophotometer. All the derived target compounds are > 95% pure proved by HPLC analysis, performed on a Agilent 1100 HPLC instrument using a Phenomenex Synergi 4 µ Hydro-RP 80A column (250mm × 4.6mm). All the target compounds were eluted with acetonitrile and water (containing 0.1% formic acid) in various proportions detected at their maximum absorption wave lengths.
(S)-methyl 4-(2-((tert-butoxycarbonyl)amino)-2-phenylacetamido)benzoate (a) have been synthesized and described in our previous work. [8b]
(S)-methyl 4-(2-amino-2-phenylacetamido)benzoate hydrochloride (b)
To a solution of compound e (3.84 g, 10 mmol) in EtOAc (30 mL), a solution of EtOAc (30 mL) saturated by dry HCl gas was added. The reaction solution was stirred at room temperature for 5 h. Precipitates appeared and were filtered, with the filter being washed with ether, to give desired compound b (2.18 g, 68% yield). ESI-MS m/z: 285.3 [M+H]+.
General Procedure for the Preparation of c1 and its Analogues. (S)-methyl 4-(2-(4-chlorobenzamido)-2-phenylacetamido)benzoate (c1)
To a solution of b (0.96 g, 3.0 mmol) in THF (50 mL) / H2O (1.0 mL), NaHCO3 (1.01 g, 12 mmol) was added. After 10 min, 4-chlorobenzoyl chloride (0.58 g, 3.3 mmol) was added. The reaction solution was stirred at room temperature for 8 h. Then, the solvent was evaporated with the residue being taken up in EtOAc (50 mL). the EtOAc solution was washed with saturated citric acid (3 × 20 mL), NaHCO3 (3 × 20 mL), and brine (3 × 20 mL), dried over MgSO4, and evaporated under vacuum. The desired compound c1 (0.89 g, 70% yield) was derived by crystallization in EtOAc as white powder. 1H NMR (DMSO-d6) δ 10.76 (s, 1H), 9.20 (d, J = 6.6Hz, 1H), 7.97 (d, J = 7.8Hz, 2H ), 7.92 (d, J = 8.4Hz, 2H ), 7.76 (d, J = 8.4Hz, 2H ), 7.58 (d, J = 7.2Hz, 2H ), 7.54 (d, J = 8.4Hz, 2H ), 7.42-7.39 (m, 2H), 7.36-7.34 (m, 1H), 5.83 (d, J = 7.2Hz, 1H ), 3.82 (s, 3H).
(S)-methyl 4-(2-(2-chlorobenzamido)-2-phenylacetamido)benzoate (c2)
White powder, 74% yield. 1H NMR (DMSO-d6) δ 10.80 (s, 1H), 9.34 (d, J = 7.2Hz, 1H), 7.93 (d, J = 7.8Hz, 2H), 7.76 (d, J = 7.8Hz, 2H), 7. 58 (d, J = 7.8Hz, 2H), 7.50-7.44 (m, 3H), 7.40-7.38 (m, 3H), 7.35-7.32 (m, 1H), 5.85 83 (d, J = 7.2Hz, 1H), 3.82 (s, 3H).
(S)-methyl 4-(2-(2-naphthamido)-2-phenylacetamido)benzoate (c3)
White powder, 77% yield. 1H NMR (DMSO-d6) δ 10.79 (s, 1H), 9.23 (d, J = 7.2Hz, 1H), 8.61 (s, 1H), 8.04 (d, J = 7.8Hz, 1H), 8.02-7.98 (m, 3H), 7.93 (d, J = 9.0Hz, 2H), 7.78 (d, J = 8.4Hz, 2H), 7.64-7.59 (m, 4H), 7.44-7.41 (m, 2H), 7.36 (t, J = 7.2Hz, 1H), 5.91 (d, J = 7.2Hz, 1H), 3.82 (s, 3H).
(S)-methyl 4-(2-(4-fluorobenzamido)-2-phenylacetamido)benzoate (c4)
White powder, 75% yield. 1H NMR (DMSO-d6) δ 10.72 (s, 1H), 9.12 (d, J = 7.2Hz, 1H), 8.03-8.01 (m, 2H), 7.92 (d, J = 9.0Hz, 2H), 7.75 (d, J = 9.0Hz, 2H), 7.57 (d, J = 7.8Hz, 2H), 7.42-7.39 (m, 2H), 7.36-7.33 (m, 1H), 7.31-7.28 (m, 2H), 5.82 (d, J = 7.2Hz, 1H), 3.82 (s, 3H).
(S)-methyl 4-(2-(naphthalene-1-sulfonamido)-2-phenylacetamido)benzoate (c5)
White powder, 79% yield. 1H NMR (DMSO-d6) δ 10.47 (s, 1H), 9.15 (d, J = 9.6Hz, 1H), 8.73 (d, J = 8.4Hz, 1H), 8.13 (d, J = 7.2Hz, 1H), 8.06 (d, J = 8.4Hz, 1H), 7.95 (d, J = 8.4Hz, 1H), 7.83 (d, J = 9.0Hz, 2H), 7.67-7.65 (m, 1H), 7.61-7.58 (m, 1H), 7.53-7.50 (m, 1H), 7.41 (d, J = 8.4Hz, 2H), 7.29-7.28 (m, 2H), 7.17-7.16 (m, 3H), 5.20 (d, J = 9.6Hz, 1H), 3.81 (s, 3H).
(S)-methyl 4-(2-(4-(tert-butyl)phenylsulfonamido)-2-phenylacetamido)benzoate (c6)
White powder, 74% yield. 1H NMR (DMSO-d6) δ 10.52 (s, 1H), 8.73 (d, J = 9.6Hz, 1H), 7.84 (d, J = 8.4Hz, 2H), 7.66 (d, J = 7.8Hz, 2H), 7.50 (d, J = 8.4Hz, 2H), 7.37-7.36 (m, 4H), 7.27-7.23 (m, 3H), 5.17 (d, J = 9.6Hz, 1H), 3.80 (s, 3H), 1.17 (s, 9H).
(S)-methyl 4-(2-benzamido-2-phenylacetamido)benzoate (c7)
White powder, 72% yield. 1H NMR (DMSO-d6) δ 10.72 (s, 1H), 9.07 (d, J = 7.2Hz, 1H), 7.94 (d, J = 9.0Hz, 2H), 7.92 (d, J = 9.0Hz, 2H), 7.75 (d, J = 9.0Hz, 2H), 7.58 (d, J = 7.8Hz, 2H), 7.56-7.54 (m, 1H), 7.47 (t, J = 7.8Hz, 2H), 7.42-7.39 (m, 2H), 7.36-7.33 (m, 1H), 5.84 (d, J = 7.2Hz, 1H), 3.82 (s, 3H).
(S)-methyl 4-(2-phenyl-2-(4-(trifluoromethyl)phenylsulfonamido)acetamido)benzoate (c8)
White powder, 75% yield. 1H NMR (DMSO-d6) δ 10.61 (s, 1H), 9.14 (d, J = 9.6Hz, 1H), 7.91 (d, J = 8.4Hz, 2H), 7.85 (d, J = 9.0Hz, 2H), 7.75 (d, J = 8.4Hz, 2H), 7.48 (d, J = 8.4Hz, 2H), 7.36 (d, J = 7.2Hz, 2H), 7.26-7.22 (m, 3H), 5.24 (d, J = 9.6Hz, 1H), 3.81 (s, 3H).
(S)-methyl 4-(2-(2,6-difluorophenylsulfonamido)-2-phenylacetamido)benzoate (c9)
White powder, 76% yield. 1H NMR (DMSO-d6) δ 10.66 (s, 1H), 9.37 (d, J = 9.6Hz, 1H), 7.88 (d, J = 9.0Hz, 2H), 7.57 (d, J = 9.0Hz, 2H), 7.55-7.53 (m, 1H), 7.46 (d, J = 9.0Hz, 2H), 7.31-7.29 (m, 2H), 7.26 (t, J = 7.2Hz, 1H), 7.11 (t, J = 9.0Hz, 2H), 5.38 (d, J = 9.6Hz, 1H), 3.81 (s, 3H).
(S)-methyl 4-(2-(4-chlorophenylsulfonamido)-2-phenylacetamido)benzoate (c10)
White powder, 77% yield. 1H NMR (DMSO-d6) δ 10.59 (s, 1H), 8.97 (d, J = 9.6Hz, 1H), 7.88 (d, J = 8.4Hz, 2H), 7.71 (d, J = 8.4Hz, 2H), 7.52 (d, J = 8.4Hz, 2H), 7.46 (d, J = 8.4Hz, 2H), 7.37 (d, J = 6.6Hz, 2H), 7.29-7.25 (m, 3H), 5.20 (d, J = 9.6Hz, 1H), 3.81 (s, 3H).
(S)-methyl 4-(2-(2-methoxybenzamido)-2-phenylacetamido)benzoate (c11)
White powder, 78% yield. 1H NMR (DMSO-d6) δ 10.83 (s, 1H), 9.18 (d, J = 7.2Hz, 1H), 7.93 (d, J = 8.4Hz, 2H), 7.74 (d, J = 8.4Hz, 2H), 7.56 (d, J = 7.8Hz, 2H), 7.54 (d, J = 8.4Hz, 1H), 7.41 (t, J = 7.8Hz, 2H), 7.33 (t, J = 7.2Hz, 1H), 7.24 (d, J = 9.0Hz, 1H), 7.08 (t, J = 7.2Hz, 1H), 5.87 (d, J = 7.2Hz, 1H), 4.00 (s, 3H), 3.82 (s, 3H).
(S)-methyl 4-(2-(4-methoxybenzamido)-2-phenylacetamido)benzoate (c12)
White powder, 82% yield. 1H NMR (DMSO-d6) δ 10.70 (s, 1H), 8.89 (d, J = 7.2Hz, 1H), 7.94 (d, J = 9.0Hz, 2H), 7.92 (d, J = 7.8Hz, 2H), 7.75 (d, J = 7.2Hz, 2H), 7.57 (d, J = 7.2Hz, 2H), 7.40 (t, J = 7.8Hz, 2H), 7.34 (t, J = 7.8Hz, 1H), 6.99 (d, J = 8.4Hz, 2H), 5.82 (d, J = 7.2Hz, 1H), 4.02 (s, 3H), 3.81 (s, 3H).
(S)-methyl 4-(2-(3-bromobenzamido)-2-phenylacetamido)benzoate (c13)
White powder, 76% yield. 1H NMR (DMSO-d6) δ 10.72 (s, 1H), 9.27 (d, J = 6.6Hz, 1H), 8.16 (s, 1H), 7.93-7.91 (m, 3H), 7.76-7.74 (m, 3H), 7.57 (d, J = 7.2Hz, 2H), 7.45-7.40 (m, 3H), 7.35 (t, J = 7.2Hz, 1H), 5.81 (d, J = 6.6Hz, 1H), 3.82 (s, 3H).
(S)-methyl 4-(2-phenyl-2-(2-propylpentanamido)acetamido)benzoate (c14)
White powder, 77% yield. 1H NMR (DMSO-d6) δ 10.66 (s, 1H), 8.65 (d, J = 7.2Hz, 1H), 7.91 (d, J = 8.4 Hz, 2H), 7.72 (d, J = 8.4 Hz, 2H), 7.48 (d, J = 7.8 Hz, 2H), 7.40-7.37 (m, 2H), 7.33-7.31 (m, 1H), 5.65 (d, J = 7.2Hz, 1H), 3.81 (s, 3H), 1.45-1.40 (m, 2H), 1.28-1.21 (m, 4H), 1.19-1.13 (m, 2H), 0.85 (m, 3H), 0.80 (t, J = 7.8 Hz, 3H).
(S)-methyl 4-(2-phenyl-2-(2-phenylacetamido)acetamido)benzoate (c15)
White powder, 73% yield. 1H NMR (DMSO-d6) δ 10.77 (s, 1H), 9.02 (d, J = 7.8Hz, 1H), 7.90 (d, J = 9.0 Hz, 2H), 7.72 (d, J = 9.0 Hz, 2H), 7.50 (d, J = 7.8 Hz, 2H), 7.40-7.37 (m, 2H), 7.33-7.31 (m, 1H), 7.30-7.28 (m, 4H), 7.22-7.19 (m, 1H), 5.63 (d, J = 7.8Hz, 1H), 3.81 (s, 3H), 3.59 (m, 2H).
(S)-methyl 4-(2-(naphthalene-2-sulfonamido)-2-phenylacetamido)benzoate (c16)
White powder, 78% yield. 1H NMR (DMSO-d6) δ 10.55 (s, 1H), 8.94 (d, J = 9.6Hz, 1H), 8.34 (s, 1H), 7.96-7.94 (m, 2H), 7.90 (d, J = 8.4Hz, 1H), 7.79-7.77 (m, 1H), 7.74 (d, J = 9.0Hz, 2H), 7.61-7.59 (m, 1H), 7.56-7.53 (m, 1H), 7.39-7.36 (m, 4H), 7.23 (t, J = 7.2Hz, 2H), 7.19-7.17 (m, 1H), 5.25 (d, J = 8.4Hz, 1H), 3.80 (s, 3H).
(S)-methyl 4-(2-(5-(dimethylamino)naphthalene-1-sulfonamido)-2-phenylacetamido)benzoate (c17)
White powder, 75% yield. 1H NMR (DMSO-d6) δ 10.40 (s, 1H), 9.06 (d, J = 9.6Hz, 1H), 8.37 (d, J = 8.4Hz, 1H), 8.28 (d, J = 7.8Hz, 1H), 8.12 (d, J = 7.2Hz, 1H), 7.81 (d, J = 8.4Hz, 2H), 7.55-7.52 (m, 1H), 7.49 (t, J = 7.8Hz, 1H), 7.40 (d, J = 8.4Hz, 2H), 7.30-7.29 (m, 2H), 7.18-7.16 (m, 4H), 5.17 (d, J = 9.6Hz, 1H), 3.80 (s, 3H), 2.71 (s, 6H).
General Procedure for the Preparation of c18 and c19. (S)-methyl 4-(2-(3-(2-methoxyphenyl)ureido)-2-phenylacetamido)benzoate (c18)
To a solution of triphosgene (1.5 g, 5.0 mmol) in dioxane (40 mL), 2-methoxyaniline (1.23 g, 10 mmol) was added. The reaction solution was stirred at 110 °C for 8 h. Then, the solvent was evaporated sufficiently without further purification.
The residue was dissolved in DCM, compound b (1.6 g, 5.0 mmol) and Et3N (0.61 g, 6.0 mmol) was added. The reaction solution was stirred at room temperature for 5 h. Then, the solvent was evaporated with the residue being taken up in EtOAc (50 mL). the EtOAc solution was washed with saturated citric acid (3 × 20 mL), NaHCO3 (3 × 20 mL), and brine (3 × 20 mL), dried over MgSO4, and evaporated under vacuum. The desired compound c18 (0.97 g, 45% yield) was derived by crystallization in EtOAc as white powder. 1H NMR (DMSO-d6) δ 10.75 (s, 1H), 8.39 (s, 1H), 8.05 (d, J = 6.6Hz, 1H), 7.91 (d, J = 8.4Hz, 2H), 7.89 (d, J = 7.8Hz, 1H), 7.74 (d, J = 8.4Hz, 2H), 7.52 (d, J = 7.2Hz, 2H), 7.42-7.39 (m, 2H), 7.34-7.32 (m, 1H), 6.97 (d, J = 7.8Hz, 1H), 6.89-6.87 (m, 1H), 6.82 (t, J = 7.8Hz, 1H), 5.56 (d, J = 7.8Hz, 1H), 4.02 (s, 3H), 3.81 (s, 3H).
(S)-methyl 4-(2-(3-(2,6-diisopropylphenyl)ureido)-2-phenylacetamido)benzoate (c19)
White powder, 43% yield. 1H NMR (DMSO-d6) δ 10.76 (s, 1H), 7.92 (d, J = 8.4Hz, 2H), 7.74 (s, 2H), 7.52 (d, J = 6.6Hz, 2H), 7.40 (t, J = 6.6Hz, 2H), 7.33-7.31 (m, 1H), 7.17 (d, J = 7.8Hz, 2H), 7.10-7.18 (m, 2H), 5.59 (d, J = 8.4Hz, 1H), 3.82 (s, 3H), 3.33 (s, 2H), 1.10 (d, J = 6.6Hz, 12H).
General Procedure for the Preparation of D1–D19. (S)-4-chloro-N-(2-((4-(hydroxycarbamoyl)phenyl)amino)-2-oxo-1-phenylethyl)benzamide (D1)
Compound g1 (0.5 g, 1.18 mmol) was dissolved in 14 mL of NH2OK (0.56 g, 24 mmol) methanol solution. After 2 h, the solvent was evaporated under vacuum. The residue was acidified with saturated citric acid, and then extracted with EtOAc (3 × 20 mL). The organic layers were combined, washed with brine (3 × 20 mL) and dried over MgSO4. The desired compound D1 (0.23 g, 54% yield) was derived by crystallization in EtOAc as white powder. Mp: 250–252 °C. [α]20D −6.88 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.10 (s, 1H), 10.58 (s, 1H), 9.15 (d, J = 7.8 Hz, 1H), 8.93 (s, 1H), 7.98 (d, J = 8.4 Hz, 2H), 7.72 (d, J = 8.4 Hz,), 7.67 (d, J = 9.0 Hz,), 7.58 (d J = 7.2 Hz, 2H), 7.55 (d, J = 7.2 Hz, 1H), 7.42-7.39 (m, 2H), 7.36-7.34 (m, 1H), 5.84 (d, J = 7.8 Hz, 1H). HRMS (AP-ESI) m/z calcd for C22H19ClN3O4 [M + H]+ 424.1064, found 424.1057. 13C NMR (75 MHz, DMSO-d6) δ: 58.56, 118.96, 127.96, 128.18, 128.34, 128.59, 128.60, 129.03, 132.88, 136.76, 137.60, 141.77, 164.26, 166.04, 169.51. HPLC retention time: 9.6 min, eluted by CH3CN: H2O = 45: 55.
(S)-2-chloro-N-(2-((4-(hydroxycarbamoyl)phenyl)amino)-2-oxo-1-phenylethyl)benzamide (D2)
White powder crystallized in EtOAc, 45% yield. Mp: 250–252 °C. [α]20D −8.20 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.11 (s, 1H), 10.62 (s, 1H), 9.29 (d, J = 7.8 Hz, 1H), 8.93 (s, 1H), 7.73 (d, J = 9 Hz, 2H), 7.67 (d, J = 8.4 Hz, 2H), 7.58 (d, J = 7.8 Hz, 2H), 7.48 (d, J = 6.6 Hz, 1H), 7.47 (d, J = 4.8 Hz, 1H), 7.45 (d, J = 8.4 Hz, 1H), 7.41-7.40 (m, 2H), 7.38 (t, J = 8.4 Hz, 1H), 7.35-7.32 (m, 1H), 5.86 (d, J = 7.8 Hz, 1H). HRMS (AP-ESI) m/z calcd for C22H19ClN3O4 [M + H]+ 424.1064, found 424.1057. 13C NMR (75 MHz, DMSO-d6) δ: 58.06, 118.97, 127.35, 127.97, 128.25, 128.46, 128.97, 129.76, 129.93, 131.34, 136.55, 137.70, 141.71, 164.24, 166.78, 169.20. HPLC retention time: 7.5 min, eluted by CH3CN: H2O = 40: 60.
(S)-N-(2-((4-(hydroxycarbamoyl)phenyl)amino)-2-oxo-1-phenylethyl)-2-naphthamide (D3)
White powder crystallized in EtOAc, 47% yield. Mp: 238–240 °C. [α]20D −7.01 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.13 (s, 1H), 10.77 (s, 1H), 9.22 (d, J = 7.2 Hz, 1H), 8.93 (s, 1H), 8.63 (s, 1H), 8.05 (d, J = 7.8 Hz, 1H), 8.03 (d, J = 9.0 Hz, 1H), 8.02 (d, J = 8.4 Hz, 1H), 8.00 (d, J = 9.0 Hz, 1H), 7.74-7.71 (m, 2H), 7.65 (d, J = 7.8 Hz, 2H), 7.63-7.62 (m, 1H), 7.62-7.60 (m, 1H), 7.44-7.41 (m, 2H), 7.37-7.35 (m, 1H), 5.95 (d, J = 7.8 Hz, 1H). HRMS (AP-ESI) m/z calcd for C26H22N3O4 [M + H]+ 440.1610, found 440.1607. 13C NMR (75 MHz, DMSO-d6) δ: 58.66, 119.01, 125.07, 127.13, 127.92, 128.04, 128.16, 128.38, 128.40, 128.65, 128.70, 128.94, 129.36, 131.41, 132.50, 134.71, 137.96, 141.91, 164.25, 166.96, 169.69. HPLC retention time: 8.8 min, eluted by CH3CN: H2O = 45: 55.
(S)-4-fluoro-N-(2-((4-(hydroxycarbamoyl)phenyl)amino)-2-oxo-1-phenylethyl)benzamide (D4)
White powder crystallized in EtOAc, 46% yield. Mp: 240–242 °C. [α]20D −5.64 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.12 (s, 1H), 10.65 (s, 1H), 9.10 (d, J = 7.2 Hz, 1H), 8.93 (s, 1H), 8.05-8.03 (m, 2H), 7.72 (d, J = 8.4, 2H), 7.69 (d, J = 9, 2H), 7.59 (d, J = 9.0 Hz, 2H), 7.42-7.39 (m, 2H), 7.35 (t, J = 7.2 Hz, 1H), 7.30 (t, J = 9.0 Hz, 1H), 5.86 (d, J = 7.2 Hz, 1H). HRMS (AP-ESI) m/z calcd for C22H19FN3O4 [M + H]+ 408.1359, found 408.1355. 13C NMR (75 MHz, DMSO-d6) δ: 58.61, 115.58, 118.98, 127.90, 128.17, 128.43, 128.91, 130.60, 130.62, 137.79, 141.86, 163.66, 164.27, 165.93, 169.61. HPLC retention time: 7.7 min, eluted by CH3CN: H2O = 40: 60.
(S)-N-hydroxy-4-(2-(naphthalene-1-sulfonamido)-2-phenylacetamido)benzamide (D5)
White powder crystallized in EtOAc and hexane (1: 1), 53% yield. Mp: 152–154 °C. [α]20D −7.60 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.07 (s, 1H), 10.33 (s, 1H), 9.09 (d, J = 9 Hz, 1H), 8.92 (s, 1H), 8.74 (d, J = 8.4 Hz, 1H), 8.14-8.13 (m, 1H), 8.08 (d, J = 7.8 Hz, 1H), 7.96 (d, J = 8.4 Hz, 1H), 7.68-7.66 (m, 1H), 7.63 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 7.8 Hz, 2H), 7.52 (t, J = 7.8 Hz, 2H), 7.34 (d, J = 9 Hz, 2H), 7.29 (d, J = 7.8 Hz, 1H), 7.18-7.16 (m, 1H), 5.21 (d, J = 8.4 Hz, 1H). HRMS (AP-ESI) m/z calcd for C25H22N3O5S [M + H]+ 476.1280, found 476.1273. HPLC retention time: 13.0 min, eluted by CH3CN: H2O = 40: 60.
(S)-4-(2-(4-(tert-butyl)phenylsulfonamido)-2-phenylacetamido)-N-hydroxybenzamide (D6)
White powder crystallized in EtOAc and hexane (1: 1), 48% yield. Mp: 155–157 °C. [α]20D −7.77 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.08 (s, 1H), 10.39 (s, 1H), 8.93 (s, 1H), 8.67 (d, J = 9.6 Hz, 1H), 7.67 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 9 Hz, 2H), 7.39-7.27(m, 4H), 7.27 (d, J = 8.4 Hz, 2H), 7.25-7.23 (m, 1H), 5.17 (d, J = 10.2, 1H), 1.14 (s, 9H). HRMS (AP-ESI) m/z calcd for C25H28N3O5S [M + H]+ 482.1749, found 482.1743. 13C NMR (75 MHz, DMSO-d6) δ: 60.19, 118.99, 125.88, 126.96, 127.46, 128.01, 128.04, 128.26, 128.76, 137.24, 138.07, 141.19, 155.62, 164.07, 168.17. HPLC retention time: 10.6 min, eluted by CH3CN: H2O = 45: 55.
(S)-4-(2-benzamido-2-phenylacetamido)-N-hydroxybenzamide (D7)
White powder crystallized in EtOAc, 42% yield. Mp: 229–230 °C. [α]20D −6.60 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.10 (s, 1H), 10.59 (s, 1H), 9.02 (d, J = 7.8 Hz, 1H), 8.9 (s, 1H), 7.95 (d, J = 7.2 Hz, 2H), 7.72 (d, J = 9 Hz, 2H), 7.67 (d, J = 9 Hz, 2H), 7.59 (d, J = 7.8 Hz, 1H), 7.56 (t, J = 7.2 Hz, 2H), 7.48 (t, J = 7.8 Hz, 2H), 7.46-7.36 (m, 2H), 7.35 (t, J = 7.2 Hz, 1H), 5.86 (d, J = 7.2 Hz, 1H). HRMS (AP-ESI) m/z calcd for C22H20N3O4 [M + H]+ 390.1454, found 390.1449. 13C NMR (75 MHz, DMSO-d6) δ: 58.49, 118.95, 127.93, 128.17, 128.27, 128.34, 128.60, 128.93, 131.94, 134.13, 137.84, 141.82, 164.27, 166.97, 169.94. HPLC retention time: 9.9 min, eluted by CH3CN: H2O = 35: 65.
(S)-N-hydroxy-4-(2-phenyl-2-(4-(trifluoromethyl)phenylsulfonamido)acetamido)benzamide (D8)
White powder crystallized in EtOAc and hexane (1: 1), 40% yield. Mp: 200–202 °C. [α]20D −7.20 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.10 (s, 1H), 10.48 (s, 1H), 9.09 (d, J = 9.6 Hz, 1H), 8.94 (s, 1H), 7.92 (d, J = 8.4 Hz, 2H), 7.76 (d, J = 8.4 Hz, 2H), 7.67 (d, J = 9 Hz, 2H), 7.41 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 7.8 Hz, 2H), 7.26 (d, J = 8.4 Hz, 2H), 7.23-7.22 (m, 1H), 5.24 (d, J = 9 Hz, 1H). HRMS (AP-ESI) m/z calcd for C22H19F3N3O5S [M + H]+ 494.0997, found 494.0993. 13C NMR (75 MHz, DMSO-d6) δ: 60.18, 119.02, 127.52, 127.54, 127.99, 128.07, 128.79, 128.82, 128.86, 136.72, 140.99, 144.96, 164.08, 167.83. HPLC retention time: 8.6 min, eluted by CH3CN: H2O = 45: 55.
(S)-4-(2-(2,6-difluorophenylsulfonamido)-2-phenylacetamido)-N-hydroxybenzamide (D9)
White powder crystallized in EtOAc and hexane (1: 1), 47% yield. Mp: 215–217 °C. [α]20D −8.40 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.09 (s, 1H), 10.52 (s, 1H), 9.29 (d, J = 9 Hz, 2H), 8.93 (s, 1H), 7.68 (d, J = 9 Hz, 2H), 7.56 (m, 1H), 7.49 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 7.8 Hz, 2H), 7.32-7.9 (m, 2H), 7.27 (d, J = 7.2 Hz, 1H), 7.12 (t, J = 9 Hz, 2H), 5.37 (d, J = 9.6 Hz, 1H). HRMS (AP-ESI) m/z calcd for C21H18F2N3O5S [M + H]+ 462.0935, found 462.0931. 13C NMR (75 MHz, DMSO-d6) δ: 60.70, 113.45, 113.60, 118.85, 127.60, 128.18, 128.53, 128.87, 135.62, 136.91, 141.25, 158.16, 159.87, 164.18, 167.92. HPLC retention time: 8.3 min, eluted by CH3CN: H2O = 40: 60.
(S)-4-(2-(4-chlorophenylsulfonamido)-2-phenylacetamido)-N-hydroxybenzamide (D10)
White powder crystallized in EtOAc and hexane (1: 1), 45% yield. Mp: 228–230 °C. [α]20D −7.60 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.10 (s, 1H), 10.46 (s, 1H), 8.93 (d, J = 7.2 Hz, 1H), 8.92 (s, 1H), 7.72 (d, J = 8.4 Hz, 2H), 7.68 (d, J = 9.0 Hz, 2H), 7.47 (d, J = 7.2 Hz, 2H), 7.44 (d, J = 8.4 Hz, 2H), 7.38 (d, J = 7.8 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 7.26-7.25 (m, 1H), 5.20 (d, J = 7.2 Hz, 1H). HRMS (AP-ESI) m/z calcd for C21H19ClN3O5S [M + H]+ 460.0734, found 460.0725. 13C NMR (75 MHz, DMSO-d6) δ: 60.20, 119.05, 128.21, 128.85, 128.98, 129.13, 129.25, 129.30, 136.97, 137.60, 140.03, 141.10, 164.14, 168.04. HPLC retention time: 10.3 min, eluted by CH3CN: H2O = 40: 60.
(S)-N-(2-((4-(hydroxycarbamoyl)phenyl)amino)-2-oxo-1-phenylethyl)-2-methoxybenzamide (D11)
White powder crystallized in EtOAc, 46% yield. Mp: 156–158 °C. [α]20D −7.50 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.11 (s, 1H), 10.69 (s, 1H), 9.17 (d, J = 7.8 Hz, 1H), 8.93 (s, 1H), 7.91 (d, J = 7.8 Hz, 1H), 7.73 (d, J = 9 Hz, 2H), 7.65 (d, J = 8.4 Hz, 2H), 7.7-7.54 (m, 3H), 7.42-7.39 (m, 2H), 7.34 (d, J = 7.2 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.09 (t, J = 7.2 Hz, 1H), 5.87 (d, J = 7.2 Hz, 1H), 4.01 (s, 3H). HRMS (AP-ESI) m/z calcd for C23H22N3O5 [M + H]+ 420.1559, found 420.1553. 13C NMR (75 MHz, DMSO-d6) δ: 56.83, 17.75, 112.89, 119.15, 121.32, 121.37, 127.30, 128.27, 128.43, 129.18, 131.44, 133.67, 138.88, 141.43, 157.94, 164.08, 164.21, 169.28. Retention time: 8.2 min, eluted by CH3CN: H2O = 40: 60.
(S)-N-hydroxy-4-(2-(4-methoxybenzamido)-2-phenylacetamido)benzamide (D12)
White powder crystallized in EtOAc, 49% yield. Mp: 240–241 °C. [α]20D −7.80 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.09 (s, 1H), 10.55 (s, 1H), 8.91 (s, 1H), 8.83 (d, J = 7.8 Hz, 1H), 7.95 (d, J = 7.2 Hz, 2H), 7.72 (d, J = 9.0 Hz, 2H), 7.67 (d, J = 9.0 Hz, 2H), 7.57 (d, J = 7.2 Hz, 2H), 7.41–7.39 (m, 2H), 7.35-7.33 (m, 2H), 7.00 (d, J = 8.4 Hz, 2H), 5.84 (d, J = 7.2 Hz, 1H), 3.82 (s, 3H). HRMS (AP-ESI) m/z calcd for C23H22N3O5 [M + H]+ 420.1559, found 420.1554. 13C NMR (75 MHz, DMSO-d6) δ: 55.73, 58.42, 113.81, 118.95, 126.31, 127.94, 128.21, 128.35, 128.92, 130.17, 137.96, 141.82, 162.24, 164.27, 166.42, 169.79. HPLC retention time: 10.1 min, eluted by CH3CN: H2O = 40: 60.
(S)-3-bromo-N-(2-((4-(hydroxycarbamoyl)phenyl)amino)-2-oxo-1-phenylethyl)benzamide (D13)
White powder crystallized in EtOAc, 48% yield. Mp: 234–236 °C. [α]20D −6.80 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.10 (s, 1H), 10.58 (s, 1H), 9.23 (d, J = 7.2 Hz, 1H), 8.93 (s, 1H), 8.17 (s, 1H), 7.94 (d, J = 7.8 Hz, 1H), 7.76 (d, J = 7.8 Hz, 1H), 7.72 (d, J = 9.0 Hz, 2H), 7.67 (d, J = 8.4 Hz, 2H), 7.58 (d, J = 7.2 Hz, 2H), 7.45 (d, J = 7.8 Hz, 1H), 7.41 (d, J = 9.0 Hz, 1H), 7.37–7.34 (m, 1H), 5.84 (d, J = 7.8 Hz, 1H). HRMS (AP-ESI) m/z calcd for C22H19BrN3O4 [M + H]+ 468.0559, found 468.0554. HPLC retention time: 11.5 min, eluted by CH3CN: H2O = 40: 60.
(S)-N-hydroxy-4-(2-phenyl-2-(2-propylpentanamido)acetamido)benzamide (D14)
White powder crystallized in EtOAc, 47% yield. Mp: 267–269 °C. [α]20D −7.20 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.09 (s, 1H), 10.52 (s, 1H), 8.92 (s, 1H), 8.61 (d, J = 7.2 Hz, 1H), 7.71 (d, J = 8.4 Hz, 1H), 7.64 (d, J = 8.4 Hz, 2H), 7.48 (d, J = 7.2 Hz, 2H), 7.40-7.37 (m, 2H), 7.33-7.30 (m, 1H), 5.67 (d, J = 7.8 Hz, 1H), 1.47-1.43 (m, 1H), 1.28-1.21 (m, 4H), 1.19-1.12 (m, 6H). HRMS (AP-ESI) m/z calcd for C23H30N3O4 [M + H]+ 412.2236, found 412.2231. HPLC retention time: 11.9 min, eluted by CH3CN: H2O = 40: 60.
(S)-N-hydroxy-4-(2-phenyl-2-(2-phenylacetamido)acetamido)benzamide (D15)
White powder crystallized in EtOAc, 46% yield. Mp: 225–226 °C. [α]20D −6.70 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.09 (s, 1H), 10.56 (s, 1H), 8.92 (s, 1H), 8.91 (d, J = 8.4 Hz, 1H), 7.71 (d, J = 9.0 Hz, 2H), 7.63 (d, J = 9.0 Hz, 2H), 7.50 (d, J = 7.2 Hz, 2H), 7.40-7.37 (m, 2H), 7.33 (d, J = 7.2 Hz, 1H), 7.29-7.28 (m, 4H), 7.22-7.20 (m, 1H), 5.65 (d, J = 7.8 Hz, 1H), 3.60 (s, 2H). HRMS (AP-ESI) m/z calcd for C23H22N3O4 [M + H]+ 404.1610, found 404.1607. 13C NMR (75 MHz, DMSO-d6) δ: 42.10, 60.20, 118.96, 126.76, 128.22, 128.38, 129.00, 129.52, 136.77, 138.26, 141.68, 163.01, 169.59, 170.61. HPLC retention time: 9.8 min, eluted by CH3CN: H2O = 36: 64.
(S)-N-hydroxy-4-(2-(naphthalene-2-sulfonamido)-2-phenylacetamido)benzamide (D16)
White powder crystallized in EtOAc and hexane (1: 1), 53% yield. Mp: 196–198 °C. [α]20D −6.90 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.07 (s, 1H), 10.43 (s, 1H), 8.94 (s, 1H), 8.33 (s, 1H), 7.96 (d, J = 8.4 Hz, 1H), 7.94 (d, J = 9.0 Hz, 1H), 7.90 (d, J = 7.8 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.60 (t, J = 7.2 Hz, 1H), 7.57 (s, 1H), 7.55 (d, J = 8.4 Hz, 2H), 7.38 (d, J = 7.2 Hz, 2H), 7.30 (d, J = 8.4 Hz, 2H), 7.22 (t, J = 7.2 Hz, 2H), 7.17 (d, J = 7.2 Hz, 1H), 5.23 (s, 1H). HRMS (AP-ESI) m/z calcd for C25H22N3O5S [M + H]+ 476.1280, found 476.1276. HPLC retention time: 9.6 min, eluted by CH3CN: H2O = 43: 57.
(S)-4-(2-(5-(dimethylamino)naphthalene-1-sulfonamido)-2-phenylacetamido)-N-hydroxybenzamide (D17)
White powder crystallized in EtOAc and hexane (1: 1), 49% yield. Mp: 152–154 °C. [α]20D −6.78 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.08 (s, 1H), 10.28 (s, 1H), 9.04 (d, J = 9.6 Hz, 1H), 8.94 (s, 1H), 8.37 (d, J = 8.4 Hz, 1H), 8.29 (d, J = 8.4 Hz, 1H), 8.12 (d, J = 7.2 Hz, 1H), 7.60 (d, J = 9.0 Hz, 2H), 7.55-7.53 (m, 1H), 7.51-7.48 (m, 1H), 7.31-7.29 (m, 4H), 7.18-7.16 (m, 4H), 5.16 (d, J = 9.6 Hz, 1H), 2.71 (s, 6H). HRMS (AP-ESI) m/z calcd for C27H27N4O5S [M + H]+ 519.1702, found 519.1699. HPLC retention time: 10.2 min, eluted by CH3CN: H2O = 40: 60.
(S)-N-hydroxy-4-(2-(3-(2-methoxyphenyl)ureido)-2-phenylacetamido)benzamide (D18)
White powder crystallized in EtOAc, 33% yield. Mp: 201–203 °C. [α]20D −6.40 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.09 (s, 1H), 10.61 (s, 1H), 8.92 (s, 1H), 8.39 (s, 1H), 8.07 (s, 1H), 8.06 (d, J = 7.8 Hz, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.71 (d, J = 9.0 Hz, 1H), 7.66 (d, J = 8.4 Hz, 2H), 7.52 (d, J = 7.8 Hz, 2H), 7.42-7.39 (m, 2H), 7.33 (t, J = 7.2 Hz, 1H), 6.97 (d, J = 7.8 Hz, 1H), 6.90-6.87 (m, 1H), 6.84-6.82 (m, 1H), 5.57 (d, J = 7.8 Hz, 1H), 3.84 (s, 3H). HRMS (AP-ESI) m/z calcd for C23H23N4O5 [M + H]+ 435.1668, found 435.1662. 13C NMR (75 MHz, DMSO-d6) δ: 57.98, 60.19, 111.09, 118.47, 120.88, 121.63, 121.72, 127.11, 127.89, 129.02, 129.06, 129.62, 126.75, 139.06, 141.73, 147.90, 154.98, 164.27, 167.60. HPLC retention time: 8.7 min, eluted by CH3CN: H2O = 40: 60.
(S)-4-(2-(3-(2,6-diisopropylphenyl)ureido)-2-phenylacetamido)-N-hydroxybenzamide (D19)
White powder crystallized in EtOAc, 42% yield. Mp: 252–254 °C. [α]20D −8.40 (c 1.0, MeOH). 1H NMR (DMSO-d6) δ 11.12 (s, 1H), 10.67 (s, 1H), 8.95 (s, 1H), 7.77 (s, 1H), 7.71 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 7.8 Hz, 2H), 7.52 (d, J = 7.8 Hz, 2H), 7.39 (d, J = 7.2 Hz, 2H), 7.32 (d, J = 7.2 Hz, 1H), 7.18 (s, 2H), 7.10 (s, 2H), 5.58 (d, J = 8.4 Hz, 1H), 3.16-3.13 (m, 2H), 1.11 (d, J = 7.2 Hz, 12H). HRMS (AP-ESI) m/z calcd for C28H33N4O4 [M + H]+ 489.2502, found 489.2497. HPLC retention time: 8.8 min, eluted by CH3CN: H2O = 48: 52.
In Vitro HDAC Enzyme Inhibition Assay
In vitro activity assay against Hela cell nucleus extract has been described in our previous work. [11] Briefly, Hela cell nucleus extract solution (10 µL) was mixed with various concentrations of compound sample (50 µL). Fluorogenic substrate (40 µL) was added, and after 30 min of incubation at 37 °C, the reaction was stopped by addition of developer (100 µL) containing trypsin and TSA. Fluorescence intensity was measured after 20 min of reaction using a microplate reader at excitation and emission wavelengths of 390 nm and 460 nm. The IC50 values were determined based on the fluorescence intensity readings. The procedures of HDAC1, 2, 3 and 6 assays were similar to that of Hela nucleus extract.
Molecular Docking Study
The docking procedure was performed using Sybyl-X 2.0 and Maestro v7.5 (schrodinger Inc, supported by Shanghai Institute of Materia Medica Chinese Academy of Sciences). All the molecules sketched and minimized by Sybyl-X were ionized and assigned OPLS 2005 force field by LigPrep. The structure of human HDAC2 derived from the protein database (PDB entry: 3MAX) was used for the docking study. The HDAC2 structure was prepared by removal of waters and ligands, assignment of OPLS 2005 force field using Maestro. The active site was defined as a cubic box centered by zinc ion by Glide dock, and the side length of the box is 20 Å. Extra precision was applied in the docking process, other parameters were set as default.
In Vitro Antiproliferative Assay
Tumor cell inhibition assay was determined using MTT method. Briefly, 2,000 cells were seeded into each well of 96-well plates which were incubated at 37 °C, 5% CO2 for overnight. Then the cells were treated with compound sample in various concentrations for 48 h. After that, a 0.5% MTT solution was added to each well. After 4 h of incubation, formazan formed from MTT was extracted by adding DMSO (200 µL) for 5 min. Optical density values were then detected at 570 nm on a microplate reader.
In Vivo Antitumor Assay against U937 Xenograft
For in vivo antitumor efficacy study, 1.8 × 107 U937 cells were inoculated subcutaneously in the right shoulder of male athymic nude mice (5–6 weeks old, Slac Laboratory Animal, Shanghai). Then days after injection, tumors were palpable and mice were randomized into treatment and control groups (5 mice per group). The treatment groups were administrated with 100 mg/kg/d intragastricly, the control group was administrated with equal volume of PBS solution. During treatment, the body weight of mice was monitored regularly. After 16 days of administration, the mice were executed and the tumor weight was measured by an electronic balance.
Western blot analysis
For western blot analysis, total protein extracts were separated on a polyacrylamide gel, transferred onto PVDF membranes and blotted as previously described.[12] Briefly, U937 and MDA-MB-231 cells with different treatment were collected and lysed with lysis buffer for 30 min, then centrifuged for 15 min at 4000 rpm at 4 °C, the supernatant was the whole-cell extracts. Total protein extracts were separated by 12% SDS polycrylamide gel electrophoresis and transferred onto PVDF membranes (Cat. IPVH00010, Millipore). Membrane was blocked with 5% milk in TBS-T for 1 h at room temperature, then incubated with a 1:1000 or 1:2000 dilution of primary antibody overnight at 4 °C including tubulin (Sigma), acetylated tubulin (Sigma), histone H3 (Sigma), acetylated histone H3 (Sigma), acetylated histone H4 (Sigma), p21 (Cell Signaling), c-Raf (Sigma), p-Erk (Sigma), Erk (Sigma), p-Akt (Sigma) and Akt (Sigma). Then, the membrane was washed three times and incubated at 1:2000 dilution of anti-mouse or anti-rabbit goat-HRP-conjugated secondary antibodies for 2 h at room temperature. Finally, the membrane was washed another three times and developed by enhanced chemiluminescence (ECL, Cat. WBKL S0050, Milipore).
Scheme 1.
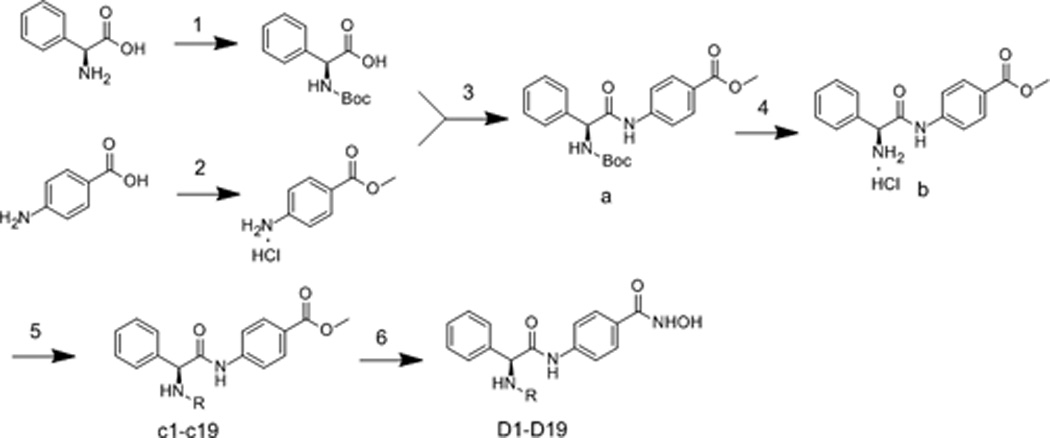
Synthesis of compounds D1-D19. Reagents and conditions: (1) (Boc)2O, Et3N, MeOH/H2O; (2) CH3COCl, CH3OH; (3) TBTU, Et3N, THF; (4) EtOAc, HCl; (5) RSO2Cl, RCOCl, NaHCO3, THF/H2O; RNH2, triphosgene, dioxane, Et3N, DCM; (6) NH2OK, MeOH.
ACKNOWLEDGMENT
This work was supported by National Nature Science Foundation of China (Grant No. 21172134), National High Technology Research and Development Program of China (Grant No. 2011ZX09401-015), Doctoral Foundation of Ministry of Education of China (Grant No. 20110131110037), Independent Innovation Foundation of Shandong University, IIFSDU (Grant No. 2013GN013) and National Cancer Institute of the National Institute of Health (USA) Under Award number R01CA163452.
REFERENCES
- 1.a Bernstein BE, Tong JK, Schreiber SL. P Natl Acad Sci USA. 2000;97:13708–13713. doi: 10.1073/pnas.250477697. [DOI] [PMC free article] [PubMed] [Google Scholar]; b De Ruijter AJM, Van Gennip AH, Caron HN, Kemp S, Van Kuilenburg ABP. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Foglietti C, Filocamo G, Cundari E, De Rinaldis E, Lahm A, Cortese R, Steinkuhler C. J Biol Chem. 2006;281:17968–17976. doi: 10.1074/jbc.M511945200. [DOI] [PubMed] [Google Scholar]
- 2.a Gronroos E, Hellman U, Heldin CH, Ericsson J. Mol Cell. 2002;10:483–493. doi: 10.1016/s1097-2765(02)00639-1. [DOI] [PubMed] [Google Scholar]; b Giandomenico V, Simonsson M, Gronroos E, Ericsson J. Mol Cell Biol. 2003;23:2587–2599. doi: 10.1128/MCB.23.7.2587-2599.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jin YH, Jeon EJ, Li QL, Lee YH, Choi JK, Kim WJ, Lee KY, Bae SC. J Biol Chem. 2004;279:29409–29417. doi: 10.1074/jbc.M313120200. [DOI] [PubMed] [Google Scholar]
- 3.a Bode AM, Dong ZG. Nat Rev Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]; b Chen LF, Greene WC. Nat Rev Mol Cell Bio. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]; c Kovacs JJ, Murphy PJM, Gaillard S, Zhao XA, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. Mol Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 4.Marks PA, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 5.a Thiagalingam SAM, Cheng K-H, Lee HJ, Mineva N, Thiagalingam A, Ponte JF. Annals of the New York Academy of Sciences. 2003;983:84–100. doi: 10.1111/j.1749-6632.2003.tb05964.x. [DOI] [PubMed] [Google Scholar]; c Marks PA, Richon VM, Rifkind RA. J Nat Cancer Insti. 2000;92:1210–1216. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- 6.Richon VM, Emiliani S, Verdin E, Webb Y, Breslow R, Rifkind RA, Marks PA. P Natl Acad Sci USA. 1998;95:3003–3007. doi: 10.1073/pnas.95.6.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ueda H, Nakajima H, Hori Y, Fujita T, Nishimura M, Goto T, Okuhara M. J Antibiot. 1994;47:301–310. doi: 10.7164/antibiotics.47.301. [DOI] [PubMed] [Google Scholar]
- 8.a Zhang L, Li MY, Feng JH, Fang H, Xu WF. Med Chem Res. 2012;21:152–156. [Google Scholar]; b Zhang L, Wang X, Li X, Xu W. J Enzym Inhibit Med Chem. 2013 [Google Scholar]
- 9.a Verbrugge I, Johnstone RW, Bots M. Epigenomics. 2011;3:547–565. doi: 10.2217/epi.11.82. [DOI] [PubMed] [Google Scholar]; b Gryder BE, Sodji QH, Oyelere AK. Future Med Chem. 2012;4:505–524. doi: 10.4155/fmc.12.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, Faderl S, Koller C, Morris G, Rosner G, Loboda A, Fantin VR, Randolph SS, Hardwick JS, Reilly JF, Chen C, Ricker JL, Secrist JP, Richon VM, Frankel SR, Kantarjian HM. Blood. 2008;111:1060–1066. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]; b Vansteenkiste J, Van Cutsem E, Dumez H, Chen C, Ricker JL, Randolph SS, Schoffski P. Invest New Drugs. 2008;26:483–488. doi: 10.1007/s10637-008-9131-6. [DOI] [PubMed] [Google Scholar]; c Munster PN, Lacevic M, Schmitt M, Bicaku E, Marchion D, Stephens A, Sullivan L, Minton S. J Clin Oncol. 2008;26 [Google Scholar]; d Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS, Reilly JF, Loboda A, Nebozhyn M, Fantin VR, Richon VM, Scheithauer B, Giannini C, Flynn PJ, Moore DF, Jr, Zwiebel J, Buckner JC. J Clin Oncol. 2009;27:2052–2058. doi: 10.1200/JCO.2008.19.0694. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hymes K, Dummer R, Sterry W, Steinhoff M, Assaf C, Kerl H, Ahern J, Rizvi S, Ricker JL, Whittaker S. J Clin Oncol. 2008;26 [Google Scholar]; f Duvic M, Olsen EA, Breneman D, Pacheco TR, Parker S, Vonderheid EC, Abuav R, Ricker JL, Rizvi S, Chen C, Boileau K, Gunchenko A, Sanz-Rodriguez C, Geskin LJ. Clinical Lymph Myelom. 2009;9:412–416. doi: 10.3816/CLM.2009.n.082. [DOI] [PubMed] [Google Scholar]; g Piekarz R, Robey R, Sandor V, Fojo AT, Bakke S, Wilson W, Kingma D, Turner M, Tucker E, Bates SE. Clin Cancer Res. 2001;7:3726S–3726S. [Google Scholar]; h Piekarz RL, Robey R, Sandor V, Bakke S, Wilson WH, Dahmoush L, Kingma DM, Turner ML, Altemus R, Bates SE. Blood. 2001;98:2865–2868. doi: 10.1182/blood.v98.9.2865. [DOI] [PubMed] [Google Scholar]; i Stadler WM, Margolin K, Ferber S, McCulloch W, Thompson JA. Clinical Genitourin Cancer. 2006;5:57–60. doi: 10.3816/CGC.2006.n.018. [DOI] [PubMed] [Google Scholar]; j Schrump DS, Fischette MR, Nguyen DM, Zhao M, Li X, Kunst TF, Hancox A, Hong JA, Chen GA, Kruchin E, Wright JJ, Rosing DR, Sparreboom A, Figg WD, Steinberg SM. Clin Cancer Res. 2008;14:188–198. doi: 10.1158/1078-0432.CCR-07-0135. [DOI] [PubMed] [Google Scholar]; k Whitehead RP, Rankin C, Hoff PMG, Gold PJ, Billingsley KG, Chapman RA, Wong L, Ward JH, Abbruzzese JL, Blanke CD. Invest New Drugs. 2009;27:469–475. doi: 10.1007/s10637-008-9190-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang YJ, Feng JH, Liu CX, Zhang L, Jiao J, Fang H, Su L, Zhang XP, Zhang J, Li MY, Wang BH, Xu WF. Bioorg Med Chem. 2010;18:1761–1772. doi: 10.1016/j.bmc.2010.01.060. [DOI] [PubMed] [Google Scholar]
- 12.Feng J, Fang H, Wang X, Jia Y, Zhang L, Jiao J, Zhang J, Gu L, Xu W. Cancer Biol Ther. 2011;11:477–489. doi: 10.4161/cbt.11.5.14529. [DOI] [PubMed] [Google Scholar]