

Abstract
Bacterial biofilms pose a significant challenge in clinical environments due to their inherent lack of susceptibility to antibiotic treatment. It is widely recognized that most pathogenic bacterial strains in the clinical setting persist in the biofilm state, and are the root cause of many recrudescent infections. Discovery and development of compounds capable of either inhibiting biofilm formation or initiating biofilm dispersal may provide new therapeutic avenues for reducing the number of hospital acquired, biofilm-mediated infections. We now report the application of our recently reported image-based, high-throughput screen to the discovery of microbially-derived natural products with biofilm inhibitory activity against Vibrio cholerae. Examination of a prefractionated library of microbially-derived marine natural products has lead to the identification of a new biofilm inhibitor that is structurally unrelated to previously reported inhibitors and is one of the most potent inhibitors reported to date against V. cholerae. Combination of this compound with sub-MIC concentrations of a number of clinically relevant antibiotics was shown to improve the biofilm inhibitory efficacy of this new compound compared to monotherapy treatments, and provides evidence for the potential therapeutic benefit of biofilm inhibitors in treating persistent biofilm-mediated infections.
Keywords: biofilm inhibitor, drug discovery, Vibrio cholerae. natural products, image-based screening
Introduction
Bacterial biofilms are assemblages of sessile cells encased in an extracellular matrix composed of exopolysaccharides (EPS), extracellular DNA, and proteins.[1] Biofilm-mediated infections are prevalent in the clinic, and infections of this nature pose a significant threat to the medical community. To give an idea of the scale of this problem, up to 17 million new biofilm-mediated infections occur each year in the United States, contributing to an estimated 550,000 deaths.[2] Biofilms also contribute to hospital-acquired infections caused by indwelling medical devices, such as catheters. This is a significant concern, considering central venous catheters alone cause over 248,000 infections per year, at a cost of $6,400-$29,000 per infection in the United States.[3] These infections result in extended hospital stays and significant increases in healthcare costs spent to resolve biofilm-mediated infections.[4]
Bacteria in the biofilm state have been shown to be 10-10,000-fold less susceptible to antibiotic treatment.[5] Estimations made by the Centers for Disease Control (CDC) and the National Institutes of Health (NIH) attribute 65% to 80% of human infections as biofilm mediated. Consequently, biofilm formation is often responsible for chronic infections due to bacterial persistence despite antibiotic treatment. Considering that the formation of biofilms undermines the utility of existing antibiotics, our research team is interested in the development of new solutions that specifically address biofilm formation. To date, very few compounds have been shown to selectively inhibit biofilm formation in a non-microbicidal manner. Scheme 1 illustrates four examples of natural products reported to display anti-biofilm activities without a bactericidal activities.[6-10]
Scheme 1.

Examples of natural products reported to possess non-microbicidal biofilm inhibitory activities. Ellagic acid is active against biofilm formation of Staphylococcus aureus with an IC50 of 50 μM.[6] Phloretin selectively inhibits Escherichia coli O157:H7 biofilm formation by 89% at 91 μM.[7] Oroidin displays a biofilm IC50 of 190 μM against Pseudomonas aeruginosa PA01 and 166 μM against P. aeruginosa PA14.[12] (-)-Ageloxime-D is reported to inhibit biofilm formation in Staphylococcus epidermidis.[10]
It has been suggested that the development of biofilm inhibitors may restore susceptibility to antibiotics in pathogenic infections, thus renewing the utility of existing therapies. Treatment strategies employing co-dosing of antibiotics and compounds disrupting biofilm formation may therefore provide a new avenue for combatting antibiotic resistance.[11]
Additionally, small molecule biofilm modulators have promising potential for development as coatings for indwelling medical devices. Medical implant devices are a major source of nosocomial infections. Compound coatings of this nature could therefore prevent initial bacterial colonization of these surfaces, and reduce the instances of persistent bacterial infections among inpatient populations.
Molecules capable of modulating biofilm formation, or inducing detachment of preformed biofilms, are already serving as powerful tools for further elucidating the pathways and regulators that control biofilm formation. For example, the discovery of autoinducer compounds lead to significant advances in the understanding of the molecular mechanisms responsible for quorum sensing.[13-15] Today, there remain a number of pathways whose investigation will contribute to a more complete picture of the molecular mechanisms that control biofilm formation. By discovering new classes of biofilm inhibitor such as the compound described below, we aim to develop a suite of tools for identifying novel targets for future drug development programs.
Results and Discussion
Image-based screening platform
We recently reported the development of a 384-well, image-based screening platform for the direct observation and quantification of biofilm formation in response to treatment with small molecule libraries using GFP-tagged V. cholerae.[16] This system uses an epifluorescence microscope to capture multiple images for each treated well (20% total well coverage) at a single focal plane, followed by identification and quantification of biofilm coverage to directly determine the test compound's ability to inhibit biofilm formation. The main benefit of this platform is the significant reduction in the time required to collect imaging data for large compound libraries compared with either confocal or electron microscopy techniques traditionally employed for this type of study.
In this new study, a 1,248-member prefractionated marine natural product library was evaluated for biofilm inhibitory activity in this image-based screening platform. Of the prefractions screened, 7 hits showed non-microcidal biofilm inhibitory activity with normalized OD600 values greater than 0.7 and normalized percent biofilm coverage values less than 20% (reported as 0.2). Of these, prefraction 1671D showed the strongest effect on biofilm formation, and was therefore subjected to one-compound-one-well ‘peak library’ fractionation using our standard protocol (See Supporting Information).[17, 18] This method automatically separates the constituents of prefractions of interest based on HPLC retention times, and connects specific constituents to observed biological activities through secondary screening. Screening of the resulting peak library from prefraction 1671D revealed a strong region of biofilm inhibition at minutes 26-28 that corresponded to a single peak in the chromatogram (Figure 1, upper trace). This activity was coupled with a striking biofilm macrocolony phenotype, providing strong justification for purification and full structural characterization of the active constituent.
Figure 1.

Peak library screening results for 1671D. A) Peak library HPLC chromatogram, B) corresponding optical density data and biofilm coverage data acquired for each two-minute time slice of the 1671D prefraction.
Characterization of compound 5
Purification of the peak highlighted in Figure 1 from large-scale liquid culture (4 L) using standard chromatographic methods (see Experimental) afforded the desired compound as a pale yellow crystalline solid (18 mg). High-resolution electrospray-ionization Fourier transform mass spectrometry identified a single [M + H]+ adduct consistent with the molecular formula of C12H11NO5 (eight degrees of unsaturation). This molecular formula was confirmed by examination of the number of resonances in the 1H and 13C NMR spectra, which were in agreement with the number of atoms predicted from the mass spectrum (Table S1). The high degree of unsaturation, coupled with the presence of 10 downfield carbons between δC 98.9 and 168.0, and two aromatic methine protons at δH 6.87 and 7.20 strongly suggested the presence of a conjugated core aromatic ring structure. gHMBC correlations from the aromatic methine signals at δH 6.87 and 7.20 to carbons at δC 109.5, 122.7, 143.5, 155.8 and δC 108.1, 122.7, 155.8, 168.0 respectively indicated that these two protons resided on the same tetrasubstituted phenyl ring (Figure 2A, substructure A). Additionally, the NMR data revealed the presence of two methoxy subunits (δH 3.79, δC 56.6 and δH 3.91, δC 53.4), one of which was present as a methyl ester with a carbonyl carbon at δC 168.0. The gHMBC correlation for the methyl singlet at δH 3.79 to the carbon at δC 155.8 indicated that this substituent existed as a pendant methyl ether on the aromatic ring. The methyl singlet at δH 3.91 showed gHMBC correlations to both a carboxylic carbon at δC 168.0 and the aromatic carbon at δC 115.1, confirming the presence of a methyl ester pendant to the aromatic ring (Figure 2A, substructure A).
Figure 2.
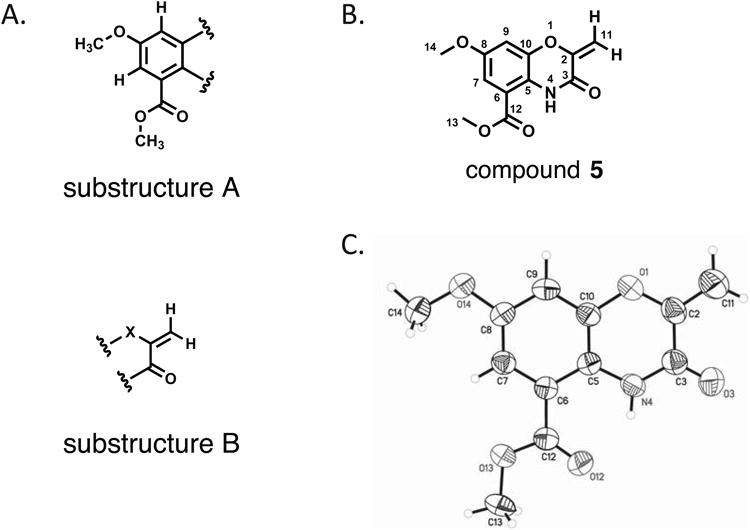
Structure for compound 5. A) substructures A and B, as determined from 1D and 2D NMR data, B) molecular structure for the choromophore of auromomycin, C) ORTEP structure of compound 5.
Two additional proton resonances at δH 5.06 and 5.51 showed strong COSY couplings to one another, and correlated to the same carbon resonance at δC 98.9 in the phase-sensitive gHSQC, indicating that these two signals were part of an exocyclic methylene moiety. This was confirmed by gHMBC correlations from both proton signals to a second olefinic carbon signal at δC 149.0, whose chemical shift suggested that it was adjacent to a heteroatom as part of a larger conjugated aromatic system. Finally, both methylene protons showed gHMBC correlations to a carbonyl carbon at δC 155.9 (Figure 2A, substructure B), suggesting the exocyclic alkene was adjacent to a carbonyl moiety.
Despite determination of subunits that accounted for the majority of the atoms in the molecular formula, unequivocal assignment of the complete structure was precluded by the low number of proton NMR resonances and the large number of heteroatoms and quaternary carbons in the molecule. To address this issue, recrystallization of compound 5 from toluene/EtOAc provided an orthorhombic yellow crystal, which was analyzed by X-ray diffraction. The crystal structure obtained from this experiment (Figure 2C) confirmed the assignment of the substructures determined by NMR analysis, and identified compound 5 as the previously reported central chromophore of the antitumor enediyne natural product auromomycin.[19, 20]
Biofilm inhibitory activity of compound 5
In order to determine the biofilm inhibitory activity (IC50) of compound 5, a two-fold dilution series was examined using our standard imaging platform to afford an IC50 of 60.1 μM against V. cholerae (Figure 3). Analysis of both the epifluorescence images obtained from this dilution series and confocal images collected from relevant individual wells from the same 384-well plate allowed evaluation of the macrocolony morphology caused by treatment with compound 5. Figures 4A and C display the images obtained from the epifluorescence and confocal platforms respectively, and show a dose-dependent reduction in biofilm macrocolony size as a function of compound concentration. COMSTAT analysis was applied to quantify the confocal images and provide a measure of the effect of compound 5 on the three-dimensional structure of the biofilm microcolonies. COMSTAT analysis uses a suite of image features to generate four shape variables that quantitatively describe biofilm structure.[21] This experiment was performed to confirm that the biofilm inhibition observed with the two-dimensional results acquired using the epifluorescence platform could be recapitulated using higher resolution confocal microscopy.
Figure 3.

Concentration curve for compound 5.
Figure 4.

Biofilm image analysis. A) Confocal laser scanning microscopic images of V. cholerae at increasing concentrations of compound 5 (Magnification 10×), B) quantitative image analysis conducted using COMSTAT analysis, C) epifluorescence images of biofilm microcolonies formed in a negative control well and wells treated with 49.4 and 74.1 μM of compound 5 (Magnification 20×).
Figure 4B illustrates the results from the COMSTAT analysis, showing a significant decrease in biomass with increasing concentration of compound 5. It is important to note that while at concentrations of 74.1 μM and 49.4 μM the COMSTAT values for biomass and average thickness are somewhat higher than those found for the negative control, the cross-sections acquired for these wells show that biofilm coverage is more evenly dispersed, and lacks the large macrocolonies present in negative control images. The epifluorescence images (Figure 4C) for these concentrations show microcolonies of similar size to the DMSO controls, but displaying a less bright, weaker biofilm phenotype, consistent with reduced cell density and lower occupancy of these macrocolonies.
Typically with previous inhibitor discovery projects,[16] we have observed inhibition phenotypes where macrocolonies remain bright, but shrink in size with increasing concentrations of inhibitor. The phenotype observed for compound 5 is unusual in that the microcolonies remain large, but the biofilm architecture becomes attenuated as the concentration of compound 5 increases. This is the first time we have observed this phenotype in our screening program, and makes this compound of high priority for further follow up studies.
In order to further validate the results obtained in the primary screen, we analyzed biofilm formation under static conditions using chambered cover glasses in 1 mL growth medium. Inhibition activity originally observed in well plates was recapitulated in this format (data not shown). The same static biofilm system was also used to evaluate the ability of compound 5 to disperse preformed biofilms, in order to determine whether the compound effects biofilm attachment and development or initiates a transition of cells in the biofilm state to the planktonic state. In this experiment, biofilms were allowed to pre-form for two hours prior to compound addition, with incubation and imaging following the standard protocol. Analysis of confocal images did not show any decrease in biomass after preformed biofilms were treated with compound 5, suggesting this compound does not disrupt biofilm formation by initiating biofilm dispersal.
n order to evaluate the selectivity of compound 5 for disruption of biofilm formation over bactericidal or bacteriostatic behaviors a number of additional assays were performed. Compound 5 was found to have no impact upon the growth of V. cholerae in liquid culture up to 250 μM, the highest concentration tested, using both OD600 and cfu evaluation methods (Figure S4). BioMAP analysis was used to evaluate the bactericidal activity of prefraction 1671D against a panel of clinically relevant bacterial pathogens.[17] Results from screening this prefraction against the 15 different bacterial stains (5 Gram-positive species and 9 Gram-negative species) revealed that compound 5 had no bactericidal activity against any of the tested strains. Additionally, compound 5 showed no mammalian cell cytotoxicity against HeLa cells, up to the highest tested concentration (250 μM), indicating that compound 5 possesses selective activity for biofilm inhibition without directly impacting either bacterial or host cell survival.
There are very few examples of agents capable of causing inhibition of biofilm formation in V. cholerae. Two different studies have reported proteins produced by Bacillus spp. capable of inhibiting V. cholerae biofilms,[22, 23] and a number of studies have reported inhibitory activity of bacterial extracts.[24, 25] However, only one other study has identified small molecules capable of inhibiting biofilm formation against this target. This compound was discovered through the design of a screening platform in order to identify small molecule inhibitors of diguanylate cyclase, an enzyme shown to be essential for the production of a cyclic di-GMP, the signaling molecule that regulates biofilm formation in V. cholerae. The most active compound was reported to inhibit biofilm formation at 26.2 μM.[26] Interestingly, this compound was found to only disrupt biofilm formation under flow-cell conditions, and not in static culture, unlike the inhibitor discussed in this study, which is capable of inhibiting initial biofilm formation under static conditions. Compound 5 is one of only a small number of biofilm inhibitors with activity against V. cholerae and is the first example of an inhibitor possessing this fused heterocyclic ring system.
Co-dosing of biofilm inhibitors with sub-MICs of antibiotics
It is well-documented that sub-MIC (minimum inhibitory concentration) doses of antibiotics are capable of inducing bacterial biofilm formation in vitro.[27] In 2005, a study reported in Nature illustrated the biofilm-inducing properties of sub-MIC concentrations of tobramycin, and the following years resulted in dozens of additional publications reporting similar results.[28] To date, tetracycline has been shown to induce biofilm formation in five different bacterial species, and rifamycin in three different bacterial species.[27] These studies raise concerns that therapeutic treatments of infections where prescribed antibiotics are not administered at sufficiently high doses may be contributing to the severity and persistence of infections by inducing biofilm formation. This is of concern for treatment of bacterial infections, since bacterial biofilms are inherently less responsive to antibiotic treatment. Thus, it has been suggested that the development of cotherapeutic agents to suppress biofilm induction could serve as a valuable solution to this problem by restoring antibiotic susceptibility.[11] Recently Melander and co-workers have demonstrated this effect using 2-aminoimidazole-derived biofilm inhibitors coupled with conventional antibiotics, as one reported example of this phenomenon.[29-31]
Three commercially available antibiotics, tetracycline, ceftazidime, and ciprofloxacin, were chosen for evaluation using this strategy due to their orthogonal bacterial targets and their utility as therapeutics against V. cholerae infections.[32] Dilution series for each antibiotic were evaluated using our image-based screen in order to determine both sub-MIC and sub-BIC (biofilm inhibitory concentration in terms of biofilm coverage) concentrations. A dilution series of compound 5 was then co-treated with a single fixed concentration of each antibiotic (a concentration qualifying as both sub-MIC and sub-BIC). This allowed us to evaluate whether the biofilm inhibition efficacy of compound 5 was affected by the addition of sub-MIC and sub-BIC levels of antibiotics. These results indicate that the addition of low concentrations of antibiotics significantly enhance the biofilm inhibitory activity of compound 5, as illustrated in Figure 5. Addition of sub-BIC quantities of tetracycline improved biofilm clearance by halving the concentration of compound 5 required to cause a 50% decrease in biofilm coverage. Sub-BIC concentrations of ceftazidime and ciprofloxacin were also found to improve the biofilm-clearing efficacy of compound 5, suggesting that this strategy is broadly applicable to antibiotics with different modes of action.
Figure 5.

Co-dosing concentration curves. IC50 curves for the inhibition of V. cholerae biofilms by compound 5 alone shown in blue. Shown in red are the IC50 curves for the inhibition of V. cholerae biofilms treated with varying concentrations of compound 5 plus treatment with a fixed concentration of A) tetracycline at 0.19 μM, B) ceftazidime at 222 μM, and C) ciprofloxacin at 0.037 μM. Ceftazidime was shown to be weakly effective against as an antibiotic against the strain of V. cholerae used in this study, reflecting the seemingly high sub-MIC and sub-BIC concentration.
Conclusion
This study reports the discovery of a new structural class of biofilm inhibitors, discovered through the application of an image-based, high-throughput screening platform to our in-house prefractionated marine natural products library. Using this target-independent phenotypic screening platform in concert with standard antibiotic and cytotoxicity assays we have demonstrated that this compound is a selective, non-bacteridical inhibitor of V. cholerae biofilms, and possesses a unique phenotype, causing diffuse microcolony formation. By shifting the focus of therapeutic discovery from antibiotic development to a more subtle inhibition of biofilm colonization we have identified a compound with potential to restore the efficacy of traditional antibiotics that is orthogonal to existing therapeutic options, and offers a unique approach to the elimination of persistent biofilm-mediated bacterial infections.
Experimental Section
Bacterial strains and growth conditions
V. cholerae cultures were grown in Luria-Bertani (LB) medium (1% tryptone, 0.5% yeast extract and 1% NaCl, pH 7.5) with aeration at 30 °C or on solid LB plates (1% agar), using GFP-tagged V. cholerae strain FY_Vc_240[33] and GFP-tagged V. cholerae strain FY_Vc_6226 (negative control).[34] Each of these strains was developed from the reference strain V. cholerae O1, El Tor A1552, rugose variant (FY_Vc_2).
Biological screening
Images were acquired as previously reported.[16] Briefly, liquid culture (prepared in LB with V. cholerae strain Fy_Vc_240 at a density of 1.28 × 106 CFU/mL) was dispensed into 384-well microtiter plates (40 μL, black-walled, clear-bottom) using a WellMate peristaltic microplate dispenser. Plates were centrifuged for 1 minute at 1200 rpm to remove air bubbles and bring the culture down from the walls of the plate. 384-well plates with DMSO stocks of test compounds were pinned into the culture plates (200 nL). Pinned culture plates were then incubated for 4.5 hours at 30 °C. Following incubation, OD600 readings were collected, followed by vigorous agitation and washing three times with 1% phosphate buffered saline (PBS). Plates were imaged in PBS by epifluorescence microscopy (ImageXpress Molecular Devices) at 20× magnification. Eight sites (distributed throughout the well) were imaged for each well in the 384-well microtiter plate. MetaXpress software (Molecular Devices) was used for calculation of biofilm coverage. Confocal images of biofilms were captured with an LSM 5 PASCAL system (Zeiss) at 488-nm excitation and 543-nm emission wavelengths. Three-dimensional images of the biofilms were reconstructed using Imaris software (Bitplane) and quantified using COMSTAT.[21] Further details on marine bacteria cultivation, natural product prefraction library generation, prefractionation strategy, and peak library preparation are available in the Supplementary Methods.
Isolation and structure elucidation for compound 5
The producing organism was isolated from a marine sediment sample collected by self-contained underwater breathing apparatus at Pinnacle Rock, Juan de Fuca Straight, Washington, from a depth of 32 ft under permit #10-395 issued from the Washington Department of Fish and Wildlife. The sediment was collected into a sterile 15 mL Falcon tube and stored at 4 °C until work-up. The supernatant was removed, and the sediment stamped on solid HVS media.[35] The isolate was repeatedly subcultured on Difco Marine Broth plates until pure, prepared in liquid culture (10 mL), and preserved as a glycerol frozen stock. Subsequent cultures were prepared from glycerol frozen stocks by plating upon solid media followed by preparation in liquid culture (1 L scale) using modified SYP medium (10 g starch, 4 g peptone, 2 g yeast extract and 31.2 g instant ocean in 1 L of distilled water). The 1 L liquid bacterial cultures were extracted with 1:1 CH2Cl2/MeOH, and concentrated to dryness in vacuo. The crude extracts were then resuspended in a MeOH/H2O solution, and further fractionated by solid phase extraction chromatography into six prefractions of increasing polarity. The active peak was purified from prefraction 1671D using RP-HPLC (Phenomenex Synergi Fusion-RP 80Å, 250 × 10.00 mm, 10 μm) applying an isocratic separation (63% MeOH, 37% H2O with 0.02% formic acid, 3 mL min-1, tR= 21 min.) to afford pure compound as a pale yellow powder (approx. 4.5 mg L-1). Solvents used for chromatographic isolation were HPLC grade and used without further purification. NMR data were acquired using a Varian Unity Inova spectrometer at 600 MHz equipped with a 5 mm HCN triple resonance cryoprobe, and referenced to residual proton signals. HRMS data for the pure compound was acquired using University of California, Berkeley QB3/Chemistry Mass Spectrometry Facility's multimode electrospray ionization (ESI) Fourier Transform mass spectrometer (FTMS).
Supplementary Material
Acknowledgments
We thank R. S. Lokey and W. Bray for access to the UCSC Chemical Screening Center, W. R. Wong for BioMAP screening, D. Winslow for assistance with data analysis, and N. Fong for culture preparation. This work was supported by NIH grant 1R21AI098836-01 (RGL and FHY).
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Hall-Stoodley L, Costerton JW, Stoodley P. Nat Rev Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 2.Wolcott R, Dowd S. Plast Reconstr Surg. 2011;127:28S–35S. doi: 10.1097/PRS.0b013e3181fca244. [DOI] [PubMed] [Google Scholar]
- 3.Calfee DP. Annu Rev Med. 2012;63:359–371. doi: 10.1146/annurev-med-081210-144458. [DOI] [PubMed] [Google Scholar]
- 4.Kirketerp-Møller K, Zulkowski K, James G. In: Biofilm Infections. Bjarnsholt T, Jensen PO, Moser C, Hoiby N, editors. Springer; New York: 2011. pp. 11–24. [Google Scholar]
- 5.Jefferson KK, Goldmann DA, Pier GB. Antimicrob Agents Ch. 2005;49:2467–2473. doi: 10.1128/AAC.49.6.2467-2473.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quave CL, Estévez-Carmona M, Compadre CM, Hobby G, Hendrickson H, Beenken KE, Smeltzer MS. PLOS ONE. 2012;7:e28737. doi: 10.1371/journal.pone.0028737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee JH, Regmi SC, Kim JA, Cho MH, Yun H, Lee CS, Lee J. Infect Immun. 2011;79:4819–4827. doi: 10.1128/IAI.05580-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelly SR, Jensen PR, Henkel TP, Fenical W, Pawlik JR. Aquat Microb Ecol. 2003;31:175–182. [Google Scholar]
- 9.Yamada A, Kitamura H, Yamaguchi K, Fukuzawa S, Kamijima C, Yazawa K, Kuramoto M, Wang GYS, Fujitani Y, Uemura D. B Chem Soc Jpn. 1997;70:3061–3069. [Google Scholar]
- 10.Hertiani T, Edrada-Ebel R, Ortlepp S, van Soest RWM, de Voogd NJ, Wray V, Hentschel U, Kozytska S, Müller WEG, Proksch P. Bioorgan Med Chem. 2010;18:1297–1311. doi: 10.1016/j.bmc.2009.12.028. [DOI] [PubMed] [Google Scholar]
- 11.Harris TL, Worthington RJ, Melander C. Angew Chem Int Edit. 2012;51:11254–11257. doi: 10.1002/anie.201206911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards J, Ballard T, Melander C. Org Biomol Chem. 2008;6:1356–1363. doi: 10.1039/b719082d. [DOI] [PubMed] [Google Scholar]
- 13.Smith KM, Bu Y, Suga H. Chem Biol. 2003;10:81–89. doi: 10.1016/s1074-5521(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 14.Rasmussen TB, Skindersoe ME, Bjarnsholt T, Phipps RK, Christensen KB, Jensen P, Andersen JB, Koch B, Larsen TO, Hentzer M, Eberl L, Høiby N, Givskov M. Microbiology. 2005;151:1325–1340. doi: 10.1099/mic.0.27715-0. [DOI] [PubMed] [Google Scholar]
- 15.Ng WL, Perez L, Cong J, Semmelhack MF, Bassler BL. PLOS Pathog. 2012;8 doi: 10.1371/journal.ppat.1002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peach KC, Bray WM, Shikuma NJ, Gassner NC, Lokey RS, Yildiz FH, Linington RG. Mol Biosyst. 2011;7:1176–1184. doi: 10.1039/c0mb00276c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong WR, Oliver AG, Linington RG. Chem Biol. 2012;19:1483–1495. doi: 10.1016/j.chembiol.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schulze CJ, Bray WM, Woerhmann MH, Stuart J, Lokey RS, Linington RG. Chem Biol. 2013 doi: 10.1016/j.chembiol.2012.12.007. in press (available at http://dx.doi.org/10.1016/j.chembiol.2012.1012.1007) [DOI] [PMC free article] [PubMed]
- 19.Kumada Y, Miwa T, Naoi N, Watanabe K, Naganawa H, Takita T, Umezawa H, Nakamura H, Iitaka Y. J Antibiot. 1983;36:200–202. doi: 10.7164/antibiotics.36.200. [DOI] [PubMed] [Google Scholar]
- 20.Shibuya M, Sakurai H, Maeda T, Nishiwaki E, Saito M. Tetrahedron Lett. 1986;27:1351–1354. [Google Scholar]
- 21.Heydorn A, Nielsen AT, Hentzer M, Sternberg C, Givskov M, Ersbøll BK, Molin S. Microbiology. 2000;146:2395–2407. doi: 10.1099/00221287-146-10-2395. [DOI] [PubMed] [Google Scholar]
- 22.Augustine N, Kumar P, Thomas S. Arch Microbiol. 2010;192:1019–1022. doi: 10.1007/s00203-010-0633-1. [DOI] [PubMed] [Google Scholar]
- 23.Kalpana BJ, Aarthy S, Pandian SK. Appl Biochem Biotech. 2012;167:1778–1794. doi: 10.1007/s12010-011-9526-2. [DOI] [PubMed] [Google Scholar]
- 24.Augustine N, Wilson PA, Kerkar S, Thomas S. Curr Microbiol. 2012;64:338–342. doi: 10.1007/s00284-011-0073-4. [DOI] [PubMed] [Google Scholar]
- 25.Nithya C, Pandian SK. Arch Microbiol. 2010;192:843–854. doi: 10.1007/s00203-010-0612-6. [DOI] [PubMed] [Google Scholar]
- 26.Sambanthamoorthy K, Sloup RE, Parashar V, Smith JM, Kim EE, Semmelhack MF, Neiditch MB, Waters CM. Antimicrob Agents Ch. 2012;56:5202–5211. doi: 10.1128/AAC.01396-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaplan JB. Int J Artif Organs. 2011;34:737–751. doi: 10.5301/ijao.5000027. [DOI] [PubMed] [Google Scholar]
- 28.Hoffman LR, D'Argenio DA, MacCosses MJ, Zhang Z, Jones RA, Miller SI. Nature. 2005;436:1171–1175. doi: 10.1038/nature03912. [DOI] [PubMed] [Google Scholar]
- 29.Rogers SA, Huigens RW, Cavanagh J, Melander C. Antimicrob Agents Ch. 2010;54:2112–2118. doi: 10.1128/AAC.01418-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pammi M, Liang R, Hicks JM, Barrish J, Versalovic J. Pediatr Res. 2011;70:578–583. doi: 10.1203/PDR.0b013e318232a984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Estrela AB, Abraham WR. Pharmaceuticals. 2010;3:1374–1393. doi: 10.3390/ph3051374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khan WA, Bennish ML, Seas C, Khan EH, Ronan A, Dhar U, Busch W, Salam MA. The Lancet. 1996;348:296–300. doi: 10.1016/s0140-6736(96)01180-4. [DOI] [PubMed] [Google Scholar]
- 33.Beyhan S, Bilecen K, Salama SR, Casper-Lindley C, Yildiz FH. J Bacteriol. 2007;189:388–402. doi: 10.1128/JB.00981-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fong J, Syed K, Klose K, Yildiz F. Microbiology. 2010;156:2757. doi: 10.1099/mic.0.040196-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hong K, Gao AH, Xie QY, Gao HG, Zhuang L, Lin HP, Yu HP, Li J, Yao XS, Goodfellow M, Ruan JS. Mar Drugs. 2009;7:24–44. doi: 10.3390/md7010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.